

ABSTRACT
Mitochondria are sentinel organelles that are impacted by various forms of cellular stress, including viral infections. While signaling events associated with mitochondria, including those activated by pathogen associated molecular patterns (PAMPs), are widely studied, alterations in mitochondrial distribution and changes in mitochondrial dynamics are also beginning to be associated with cellular insult. Cells of neuronal origin have been demonstrated to display remarkable alterations in several instances, including neurodegenerative disorders. Venezuelan Equine Encephalitis Virus (VEEV) is a New World alphavirus that infects neuronal cells and contributes to an encephalitic phenotype. We demonstrate that upon infection by the vaccine strain of VEEV (TC-83), astrocytoma cells experience a robust drop in mitochondrial activity, which corresponds with an increased accumulation of reactive oxygen species (ROS) in an infection-dependent manner. Infection status also corresponds with a prominent perinuclear accumulation of mitochondria. Cellular enzymatic machinery, including PINK1 and Parkin, appears to be enriched in mitochondrial fractions as compared with uninfected cells, which is indicative of mitochondrial damage. Dynamin related protein 1 (Drp1), a protein that is associated with mitochondrial fission, demonstrated a modest enrichment in mitochondrial fractions of infected cells. Treatment with an inhibitor of mitochondrial fission, Mdivi-1, led to a decrease in caspase cleavage, suggesting that mitochondrial fission was likely to contribute to apoptosis of infected cells. Finally, our data demonstrate that mitophagy ensues in infected cells. In combination, our data suggest that VEEV infection results in significant changes in the mitochondrial landscape that may influence pathological outcomes in the infected cell.
KEYWORDS: membrane depolarization, mitochondrial dynamics, mitophagy, reactive oxygen species, Venezuelan equine encephalitis
Introduction
Venezuelan Equine Encephalitis Virus (VEEV) is a single-stranded RNA virus belonging to the genus Alphavirus, family Togaviridae. This New World alphavirus is an emerging infectious agent that causes natural outbreaks in many parts of the world.1-3 VEEV was weaponized in the past and continues to be classified as a bio-threat agent, as well as a category B select agent, owing to the retention of stability and infectivity in aerosol form.4 New World alphaviruses are arboviruses (arthropod-borne viruses) that infect equines and humans. Equines typically develop generalized symptoms within 2–5 days of VEEV infection, including fever, tachycardia, depression, and anorexia. However, more serious complications, including hyper-excitability and encephalitis sometimes occur within 5–10 days of infection, often resulting in death of the animal within one week. If humans are infected with VEEV, symptoms appearing within 2–5 days can range from febrile, or flu-like symptoms, such as malaise, fever, chills and myalgia, to coma and death in ∼1% of cases. Currently, no United States Food and Drug Administration (FDA) approved vaccines or therapeutics are available as countermeasure strategies for VEEV infections. There are two investigational vaccines for VEEV that are offered to at-risk personnel.5 These include the VEEV TC-83 strain, which is a live-attenuated virus and is known to induce a fairly robust primary immune response. The second candidate is VEEV C-84, which is a formalin-inactivated vaccine and induces relatively weaker immune responses as compared to TC-83.
Events that occur in the infected host cell, including activation of phospho-signaling events such as NFκB signaling cascade, MEK-ERK signaling cascade and the ubiquitin proteasome pathway, play important roles in the establishment of a productive infection.6-8 Many host proteins have been shown to interact with VEEV proteins that critically influence viral multiplication.9,10 Thus, multiple lines of evidence allude to the importance of host components in viral multiplication and host pathology.
The role of mitochondrial dynamics during acute viral infections is beginning to be appreciated, and the list of viruses that impact mitochondria and innate immune responses is growing. Mitochondria are important to many cellular processes, including energy production, aging, innate immunity and cell survival.11-14 Mitochondrial size, structure and motility influence the maintenance of cellular homeostasis, as has been demonstrated in the case of neurodegenerative disorders including Parkinson's, Alzheimer's and Huntington's disease.15,16 Many viral proteins target the mitochondria and interfere with their functionality. The hepatitis B virus (HBV) encoded regulatory protein HBV X (HBx) localizes to the mitochondrial membrane where it alters membrane potential and elevates the levels of calcium ions and Reactive Oxygen Species (ROS), thereby causing damage to the mitochondria and activating latent transcription factors.17-20 Similarly, Hepatitis C virus (HCV), a positive-strand RNA virus in the Flaviviridae family, has also been shown to damage the mitochondria in the liver during infection due to an increase in ROS resulting in membrane depolarization and dysfunction.20 HCV infection also induces a perinuclear phenotype, wherein mitochondria cluster in the perinuclear space.21 In addition, HCV infection and altered mitochondrial dynamics were demonstrated to result in changes to the mitochondrial membrane proteome and mitochondrial localization of a host kinase, PINK1, and ubiquitin ligase, Parkin.21,22 Such alterations in enzymes that can mediate phosphorylation and ubiquitination of mitochondrial and associated proteins can influence interactions of mitochondria with motor proteins, such as kinesin, that disrupt mitochondrial mobility in neurons infected with Herpes simplex virus (HSV). In the case of Human Immunodeficiency Virus (HIV), viral protein R (Vpr) is known to associate with the mitochondrial outer membrane, leading to a decrease in expression of mitofusin 2. This induces mitochondrial fragmentation, ultimately affecting T lymphocyte viability. Respiratory Syncytial Virus (RSV) encodes a nonstructural protein, NS1, that has been shown to interfere with antiviral signaling originating from the mitochondria.23 The virulence factor of Rift Valley fever virus (RVFV), a non-structural S protein (NSs), associates with the mitochondria to disrupt the redox balance of the cell, leading to apoptosis.24 RVFV also encodes a non-structural M protein (NSm) that localizes to the mitochondria and has been associated with the onset of apoptosis in infected cells.25
In the case of New World alphaviruses, infection is known to cause lethal outcomes with cytopathic effects in cells of neuronal origin.10,26,27 There is little information regarding the mechanisms that underlie cell death, aside from caspase activation, in a manner that is dependent on infection. The field of New World alphaviruses has not, thus far, focused on the mitochondria and the influence of infection on mitochondrial dynamics. Our studies initiated with the hypothesis that VEEV infection will result in alterations in mitochondrial dynamics and disruption of membrane potential. We additionally hypothesized that mitochondrial changes will be manifested at the level of intracellular distribution and proteomic composition. To address the impact of VEEV infection on the mitochondria of infected cells, we adopted a combination strategy that included biochemical analysis of mitochondrial membrane potential and mitochondrial composition. We also utilized confocal and electron microscopic methods to study the localization of host and viral components to the mitochondria and alterations in mitochondrial structure. We utilized the TC-83 virus to study the consequences of infection on astrocytoma cells (U87MG cells), as we have demonstrated in the past that TC-83 infection induces several innate immune signaling events and leads to death of infected cells.6,7 Cumulatively, our studies indicate that VEEV infection disrupts mitochondrial structure and function. We demonstrate that mitochondrial fission may partially contribute to apoptosis that occurs in infected cells. Finally, our data suggest that mitophagy is a consequence of VEEV infection in cells that may contribute in an additive manner to the pathology associated with the infection. The outcomes of this study will aid in the understanding of the influence of mitochondria on disease progression and neuronal outcomes such as death of neurons in VEEV infection and in the design of appropriate combinatorial therapeutic strategies that may protect neuronal cells.
Results
VEEV infection results in loss of mitochondrial membrane potential
Viruses including HCV, HIV, RSV and RVFV have an impact on mitochondrial function in infected cells as reflected by an increase in ROS levels that correlate with infection status.17-20,24,28 ROS and malfunctioning mitochondria are important contributors to apoptosis in these infected cells. We hypothesized that VEEV infection would disrupt mitochondrial function which would contribute to apoptosis. We have utilized the human astrocytoma cell line, U87MG, as a model system to understand mitochondrial events that occur in VEEV infected cells. U87MG cells are readily infected by VEEV and display robust viral replication kinetics.6-8,28-30 U87MG cells were infected with the TC-83 strain of VEEV at increasing multiplicities of infection (MOIs). Changes in mitochondrial membrane potential were quantified by labeling active mitochondria with TMRE reagent (TetraMethyl Rhodamine, Ethyl ester). The data demonstrated that TC-83 infection resulted in a decrease in membrane potential at all time points tested, with significant decreases occurring at 6 hpi in the higher MOIs (10 and 20) and at all MOIs tested at 24 hpi when compared with the respective mock controls (Fig. 1A). U87MG cells were infected with RVFV as a virological control (RVFV). It has already been demonstrated that RVFV infection of human lung cells leads to increase in ROS and apoptosis.24 As it was not demonstrated before that RVFV could establish an infection in U87MG cells, we verified that the MP-12 strain of RVFV could infect U87MG cells by quantifying infectious titer (Fig. 1B). This indicates that our virological control can effectively replicate in our U87MG model system. Overall, the results illustrated that TC-83 infection of U87MG cells leads to a drop in mitochondrial membrane potential in an infectious dose-dependent and time-dependent manner.
