

ABSTRACT
Elucidation of mechanisms underlying the establishment, maintenance of and reactivation from HIV-1 latency is essential for the development of therapeutic strategies aimed at eliminating HIV-1 reservoirs. Microbial translocation, as a consequence of HIV-1-induced deterioration of host immune system, is known to result in a systemic immune activation and transient outbursts of HIV-1 viremia in chronic HIV-1 infection. How these microbes cause the robust HIV-1 reactivation remains elusive. Dendritic cells (DCs) have previously been shown to reactivate HIV-1 from latency; however, the precise role of DCs in reactivating HIV-1 from latently infected T-cell remains obscure. In this study, by using HIV-1 latently infected Jurkat T cells, we demonstrated that AIDS-associated pathogens as represented by Mycobacterium bovis (M. bovis) Bacillus Calmette–Guérin (BCG) and bacterial component lipopolysaccharide (LPS) were unable to directly reactivate HIV-1 from Jurkat T cells; instead, they mature DCs to secrete TNF-α to accomplish this goal. Moreover, we found that HIV-1 latently infected Jurkat T cells could also mature DCs and enhance their TNF-α production during co-culture in a CD40-CD40L-signaling-dependent manner. This in turn led to viral reactivation from Jurkat T cells. Our results reveal how DCs help AIDS-associated pathogens to trigger HIV-1 reactivation from latency.
KEYWORDS: CD40-CD40L signaling, dendritic cells, HIV-1, TNF-α, viral latency
Introduction
Although combination antiretroviral therapy (cART) can suppress plasma HIV-1 viral load to undetectable levels, it cannot eradicate the virus because of the existence of cellular reservoirs of latently infected cells.1-3 The interruption of cART-treatment inevitably leads to a rapid and robust viral rebound.4-7 Despite therapeutic approaches aimed at purging the latent reservoirs are being explored, HIV-1 latency remains a major obstacle toward a functional HIV cure.8
HIV-1 integrates into host genome early during infection to form a pool of transcription-silent proviruses, by which HIV-1 effectively evades host immune responses and establishes persistent infection.9-11 HIV-1 can hide into the anatomic sites of gastrointestinal mucosa, lymph nodes, genital tract and the central nervous system for persistency,12 and the memory and transitional CD4+ T memory cells are the main cellular reservoirs.13,14 In addition, macrophages and DCs also harbor latent HIV-1 proviruses.15-19 Elucidation of mechanisms underlying the establishment, maintenance of and reactivation from HIV-1 latency is essential for the development of therapeutic strategies aimed at eliminating HIV-1 reservoirs.
HIV-1 infection causes the deterioration of the host immune system and leads to the loss of control of microbial translocation.20 As a result, AIDS-associated opportunistic pathogens, including fungi, bacteria and viruses, cause systemic immune activation, which is usually associated with transient outbursts of HIV-1 viremia and accelerated disease progression.20-30 How these co-infected pathogens accelerate the deterioration of the immune response and cause the robust HIV-1 reactivation remain largely unknown.
The LPS-treated macrophages efficiently induce an in vitro reactivation of HIV-1 from resting CD4+ T cells isolated from cART-treated patients. Proinflammatory cytokines produced by LPS-treated macrophages have been implicated in the reactivation of HIV-1, as the depletion of these cytokines from the culture supernatant of LPS-treated macrophages markedly reduced the amount of reactivated HIV-1.31
DCs have previously been shown to reactivate HIV-1 from latency.32-34 As professional antigen presenting cells, DCs capture, process antigens and migrate from peripheral tissues to lymphoid nodes where they interact closely with T cells to activate them.35-37 The intimate interactions between DCs and T cells provide an environment for DCs to activate and purge HIV-1 from latently infected T cells. Lymphoid tissues- and intestine- residing DCs as well as blood derived CD1c+ and CD141+ myeloid DCs can induce viral production from HIV-1 latently infected effector T cells.33 Monocyte-derived DCs (MDDCs) can also purge latent HIV-1 from resting memory CD4+ T cells.32,33 A recent study has demonstrated that interaction between DCs and T lymphocytes triggers MDDCs to secrete unknown components that were capable of activating provirus from actively proliferating primary T lymphocytes.34 However, the precise role and the underlying mechanism of DCs in reactivating HIV-1 from latently infected T-cells remain obscure.
In this study, by using HIV-1 latently infected Jurkat T cells, we demonstrated the crucial role of DCs in promoting HIV-1 reactivation from latency. AIDS-associated pathogens as represented by M. bovis BCG and bacterial component LPS were unable to directly reactivate HIV-1 from latency; instead, they mature MDDCs to secrete TNF-α. Also, we found that the HIV-1 latently infected Jurkat T cells could mature MDDCs during co-culture in a CD40-CD40L-signaling-dependent manner, and this led to enhanced TNF-α production and consequently augmented viral reactivation from T cells.
Results
Matured DCs secrete TNF-α to reactivate HIV-1 from latency
Microbial translocation was discovered as a cause of systemic immune activation in HIV-1 chronic infection, leading to transient bursts of HIV-1 viremia in patients. The elevated level of bacterial product LPS in plasma was defined as a marker of microbial translocation.20 To investigate whether AIDS-associated pathogens could directly reactivate HIV-1 from latently infected cells, we stimulated HIV-1 latently infected Jurkat T-cell clone (C11) with M. bovis BCG or LPS. C11 cells harbor an HIV-1 proviral DNA encoding enhanced green-fluorescent protein (EGFP), and can be reactivated upon stimulation with TNF-α, SAHA (Vorinostat) or trichostatin A. The expression of GFP can be used as an indicator of HIV-1 reactivation from latency.38,39 However, the direct stimulation with these pathogens or components did not reactivate HIV-1 from C11 cells, in contrast to positive control using TNF-α stimulation (Fig. 1A), implying an indirect mechanism was at play.
Figure 1.
