

ABSTRACT
Streptococcus pneumoniae is a bacterial pathogen that commonly resides in the human nasopharynx, typically without causing any disease. However, in some cases these bacteria migrate from the nasopharynx to other sites of the body such as the lungs and bloodstream causing pneumonia and sepsis, respectively. This study used a mouse model of infection to investigate the potential role of Mucin 1 (MUC1), a cell membrane-associated glycoprotein known for playing a key barrier role at mucosal surfaces, in regulating this process. Wildtype (WT) and MUC1-deficient (Muc1−/−) mice were infected intranasally with an invasive strain of S. pneumoniae and bacterial loads in the nasopharynx, lungs, and blood were analyzed. Lungs were graded histologically for inflammation and cytokine profiles in the lungs analyzed by ELISA. While there was no difference in pneumococcal colonization of the nasopharynx between WT and Muc1−/− mice, infected Muc1−/− mice showed high pneumococcal loads in their lungs 16 hours post-infection, as well as bacteremia. In contrast, infected WT mice cleared the pneumococci from their lungs and remained asymptomatic. Infection in Muc1−/− mice was associated with an elevation in lung inflammation, with cellular recruitment especially of monocytes/macrophages. While MUC1-deficiency has been shown to increase phagocytosis of Pseudomonas aeruginosa, macrophages from Muc1−/− mice exhibited a reduced capacity to phagocytose S. pneumoniae indicating diverse and bacterial-specific effects. In conclusion, these findings indicate that MUC1 plays an important role in protection against severe pneumococcal disease, potentially mediated by facilitating macrophage phagocytosis.
KEYWORDS: bacteremia, inflammation, macrophages, Muc1, phagocytosis, Streptococcus pneumoniae
Introduction
Streptococcus pneumoniae (commonly referred to as pneumococci) are Gram positive bacteria that are frequent asymptomatic colonizers of the mucosal surface of the human nasopharynx and upper airways, especially in children.1,2 Their nasopharyngeal carriage rates in young children range from approximately 25% in developed countries to over 80% in developing countries.3 A key step in the pathogenic progression of this infection is that, in some cases, pneumococci transmit from the nasopharynx to other sites of the body, causing mucosal infections such as otitis media (middle ear) and sinusitis (sinuses) as well as life-threatening diseases including pneumonia (lung), bacteremia (blood) and meningitis (brain).1 Despite the availability of a pneumococcal conjugate vaccine and antibiotics, pneumococcal pneumonia and invasive pneumococcal disease (sepsis and meningitis) continue to be a major cause of childhood morbidity and mortality worldwide.4
The transition from commensal pneumococcal colonization to pneumococcal disease involves a range of host, environmental, and bacterial factors. Research over the years has established that several pneumococcal virulence factors, including the capsule, adhesins and pneumolysin (a toxin) affect disease progression and invasion.5 The host possesses a range of defenses which prevent asymptomatic carriage from progressing to invasive pneumococcal disease. The respiratory epithelium at the nasopharynx along with mucociliary clearance offers the first barrier to the invading pneumococci.6 If the pneumococci pass this initial defense mechanism, they reach the lower respiratory tract, where resident alveolar macrophages phagocytose and kill these bacteria, limiting their numbers.6,7 In parallel, pneumococci activate innate immune responses, including the production of cytokines, chemokines and antimicrobial peptides, aimed at clearing the infection.6 Most of these innate defenses are triggered by the recognition of the pneumococci (or its ligands) by toll-like receptors (TLR) and cytosolic inflammasomes. Lipoteichoic acid of the S. pneumoniae cell wall is a TLR2 activator,8 while pneumolysin and pneumococcal DNA are agonists of TLR4 and TLR9 respectively.9,10 Pneumococci also activate the cytosolic receptors AIM2 and NLRP3,11,12 resulting in the assembly of multiprotein enzyme complexes (inflammasomes) that serve as platforms for maturation of the inflammatory cytokines IL-1β and IL-18. When this inflammatory response fails to eradicate pneumococci from the lungs, pneumonia and occasionally sepsis can ensue. The effectiveness of these host regulatory processes at conferring protection is demonstrated by the observation that most people who carry pneumococci in the nasopharynx do not develop pneumococcal disease.
Mucin 1 (MUC1) is one host factor known to protect the human body against an array of bacterial pathogens that invade mucosal surfaces of the respiratory, gastrointestinal and urogenital tract.13,14 This cell membrane-associated, extensively O-glycosylated protein is expressed on the epithelial cells that line all the major tracts of the human body, as well as by immune cells.13 The 2 primary mechanisms by which MUC1 has been shown to protect against disease progression is by i) acting as a releasable decoy molecule,15 thereby restricting pathogen adherence to the epithelial surface and ii) by modulating cell signaling pathways, most notably, suppression of the TLR signaling pathway.16
We have previously demonstrated that while epithelial MUC1 protects against colonization with the gastric pathogenic bacterium, Helicobacter pylori,17 we later found that it is MUC1 expressed by immune cells that controlled H. pylori-induced inflammation by suppressing activation of the NLRP3 inflammasome and IL-1β production.18 As numerous bacteria activate the NLRP3 inflammasome, we questioned whether NLRP3 inflammasome regulation by MUC1 was a key modulatory mechanism common for other bacterial infections. To investigate this theory, S. pneumoniae was chosen; MUC1 is known to suppress inflammation in response to other lung bacterial infections,,19,20 and S. pneumoniae also activates the NLRP3 inflammasome.12
Results
MUC1 protects mice from invasive S. pneumoniae infection
Wildtype (WT) and Muc1−/− mice were infected intranasally with S. pneumoniae strain D39. At 16 hours post-infection, nasopharynx, lungs and blood were extracted from the infected mice and pneumococcal burden determined by colony-forming assay. In a pilot study, Muc1−/− mice became ill and had to be killed after 24 hours of infection; therefore infections were not continued beyond 16 hours due to animal welfare considerations. There was no difference in the number of S. pneumoniae recovered from the nasopharynx of infected WT and Muc1−/− mice (Fig. 1A), showing that nasopharyngeal carriage was not affected by MUC1. In contrast, 4 of 12 WT mice had pneumococci recovered from the lungs compared with 11 of 12 Muc1−/− mice (p = 0.009, Fisher's exact test) and pneumococcal loads were significantly higher in the Muc1−/− mice (Fig. 1B). Similarly, only 1 of 6 infected WT mice had pneumococci recovered from the broncho-alveolar lavage fluid (BALF) compared with 4 of 6 Muc1−/ - mice, although this did not reach significance (Fig. 1C).