Figure 1.
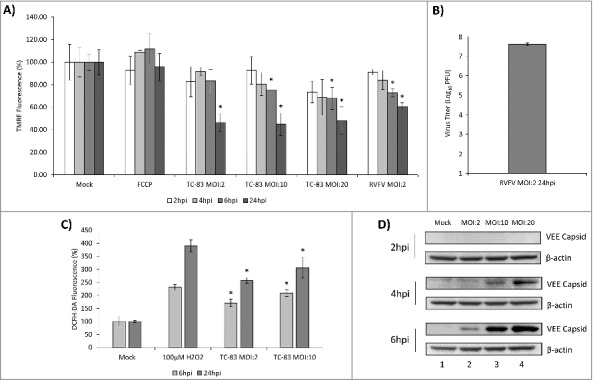
TC-83 infection of U87MG cells resulted in loss of membrane potential and an increase in reactive oxygen species. (A) TMRE fluorescence analysis of uninfected (mock) and TC-83 infected cells indicating increased mitochondrial damage as the result of infection. (B) Validation of the virological control (RVFV) used in (A). Quantitation of extracellular virus via plaque assay indicates successful replication of RVFV in U87MG cells. (C) Accumulation of ROS detected in infected cells using fluorescent DCFH-DA reagent. Data represents an average of two independent experiments performed with triplicate samples. (D) Western blot analysis from uninfected (mock) and TC-83 infected whole cell lysates, probed for VEEV capsid. β-actin, protein-loading control. (A, C) * indicates significance between infected and time matched mock control.
ROS accumulation in cells is a direct reflection of disruption in mitochondrial function. We wanted to determine if TC-83 infection results in accumulation of ROS in the infected cells. In order to obtain a quantitative assessment of ROS levels in the TC-83 infected cells, we conducted an intracellular ROS quantification assay that is based on the oxidation of 2′,7′-dichlorodihydrofluorescein (DCFH-DA) by ROS. The data indicated significant increases in ROS in the TC-83 infected cells as compared to the uninfected cells at all tested MOIs and time points (Fig. 1C). U87MG cells treated with H2O2 demonstrated a robust accumulation of ROS as expected. As an additional control for infection, we determined the abundance of VEEV capsid protein at the MOIs tested (Fig. 1D). Our results indicate an MOI-dependent and time-dependent accumulation of VEEV capsid, which can be detected as early as 4hpi in U87MG cells infected with an MOI of 10 or 20 and is detectable at all MOIs at 6hpi. Cumulatively, our data supported a time-dependent loss of membrane potential and accumulation of ROS in TC-83 infected cells.
Mosquito cells may be less susceptible to mitochondrial dysfunction in the context of TC-83 infection
In the context of mosquito-transmitted virus infections, it has been observed that insect cells possess distinct mechanisms to regulate ROS levels which actually contribute to decreased apoptosis.31-33 It is therefore of relevance to determine if VEEV infection will impact insect cells and lead to accumulation of ROS in a similar manner as we observed in U87MG cells. We studied whether infection of mosquito cells with TC-83 would lead to a disruption of mitochondrial function. C6/36 cells and U87MG cells were infected with TC-83 (MOI:10), and mitochondrial membrane potential was measured at 6 hpi. Measurement of TMRE fluorescence indicated that the mosquito cells did not experience as much of a drop in membrane potential as the U87MG cells (Fig. 2A). Using the DCFH-DA assay, we measured ROS accumulation in the infected C6/36 cell line and determined that unlike the U87MG cell line, there was not a significant increase in ROS (Fig. 2B). We quantified relative viral load in both the C6/36 cells and U87MG cells. While the relative viral load in the C6/36 cells is approximately 1.5 logs lower than the U87MG cells at the early 6-hour time point, the cells support a progressive viral infection as reflected by the increasing viral load at 24 hpi (Fig. 2C). Therefore, our data are suggestive of the idea that mosquito cells may be less susceptible to TC-83 infection-induced disruption of mitochondrial function in comparison to human cells.
Figure 2.
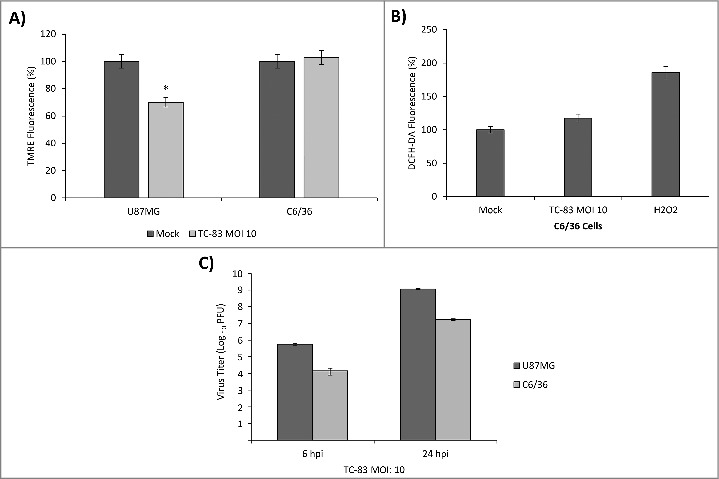
TC-83 infection of C6/36 mosquito cells resulted in no significant loss of membrane potential or increase in reactive oxygen species. (A) TMRE fluorescence analysis of uninfected (mock) and TC-83 infected U87MG cells, and C6/36 mosquito cells indicating no increased mitochondrial damage in the C6/36 cell line. *indicates significance between U87MG infected and mock fluorescence. (B) Non-significant accumulation in ROS detected in infected C6/36 cells using DCFH-DA reagent. Data represents an average of two independent experiments performed with triplicate samples. (C) Extracellular viral titers as determined by plaque assay in U87MG and C6/36 cells.
Mitochondrial distribution is altered in TC-83 infected U87MG cells
Viral infections, such as those caused by HBV, have been shown to cause a perinuclear clustering of mitochondria in an infection-dependent manner.21 A similar redistribution of mitochondria around the nucleus of infected neurons was demonstrated in the case of HSV infection accompanied by increased concentration of viral tegument proteins around the nucleus.34 Interestingly, such a perinuclear clustering of mitochondria was also observed in fibroblasts obtained from Alzheimer's patients, suggestive of a relevance to neurodegenerative conditions.35 We wanted to determine whether TC-83 infection led to alterations in the distribution of mitochondria in infected cells. TC-83 infected U87MG cells were processed for immunofluorescence analysis using anti-TOMM20 (mitochondrial membrane protein) and anti-VEEV capsid antibodies. We observed that in the cells positive for VEEV capsid, the mitochondria appeared to accumulate around the nucleus (Fig. 3A). This was in contrast to the filamentous, disperse localization that was evident in the uninfected cells. We determined that 62% and 69% of infected cells, at 2hpi and 6hpi respectively, displayed the perinuclear clustering phenotype (Fig. 3B). The fragmented, perinuclear phenotype is shown at higher magnification in Fig. 3C to contrast the mitochondrial intracellular distribution in the infected cells with the tubular network in uninfected cells. As part of this analysis, we observed that the VEEV capsid signal partially co-localized with the TOMM20 signal in the perinuclear region, suggesting that the viral capsid protein may also be a component of dysfunctional mitochondria in TC-83 infected cells (Fig. 3D). As an added control, the distribution of VEEV capsid protein relevant to the endoplasmic reticulum (ER) was determined using antibodies to calnexin (Fig. 3E). Our analysis did not reveal any localization of capsid protein in the ER at this time point.
Figure 3.