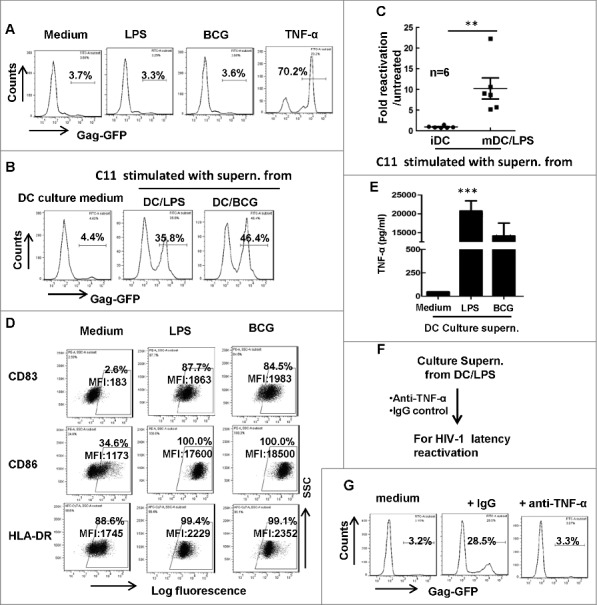
Matured MDDCs secrete TNF-α in supernatant to reactivate HIV-1 from latency. (A) The direct stimulation with BCG or LPS does not reactivate HIV-1 from C11 cells. C11 cells (4 × 105) were treated with or without LPS (10 ng/ml), M. bovis BCG (108 CFU) or TNF-α (20 ng/ml) for 24 h and viral reactivation was detected with flow cytometry by quantifying GFP expression. (B, C) The culture supernatant from treated-MDDCs activated HIV-1 from latency. MDDCs (2 × 105) were treated with LPS (10 ng/ml) or M. bovis BCG (108 CFU) for 2 days, then culture supernatant was harvested to stimulate C11 cells for additional 24 h, and viral reactivation was detected with flow cytometry. The percentage of GFP-positive cells was labeled, (C) Six independent repeats were summarized and the relative HIV-1 reactivation was analyzed. (D) MDDCs phenotype. MDDCs were treated with or without LPS or M. bovis BCG as above, and the expressions of CD83, CD86 and HLA-DR were detected with flow cytometry. The percentage of positive cells was labeled and the mean fluorescence intensity (MFI) was calculated. (E) TNF-α production in supernatant from LPS- or BCG-stimulated DCs was quantified with specific ELISA kit. Data were presented as mean ± standard deviation (SD). (F, G) Neutralization of TNF-α in culture supernatant from LPS-treated MDDCs abolishes the stimulation to reactivate HIV-1 from C11 cells. Culture supernatant collected from LPS-stimulated MDDCs was prior-treated with anti-TNF-α neutralizing antibodies (20 μg/ml) or control IgG for 1 h, and then was used for C11 stimulation (F), and viral reactivation was detected with flow cytometry (G). One representative result from at least 3 repeats was shown. *** P < 0.001 were considered as significant difference in ANOVA analysis.
As professional antigen presenting cells that processing and presenting antigen to T-cells, DCs initiate host innate immunity and bridge the adaptive immunity including stimulating T-cell differentiation.35-37 We sought to determine the potential role of DCs in HIV-1 reactivation boosted by AIDS-associated pathogens or components. We treated MDDCs with BCG or LPS for 24 h, and harvested culture supernatant to stimulate C11 cells. Intriguingly, these DC culture supernatant could efficiently reactivate HIV-1 from latency, as demonstrated by detecting the GFP expression in C11 cells (Fig. 1B, 1C).
The stimulation with BCG and LPS could mature DCs, as indicated by the increased expression of HLA-DR and the co-stimulatory molecules CD83 and CD86 (Fig. 1D), and the increased TNF-α secretion in the supernatant (Fig. 1E). To determine the role of secreted TNF-α in activating HIV-1 from latency, the culture supernatant was treated with specific anti-TNF-α antibody before culturing with C11 cells (Fig. 1F). The depletion of TNF-α abolished the supernatant-driven HIV-1 reactivation from latently infected cells (Fig. 1G), suggesting that the secreted TNF-α by matured DCs caused reactivation of HIV-1 from C11 cells. Taken together, these data demonstrate that AIDS-associated pathogens or their components, while unable to directly reactivate HIV-1 from latency, can mature DCs to secrete TNF-α to accomplish the task.
Immature DCs display higher efficiency in reactivating HIV-1 compared with LPS-matured DCs through direct-contact with T cells
Having demonstrated the role of culture supernatant from treated-DCs in reactivating HIV-1, we next investigated the role of DC-T cell direct-contact for viral reactivation. LPS-treated or untreated DCs were harvested and co-cultured with C11 cells for 24 h (Fig. 2A). Intriguingly, compared with LPS-matured DCs, untreated immature DCs (iDCs) displayed higher efficiency in reactivating HIV-1 during co-culture with C11 cells (the ratio of C11: DCs was 2:1) (Fig. 2B and C). When a transwell culture plate with a 0.4-μm insert membrane was used to separate C11 cells from MDDCs (Fig. 2D), HIV-1 reactivation disappeared (Fig. 2E), demonstrating that the direct-contact between DCs, particularly iDCs, with T cells, was essential for reactivating HIV-1 from T cells. Taken together, these data demonstrate that iDCs display higher efficiency in reactivating HIV-1 compared with LPS-matured DCs through direct-contact with T cells.
Figure 2.
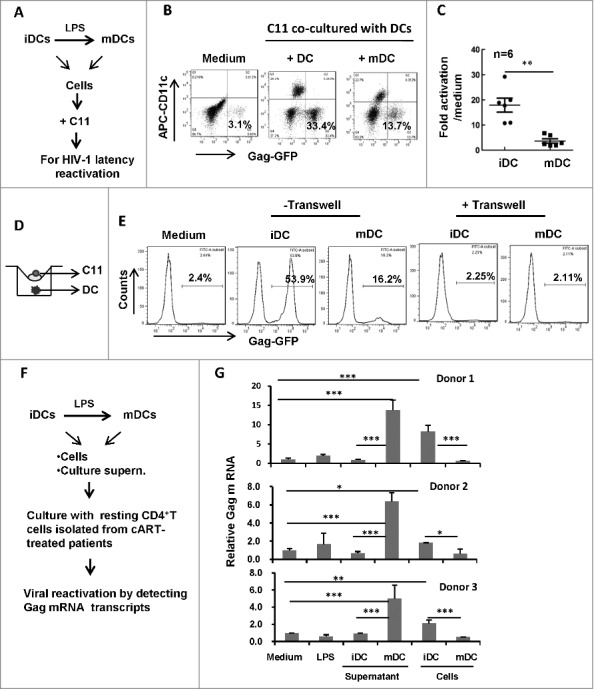
HIV-1 reactivation during the co-culture with MDDCs. (A) Scheme for HIV latency reactivation assay. (B, C) MDDCs-triggered HIV-1 reactivation from C11 during co-culture. C11 cells (4 × 105) were co-cultured or not with immature MDDCs or LPS-treated MDDCs (ratio C11:DC, 2:1) for 24 h. C11 cells (CD11c−) were distinguished from MDDCs with CD11c immunostaining (B), and 6 independent repeats were summarized and analyzed (C). (D, E) Separation of C11 from MDDCs abolishes HIV-1 reactivation. A transwell culture plate with a 0.4-μm insert membrane was used or not to separate the C11 from MDDCs (D), and HIV-1 reactivation in C11 cells was detected with flow cytometry and analyzed as above (E). (F,G) HIV-1 reactivation from the resting primary CD4+ T cells. Resting CD4+ T-cells were isolated from cART-treated patients and further treated with LPS or cultured with immature MDDCs or LPS-treated MDDCs or with their related culture medium, respectively, for 3 d. Viral reactivation was detected by semi-quantifying gag mRNA transcripts and normalized with GAPDH mRNA. The relative gag mRNA products were calculated. * P < 0.05, ** P < 0.01 *** P < 0.001, were considered as significant difference in ANOVA analysis.