Figure 1.

MUC1 controls pneumococcal levels in the lungs and blood of S. pneumoniae infected mice. Wildtype (WT) and Muc1−/− mice were infected intranasally with S. pneumoniae D39 strain. Pneumococcal levels were determined 16 hours (A, B, C and D) and 1 hour (E) after infection by colony-forming assay. (A) No significance difference in S. pneumoniae loads in the nasopharynx of WT and Muc1−/− mice. (B) Muc1−/− display significantly higher levels of S. pneumoniae in the lungs as compared with WT mice; data pooled from 2 independent experiments (***p < 0.001, 2-tailed Student's t-test) (C) S. pneumoniae detected in the broncho-alveolar lavage fluid (BALF) from infected WT and Muc1−/− mice. (D) A significantly higher number of S. pneumoniae-infected Muc1−/− mice developed bacteremia as compared with infected WT mice; data pooled from 3 independent experiments (#p < 0.05, Fisher's exact test). (E) After 1 hour of infection, similar levels of pneumococci were recovered from the lungs of WT and Muc1−/− mice. Graphs present individual mice (points) and group medians (horizontal bar). The limit of detection (LD, 100 CFU/ml) is shown as a dotted line. BALF- broncho-alveolar lavage fluid.
During S. pneumoniae infections, high bacterial loads in the lungs can result in leakage or transcytosis of pneumococci from the lung into the bloodstream, leading to bacteremia, a severe form of pneumococcal disease. To evaluate if the infected mice had bacteremia, a colony-forming assay was performed on blood collected from mice infected with S. pneumoniae for 16 hours. Analyses revealed that while only 1 out of 16 infected WT mice examined had S. pneumoniae-bacteremia, 9 of 16 infected Muc1−/− mice showed bacteremia (p = 0.006, Fisher's exact test) and pneumococcal loads in the blood were significantly higher in the Muc1−/− mice (Fig. 1D), indicative of an invasive infection in the absence of MUC1. Mice showing the highest bacteremia had an early onset of disease symptoms including ruffled fur, hunched posture and inactivity.
To determine whether MUC1 was affecting the ability of S. pneumoniae to colonize or survive in the lung, we quantified bacterial levels in the lungs of mice infected for one hour. This revealed no difference in S. pneumoniae numbers in the lungs of WT and Muc1−/− mice 1 hour post-infection (Fig. 1E), indicating that the bacteria were reaching the lungs but that MUC1 was playing a critical role in the clearance of the pneumococci. MUC1 on epithelial cells has been previously shown to serve as a barrier and releasable decoy receptor during bacterial infections.15,21,22 To test if a deficiency in MUC1 on lung epithelial surface could potentially contribute to the higher S. pneumoniae colonization levels in the infected mouse lungs, an in vitro adherence assay with a lung epithelial cell line (A549) was performed. Wildtype or A549 cells with reduced expression of MUC1 (about 75% reduction; Fig. 2A) were co-cultured with pneumococci for 1, 2 and 6 hours. The level of S. pneumoniae adherence to these cells correlated with MUC1 expression; within 1 hour, the adherence of pneumococci to A549 cells with reduced MUC1 expression was significantly lower as compared with control A549 cells (Fig. 2B). This provides evidence that MUC1 actually supports attachment of S. pneumoniae to epithelial cells, and therefore that the loss of this mucin on the lung epithelium may not be the mechanism behind the increased bacterial numbers observed in the lungs and blood of Muc1−/− mice.
Figure 2.

Knockdown of MUC1 expression is associated with reduced pneumococcal adherence to A549 cells. (A) MUC1 Expression in wildtype (WT) and mutated clone (MUC1 clone) of A549 cells was quantified using qPCR. There was nearly 75% knockdown of MUC1 expression in MUC1 clone as compared with WT. (B) WT and MUC1 clone of A549 cells (n = 5) were co-cultured with S. pneumoniae D39 strain at a multiplicity of infection of 50: 1 and adherence assessed following 1, 2 and 6 hours of incubation. MUC1 knockdown in A549 cells lead to significantly lower binding of pneumococci (*p < 0.05, **p < 0.01, Student's t-test).
MUC1 protects against S. pneumoniae-induced lung inflammation
Histological examinations were performed to assess the impact of S. pneumoniae infection on lung pathology. Representative photomicrographs of H&E stained lung sections of control and infected miceare presented in Fig. 3A–D. Blinded lung sections were graded on a scale of 1–4 for cellular infiltration (CI) to assess pathology. While there was no significant difference in CI score between uninfected and infected WT mice, S. pneumoniae infection resulted in a significant increase in CI in Muc1−/− mice (Fig. 3D and E).
Figure 3.

MUC1 prevents lung inflammation in S. pneumoniae infected mice. Representative H&E stained lung photomicrographs in (A) uninfected WT (B) uninfected Muc1−/− (C) infected WT (D) infected Muc1−/− mice. (E) Blinded H&E stained lung sections were graded for cellular infiltration. Infected Muc1−/− displayed significantly higher cellular infiltrate in the lungs as compared with uninfected Muc1−/− mice. (*p < 0.05, Mann-Whitney). Graph presents individual mice (points) and group medians (horizontal bar). Arrows indicate cellular infiltrate. Uninf, uninfected; Inf, infected. Scale bar = 100 µm.