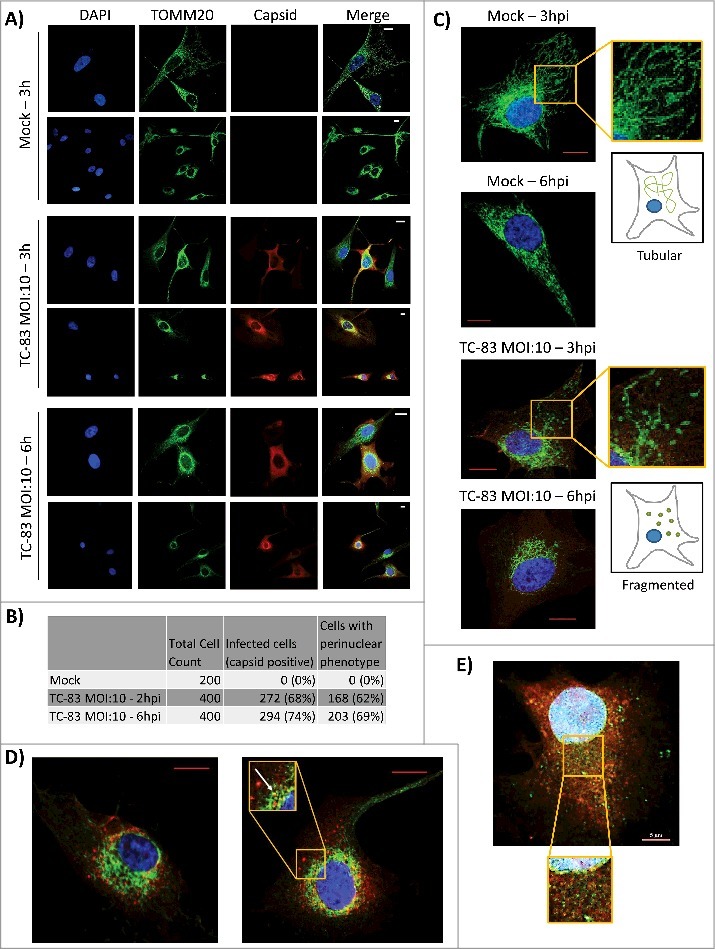
VEEV capsid localized in the mitochondria of infected cells. (A) Confocal images showing the co-localization of VEEV capsid (red) with mitochondrial marker TOMM20 (green). (B) Cell count summary of perinuclear phenotype observed in panel (A). (C) Magnified images of uninfected U87MGs displaying healthy, tubular mitochondria networks, and infected cells displaying a perinuclear clustering of mitochondria. (D). Confocal images showing perinuclear clustering of TOMM20 (green), and partial co-localization with VEEV capsid (red) (see arrow). E) Confocal images showing no co-localization between ER marker calnexin (green) and VEEV capsid (red). Scale bar indicates 5µm. A, C-E) Nuclei stained with DAPI (blue). (A, C-D) Scale bars indicate 10µm.
We adopted a biochemical fractionation method to enrich for mitochondrial membranes as an alternate strategy to determine VEEV capsid association with mitochondria. U87MG cells were infected at increasing MOIs and separated into mitochondrial membrane and cytosolic fractions. As expected, TOMM20 was detected in the WCE (lanes 1–3), enriched in the mitochondrial membrane fractions (lanes 4–6) and absent from the cytosolic fraction (lanes 7–9). VEEV capsid protein was enriched in the mitochondrial fraction obtained from infected cells with a minor 7% increase in the mitochondrial fraction at MOI: 20 versus MOI: 10 (Fig. 4B, lanes 5 and 6). Thus, microscopic and biochemical fraction methods indicate that intracellular VEEV capsid protein could be detected in the mitochondria in TC-83 infected cells.
Figure 4.
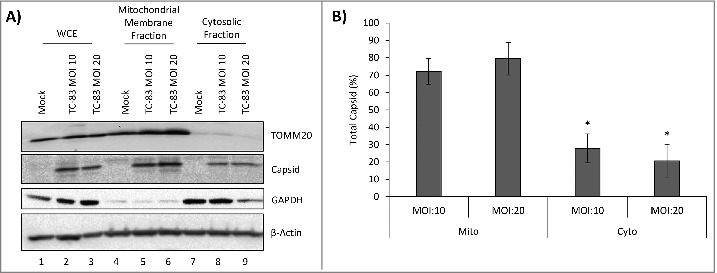
VEEV capsid localized to the mitochondrial membranes of infected cells in an infection dependent manner. (A) Western blot analysis of whole cell extract (WCE) and subcellular fractions from uninfected (mock) and TC-83 infected cells. Mitochondrial marker: TOMM20; cytosolic marker: GAPDH; β-actin: protein loading control. (B) Capsid bands intensities analyzed by Quantity One software. Distribution of VEEV capsid in subcellular fractions graphed as a percentage of total capsid. *indicates significance between mitochondrial and cytosolic quantities.
PINK1 and Parkin localized in mitochondria in TC-83 infected cells
Mitochondrial dysfunction, as observed with viral infection such as HBV and neurodegenerative disorders, is coincident with re-localization of specific host enzymes to the mitochondria. PINK1 is a host kinase that localizes to the mitochondria, where it is stabilized in the context of mitochondrial dysfunction. Localization of PINK1 in the mitochondria leads to the increased retention of Parkin, a ubiquitin ligase, in the mitochondria. Thus, localization of PINK1 and Parkin enzymes in the mitochondria are markers of damaged mitochondria that correspond with elimination of defective mitochondria by mitophagy.21,36-38 We hypothesized that TC-83 infection will induce re-localization of PINK1 and Parkin to the mitochondria. WCE from all samples indicated that TOMM20, PINK1 and Parkin levels were comparable between uninfected and infected cells (Fig. 5A, lanes 1–3). This suggested that if there were differential enrichment of any of these target proteins in the mitochondria of infected cells, it was not the result of differential protein expression. We also independently determined that TC-83 infection did not result in an increase in the total protein levels of PINK1 and Parkin in infected cells by western blot analysis (data not shown). We observed that PINK1 was significantly enriched in the mitochondrial membrane fractions (Figures 5A lanes 4–6, and 5B). Additionally, Parkin recruitment to the mitochondrial membranes was also increased in infected cells (Figures 5A lanes 4–6, and 5B). In contrast to PINK1, Parkin could still be detected in the cytosolic fractions of infected cells (Fig. 5A lanes 7–9). We also studied whether PINK1 and Parkin co-localized with VEEV capsid in infected cells. Immunofluorescent analysis of infected cells revealed that PINK1 co-localized with VEEV capsid in 47% of TC-83 infected cells (Fig. 5C), while Parkin co-localized with VEEV capsid in 13% of infected cells (Fig. 5D). Cumulatively, our microscopic and biochemical fractionation studies demonstrated that TC-83 infection resulted in re-localization of PINK1 and Parkin to mitochondrial membranes, with a partial co-localization of these host proteins with VEEV capsid.
Figure 5.