Resting CD4+ T-cells are the major source of HIV-1 latency and their activation leads to HIV-1 reactivation from viral latency.19 To further confirm the distinguish effects of cells or culture medium from immature and LPS-treated mature DCs on HIV-1 reactivation, the resting CD4+ T-cells were isolated from cART-treated patients and further treated with LPS or cultured with immature MDDCs or LPS-treated MDDCs or with their related culture medium, respectively, for 3 d. Viral reactivation was detected by semi-quantifying gag message RNA (mRNA) transcripts and normalized with GAPDH mRNA (Fig. 2F). The harvested culture supernatant from LPS-treated mature MDDCs and the co-culture with immature MDDCs could efficiently reactivate HIV-1 from CD4+ T-cells isolated from cART-treated patients, while the treatments with either LPS or culture supernatant from immature MDDCs, or the co-culture with LPS-treated MDDCs, could not reactivate HIV-1 (Fig. 2G).
Co-culture with C11 cells mature iDCs and promote TNF-α secretion
To investigate the mechanism of iDC-mediated reactivation of HIV-1 from co-cultured T cells, we analyzed the phenotype of DCs after co-culture them with T cells for 24 h. The expressions of CD83, CD86 and HLA-DR were increased both on iDCs and LPS-treated DCs after co-culture with C11 cells (Fig. 3A), indicating maturation (or further maturation) of DCs. Notably, the phenotypic change was more pronounced on iDCs, as demonstrated by the elevated CD83 and CD86 expression (Fig. 3A). When detecting TNF-α expression at the mRNA level, the co-culture with C11 cells stimulated more TNF-α expression from iDCs, whereas C11 cells alone did not produce TNF-α (Fig. 3B). Similarly, the addition of anti-TNF-α antibody during DC-T cell co-culture abolished HIV-1 reactivation from C11 cells (Fig. 3C and D), implying that TNF-α production from DCs during co-culture with T cells is responsible for reactivating HIV-1. Taken together, these data demonstrate that co-culture with C11 cells lead to iDCs maturation and TNF-α secretion, which in turn reactivate HIV-1.
Figure 3.
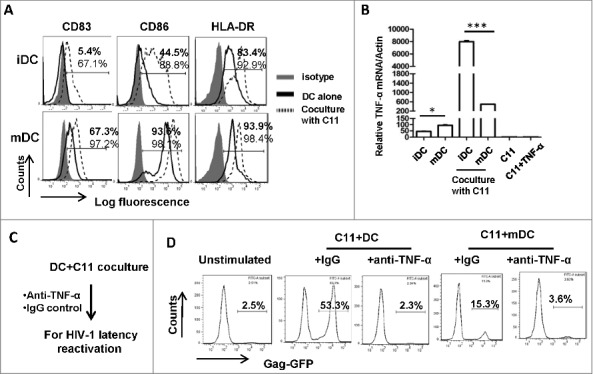
Co-cultured C11 cells mature MDDCs and promote TNF-α secretion for reactivating HIV-1. (A) Phenotype characterization of MDDCs. immature MDDCs or LPS-treated MDDCs were co-cultured with or without C11 cells as above, and MDDCs were distinguished with CD11c+ immunostaining and the expressions of CD83, CD86 and HLA-DR were detected with flow cytometry. The percentage of positive cells was labeled. (B) Assay for TNF-α expression. MDDCs were co-cultured with or without C11 cells for 6 h, and C11 cells stimulated with or without TNF-α (20 ng/ml) for 6 h were also prepared. TNF-α expression was detected at mRNA level by qRT-PCR and normalized with β-actin. Data were presented as mean ± standard deviation (SD). (C, D) The addition of anti-TNF-α neutralizing antibodies during C11-MDDCs co-culture abolishes HIV-1 reactivation. 20 μg/ml anti-TNF-α neutralizing antibodies or IgG control were added during C11-MDDCs co-culture for 24 h (C), and viral reactivation was detected with flow cytometry (D). One representative result from at least 3 independent repeats was shown. * P < 0.05 and *** P < 0.001 were considered as significant difference in ANOVA analysis.
Co-culture with C11 cells matures iDCs through the CD40-CD40L signaling pathway
The interactions between multiple pairs of co-stimulatory molecules, such as CD40-CD40L and B7.1/B7.2-CD28, transmit second and third signals during APC-mediated T-cell activation.37,40 CD40 belongs to the TNF-receptor super-family and is constitutively expressed by DCs, B cells and macrophages, and the CD40-CD40L engagement on DC surface permits DCs maturation and the enhanced production of certain pro-inflammatory cytokines such as TNF-α, IL-8, MIP-1α and IL-12. Some of these cytokines help to effectively trigger T-cell activation and differentiation.41-44 Thus, we investigated whether CD40-CD40L signaling was involved in reactivating DCs during co-culture with HIV-1 latently infected Jurkat T cells. CD40 showed higher expression on LPS-treated DC than untreated iDCs (Fig. 4A), and co-culture with C11 cells significantly increased CD40 expression on iDCs, but decreased CD40 expression on LPS-treated DCs, at both transcription and translation levels (Fig. 4A and B).
Figure 4.