Analysis of the cytokine profile in the nasopharynx revealed no significant differences in the levels of pro-inflammatory cytokines between uninfected and infected mice although a trend toward increased levels of pro-inflammatory cytokines in infected Muc1−/− mice compared with uninfected Muc1−/− mice was observed (Fig. 4). However, in the lung there was significant upregulation of all the pro-inflammatory cytokines assayed (IL-6, MIP-2, KC, TNF-α, IL-1β, IL-17A, IL-17F and IFN-γ) after S. pneumoniae infection in Muc1−/− mice (Fig. 5). In contrast, pro-inflammatory cytokines were not significantly increased in the lungs of S. pneumoniae infected WT mice (Fig. 5). This complete lack of a detectable cytokine response to S. pneumoniae infection in the WT mice was surprising, but is possibly related to the relatively low infection dose required used in these studies due to the high susceptibility of the Muc1−/− mice.
Figure 4.

Pro-inflammatory cytokine profile of the nasopharynx. Cytokine levels from nasopharyngeal homogenates of Wildtype (WT) and Muc1−/ mice (n = 7–8) infected with S. pneumoniae D39 strains for 16 hours were measured by ELISA. Graphs present the median (horizontal bar), interquartile range (box) and 10th and 90th percentiles (error bars). Uninf, uninfected; Inf, infected. Data analyzed using Student's t-test.
Figure 5.
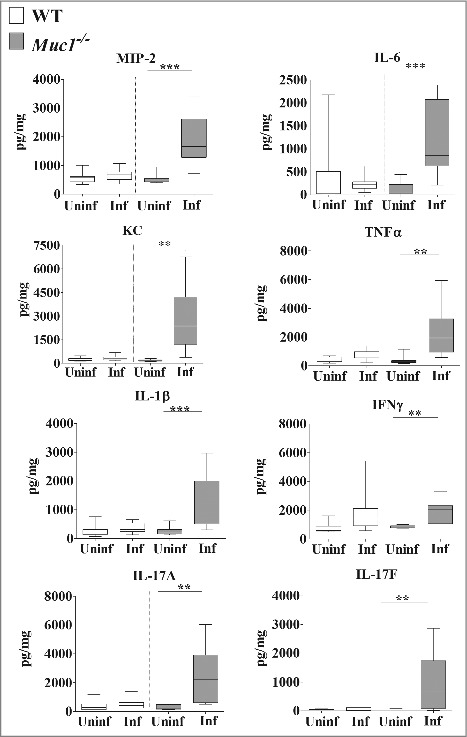
MUC1 deficiency is associated with elevated inflammatory cytokines responses in the lungs during S. pneumoniae infections. Cytokine levels from lung homogenates of wildtype (WT) and Muc1−/− mice (n = 13–14) infected with S. pneumoniae D39 strains for 16 hours were measured by ELISA. Infected Muc1−/− mice produced significantly higher amounts of all pro-inflammatory cytokines assayed, as compared with uninfected Muc1−/− mice (*p < 0.05, **p < 0.01,Student's t-test). Graphs present the median (horizontal bar), interquartile range (box) and 10th and 90th percentiles (bars). Uninf, uninfected; Inf, infected.
To further characterize the immune response in the lungs of S. pneumoniae infected mice, lung infiltrating immune cells were analyzed by multicolor flow cytometry using previously published methods.33,34 This analysis revealed significantly higher numbers of alveolar macrophages in the lungs of both WT and Muc1−/− mice after S. pneumoniae infection (Fig. 6). Additionally, there was elevated recruitment of Ly6C− monocytes and interstitial macrophages, only in the lungs of infected Muc1−/− mice as compared with uninfected Muc1−/−, but not in infected WT mice. These results indicated that cells of the monocyte-macrophage lineage are the main types recruited during S. pneumoniae infection in Muc1−/− mice.
Figure 6.
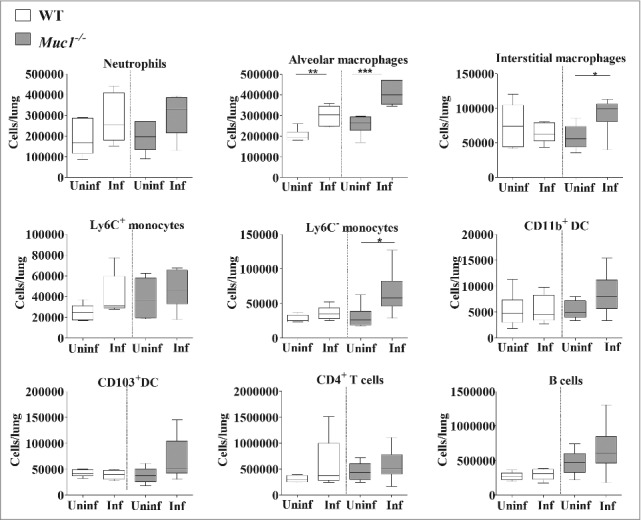
Immunophenotyping of cells infiltrating the lungs shows increased levels of monocytes and macrophages in S. pneumoniae infected mice. Flow cytometry was used to characterize the immune cells recruited to the lungs of S. pneumoniae infected mice and uninfected wildtype (WT) and Muc1−/− mice (n = 5–6). There were significantly more alveolar macrophages, interstitial macrophages and Ly6c- monocytes in the lungs of infected Muc1−/− mice as compared with uninfected Muc1−/− mice. For WT mice, there were significantly more alveolar macrophages in infected mice compared with uninfected mice (*p < 0.05, **p < 0.01, Student's t-test).Graphs present the median (horizontal bar), interquartile range (box) and 10th and 90th percentiles (bars). DC-dendritic cells; Uninf, uninfected; Inf, infected.