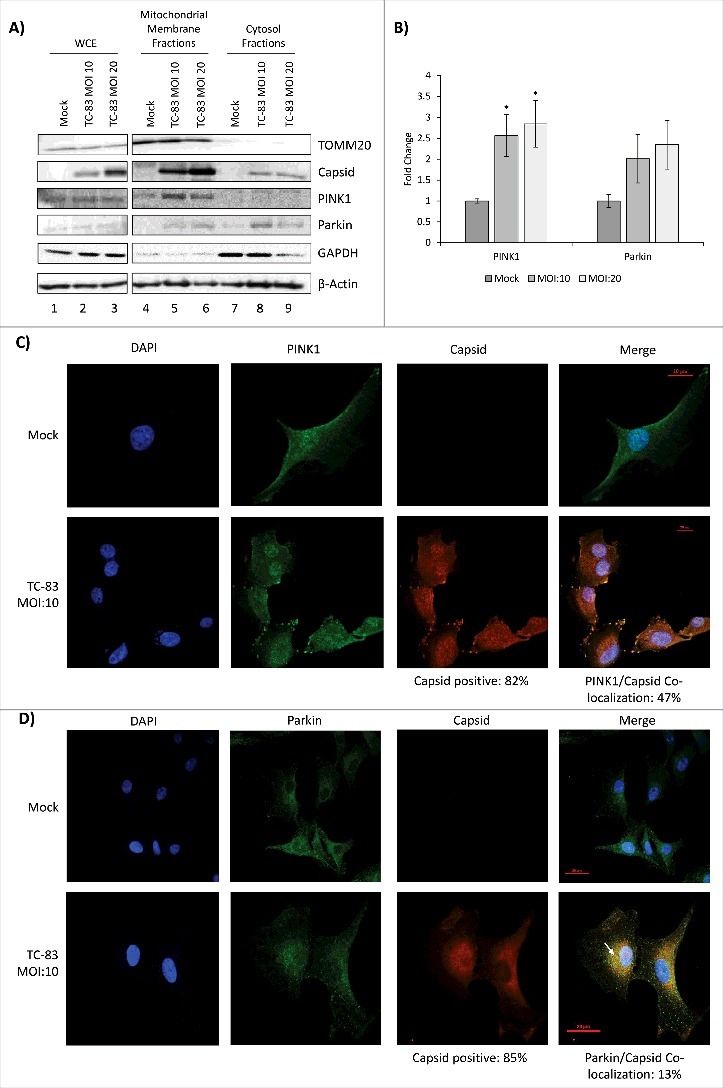
PINK1 and Parkin co-localize in the mitochondrial membrane of TC-83 infected cells. (A) Western blot analysis of whole cell extract (WCE) and subcellular fractions from uninfected (mock) and TC-83 infected cells. Infection control: capsid; mitochondrial marker: TOMM20; cytosolic marker: GAPDH; β-actin: protein loading control. (B) PINK1 and Parkin mitochondrial membrane band intensities analyzed by Quantity One software. * indicates significance between infected and mock PINK1 quantities. (C) Confocal images showing co-localization of PINK1 (green) and VEEV capsid (red). (D) Confocal images showing partial co-localization of Parkin and VEEV capsid (red) (C-D) Nuclei stained with DAPI (blue). Scale bars indicate 20µm.
Increased phosphorylation of Drp1 protein can be detected in infected cells
An important marker of mitochondrial fission is an increase in phosphorylation of Drp1 protein on Serine 616 (pDrp1-Ser616), as demonstrated in HBV infection.21 We wanted to determine if Drp1 protein displayed phosphorylation of Serine 616 in TC-83 infected cells. Analysis of total protein extracts revealed a modest increase in pDrp1-Ser616 in TC-83 infected cells (Fig. 6A). This increase was noted as early as 2hpi, suggesting that very early changes in mitochondrial architecture occur in infected cells. We also wanted to determine if Drp1 phosphorylation was a dynamic feature that changed with MOI. To that end, we observed a progressive increase in Drp1-Ser616 phosphorylation between MOI: 5 and 10, while at MOI: 20, phosphorylation of Drp1 was slightly decreased (Fig. 6B). This behavior of phosphorylated residues as noted in Fig. 6A and 6B is consistent with transient phosphorylation modification of target proteins by host kinases, which follow temporal patterns of phosphorylation and dephosphorylation and may reflect the possibility of a downward trend in a bell shaped curve. The phosphorylation of Drp1-Ser616 in infected cells was quantified based on three independent experiments which suggested a statistically significant increase in infected cells (Fig. 6C). We also studied if any increase in mitochondrial localization of pDrp1-Ser616 could be observed in TC-83 infected cells. While no significant difference could be detected in the mitochondrial localization of total Drp1 between infected and uninfected cells, there was a modest 51% increase observed in the mitochondrial localization of pDrp1-Ser616 in infected cells (Fig. 6D, lanes 3–4).
Figure 6.
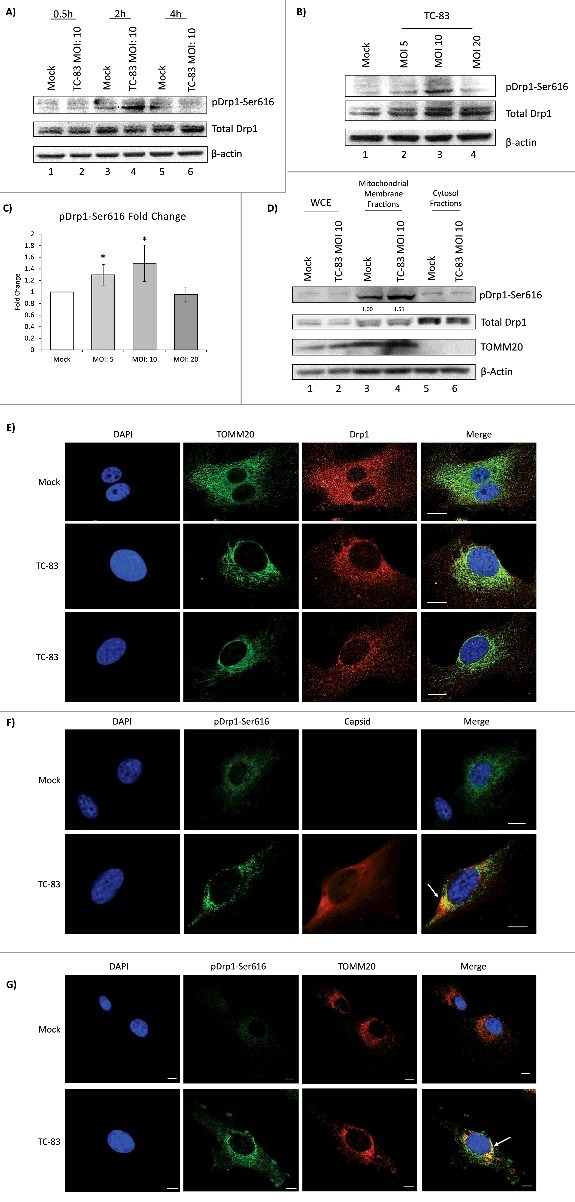
Increased phosphorylation of Drp1 protein in TC-83 infected cells. (A) Western blot analysis of time course investigating pDrp1-Ser616 and total Drp1 in uninfected (mock) and TC-83 infected cells. (B) Western blot analysis of whole cell extracts collected at 2hpi and probed for pDrp1-Ser616 and total Drp1. (C) pDrp1-Ser616 band intensities from (B). * indicates significance between infected and mock pDrp1-Ser616 quantities. (D) Western blot analysis of whole cell extract (WCE) and subcellular fractions from uninfected (mock) and TC-83 infected cells. Mitochondrial marker: TOMM20. A-B, (D) β-actin: protein loading control. (C-D) Band intensities were analyzed by Quantity One software. (E) Confocal images showing perinuclear clustering of TOMM20 (green), is associated with similar perinuclear redistribution of total Drp1 (red). (F) Confocal images showing partial co-localization of pDrp1-Ser616 (green) with VEEV capsid (red). (G) Confocal images showing co-localization of pDrp1-Ser616 (green) with TOMM20 (red). (E-G) Nuclei stained with DAPI (blue). Scale bars indicate 10µm.
We hypothesized that the Drp1 localization pattern in infected cells would change in a manner that parallels the perinuclear clustering of mitochondria. Using confocal microscopy, we observed that in uninfected cells, Drp1 showed a diffuse cytoplasmic localization pattern (Fig. 6E, top panel). In TC-83 infected cells, more Drp1 was localized to the perinuclear regions in a pattern similar to TOMM20 (Fig. 6E, bottom panels). A similar study was conducted to investigate the intracellular distribution of pDrp1-Ser616 (Fig. 6F-G). Uninfected cells displayed a diffuse pattern of pDrp1-Ser616 similar to total Drp1 in Fig. 6E (top panel). In the case of TC-83 infected cells, phosphorylated Drp1-Ser616 formed more punctate foci, partially co-localizing with VEEV capsid (Fig. 6F, bottom panel) and TOMM20 (Fig. 6G, bottom panel). Thus, our data suggest that TC-83 infection results in an early increase in phosphorylation of Drp1 on Serine 616, leading to a modest increase in pDrp1-Ser616 at the mitochondrial membrane. This event most likely indicates an increase in mitochondrial fission in TC-83 infected cells.