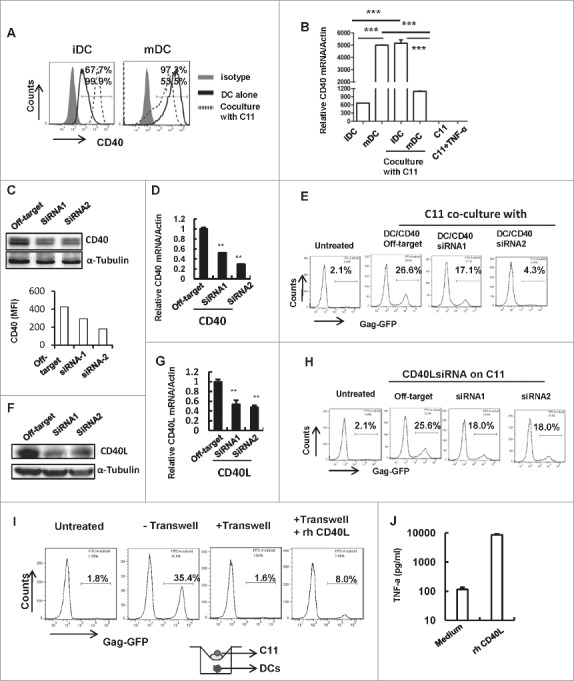
Co-cultured C11 cells mature iDCs through CD40-CD40L signaling pathway. (A) CD40 expression on MDDCs. Immature MDDCs or LPS-treated MDDCs were co-cultured with or without C11 cells for 24 h, and MDDCs were distinguished with CD11c+ immunostaining and the expression of CD40 was detected with flow cytometry, and the positive percentage was shown. (B) CD40 expression detected at mRNA level. MDDCs were co-cultured with or without C11 cells for 6 h, and C11 cells stimulated with or without TNF-α (20 ng/ml) for 6 h were also prepared. CD40 expression was detected at mRNA level by qRT-PCR and normalized with β-actin. Data were presented as mean ± SD. (C, D, E) CD40 knockdown impairs iMDDCs-purged HIV-1 reactivation from latency. IMDDCs were transfected with specific CD40 siRNAs or off-target control for 48 h, and CD40 expression was detected by Western blotting (upper panel) and flow cytometry (lower panel, MFI for CD40-positive staining was calculated) at protein level (C), and by mRNA quantification at transcription level (D). These transfected MDDCs were further co-cultured with C11 cells for additional 24 h and HIV-1 reactivation was assessed with flow cytometry (E). (F, G, H) CD40L knockdown on C11 diminishes the response to MDDCs stimulation for HIV-1 reactivation. C11 cells were nucleofected with CD40L specific siRNAs or off-target control for 48 h, and CD40L expression was detected by Western blotting at protein level (F) and mRNA quantification at transcription level (G), and these nucleofected C11 cells were co-cultured with immature MDDCs for additional 24 h, and HIV-1 reactivation was assessed with flow cytometry (H). (I, J) The supply with recombinant CD40Ligand protein complements HIV-1 reactivation from C11 cells. Recombinant CD40 ligand protein (1 μg/ml) was supplied during transwell-culture (pore size, 0.4 μm) of C11 cells with iDCs for 24 h, and C11 cells were harvested and GFP expression was detected with flow cytometry (I), and the supernatant was collected for detecting TNF-α with ELISA (J). One representative result from at least 3 independent repeats was shown. ** P < 0.01 and *** P < 0.001 were considered as significant difference in ANOVA analysis.
To investigate the potential role of CD40-CD40L signaling in mediating iDC activation, we knocked down CD40 expression on DCs with specific interference RNAs (siRNAs), as demonstrated by the reduced CD40 expression at both mRNA transcript and protein levels (Fig. 4C and D). The CD4 decrease impaired the capacity of iDCs to reactivate HIV-1 from co-cultured C11 cells (Fig. 4E). On the other hand, when CD40L expression on C11 was interfered with siRNA (Fig. 4F and G), these T cells were less likely to be induced for HIV-1 reactivation upon co-culture with iDCs (Fig. 4H). Moreover, when we separated C11 cells from DCs with the insert of transwell plate but supplied with recombinant CD40L protein, HIV-1 reactivation from C11 cells could be restored (Fig. 4I). The supplement of CD40L protein induced TNF-α production from DCs (Fig. 4J). Taken together, these data demonstrate that co-culture with C11 cells activate iDCs through CD40-CD40L signaling pathway, and these matured DCs in turn activate T cells for HIV-1 reactivation.
Discussion
Microbial translocations or co-infections are involved in AIDS disease progression and the reactivation of HIV-1 in latently infected cells in vivo,21,31,45-47 but the underlying mechanisms by which microbes activate HIV-1 from latency to elevate viral burden in vivo remain elusive. In this study, we demonstrated that representative pathogen M. bovis BCG and bacterial component LPS were unable to directly reactivate HIV-1 from latently infected Jurkat T cells; instead, they matured DC to secrete TNF-α to accomplish this task. Our results uncover a bridging role of DCs in microbes-triggered robust HIV-1 replication. Moreover, we found that the co-culture with HIV-1 latently infected Jurkat T cells led to the maturation of DCs and enhanced TNF-α production in a CD40-CD40L-signaling-dependent manner (Fig. 5).
Figure 5.
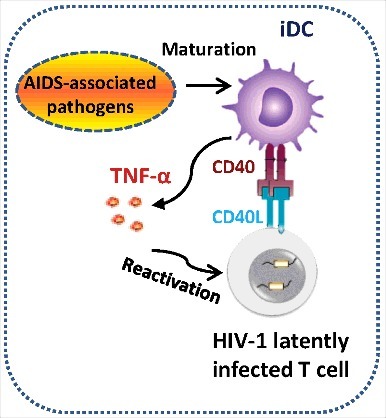
Schematics illustrating the role of DCs in triggering HIV-1 reactivation. AIDS-associated pathogens (such as M. bovis BCG) or bacterial compound LPS mature DCs to secrete TNF-α into the supernatant to reactivate HIV-1 from latency. Alternatively, co-culture with HIV-1 latently infected T cells mature DCs through CD40-CD40L signaling pathway, and these matured DCs secrete TNF-α to activate T cells, allowing HIV-1 reactivation.
DC subsets reside in various tissues may have differential latency reactivation properties. Blood-borne CD1c+ and CD141+ myeloid DCs as well as blood CD14+ monocyte-derived DCs, intestine- and lymphoid tissues- residing DCs, but not skin- or genital tract- derived DCs, can induce viral production from HIV-1 latently infected effector T cells.33 The stimulation with different Toll like receptor (TLR) agonists may impart distinct capacity on myeloid DCs or MDDCs for activating HIV-1 from latency. Poly I: C stimulation through TLR3 induced type I interferon production in both CD1c+-myeloid DCs and MDDCs and abolished their capacity for reactivating HIV-1 from latency. In contrast, stimulation with TLR 1, 2, 7 and 8 agonists enhanced DCs′ capacity for reactivating HIV-1 from latency.33 The stimulations with LPS or Flagellin through TLR4 or TLR5, respectively, impaired the ability of MDDCs to reactivate HIV.33 Consistent with the previous finding,33 in our study, we found that LPS-treated MDDCs had reduced capacity to reactivate HIV-1 from latently infected Jurkat T cells through cell-to-cell contact. Elevated co-inhibitory signaling may be an explanation. Co-inhibitory molecules PD-L1/PD-L2 on mature DCs were upregulated,48 and the PD-L1/PD-L2-PD1 signaling could restrain T cells from hyperactivation.