MUC1 promotes S. pneumoniae phagocytosis by macrophages
Macrophages are the initial responders in the lungs, and the main phagocytic cells during initial stages of S. pneumoniae infection, responsible for the killing and clearing of S. pneumoniae.7 We therefore tested whether the increased pneumococcal loads present in the lungs of Muc1−/− mice, despite increased numbers of alveolar macrophages, might be at least partially due to inefficient phagocytosis. Macrophages from WT and Muc1−/− mice were co-cultured with 4 CFSE-labeled strains of S. pneumoniae and the levels of internalized bacteria analyzed by fluorometry. There was a significant reduction in the ability of Muc1−/− macrophages to phagocytose 3 strains of S. pneumoniae compared with WT cells, with the fourth trending the same way but not quite reaching significance (Fig. 7A). To study the effect that reduced phagocytosis has on bacterial killing, the levels of live pneumococci in the culture supernatants of D39 bacteria co-cultured with macrophages was determined by colony-forming assay. This showed significantly higher levels of live pneumococci when these bacteria were co-cultured with Muc1−/− as compared with wildtype macrophages, indicating reduced bacterial killing in the absence of MUC1 (Fig. 7B).
Figure 7.

MUC1 facilitates the phagocytosis and killing of S. pneumoniae. Macrophages from wildtype (WT) and Muc1−/− mice (n = 5) were co-cultured in vitro with CFSE-labeled S. pneumoniae strain D39, TIGR4A, PMP1 and PMP1287 and P. aeruginosa strain NCTC10662 (multiplicity of infection 50:1) for time-points shown. (A) Muc1−/− macrophages have significantly decreased intracellular S. pneumoniae D39, TIGR4A and PMP1278 strains as compared with WT cells (*p < 0.05, **p < 0.01, Student's t-test). (B) Significantly reduced killing of S. pneumoniae D39 strain by Muc1−/− macrophages compared with WT macrophages was observed after 120 min of co-culture (**p < 0.01, Student's t-test). (C) While intracellular S. pneumoniae was significantly higher in WT macrophages as compared with Muc1−/− macrophages, Muc1−/− macrophages had significantly increased phagocytosis of P. aeruginosa 60 mins after co-culture (**p < 0.01, ***p < 0.001, Student's t-test). Data shown are representative of 4 experiments for S. pneumoniae D39 and 1 for other bacterial strains. Graphs present the median (horizontal bar), interquartile range (box) and 10th and 90th percentiles (bars).
It has been previously published that loss of MUC1 improved the phagocytic efficiency of P. aeruginosa, another respiratory pathogen. To confirm our results, we repeated the phagocytosis assay using these bacterial pathogens. We observed that, consistent with previous literature,23 Muc1−/− macrophages exhibited increased phagocytosis of P. aeruginosa compared with WT, Muc1−/− macrophages within the same study internalized reduced levels of S. pneumoniae (Fig. 7C). This indicates that MUC1 exerts opposite effects on the phagocytosis of P. aeruginosa and S. pneumoniae by macrophages.
Discussion
S. pneumoniae infections are responsible for life threatening diseases including pneumonia, bacteremia and meningitis, especially in children under the age of 5 y, the elderly and immunocompromised individuals.1 In the current study, MUC1 was found to provide protection against pneumococcal pneumonia and bacteremia most likely by facilitating the phagocytosis of S. pneumoniae by macrophages.
Our initial hypothesis was that MUC1 might regulate S. pneumoniae-associated disease severity by suppressing the activation of NLRP3 inflammasome. However, given the above effects that resulted in the rapid appearance of S. pneumoniae in blood, it was not possible to examine the role of MUC1 at regulating the NLRP3 inflammasome in S. pneumoniae infection using this model. In pilot studies, sub-lethal doses of pneumococci were tested. However, at these low infectious doses, the pneumococci were rapidly cleared from the nasopharynx and lungs of the infected WT and Muc1−/− mice, with no detectable immune responses within 24 hours of infection. While IL-1β levels were increased in the lungs of S. pneumoniae infected Muc1−/− mice after 16 hours of infection, it was not possible to determine whether this was due to loss of regulation of the NLRP3 inflammasome in this tissue or resulted from the increased bacterial colonization that arose due to inefficient macrophage phagocytosis due to loss of MUC1.
Infected WT and Muc1−/− mice had no difference in pneumococcal colonization of the nasopharynx, suggesting MUC1 does not affect nasopharyngeal carriage, at least within the relatively short time frame of the current model. It did however play an important role in reducing the bacterial burden in the lungs. While WT and Muc1−/− mice had similar bacterial levels in the lungs 1 hour after infection, by 16 hours post-infection, pneumococci were only detectable in the lungs of mice that lacked MUC1. Infection in both WT and Muc1−/− mice was associated with elevated number of alveolar macrophages in the lung, which was expected as macrophages are the first responders to invading pneumococci.7 In WT mice, pneumococci that reached the lung were subsequently cleared by these alveolar macrophages, preventing lung inflammation. In the absence of MUC1 however, despite an increased recruitment of monocytes and macrophages, the pulmonary infection was not cleared, leading to elevated inflammation, bacteremia and disease.