Mitochondria display structural abnormalities in TC-83 infected cells
We performed Transmission Electron Microscopy (TEM) analysis of U87MG cells infected with TC-83 to determine whether infection could lead to changes in the mitochondrial structure. Uninfected cells displayed what appeared to be normal mitochondria with intact internal membranes and cristae (Fig. 7A). In infected cells, mitochondria displayed a range of abnormalities in structure, including partially swollen mitochondria (Fig. 7B), internal membrane compacting (Fig. 7C) and congregated mitochondria (Fig. 7D). In the case of internal membrane compacting, we observed membranous structures that had no apparent connection to the outer membranes. These compact inner structures, which had a blebbed, membrane-like architecture, were not detected in uninfected cells and were larger than traditional granules. Collectively, our TEM studies revealed alterations in the structures of mitochondria in TC-83 infected cells.
Figure 7.
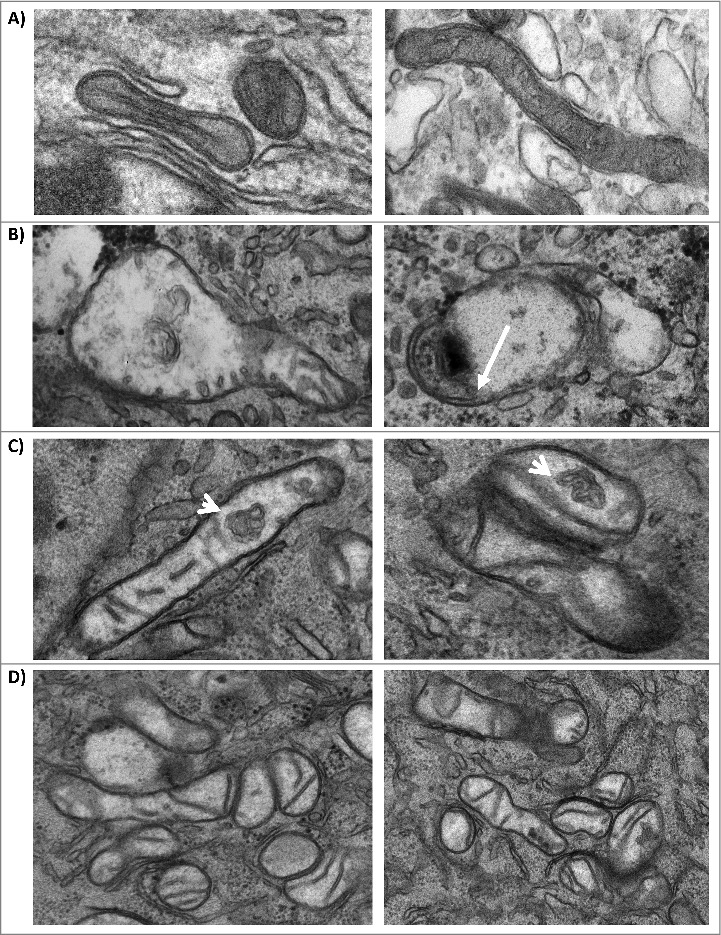
Mitochondria display structural alterations in infected cells. (A) TEM images of uninfected U87MG cells. TEM images of TC-83 infected cells displaying: (B) heterogeneously swollen mitochondria; (C) structural alterations, including membranous structures in the interior of mitochondria (arrows) that did not appear to have connections with the inner cristae; and (D) mitochondrial clusters.
Mitochondrial fission contributes to apoptosis in TC-83 infected cells
We questioned whether mitochondrial fission could play a role in the establishment of a productive TC-83 infection in U87MG cells. To that end, we tested whether Mdivi-1, an inhibitor of mitochondrial fission, had an inhibitory effect on TC-83 multiplication. At concentrations up to 10µM, Mdivi-1 treatment did not induce U87MG cell death (Fig. 8A). To determine if Mdivi-1 could inhibit TC-83 multiplication, U87MG cells were pre-treated with Mdivi-1 prior to infection. Bortezomib was included as a positive control of TC-83 multiplication inhibition, as we previously demonstrated that Bortezomib exerts a robust inhibitory effect.8 Our data indicated that Mdivi-1, an inhibitor of mitochondrial fission, did not have any inhibitory effect on TC-83 multiplication (Fig. 8B).
Figure 8.
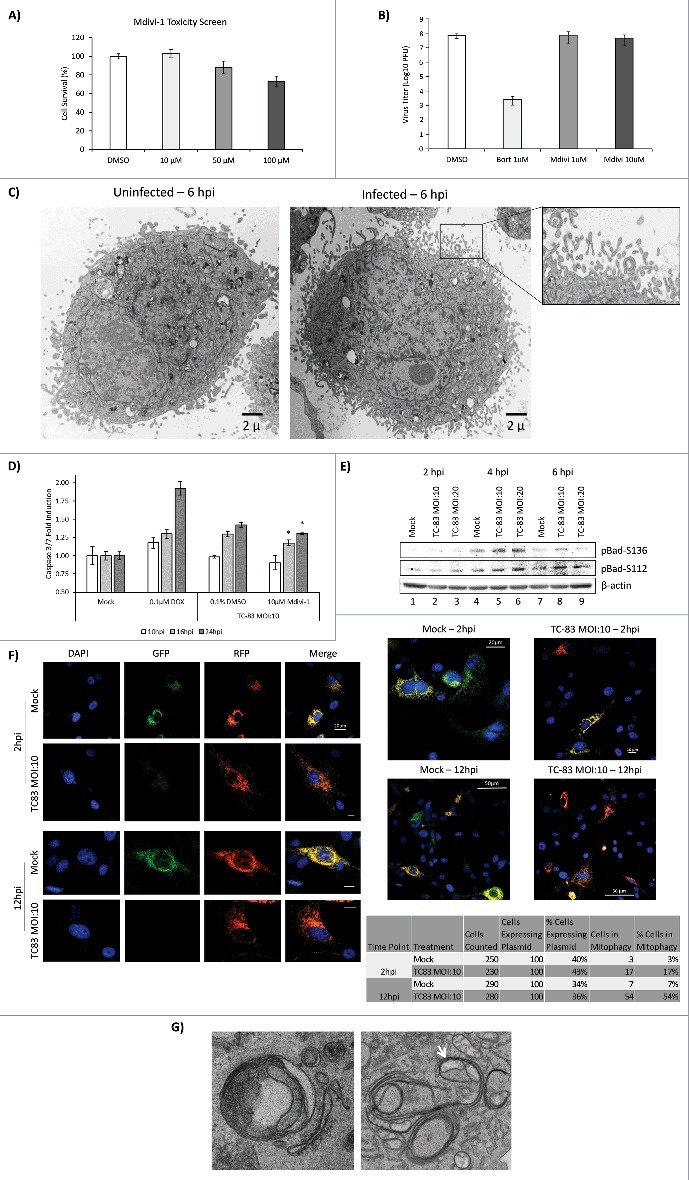
Mitochondrial fission contributes to apoptosis in TC-83 infected cells. (A) Cell survival, as determined by CellTiter Glo luminescence, following treatment with Mdivi-1. (B) Plaque assay quantitation of extracellular virus following treatment of cells with either Bortezomib (Bort) or Mdivi-1. (C) TEM images uninfected and TC-83 infected cells showing blebbing of the plasma membrane only in infected cells. (D) Induction of Caspase 3/7 measured in uninfected (mock), doxorubicin (DOX) treated, and TC-83 infected cells. * indicates Mdivi-1 treatment result in a statistically significant decrease in Caspase 3/7 induction vs. the untreated TC-83 control. (E) Western blot analysis of whole cell extracts obtained from uninfected (mock) and TC-83 infected cells. β-actin: protein loading control. (F) Confocal images showing TC-83-induced mitophagy. Fluorescence signals indicate the expression of mito-mRFP-EGFP targeting mitochondria: yellow, no mitophagy; red, mitophagy. Scale bars indicate 10µm, unless otherwise noted. (G) TEM images of mitochondria in TC-83 infected cells showing enclosure of mitochondria in bilayer membrane structures within the cell.