Intriguingly, we found that LPS-treated MDDCs adopted an alternative mode to reactivate HIV-1 from latency, through enhanced TNF-α secretion. Besides LPS stimulation, the direct contact between MDDCs and latently infected T cells could also trigger soluble factor secretion to reactivate HIV-1. Cell-to-cell interaction between DC and T lymphocyte triggers DC to secrete unknown components to activate proviruses from latency. This kind of activation did not involve DC-mediated C-type lectin dendritic cell-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) signaling or T cell receptor (TCR)-stimulation,34 but relied on ICAM-1-depended cell-cell adhesion, because the blocking with specific anti-ICAM-1 antibody significantly reduced the HIV-1 reactivation.34 In our study, we demonstrated that the direct cell-to-cell contact between latently infected Jurkat T cells and MDDCs triggered the maturation of MDDCs and elevated their TNF-α secretion for reactivating HIV-1, and the signaling through the interaction between CD40L expressed on Jurkat T cells and CD40 on DCs plays a notable role in modulating DC′s capacity for reactivating HIV-1 from Jurkat T cells.
Several transformed cell lines or primary T-cells have been used for establishment of HIV-1 latency.49-55 In these models, different T-cell subtypes, viral genetic composition and cellular pathways for reactivation were adopted.56 TNF-α shows remarkable activity to revert HIV-1 latency by enhancing NF-κB-dependent gene transcription in Jurkat T-cell-based models.56-58 The transformed Jurkat CD4+ T cells were mainly used in this study. In this model, upon stimulation with TNF-α, SAHA or trichostatin A, GFP is expressed as an indication of HIV-1 reactivation from latency.38,39 However, TNF-α is inactive or have low reactivity in reactivating HIV-1 from latently infected primary CD4+ T cells.56 This may hinder the usage of these cell models to validate our results. The resting CD4+ T cell isolated from cART-treated patients provides an alternative cell model for studying HIV-1 latency,59 and these patient′s cells show robust response to TNF-α in viral outgrowth assay for quantifying the size of viral reservoir,56 we thus used these patients-isolated resting CD4+T cells to validate our results.
Taken together, our data uncover the crucial role of DCs in stimulating HIV-1 reactivation from latency, and facilitate a better understanding of host-modulation on HIV-1 latency.
Methods and materials
Ethics statement
HIV-1-infected individuals underwent combination antiretroviral therapy (cATR) for more than 3 years were recruited from Peking Union Medical College Hospital, Beijing, China. These patients have the undetected plasma viral load by using a standard (RT-) PCR-based assay (Roche). The signed informed consent was obtained from each of the research participant, and the Medical Ethics Review Committee of the Institut Pasteur of Shanghai, Chinese Academy of Sciences, China, has approved the study and the usage of human cells. The resting CD4+ T cells were purified from the peripheral blood mononuclear cells of patients by anti-CD4 specific antibody-coated microbeads (MiltenyiBiotec) as described previously.60
Cells
Human peripheral blood mononuclear cells (PBMCs) were purchased from the Changhai Hospital, Shanghai, China. The anti-CD14 specific antibody-coated microbeads (Miltenyi Biotech) were used to purify CD14+ monocytes from PBMCs, and the CD14+ monocytes were stimulated with 50 ng/ml granulocyte–macrophage colony-stimulating factor (GM-CSF) (R&D Systems) and interleukin (IL)-4 (R&D Systems) for 5 d to generate monocyte-derived DCs (MDDCs). Mature DCs (mDCs) were obtained by stimulation of MDDCs with 10 ng/ml LPS (R&D Systems) for 2 d.
HIV-1 latently infected Jurkat T cell C11 clone was provided by Prof. Huan-Zhang Zhu (Fudan University, Shanghai, China) and have been described previously.39 C11 cells were maintained in RPMI 1640 medium supplemented with 10% FBS (Gibco), 100 U/ml of penicillin and 100 μg/ml of streptomycin at 37°C under 5% CO2.
HIV-1 latency reactivation assay
HIV-1 latently infected Jurkat T C11 cells (4 × 105/well) were treated with or without TNF-α (20 ng/ml) (PHC3015, Invitrogen), 108 CFU of M. bovis BCG Danish strain (ATCC 35733)(a kind gift from Zheng W. Chen Laboratory, Institut Pasteur of Shanghai, Chinese Academy of Sciences, China) or MDDCs (2 × 105/well) culture supernatant for 24 h, and cells were harvested and GFP expression was detected with flow cytometry. TNF-α production was detected by using human TNF-α high sensitivity ELISA (eBioscience), according to the manufacturer′s instructions. In some experiments, the purified anti-human TNF-α monoclonal antibody (20 μg/ml) (eBioscience) was used to pretreat DC culture supernatant for 1 h at room temperature before adding to C11 cells for stimulation, the mouse IgG1kappa was used as the isotype control. When indicated, a transwell plate with a 0.4 μm size insert-membrane was used to separate C11 cells from MDDCs during culture. Recombinant human CD40 ligand protein (1 μg/ml) (R&D Systems) was used in the indicated MDDC-C11 co-culture. To distinguish between cell types in the DC-C11 co-culture experiment, an allophycocyanin (APC)-Alexa Fluor750-labeled anti-human CD11c antibody (B-ly6; BD Biosciences PharMingen) was used. MDDCs show a CD11c+ phenotype and C11 cells show a CD11c− phenotype.
For viral reactivation from the resting CD4+ T cells isolated from cART-treated patients, these purified resting CD4+ T cells (1 × 106) were co-cultured with immature MDDCs or LPS-treated MDDCs (0.5 × 106 for each type of cells) or with their related culture medium, respectively, for 3 d. Viral reactivation was detected by semi-quantifying gag mRNA transcripts and normalized with GAPDH mRNA, and the relative gag mRNA products were calculated. These primers were used for (RT-) PCR, Gag: Forward, 5′-GTG TGG AAA ATC TCT AGC AGT GG -3′ and Reverse, 5′-CGC TCT CGC ACC CAT CTC-3′. GAPDH: forward primer-5′-ATC CCA TCA CCA TCT TCC AGG-3′ and reverse primer-5′-CCT TCT CCA TGG TGG TGA AGA C-3′.