Factors that prevent the translocation of bacteria into the bloodstream are of utmost importance in protecting against invasive pneumococcal infection. In the current study, the correlation between pneumococci levels in the lungs and blood of Muc1−/− mice suggests that transmission to the bloodstream probably occurred via the lung epithelium. Other studies looking at the invasive potential of pneumococcal strains in mice have made similar observations, suggesting that pneumococci migrate via the pleural cavity into the blood stream.24,25
This finding is probably related to the novel observation presented here, that MUC1 facilitates the efficient phagocytosis of S. pneumoniae by macrophages. All 4 strains of pneumococci tested, selected to examine a wide range of serotypes, showed reduced phagocytosis by MUC1-deficient macrophages compared with wildtype cells, although one strain (PMP1) did not reach significance. The PMP1 strain was poorly phagocytosed by wildtype macrophages as well, possibly explaining this discrepancy with the other strains. PMP1 is from a serotype that is commonly associated with invasive disease and is known to be resistant to phagocytosis, a feature primarily attributed to the different composition of the capsule.26 Macrophages are the primary phagocytic cells present during acute pneumococcal infection,7 and depletion of macrophages in mice has been shown to significantly reduce pneumococcal clearance from lungs,27 as well as reduce survival after pneumococcal infection, an affect attributed to exacerbated inflammation.28 Macrophage phagocytosis is therefore one of the key host responses that controls pneumococcal levels either via the formation of lysosomal complexes with reactive oxygen species and nitric oxide or by the phagocytic cell undergo apoptosis.7,27,28
It was recently shown that the mechanism of protection by which mice survive the first few days of systemic S. pneumoniae infection involves neutrophils killing bacteria attached to the surface of macrophages.29 As our data indicate that MUC1 supports S. pneumoniae phagocytosis (suggesting these bacteria bind this mucin on the macrophage surface), a lack of MUC1 would likely reduce the binding of S. pneumoniae to the macrophage surface, thereby reducing their availability for killing by neutrophils; this would explain the acute morbidity and mortality observed in the acutely infected Muc1−/− mice.
Another recent finding is that MUC1 hinders the phagocytosis of Escherichia coli and the respiratory pathogen P. aeruginosa, with MUC1 deficiency being linked to loss of a steric barrier around macrophages, allowing better access of P. aeruginosa to cell-membrane receptors involved in phagocytosis, which therefore occurs with increased efficiency.23 Thus, MUC1 has not previously been reported to have a direct role in the process of phagocytosis. This contradictory role of MUC1 in facilitating or restricting phagocytosis of different pathogenic bacteria is intriguing and warrants further investigation. One possible explanation relates to their surface structures; S. pneumoniae are Gram positive bacteria while E. coli and P. aeruginosa are Gram negative, raising the possibility that this phenomenon is linked to the presence or absence of LPS. However differences in other surface molecules, such as the presence of flagellin on P. aeruginosa, cannot be ruled out.
The current study illustrates the presence of a novel mechanism via which MUC1 can regulate bacterial virulence, i.e. by promoting macrophage phagocytosis, thereby assisting in reducing bacterial loads in the lung and subsequent invasion into the bloodstream. MUC1 has been previously demonstrated to have diverse roles both on the epithelial surface and on immune cells. It is well described for its role on epithelial cells where it functions as a barrier on the epithelial surface by sterically inhibiting the binding of pathogens to host epithelium, along with acting as a releasable decoy, limiting both bacterial colonization and associated inflammation.15,17,30 MUC1 has been previously shown to serve as an adhesion site for P. aeruginosa (via flagellin) and H. pylori (via BabA and SabA adhesins).15,21 In the current study, results from an in vitro assay indicate that MUC1 on epithelial cells may also be a binding ligand for S. pneumoniae, as knockdown of MUC1 expression on a lung epithelial cell line was associated with lower adhesion of pneumococci. It is thus possible that MUC1 on the lung epithelium plays some role in protecting against invasion of the host by S. pneumoniae. However, new data in this study suggest that MUC1 on macrophages plays a key role in controlling the levels of these bacteria in the respiratory tract by facilitating phagocytosis and thereby limit invasion. As lung colonization and dissemination into other body sites such as blood and brain are key steps in pneumococcal pathogenesis, elucidating the role of MUC1 in limiting these processes is critical to understanding how the innate immune system protects against pneumococcal disease. The current study therefore adds to the well-documented heterogeneity of MUC1 functions at different sites and in response to different pathogens.
Despite the high prevalence of nasopharyngeal carriage of pneumococcal bacteria, invasive disease is a rare consequence. This indicates that host (and bacterial) processes are in place that maintain the balance between asymptomatic colonization and disease. The work presented here demonstrates the critical role the important host factor MUC1 plays in protecting against pneumococcal diseases, and suggests that the mechanism by which this occurs likely involves facilitating efficient phagocytosis and clearance of S. pneumoniae by macrophages. Further studies characterizing the interaction between MUC1 and S. pneumoniae in the respiratory tract would help in understanding how MUC1 prevents pneumococci from breaching tissue-blood barriers, a fundamental process in pneumococcal pathogenesis, and might help explain why only some colonized individuals develop invasive diseases.
Methods
Bacterial culture
S. pneumoniae D39 strain (serotype 2) was a kind gift from Prof. James C. Paton, University of Adelaide. S. pneumoniae TIGR4A (serotype 4) was obtained from Rick Malley at Children's Hospital Boston; PMP1 (serotype 1) was sourced from Janet Strachan at Microbiological Diagnostic Unit, Melbourne and PMP1278 (serotype 16F) was sourced from the Pneumococcal Research culture collection at Murdoch Childrens Research Institute. S. pneumoniae were grown overnight on horse blood agar (HBA) plates (ThermoFisher Scientific, PB0114) at 37°C, 5% CO2. The next day, bacteria were grown until log phase in brain heart infusion (BHI; Oxoid, Hampshire, UK) broth, supplemented with 10% horse serum (Sigma, St Louis, Missouri, USA) at 37°C, 5% CO2. Bacteria were washed twice in phosphate buffered saline (PBS) before use and resuspended in PBS or Roswell Park Memorial Institute 1640 medium (RPMI, Life Technologies, 11875093) supplemented with 10% fetal calf serum (FCS, Life Technologies, 16140071). Pseudomonas aeruginosa (NCTC10662) was grown overnight in BHI broth supplemented with 5% horse serum at 37°C.
Mice
Specific pathogen-free age-matched female Muc1+/+ and Muc1−/− 129/SvJ mice (kindly provided by Sandra Gendler)31 were bred and housed in the Parkville Veterinary Science animal facility, University of Melbourne. Unanesthetized, 5–7 week old, female mice were infected by intranasal delivery of 106 S. pneumoniae in 30 µl of PBS, or sham dosed with PBS alone. The challenge dose was confirmed retrospectively by plating of the inocula on HBA plates. All mouse experiments were performed with approval from the Animal Ethics Committee, University of Melbourne, Australia.