The New and Old World Alphaviruses have previously been reported to increase apoptosis in an infection-dependent manner.39-41 In the case of VEEV infection, apoptosis is partially mediated by an increase in caspase 3/7 cleavage.29,42,43 As part of our TEM analysis, we observed increased plasma membrane blebbing in TC-83 infected cells, which is consistent with increase in apoptosis (Fig. 8C).44 Therefore, we wanted to test whether an inhibition of mitochondrial fission would decrease caspase 3/7 cleavage and thus decrease apoptosis. We then performed a fluorometric analysis of caspase 3/7 cleavage in TC-83 infected cells in the context of Mdivi-1 treatment (Fig. 8D). Doxorubicin (DOX), a robust inducer of caspase 3/7 cleavage, was used as a positive control. We observed a statistically significant decrease in caspase cleavage in Mdivi-1 treated cells at 16hpi and 24hpi, indicating that mitochondrial fission may, in part, be contributing to VEEV-mediated apoptosis. To further investigate apoptosis driven by mitochondrial events in the context of TC-83 infection, we probed whole cell lysates from infected cells for two phosphorylated members of the B-cell leukemia 2 (Bcl-2) pro-apoptotic family: pBad-Ser112 and pBad-Ser136 (Fig. 8E). We observed an increase in Bad-Ser136 phosphorylation at 4hpi, with an apparent bell-curve, MOI-dependent decrease in phosphorylation noted at 6hpi (Fig. 8E, compare lanes 4–6 and 7–9). We also observed a modest increase in Bad-Ser112 phosphorylation at 6hpi (Fig. 8E, lanes 7–9).
Finally, we wanted to evaluate if TC-83 infection would result in increased mitophagy in infected cells. We reasoned that with the extent of mitochondrial alterations that we have observed in infected cells, including recruitment of PINK1 and the disruption of mitochondrial function, it is likely that mitophagy will ensue.45 To evaluate if mitochondria go through mitophagy, we used a fluorescence-based reporter assay that was previously used to demonstrate that mitophagy occurs in HCV infected cells.22 This reporter plasmid, pAT016, is a monomeric red fluorescent protein (mRFP)-enhanced green fluorescent protein (EGFP) chimeric fluorescence reporter that encodes a mitochondrial targeting signal sequence fused in-frame with mRFP and EGFP genes. The rationale behind the utility of this plasmid is that EGFP is less stable in the acidic environment of the phagosomes that engulf mitochondria than the mRFP. Therefore, in the event that mitophagy occurs, the EGFP signal would be lost or attenuated, resulting in only the mRFP signal being observed via fluorescent microscopy. Our data indicate a powerful onset of mitophagy with 54% of infected, plasmid expressing cells displaying a remarkable reduction in EGFP fluorescence and unaffected mRFP signals (Fig. 8F). To quantify relative transfection rates of the plasmid, we counted the number of cells that displayed mRFP signal. We determined the average transfection efficiency to be 37% in uninfected cells and 39% in infected cells, thus indicating a relatively equivalent representation of the reporter plasmid. As part of our TEM studies, we also observed instances where mitochondria were enclosed within membranes in infected cells, which could be indicative of mitophagy (Fig. 8G). Thus, our data cumulatively indicate that mitochondrial fission is a preliminary event that subsequently contributes to apoptosis in the infected cell without having a direct impact on the establishment of a productive infection. Our data also support the potential of mitophagy being a downstream consequence of altered mitochondrial dynamics and functionality in TC-83 infected cells.
Discussion
New World alphaviruses including VEEV are encephalitic viruses that infect humans as a result of bites by infected mosquitoes. In cases where VEEV infection leads to encephalitis, the encephalitic outcome is accompanied by death of neurons and supporting cells including astrocytes. VEEV infection is well documented to result in cytopathic effects (CPE) in a capsid protein-dependent manner. CPE outcomes in alphavirus infected cells may also be a consequence of transcriptional and translational inhibition induced by the capsid protein.10,27 The mitochondria are sentinel organelles involved in many essential cellular functions including production of energy, innate immune signaling and deciding cellular fate. Mitochondrial membrane potential is a cell type independent indicator of mitochondrial integrity and functional competence. In a healthy cell, membrane potential is maintained due to a functional mitochondrial respiratory chain and oxidative phosphorylation. As a part of the energy production process, ROS is produced in normal cells as well. However, pathogenic conditions that may result in disruption of mitochondrial membrane potential will impact the integrity of the electron transport chain and result in an abnormal accumulation of ROS. Thus, a deviation in the mitochondrial membrane potential and accumulated ROS are reliable indicators of interruption of mitochondrial function. We have observed a decrease in mitochondrial membrane potential that is also accompanied by a prominent increase in ROS in human astrocytoma cells infected with the TC-83 strain of VEEV (Fig. 1).
Many viral infections including HIV and HSV have been recognized as being causative of oxidative stress phenotypes.46,47 The observed changes in mitochondrial membrane potential and ROS accumulation may be early events that set the stage for apoptosis in VEEV infections through the intrinsic pathway. Notably, this phenotype was observed when cells were infected with the TC-83 strain of VEEV, which is an attenuated strain provided as a vaccine to at-risk personnel. TC-83 is plagued by reactogenicity concerns because it is not approved for public use. It may be an important safety consideration to ensure that disruption of mitochondrial function is not associated with the reactogenic phenotype. It will also be of interest that the virulent strains of VEEV such as Trinidad Donkey (TrD) strain may result in an exaggerated oxidative stress with different kinetics in comparison to the TC-83 strain. Such differences in oxidative stress mechanisms may be early discriminators of virulence that establish kinetics of host inflammatory responses in early stages of post-exposure.
We observed that the production of ROS and impact of infection on mitochondrial membrane potential was lower in mosquito cells when compared to human cells in a manner that corresponded with viral load in the infected cell (Fig. 2). This data may indicate that mosquito cells may be relatively more protected from mitochondrial dysfunction due to VEEV infection when compared to human cells. It has been demonstrated that in the case of flavivirus infections, infected insect cells show changes in antioxidant mechanisms that decrease the extent of cell death.31-33 It has been demonstrated for Dengue virus that infected mosquito cells mounted a stronger antioxidant response by synthesising antioxidant enzymes (elevation of Glutathione S-transferase activity).48
Mitochondrial distribution in cells and mitochondrial mobility are essential features of healthy cells. Neuronal cells in particular are notably impacted by mitochondrial distribution between the cell body and axon. Anterograde transport of mitochondria in neurons is mediated by kinesin motors on microtubules, while retrograde transport is mediated by dynein motors. In HSV infections, it was demonstrated that anterograde transport of mitochondria was disrupted due to impaired association of mitochondria with the kinesin motor.49 This resulted in severe impairment of mitochondrial mobility in an infection-dependent manner. Mitochondrial congregation in a perinuclear manner has been indicated to correspond with oxidative stress in many neurodegenerative states.50 We have observed a prominent perinuclear clustering of mitochondria in TC-83 infected cells (Fig. 3). In the context of neurons, this will have a direct impact on neuronal functionality, which may also contribute to neuronal death and encephalitis in VEEV infection. Viral protein localization in infected cells is a well-documented process for many viruses that directly influence innate immune responses of the host. An excellent example of this phenomenon is RSV nonstructural protein 1 (NS1) which interacted with mitochondrial antiviral signaling protein (MAVS) and interrupted RIG-I based signaling.23 Rubella virus is an RNA virus belonging to the family Togaviridae whose capsid protein localized to the mitochondria in a manner based on post translational modification.51 During the course of our studies on mitochondrial distribution in infected cells, we observed that VEEV capsid partially localized in the mitochondria as confirmed by microscopic and biochemical methodologies (Figs. 3 and 4). Our preliminary studies have suggested that while transfection of a plasmid that encodes VEEV capsid can produce similar phenotypes in astrocytoma cells, the extent of mitochondrial dysfunction is lesser than that observed in infected cells, indicating that multiple viral components may contribute to the observed mitochondrial events (data not shown). Similar outcomes were demonstrated for HCV where the viral core protein was shown to indicate with host enzymatic components that localized to mitochondria.21
Of interest to us was the observation that the host kinase PINK1 and ubiquitin ligase Parkin localized in mitochondria of infected cells (Figs. 5 and 6). PINK1 and Parkin have generated significant interest in recent years for their association with mitochondrial dysfunction in neurodegeneration. Of direct relevance to mitochondrial motility, PINK1 activity is known to phosphorylate Miro, a connecting link between mitochondria and kinesin. In addition, Parkin ubiquitinates Miro, which ultimately leads to its degradation. PINK1 is also known to phosphorylate Parkin and contribute to its ubiquitination function.52 Thus, a combined action of phosphorylation and ubiquitination of inherent mitochondrial proteins such as Miro will have a direct impact on mitochondrial intracellular distribution.53,54 We have recently demonstrated that VEEV capsid exists in an ubiquitinated state in U87MG cells.8 It would be interesting to determine if capsid protein in mitochondria exists in an ubiquitinated state and if such an event is mediated by Parkin. Indeed, this has been demonstrated in the case of Rubella virus capsid protein, a post-translationally modified viral core protein, specifically the phosphorylated form, associated with mitochondrial p32 protein.51,55
Such indications as described thus far usually lead to downstream compensatory mechanisms initiated by the host cell, directed toward removing damaged mitochondria. This is an integral part of maintaining cell survival while the host contends with cellular stress. These mechanisms may include mitochondrial fission or fusion, in which damaged parts of mitochondria are excised and smaller mitochondria fuse to create larger mitochondria.56,57 Drp1 is a pivotal protein in mediating mitochondrial fission as it has the capacity to constrict mitochondria. In the context of HCV infection, it has been demonstrated that mitochondrial localization of phosphorylated form of Drp1 protein (Drp1-Ser616) was a prelude to mitochondrial fission.22 Not only did we observe phosphorylated Drp1 localizing to mitochondrial membranes, but we also noticed that Drp1 was redistributed in a manner that mirrored mitochondrial accumulation around the nucleus (Fig. 6). Our observation that phosphorylated Drp1-Ser616 localized to mitochondria in a transient manner is suggestive of a dynamic relationship between Drp1 localization, mitochondrial fission and VEEV infection.