Flow cytometry
To characterize cell phenotype, the following antibodies or isotype-matched IgG were used for flow cytometry (clone numbers and sources were given in parentheses): APC-Alexa Fluor750-CD11c (B-ly6; BD Biosciences PharMingen); phycoerythrin (PE)-CD14 (61D3; eBioscience); PE-CD83 (H1B19; eBioscience); PE-CD86 (IT2.2; eBioscience), APC-cy7-HLA-DR (LN3; eBioscience) and PE-CD40 (KPL-1; eBioscience). Cells were fixed in 4% formaldehyde for 10 min at room temperature and subsequently washed with buffer (PBS supplemented with 1% FBS). Immunostaining was performed for 1 h at 4°C. Cells were analyzed with a BD LSRFortessa flow cytometer (BD Biosciences) and FlowJo 7.6.1 software (TreeStar Inc.).
siRNA interference
The CD40 specific siRNA duplex and off-target control (GenePharma, China) were used to transfect MDDCs, respectively, using the Amaxa human dendritic cell nucleofector kit (Amaxa). The knockdown of target genes was confirmed with flow cytometry and Western blotting with polyclonal antibodies against human CD40 or CD40 ligand (Abcam). The sequences of siRNA duplex were listed as follows: Off-target siRNA: 5′-UUC UCC GAA CGU GUC ACG UTT-3′ (sense), 5′-ACG UGA CAC GUU CGG AGA AdTdT-3′ (antisense); CD40 siRNA-1: 5′-CAA GAC UGA UGU UGU CUG UTT-3′ (sense), 5′-ACA GAC AAC AUC AGU CUU GTT-3′ (antisense); CD40 siRNA-2: 5′-CAG GCA GGC ACA AAC AAG ATT-3′ (sense), 5′-UCU UGU UUG UGC CUG CCU GTT-3′ (antisense). CD40 Ligand siRNA-1: 5′-GCC AGU UUG AAG GCU UUG UTT-3′ (sense); 5′-ACA AAG CCU UCA AAC UGG CTT-3′ (antisense); CD40 Ligand siRNA-2: 5′-GGU UGG ACA AGA UAG AAG ATT-3′ (sense); 5′-UCU UCU AUC UUG UCC AAC CT-3′ (antisense).
Real-time PCR
Total RNAs from differentially treated cell samples were extracted by TRIzol Reagent (Invitrogen), and reversly transcribed into cDNA by using the reverse-aid first strand cDNA synthesis kit (Fermentas). 2 μl cDNA reaction was amplified using forward and reverse primers for TNF-α, CD40, CD40L or β-actin. Primers used are as follows: TNF-α-F, 5′-CCC AGG CAG TCA GAT CT CTT C-3′, and TNF-α-R, 5′-GTG AGG AGC ACA TGG GTG GAG-3′; CD40-F, 5′-TGC TTG CTG ACC GCT GTC CT-3′, and CD40-R, 5′-CAG TGA ACT CTG TGC AGT CAC T-3′; CD40L-F, 5′-CAG ATG ATT GGG TCA GCA CTT-3′, and CD40L-R, 5′-CCT TCA CAA AGC CTT CAA ACT G-3′; β-Actin-F, 5′-GGG AAA TCG TGC GTG ACA T-3′, and β-Actin-R, 5′-GTC AGG CAG CTC GTA GCT CTT-3′. Real-time PCR was performed by using the thunderbird SYBR qPCR mix (TOYOBO), and the thermal cycling conditions were: 40 cycles of denaturation at 95°C for 15 s after the initial denaturation for 10 min, followed by primer annealing at 60°C for 15 s and extension at 72°C for 30 s. The final extension was at 72°C for 6 min. The levels of TNF-α, CD40 or CD40L RNA were calculated as fold change relative to β-actin.
Statistical analysis
SigmaStat 2.0 software (Systat Software, San Jose, CA, USA) was used to perform ANOVA to analyze the significant difference.
Disclosure of potential vonflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Huan-Zhang Zhu, Zheng W. Chen for cells or reagents.
Funding
This work was supported by grant to J.H.W from the National Grant Program on Key Infectious Disease (2014ZX10001003), the Natural Science Foundation of China (No. 81572001) and the NSFC- National Institutes of Health joint grant (81561128009), and the Instrument Developing Project (YZ201649) and the key project (QYZDB-SSW-SMC059), from the Chinese Academy of Sciences. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
References
- [1].Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity, 2012;37:377-88. https://doi.org/ 10.1016/j.immuni.2012.08.010. PMID:22999944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell, 2013;155:540-51. https://doi.org/ 10.1016/j.cell.2013.09.020. PMID:24243014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al.. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science, 1997;278:1295-300. https://doi.org/ 10.1126/science.278.5341.1295. PMID:9360927 [DOI] [PubMed] [Google Scholar]
- [4].Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science, 1997;278:1291-5. https://doi.org/ 10.1126/science.278.5341.1291. PMID:9360926 [DOI] [PubMed] [Google Scholar]
- [5].Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A, 1997;94:13193-7. https://doi.org/ 10.1073/pnas.94.24.13193. PMID:9371822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Joos B, Fischer M, Kuster H, Pillai SK, Wong JK, Boni J, Hirschel B, Weber R, Trkola A, Günthard HF, et al.. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A, 2008;105:16725-30. https://doi.org/ 10.1073/pnas.0804192105. PMID:18936487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chun TW, Davey RT Jr., Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature, 1999;401:874-5. https://doi.org/ 10.1038/44755. PMID:10553903 [DOI] [PubMed] [Google Scholar]
- [8].Deeks SG, Autran B, Berkhout B, Benkirane M, Cairns S, Chomont N, Chun TW, Churchill M, Di Mascio M, Katlama C, et al.. Towards an HIV cure: A global scientific strategy. Nat Rev Immunol, 2012;12:607-14. https://doi.org/ 10.1038/nri3262. PMID:22814509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci U S A, 1998;95:8869-73. https://doi.org/ 10.1073/pnas.95.15.8869. PMID:9671771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Archin NM, Vaidya NK, Kuruc JD, Liberty AL, Wiegand A, Kearney MF, Cohen MS, Coffin JM, Bosch RJ, Gay CL, et al.. Immediate antiviral therapy appears to restrict resting CD4+ cell HIV-1 infection without accelerating the decay of latent infection. Proc Natl Acad Sci U S A, 2012;109:9523-8. https://doi.org/ 10.1073/pnas.1120248109. PMID:22645358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dahl V, Josefsson L, Palmer S. HIV reservoirs, latency, and reactivation: Prospects for eradication. Antiviral Res, 2010;85:286-94. https://doi.org/ 10.1016/j.antiviral.2009.09.016. PMID:19808057 [DOI] [PubMed] [Google Scholar]