Viable counting of bacteria (colony-forming assay)
Blood was collected by cardiac puncture and mixed with 0.5% EDTA to prevent coagulation. Nasopharyngeal and lung tissues were collected from mice and homogenized in 2 ml BHI using a Polytron PT2100 (Radnor, Pennsylvania, USA). Serial dilutions were prepared in PBS, and 10 µl plated on HBA- plates supplemented with 5 µg/ml gentamycin (ThermoFisher Scientific). Plates were incubated overnight at 37°C, 5% CO2 then colonies counted.
Histology
Lungs were perfused through the trachea with 50% OCT compound (Scigen, Gardenia, California, USA) to preserve lung architecture. Perfused lungs were embedded in OCT and stored at -80°C. 10 µm sections were cut, mounted on Superfrost slides, fixed in 100% ice-cold ethanol and stained with hematoxylin and eosin (H&E). Blinded lung sections were graded for cellular infiltration as follows: Grade 0 (none) - no immune cells found in the lungs; Grade 1 (mild) - focal aggregates of immune cells, usually around bronchioles and blood vessels; Grade 2 (moderate) - dense cuffs of lymphocytes surrounding blood vessels and airways; Grade 3 (marked) - moderate infiltrate, extending into surrounding tissue, mostly macrophages in alveoli and variable neutrophils and lymphocytes; Grade 4 (severe) - marked infiltrate with extensive areas of lung tissue affected.
Quantification of cytokines by ELISA
Nasopharyngeal homogenates were centrifuged at 13000 g for 5 mins to pellet the debris, then supernatants collected. Five 10 µm cryosections from each lung were homogenized in 2 ml PBS, total protein quantified using a Pierce bicinchoninic acid (BCA) protein assay kit (ThermoFisher Scientific, 23225) and protein concentrations normalized to 100 µg/ml.
Cytokine levels were quantified by ELISA as described previously.32 Primary antibodies: anti-mouse tumor necrosis factor (TNF)-α (0.1 μg/well; BioLegend, San Diego, California, USA), macrophage inflammatory protein (MIP-2) (0.1 μg/well; R&D Systems), KC (0.2 µg/well) interferon (IFN-γ) (0.1 μg/well; BD Biosciences, San Jose, California, USA), IL-17A (0.5 μg/well; eBioscience, Cleveland, Ohio, USA), IL-6 (0.05 μg/well; eBioscience), IL-1β (0.2 μg/well; R&D Systems) IL-17F (0.04 µg/well; R&D Systems). Secondary antibodies: biotinylated anti-mouse TNF-α (0.025 μg/well), MIP-2 (3.7 ng/well), KC (0.01 µg/well), IFN-γ (0.05 μg/well), IL-17A (0.025 μg/well), IL-17F (0.01 µg/well), IL-6 (0.025 μg/well), IL-1β (0.03 μg/well), IL-17F; (same manufacturers as capture antibody). Sample concentration was determined against a standard curve of recombinant cytokine (same manufacturers as antibodies).
Flow cytometric analysis of lung cells
Lungs were processed for flow cytometry as described previously.33,34 Briefly, lungs were cleared of blood by perfusing via the right ventricle with PBS. Lungs were chopped with a scalpel blade and digested in RPMI media with 2% FCS and 1.5 mg/ml Collagenase D (Roche, 11088866001) at 37°C, 90 mins at 80 rpm. Digested lungs were then passed through a 70µm strainer. After lysis of erythrocytes, cell suspensions were washed twice in 2% FCS in PBS and blocked in 100 μl of PBS containing 2% FCS (blocker) with 20% mouse serum and 1 μg/ml anti-FcγII/FcγIII (2.4G2, Walter and Eliza Hall Institute, Melbourne, Australia) on ice for 30 min.
Cells were stained in 100 µl blocker with the following antibodies: anti-CD45-Alexa 700 (eBioscience), anti-F4/80-Brilliant violet 421 (eBioscience), anti-CD19-Brilliant violet 510 (BioLegend), anti-CD4-Brilliant violet 650 (BioLegend), anti-CD103-Brilliant violet 786 (BD Horizon), anti CD11b-FITC (BD PharMingen), anti-Ly6C-PerCP (BioLegend), anti-CD64-PE (BioLegend), anti-MHCII-PeCy7 (BioLegend), anti-CD11c-APC (eBioscience), anti-Ly6G-APC-Cy7 (BioLegend). Cells were washed twice, then resuspended in 0.25 mg/ml propidium iodide (ImmunoChemistry Technologies, 98) to stain dead cells which were excluded during the gating strategy. Cells were acquired on an X-20 LSR Fortessa (BD Bioscience, San Jose, California, USA) and counts normalized using AccuCount Fluorescent Particles (Spherotech, Lake forest, Illinois, USA). Data were analyzed using BD FACS Diva software. The gating strategy used to characterize different immune cells is shown in Fig. 1.
In vitro epithelial cell adherence assay
CRISPR/Cas9 system was used to knockdown MUC1 expression in A549 cells. Using CRISPR design tool ,35 2 small guide RNA (sgRNA) that target the open reading frame of MUC1 gene were designed. A549 cells were grown in complete Dulbecco's Modified Eagle Media (DMEM; without antibiotics) and incubated in 24 well plate with 5 × 105 cells/well, overnight at 37°C with 5% CO2. sgRNA containing plasmids (GFP+) were transfected into A549 cells using Lipofectamine LTX reagent (ThermoFisher Scientific) as per manufacturer's instruction. Using an Influx cell sorter (BD Bioscience), single cell clones were sorted into 96 well plates and grown (as above) to get pure cultures. Mutation of the MUC1 gene was confirmed by gel shift assay and MUC1 knockdown was confirmed by qPCR.