Transmission electron microscopic analysis of TC-83 infected cells showed changes in mitochondrial structure (Fig. 7). In a recent publication on mitochondrial changes induced in parvoviral infection, the authors include similar observations on mitochondrial dysfunction including drop in membrane potential and accumulation of ROS as we have indicated here. The authors provide electron microscopic visuals of abnormal mitochondrial phenotypes that are similar to the ones we have included in our results including heterogeneously swollen mitochondria, membrane blebbing and disappearance of cristae.58 Notably, the authors indicate activation of MEK/ERK signaling resulting from mitochondrial dysfunction in Parvovirus infection. We have previously demonstrated that VEEV infection leads to robust activation of the MEK/ERK signaling cascade, which is important for the virus to establish a productive infection.7 In the case of HCV infected cells, similar clustering of mitochondria was demonstrated.59 In HIV infected cells, mitochondria displayed morphological alterations including disappearance of cristae.60
Our studies led us to question whether mitochondrial fission was relevant to viral multiplication. Our studies with the inhibitor Mdivi-1 indicated that mitochondrial fission does not contribute to viral load in infected cells (Fig. 8B). It has been reported that viruses do, in fact, utilize the ROS rich environment of infected cells in a manner that may favor aspects of viral multiplication including RNA synthesis. In the case of flaviviruses, it was shown that the guanylyltransferase activity of NS5 encoded polymerase was enhanced by oxidative conditions.61 It remains to be determined if there are specific aspects of mitochondrial dysfunction that may be controlled by antioxidant treatment, which may have important implications in protecting terminally differentiated neurons in the infected host. Our follow up hypothesis was that mitochondrial fission may contribute to the ensuing apoptosis in infected cells. Our TEM studies revealed cellular morphology that was consistent with exaggerated membrane blebbing and apoptosis. When treated with an inhibitor of mitochondrial fission, we observed a statistically significant reduction in caspase cleavage, thus adding support to our suggestion that mitochondrial fission was a contributor to apoptosis of VEEV-infected cells.
Mitophagy is a mitochondrial elimination process in which defective mitochondria are enveloped in lipid bilayers to form autophagosomes that contain whole mitochondria. Such mitochondrial engulfment by the phagosomes leads to selective elimination of damaged mitochondria, thus decreasing the mitochondrial dysfunction load as a means of restoring cellular health. We observed that mitochondrial alterations in VEEV-infected cells contributed to mitophagic elimination of mitochondria. In our TEM images, we also observed that there were multiple mitochondria in infected cells that were associated with encircling membranes.
Taken together, our data suggest that VEEV infection produces a significant impact on mitochondrial dynamics in infected cells. The onset of mitochondrial dysfunction during early stages of VEEV infection may set the stage for downstream events that culminate in neuronal death. Mitochondrial dysfunction may be associated in a cause and effect manner with pronounced alterations to the mitochondrial proteome; a deeper understanding of which will shed more light on the mitochondrial influence on disease progression in New World alphavirus infections. Notably, mitochondria as a therapeutic target is being explored for different neurodegenerative disorders including optic neurodegeneration.62,63 Targeted, rationally designed combinatorial therapeutic strategies that can target infected cells and deplete viral load while rescuing mitochondrial health will be essential steps in ensuring recovery from New World alphavirus infections.
Materials and methods
Viruses and cell lines
The live-attenuated virus (TC-83) and C6/36 cells used in this study were obtained from BEI Resources. TC-83 attenuation from the fully virulent Trinidad Donkey (TrD) strain has been previously characterized.8,64 The MP-12 strain of RVFV was obtained by 12 serial passages of the virulent ZH548 virus in the presence of 5-fluorouracil, resulting in a total of 25 mutations that have been previously characterized.65,66 Human astrocytoma cells (U87MG cells) and African green monkey kidney cells (Veros) were maintained in DMEM supplemented with 10% Fetal Bovine Serum (FBS), 1% Penicillin/Streptomycin, and 1% L-Glutamine at 37°C and 5% CO2.
Viral infections and plaque assays
Cells were seeded in either a 96-well plate or an 8-well chamber slide, at the indicated concentration for each assay, in order to attain confluency within 24 hours. The media was removed and saved, and thereafter referred to as conditioned media. The cells were infected for 1 hour to allow for viral adsorption at 37°C, 5% CO2. The viral inoculum was then removed and replaced with the conditioned media. The cells were incubated at 37°C, 5% CO2 for the time period indicated in each experiment. Plaque assays were performed as previously described.8
TMRE assay
TMRE is a cell-permeable reagent that preferentially accumulates in active mitochondria. The TMRE assay was obtained from Abcam and performed according to manufacturer's instructions. TMRE experimental controls included carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (Abcam), a potent mitochondrial oxidative phosphorylation un-coupler, and RVFV, a virus previously shown to induce mitochondrial stress.24 U87MG cells were seeded in a black 96-well plate at a density of 10,000 cells per well. Cells were either uninfected (mock), infected with TC-83 (MOI: 2, 10 or 20), or infected with MP-12 (MOI: 2). FCCP (50µM) was diluted in complete medium and added to mock cells which where incubated at 37°C, 5% CO2 for 15 minutes. At 2, 4, 6, and 24 hpi, the media was removed and cells were treated with 1µM TMRE reagent diluted in complete medium. Cells incubated for 20 minutes at 37°C, 5% CO2 prior to reading fluorescence (Ex/Em 549/575 nm) using a DTX 880 multimode detector (Beckman Coulter).
Intracellular reactive oxygen species quantitation assay
The OxiSelect Intracellular ROS Assay Kit (Cell Bio Labs) was used according to the manufacturer's instructions. This assay utilizes 2′,7′-dichlorodihydrofluorescein (DCFH-DA), which is rapidly oxidized to the fluorescent DCF that can be measured by spectrophotometric methods. U87MG cells were seeded at 10,000 cells per well in a black 96-well plate. The cells were then either left uninfected, infected with TC-83 (MOI: 2 or 10), or treated with 100μM H2O2. Fluorescence intensity (Ex/Em 480/530nm) was measured at 6 and 24 hpi using a DTX 880 multimode detector (Beckman Coulter).
Mitochondrial and protein extractions, western blotting
Mitochondrial membrane and cytosolic fractions were obtained using the Mitochondrial Extraction Kit for Cultured Cells (Thermo Scientific) according to the manufacturer's instructions. Preparation of whole cell lysates and western blot have been previously described.6 Primary antibodies to VEEV Capsid (BEI Resources), TOMM20 (Abcam), PINK1 (Santa Cruz Biotechnology), Parkin (Abcam), pDrp1-Ser616 (Cell Signaling), total Drp1 (Cell Signaling), GAPDH (Cell Signaling) and Horseradish Peroxidase (HRP)-conjugated β-actin (Thermo Scientific) were used according to the manufacturer's instructions. Bands of interest were quantitated using Quantity One software (Bio-Rad), a ChemiDoc XRS system (Bio-Rad) and normalized to β-actin signals.