- [12].Svicher V, Ceccherini-Silberstein F, Antinori A, Aquaro S, Perno CF. Understanding HIV compartments and reservoirs. Curr HIV/AIDS Rep, 2014;11:186-94. https://doi.org/ 10.1007/s11904-014-0207-y. PMID:24729094 [DOI] [PubMed] [Google Scholar]
- [13].Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, et al.. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med, 1999;5:512-7. https://doi.org/ 10.1038/8394. PMID:10229227 [DOI] [PubMed] [Google Scholar]
- [14].Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, et al.. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med, 2009;15:893-900. https://doi.org/ 10.1038/nm.1972. PMID:19543283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature, 1995;373:123-6. https://doi.org/ 10.1038/373123a0. PMID:7816094 [DOI] [PubMed] [Google Scholar]
- [16].Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses, 2014;6:1837-60. https://doi.org/ 10.3390/v6041837. PMID:24759213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brown A, Zhang H, Lopez P, Pardo CA, Gartner S. In vitro modeling of the HIV-macrophage reservoir. J Leukoc Biol, 2006;80:1127-35. https://doi.org/ 10.1189/jlb.0206126. PMID:16923921 [DOI] [PubMed] [Google Scholar]
- [18].Haase AT, Henry K, Zupancic M, Sedgewick G, Faust RA, Melroe H, Cavert W, Gebhard K, Staskus K, Zhang ZQ, et al.. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science, 1996;274:985-9. https://doi.org/ 10.1126/science.274.5289.985. PMID:8875941 [DOI] [PubMed] [Google Scholar]
- [19].Coleman CM, Wu L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology, 2009;6:51. https://doi.org/ 10.1186/1742-4690-6-51. PMID:19486514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al.. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med, 2006;12:1365-71. https://doi.org/ 10.1038/nm1511. PMID:17115046 [DOI] [PubMed] [Google Scholar]
- [21].Blanchard A, Montagnier L, Gougeon ML. Influence of microbial infections on the progression of HIV disease. Trends Microbiol, 1997;5:326-31. https://doi.org/ 10.1016/S0966-842X(97)01089-5. PMID:9263412 [DOI] [PubMed] [Google Scholar]
- [22].Vijayakumar S, Finney John S, Nusbaum RJ, Ferguson MR, Cirillo JD, Olaleye O, Endsley JJ. In vitro model of mycobacteria and HIV-1 co-infection for drug discovery. Tuberculosis (Edinb), 2013;93 Suppl:S66-70. https://doi.org/ 10.1016/S1472-9792(13)70013-1. PMID:24388652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Steele AK, Lee EJ, Manuzak JA, Dillon SM, Beckham JD, McCarter MD, Santiago ML, Wilson CC. Microbial exposure alters HIV-1-induced mucosal CD4+ T cell death pathways Ex vivo. Retrovirology, 2014;11:14. https://doi.org/ 10.1186/1742-4690-11-14. PMID:24495380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gonzalez VD, Landay AL, Sandberg JK. Innate immunity and chronic immune activation in HCV/HIV-1 co-infection. Clin Immunol, 2010;135:12-25. https://doi.org/ 10.1016/j.clim.2009.12.005. PMID:20100670 [DOI] [PubMed] [Google Scholar]
- [25].Cavallin LE, Goldschmidt-Clermont P, Mesri EA. Molecular and cellular mechanisms of KSHV oncogenesis of Kaposi's sarcoma associated with HIV/AIDS. PLoS Pathog, 2014;10:e1004154. https://doi.org/ 10.1371/journal.ppat.1004154. PMID:25010730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Qin Y, Li Y, Liu W, Tian R, Guo Q, Li S, Li H, Zhang D, Zheng Y, Wu L, et al.. Penicillium marneffei-stimulated dendritic cells enhance HIV-1 trans-infection and promote viral infection by activating primary CD4+ T cells. PLoS One, 2011;6:e27609. https://doi.org/ 10.1371/journal.pone.0027609. PMID:22110688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dillon SM, Lee EJ, Donovan AM, Guo K, Harper MS, Frank DN, McCarter MD, Santiago ML, Wilson CC. Enhancement of HIV-1 infection and intestinal CD4+ T cell depletion ex vivo by gut microbes altered during chronic HIV-1 infection. Retrovirology, 2016;13:5. https://doi.org/ 10.1186/s12977-016-0237-1. PMID:26762145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wahl SM, Orenstein JM. Immune stimulation and HIV-1 viral replication. J Leukoc Biol, 1997;62:67-71. PMID:9225995 [DOI] [PubMed] [Google Scholar]
- [29].Shen Y, Shen L, Sehgal P, Huang D, Qiu L, Du G, Letvin NL, Chen ZW. Clinical latency and reactivation of AIDS-related mycobacterial infections. J Virol, 2004;78:14023-32. https://doi.org/ 10.1128/JVI.78.24.14023-14032.2004. PMID:15564509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Qin Y, Li YY, Jiang AP, Jiang JF, Wang JH. Stimulation of Cryptococcus neoformans isolated from skin lesion of AIDS patient matures dendritic cells and promotes HIV-1 trans-infection. Biochem Biophys Res Commun, 2012;423:709-14. https://doi.org/ 10.1016/j.bbrc.2012.06.020. PMID:22704932 [DOI] [PubMed] [Google Scholar]
- [31].Moriuchi H, Moriuchi M, Mizell SB, Ehler LA, Fauci AS. In vitro reactivation of human immunodeficiency virus 1 from latently infected, resting CD4+ T cells after bacterial stimulation. J Infect Dis, 2000;181:2041-4. https://doi.org/ 10.1086/315496. PMID:10837189 [DOI] [PubMed] [Google Scholar]
- [32].Marini A, Harper JM, Romerio F. An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol, 2008;181:7713-20. https://doi.org/ 10.4049/jimmunol.181.11.7713. PMID:19017960 [DOI] [PubMed] [Google Scholar]
- [33].van der Sluis RM, van Capel TM, Speijer D, Sanders RW, Berkhout B, de Jong EC, Jeeninga RE, van Montfort T. Dendritic cell type-specific HIV-1 activation in effector T cells: Implications for latent HIV-1 reservoir establishment. AIDS, 2015;29:1003-14. https://doi.org/ 10.1097/QAD.0000000000000637. PMID:25768834 [DOI] [PubMed] [Google Scholar]
- [34].van der Sluis RM, van Montfort T, Pollakis G, Sanders RW, Speijer D, Berkhout B, Jeeninga RE. Dendritic cell-induced activation of latent HIV-1 provirus in actively proliferating primary T lymphocytes. PLoS Pathog, 2013;9:e1003259. https://doi.org/ 10.1371/journal.ppat.1003259. PMID:23555263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wilkinson J, Cunningham AL. Mucosal transmission of HIV-1: First stop dendritic cells. Curr Drug Targets, 2006;7:1563-9. https://doi.org/ 10.2174/138945006779025482; PMID:17168831 [DOI] [PubMed] [Google Scholar]