A549 cells (WT and a mutated MUC1 clone) were incubated in a 96 well plate with 1 × 105 cells/well and grown to a confluent monolayer. Cells were co-cultured with 5 × 106 pneumococci/well and plates were centrifuged at 1200 g for 2 mins to facilitate bacterial contact with the cell monolayer. After the incubation period, culture supernatant (with the unattached bacteria) was removed and cells were washed once. Adherent bacteria and epithelial cells were detached from the wells by treatment with trypsin (Sigma). The number of bound bacteria was determined by colony forming assay.
In vitro phagocytosis and killing assay
Non-induced peritoneal macrophages were collected from WT and Muc1−/− mice by flushing the peritoneal cavity with 10 ml of ice-cold PBS containing 2% FCS. Cells were resuspended in complete RPMI media (without antibiotics) and incubated in 96 well plates with 5 × 105 cells/well, overnight at 37°C with 5% CO2.
Pelleted cultures of S. pneumoniae and P. aeruginosa were resuspended in 10 ml RPMI media with 5 µM carboxyfluorescein succinimidyl ester (CFSE; ThermoFisher Scientific, C34554). Bacterial suspensions were incubated in the dark at 37°C in a shaker incubator for 30 mins. The volume was doubled using RPMI containing 10% FCS to quench extracellular CFSE. Bacteria were washed twice by centrifugation at 3750 g for 15 mins in PBS, then counted using a hemocytometer. Macrophages were co-cultured with CFSE-labeled bacteria then extracellular CFSE quenched by incubating cells in 1.5 µg/ml trypan blue for 5 mins at 37°C. Culture supernatant was removed, serially diluted onto HBA plates for estimating viable counts for killing assay. Cells were washed twice in PBS and intracellular fluorescence analyzed by reading plates at Em488/Ex533 using an Infinite M200PRO (Tecan, Mannedorf, Switzerland) plate reader.
Statistics
Fisher's exact test was used to assess the absence or presence of bacteria in lungs and blood. The non-parametric Mann-Whitney test was used to assess bacterial levels at different sites and histological grading scores. All other data were analyzed using 2-tailed Student t-test. All statistical analyses were performed using GraphPad Prism software.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Victorian Government's Operational Infrastructure Support Program and National Health and Medical Research Council project grant GNT1046254. PS was supported by a Senior Research Fellowship from the National Health and Medical Research Council of Australia. PD was supported by a Victoria India Doctoral Scholarship.
References
- [1].van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 2009; 374:1543-56; PMID:19880020; https://doi.org/ 10.1016/S0140-6736(09)61114-4 [DOI] [PubMed] [Google Scholar]
- [2].Bogaert D, De Groot R, Hermans PW. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 2004; 4:144-54; PMID:14998500; https://doi.org/ 10.1016/S1473-3099(04)00938-7 [DOI] [PubMed] [Google Scholar]
- [3].World Health Organization Pneumococcal vaccines WHO position paper - 2012. Wkly Epidemiol Rec 2012; 87:129-44; PMID:24340399 [PubMed] [Google Scholar]
- [4].O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T, et al.. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 2009; 374:893-902; PMID:19748398; https://doi.org/ 10.1016/S0140-6736(09)61204-6 [DOI] [PubMed] [Google Scholar]
- [5].Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol 2008; 6:288-301; PMID:18340341; https://doi.org/ 10.1038/nrmicro1871 [DOI] [PubMed] [Google Scholar]
- [6].Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol 2012; 14:460-6; PMID:22212419; https://doi.org/ 10.1111/j.1462-5822.2011.01746.x [DOI] [PubMed] [Google Scholar]
- [7].Aberdein JD, Cole J, Bewley MA, Marriott HM, Dockrell DH. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin Exp Immunol 2013; 174:193-202; PMID:23841514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].van Rossum AM, Lysenko ES, Weiser JN. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infect Immun 2005; 73:7718-26; PMID:16239576; https://doi.org/ 10.1128/IAI.73.11.7718-7726.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A 2003; 100:1966-71; PMID:12569171; https://doi.org/ 10.1073/pnas.0435928100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, Akira S, Normark S, Henriques-Normark B. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol 2007; 9:633-44; PMID:17004992; https://doi.org/ 10.1111/j.1462-5822.2006.00814.x [DOI] [PubMed] [Google Scholar]
- [11].Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, Yamamoto T, Fernandes-Alnemri T, Yang R, Hernandez-Cuellar E, et al.. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J Immunol (Baltimore, Md: 1950) 2011; 187:4890-9; PMID:21957143; https://doi.org/ 10.4049/jimmunol.1100381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, et al.. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog 2010; 6:e1001191; PMID:21085613; https://doi.org/ 10.1371/journal.ppat.1001191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunol 2008; 1:183-97; PMID:19079178; https://doi.org/ 10.1038/mi.2008.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McGuckin MA, Lindén SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Micro 2011; 9:265-78; https://doi.org/ 10.1038/nrmicro2538 [DOI] [PubMed] [Google Scholar]
- [15].Linden SK, Sheng YH, Every AL, Miles KM, Skoog EC, Florin TH, Sutton P, McGuckin MA. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathogens 2009; 5:e1000617; PMID:19816567; https://doi.org/ 10.1371/journal.ppat.1000617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ueno K, Koga T, Kato K, Golenbock DT, Gendler SJ, Kai H, Kim KC. MUC1 mucin is a negative regulator of toll-like receptor signaling. Am J Respir Cell Mol Biol 2008; 38:263-8; PMID:18079492; https://doi.org/ 10.1165/rcmb.2007-0336RC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McGuckin MA, Every AL, Skene CD, Linden SK, Chionh YT, Swierczak A, McAuley J, Harbour S, Kaparakis M, Ferrero R, et al.. Muc1 mucin limits both Helicobacter pylori colonization of the murine gastric mucosa and associated gastritis. Gastroenterology 2007; 133:1210-8; PMID:17919495; https://doi.org/ 10.1053/j.gastro.2007.07.003 [DOI] [PubMed] [Google Scholar]
- [18].Ng GZ, Menheniott TR, Every AL, Stent A, Judd LM, Chionh YT, Dhar P, Komen JC, Giraud AS, Wang TC, et al.. The MUC1 mucin protects against Helicobacter pylori pathogenesis in mice by regulation of the NLRP3 inflammasome. Gut 2016; 65:1087-99; PMID:26079943; https://doi.org/ 10.1136/gutjnl-2014-307175 [DOI] [PubMed] [Google Scholar]
- [19].Lu W, Hisatsune A, Koga T, Kato K, Kuwahara I, Lillehoj EP, Chen W, Cross AS, Gendler SJ, Gewirtz AT, et al.. Cutting edge: enhanced pulmonary clearance of Pseudomonas aeruginosa by Muc1 knockout mice. J Immunol 2006; 176:3890-4; https://doi.org/ 10.4049/jimmunol.176.7.3890 [DOI] [PubMed] [Google Scholar]
- [20].Kyo Y, Kato K, Park YS, Gajghate S, Umehara T, Lillehoj EP, Suzaki H, Kim KC. Antiinflammatory role of MUC1 mucin during infection with nontypeable Haemophilus influenzae. Am J Respir Cell Mol Biol 2012; 46:149-56; PMID:22298528; https://doi.org/ 10.1165/rcmb.2011-0142OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lillehoj EP, Kim BT, Kim KC. Identification of Pseudomonas aeruginosa flagellin as an adhesin for Muc1 mucin. Am J Physiol Lung Cell Mol Physiol 2002; 282:L751-6; PMID:11880301; https://doi.org/ 10.1152/ajplung.00383.2001 [DOI] [PubMed] [Google Scholar]
- [22].Kato K, Lillehoj EP, Kai H, Kim KC. MUC1 expression by human airway epithelial cells mediates pseudomonas aeruginosa adhesion. Front Biosci (Elite edition) 2010; 2:68-77; PMID:20036855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kato K, Uchino R, Lillehoj EP, Knox K, Lin Y, Kim KC. Membrane-Tethered MUC1 Mucin Counter-Regulates the Phagocytic Activity of Macrophages. Am J Respir Cell Mol Biol 2016; 54:515-23; PMID:26393683; https://doi.org/ 10.1165/rcmb.2015-0177OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Harvey RM, Stroeher UH, Ogunniyi AD, Smith-Vaughan HC, Leach AJ, Paton JC. A variable region within the genome of Streptococcus pneumoniae contributes to strain-strain variation in virulence. PLoS One 2011; 6:e19650; PMID:21573186; https://doi.org/ 10.1371/journal.pone.0019650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hughes CE, Harvey RM, Plumptre CD, Paton JC. Development of primary invasive pneumococcal disease caused by serotype 1 pneumococci is driven by early increased type I interferon response in the lung. Infect Immun 2014; 82:3919-26; PMID:25001606; https://doi.org/ 10.1128/IAI.02067-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Melin M, Trzciński K, Antonio M, Meri S, Adegbola R, Kaijalainen T, Käyhty H, Väkeväinen M. Serotype-Related Variation in Susceptibility to Complement Deposition and Opsonophagocytosis among Clinical Isolates of Streptococcus pneumoniae. Infect Immun 2010; 78:5252-61; PMID:20855517; https://doi.org/ 10.1128/IAI.00739-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dockrell DH, Marriott HM, Prince LR, Ridger VC, Ince PG, Hellewell PG, Whyte MK. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol (Baltimore, Md: 1950) 2003; 171:5380-8; PMID:14607941; https://doi.org/ 10.4049/jimmunol.171.10.5380 [DOI] [PubMed] [Google Scholar]
- [28].Knapp S, Leemans JC, Florquin S, Branger J, Maris NA, Pater J, van Rooijen N, van der Poll T. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Critical Care Med 2003; 167:171-9; PMID:12406830; https://doi.org/ 10.1164/rccm.200207-698OC [DOI] [PubMed] [Google Scholar]
- [29].Deniset JF, Surewaard BG, Lee WY, Kubes P. Splenic Ly6Ghigh mature and Ly6Gint immature neutrophils contribute to eradication of S. pneumoniae. J Exp Med 2017; 214:1333-50; PMID:28424248; https://doi.org/ 10.1084/jem.20161621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McAuley JL, Linden SK, Png CW, King RM, Pennington HL, Gendler SJ, Florin TH, Hill GR, Korolik V, McGuckin MA. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J Clin Invest 2007; 117:2313-24; PMID:17641781; https://doi.org/ 10.1172/JCI26705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Spicer AP, Rowse GJ, Lidner TK, Gendler SJ. Delayed mammary tumor progression in Muc-1 null mice. J Biol Chem 1995; 270:30093-101; PMID:8530414; https://doi.org/ 10.1074/jbc.270.50.30093 [DOI] [PubMed] [Google Scholar]
- [32].Chionh YT, Ng GZ, Ong L, Arulmuruganar A, Stent A, Saeed MA, Wee JL, Sutton P. Protease-activated receptor 1 suppresses Helicobacter pylori gastritis via the inhibition of macrophage cytokine secretion and interferon regulatory factor 5. Mucosal Immunol 2015; 8:68-79; PMID:24866378; https://doi.org/ 10.1038/mi.2014.43 [DOI] [PubMed] [Google Scholar]
- [33].Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 2013; 49:503-10; PMID:23672262; https://doi.org/ 10.1165/rcmb.2013-0086MA [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP, Tighe RM, Blackwell TS. Identification of myeloid cell subsets in murine lungs using flow cytometry. Am J Respir Cell Mol Biol 2013; 49:180-9; PMID:23492192; https://doi.org/ 10.1165/rcmb.2012-0366MA [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al.. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 2013; 31:827-32; PMID:23873081; https://doi.org/ 10.1038/nbt.2647 [DOI] [PMC free article] [PubMed] [Google Scholar]