- [36].Hansasuta P, Rowland-Jones SL. HIV-1 transmission and acute HIV-1 infection. Br Med Bull, 2001;58:109-27. https://doi.org/ 10.1093/bmb/58.1.109. PMID:11714627 [DOI] [PubMed] [Google Scholar]
- [37].Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature, 1998;392:245-52. https://doi.org/ 10.1038/32588. PMID:9521319 [DOI] [PubMed] [Google Scholar]
- [38].Ding D, Qu X, Li L, Zhou X, Liu S, Lin S, Wang P, Liu S, Kong C, Wang X, et al.. Involvement of histone methyltransferase GLP in HIV-1 latency through catalysis of H3K9 dimethylation. Virology, 2013;440:182-9. https://doi.org/ 10.1016/j.virol.2013.02.022. PMID:23541084 [DOI] [PubMed] [Google Scholar]
- [39].Qu X, Wang P, Ding D, Li L, Wang H, Ma L, Zhou X, Liu S, Lin S, Wang X, et al.. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res, 2013;41:7771-82. https://doi.org/ 10.1093/nar/gkt571. PMID:23804764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Inaba K, Metlay JP, Crowley MT, Witmer-Pack M, Steinman RM. Dendritic cells as antigen presenting cells in vivo. Int Rev Immunol, 1990;6:197-206. https://doi.org/ 10.3109/08830189009056630. PMID:2152503 [DOI] [PubMed] [Google Scholar]
- [41].Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J Exp Med, 1994;180:1263-72. https://doi.org/ 10.1084/jem.180.4.1263. PMID:7523569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pinchuk LM, Polacino PS, Agy MB, Klaus SJ, Clark EA. The role of CD40 and CD80 accessory cell molecules in dendritic cell-dependent HIV-1 infection. Immunity, 1994;1:317-25. https://doi.org/ 10.1016/1074-7613(94)90083-3. PMID:7534204 [DOI] [PubMed] [Google Scholar]
- [43].Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol, 2009;21:265-72. https://doi.org/ 10.1016/j.smim.2009.05.010. PMID:19524453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med, 1994;179:1109-18. https://doi.org/ 10.1084/jem.179.4.1109. PMID:8145033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gonzalez OA, Li M, Ebersole JL, Huang CB. HIV-1 reactivation induced by the periodontal pathogens Fusobacterium nucleatum and Porphyromonas gingivalis involves Toll-like receptor 2 [corrected] and 9 activation in monocytes/macrophages. Clin Vaccine Immunol, 2010;17:1417-27. https://doi.org/ 10.1128/CVI.00009-10. PMID:20610663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Huang CB, Alimova YV, Strange S, Ebersole JL. Polybacterial challenge enhances HIV reactivation in latently infected macrophages and dendritic cells. Immunology, 2011;132:401-9. https://doi.org/ 10.1111/j.1365-2567.2010.03375.x. PMID:21073452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gonzalez OA, Ebersole JL, Huang CB. Oral infectious diseases: A potential risk factor for HIV virus recrudescence? Oral Dis, 2009;15:313-27. https://doi.org/ 10.1111/j.1601-0825.2009.01533.x. PMID:19364391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Meier A, Bagchi A, Sidhu HK, Alter G, Suscovich TJ, Kavanagh DG, Streeck H, Brockman MA, LeGall S, Hellman J, et al.. Upregulation of PD-L1 on monocytes and dendritic cells by HIV-1 derived TLR ligands. AIDS, 2008;22:655-8. https://doi.org/ 10.1097/QAD.0b013e3282f4de23. PMID:18317010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, Greene WC. A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One, 2012;7:e30176. https://doi.org/ 10.1371/journal.pone.0030176. PMID:22291913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bosque A, Planelles V. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods, 2011;53:54-61. https://doi.org/ 10.1016/j.ymeth.2010.10.002. PMID:20970502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cameron PU, Saleh S, Sallmann G, Solomon A, Wightman F, Evans VA, Boucher G, Haddad EK, Sekaly RP, Harman AN, et al.. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc Natl Acad Sci U S A, 2010;107:16934-9. https://doi.org/ 10.1073/pnas.1002894107. PMID:20837531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang HC, Xing S, Shan L, O'Connell K, Dinoso J, Shen A, Zhou Y, Shrum CK, Han Y, Liu JO, et al.. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest, 2009;119:3473-86. https://doi.org/ 10.1172/JCI39199. PMID:19805909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood, 2009;113:58-65. https://doi.org/ 10.1182/blood-2008-07-168393. PMID:18849485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood, 2007;110:4161-4. https://doi.org/ 10.1182/blood-2007-06-097907. PMID:17881634 [DOI] [PubMed] [Google Scholar]
- [55].Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J, 2003;22:1868-77. https://doi.org/ 10.1093/emboj/cdg188. PMID:12682019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, et al.. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog, 2013;9:e1003834. https://doi.org/ 10.1371/journal.ppat.1003834. PMID:24385908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fernandez G, Zaikos TD, Khan SZ, Jacobi AM, Behlke MA, Zeichner SL. Targeting IkappaB proteins for HIV latency activation: The role of individual IkappaB and NF-kappaB proteins. J Virol, 2013;87:3966-78. https://doi.org/ 10.1128/JVI.03251-12. PMID:23365428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McNamara LA, Ganesh JA, Collins KL. Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-kappaB activation. J Virol, 2012;86:9337-50. https://doi.org/ 10.1128/JVI.00895-12. PMID:22718820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Archin NM, Eron JJ, Palmer S, Hartmann-Duff A, Martinson JA, Wiegand A, Bandarenko N, Schmitz JL, Bosch RJ, Landay AL, et al.. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. AIDS, 2008;22:1131-5. https://doi.org/ 10.1097/QAD.0b013e3282fd6df4. PMID:18525258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li C, Wang HB, Kuang WD, Ren XX, Song ST, Zhu HZ, et al.. Naf1 Regulates HIV-1 latency by suppressing viral promoter-driven gene expression in primary CD4+ T cells. J Virol, 2017; 91(1): e01830-16. https://doi.org/ 10.1128/JVI.01830-16. [DOI] [PMC free article] [PubMed] [Google Scholar]