Transfections and DNA constructs
Transfections were performed using Attractene Transfection Reagent (Qiagen) as per the manufacturer's instructions. The p-mito-mRFP-EGFP plasmid was a kind gift of Dr. Siddiqui (University of California, San Diego), which was generated as previously described.67
Confocal microscopy
Immunofluorescence assays were performed as previously described.8 Paraformaldehyde fixed cells were mounted using Fluoromount-G, containing 4′,6-diamidino-2-phenylindole (DAPI) (Southern Biotech) to visualize the nucleus. Fluorescent secondary antibodies labeled with Alexa Fluor 488 and 568 (Thermo Fisher) were used to visualize proteins of interest.
Electron microscopy
After infection, samples were washed three times with 0.5M 2-(N-morpholino) ethanesulfonic acid (MES) buffer. Subsequently, samples were fixed with 2.5% glutaraldehyde /2% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 hours at room temperature. The fixative was replaced with a 0.1M cacodylate buffer and samples were post-fixed with 1% Osmiumtetroxide (OsO4)/1.5% Potassiumferrocyanide (KFeCN6) for 1 hour. Samples were then washed with ultra-pure water three times. Samples were incubated in 1% aqueous uranyl acetate for 1 hour, followed by 2 water washes and subsequent dehydration in grades of alcohol (10 min each: 50%, 70%, 90%; 2, 10 min washes: 100%). The samples were then treated with propyleneoxide for 1 hour and infiltrated in a 1:1 mixture of propyleneoxide and TAAB Epon. The following day the samples were embedded in TAAB Epon and polymerized at 60°C for 48 hours. Ultrathin sections (60nm) were cut on a Reichert Ultracut-S microtome, adhered to copper grids stained with lead citrate and examined in a JEOL 1200EX Transmission electron microscope. Images were recorded with an AMT 2k CCD camera. Mitochondrial images were obtained at a magnification of 25,000X, while whole cell images were obtained at a magnification of 2,500X.
Cell viability assay
Viability for cells treated with Mdivi-1 was determined using the CellTiter Glo assay (Promega). Mdivi-1 was dissolved in dimethyl sulfoxide (DMSO) prior to dilution in complete media. Treated media was overlaid on U87MG cells in a white-walled 96-well plate, which was incubated at 37°C, 5% CO2 for 24 hours, prior to addition of the CellTiter Glo substrate according to the manufacturer's instructions. Luminescence was determined using the DTX 880 multimode detector (Beckman Coulter) with an integration time of 100ms/well.
Caspase 3/7 Glo assay
U87MG cells were pre-treated with either 0.1µM Doxorubicin (DOX), 0.1% DMSO, or 10µM Mdivi-1 for 2 hours prior to infection with TC-83 at MOI: 10. Caspase activation was measured at 10, 16, or 24 hours using the Caspase 3/7 Glo assay (Promega) according to the manufacturer's protocol. Caspase 3/7 fluorescence values were adjusted for cell survival, as determined by CellTiter Glo assay (Promega). Luminescence was determined using the DTX 880 multimode detector (Beckman Coulter) with an integration time of 100ms/well.
Statistical analysis
Unless otherwise stated, graphs and images are the average of three biologically independent experiments. Standard deviations were calculated using Microsoft Excel and represented where applicable. Statistical significance was tested by unpaired, two-tailed Student's t-test between the sample data and time-matched control. The limit for statistical significance was set at P < 0.05, unless otherwise indicated.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The following reagents were obtained through the National Institutes of Health (NIH) Biodefense and Emerging Infections Research Resources Repository, National Institute of Allergy and Infectious Diseases (NIAID), NIH: (i) Venezuelan Equine Encephalitis Virus, TC-83 (Subtype IAB), NR63, (ii) Polyclonal Anti-Venezuelan Equine Encephalitis Virus, TC-83 (Subtype IA/B) Capsid Protein (antiserum, Goat), NR-9403. The mitophagy reporter plasmid pAT016 was generously provided by Dr. Aleem Siddiqui.
Funding
The microscopy studies were supported by CSM start-up fund (# 162904). The phosphorylated Bad antibodies were a kindly provided by Dr. Emanuel Petricoin III.
References
- [1].Weaver SC. Host range, amplification and arboviral disease emergence. Arch Virol Suppl 2005; 19(19):33-44; PMID:16358422; https://doi.org/ 10.1007/3-211-29981-5_4 [DOI] [PubMed] [Google Scholar]
- [2].Taylor Katherine G, Paessler S. Pathogenesis of venezuelan equine encephalitis. Vet Microbiol 2013; 167(1–2):145-50; PMID:23968890; https://doi.org/ 10.1016/j.vetmic.2013.07.012 [DOI] [PubMed] [Google Scholar]
- [3].Weaver SC, Salas R, Rico-Hesse R, Ludwig GV, Oberste MS, Boshell J, Tesh RB. Re-emergence of epidemic venezuelan equine encephalomyelitis in south america. The Lancet 1996; 348(9025):436-40; https://doi.org/ 10.1016/S0140-6736(96)02275-1 [DOI] [PubMed] [Google Scholar]
- [4].Weaver Scott C, Ferro C, Barrera R, Boshell J, Navarro J-C. Venezuelan equine encephalitis. Annu Rev Entomol 2004; 49:141-74; PMID:14651460; https://doi.org/ 10.1146/annurev.ento.49.061802.123422 [DOI] [PubMed] [Google Scholar]
- [5].Paessler S, Weaver SC. Vaccines for venezuelan equine encephalitis. Vaccine 2009; 27(4):D80-85; PMID:19837294; https://doi.org/ 10.1016/j.vaccine.2009.07.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Amaya M, Voss K, Sampey G, Senina S, de la Fuente C, Mueller C, Calvert V, Kehn-Hall K, Carpenter C, Kashanchi F, et al.. The role of IKKβ in venezuelan equine encephalitis virus infection. PloS One 2014; 9(2):e86745; PMID:24586253; https://doi.org/ 10.1371/journal.pone.0086745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Voss K, Amaya M, Mueller C, Roberts B, Kehn-Hall K, Bailey C, Petricoin E 3rd, Narayanan A. Inhibition of host extracellular signal-regulated kinase (ERK) activation decreases new world alphavirus multiplication in infected cells. Virology 2014; 468–470:490-503; PMID:25261871; https://doi.org/ 10.1016/j.virol.2014.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Amaya M, Keck F, Lindquist M, Voss K, Scavone L, Kehn-Hall K, Roberts B, Bailey C, Schmaljohn C, Narayanan A. The ubiquitin proteasome system plays a role in venezuelan equine encephalitis virus infection. PloS One 2015; 10(4):e0124792; PMID:25927990; https://doi.org/ 10.1371/journal.pone.0124792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Amaya M, Brooks-Faulconer T, Lark T, Keck F, Bailey C, Raman V, Narayanan A. Venezuelan equine encephalitis virus non-structural protein 3 (nsP3) interacts with RNA helicases DDX1 and DDX3 in infected cells. Antiviral Res 2016; 131:49-60; PMID:27105836; https://doi.org/ 10.1016/j.antiviral.2016.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Atasheva S, Krendelchtchikova V, Liopo A, Frolova E, Frolov I. Interplay of acute and persistent infections caused by venezuelan equine encephalitis virus encoding mutated capsid protein. J Virol 2010; 84(19):10004-15; PMID:20668087; https://doi.org/ 10.1128/JVI.01151-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ernster L, Schatz G. Mitochondria: a historical review. J Cell Biol 1981; 91(3):227s-255s; PMID:7033239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 2006; 125(7):1241-52; PMID:16814712; https://doi.org/ 10.1016/j.cell.2006.06.010 [DOI] [PubMed] [Google Scholar]
- [13].McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16(14):R551-60; PMID:16860735; https://doi.org/ 10.1016/j.cub.2006.06.054 [DOI] [PubMed] [Google Scholar]
- [14].Khan M, Syed GH, Kim S-J, Siddiqui A. Mitochondrial dynamics and viral infections: a close nexus. Biochim Biophys Acta BBA - Mol Cell Res 2015; 1853(10, Part B):2822-33; https://doi.org/ 10.1016/j.bbamcr.2014.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Detmer Scott A, Chan David C. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 2007; 8(11):870-9; PMID:17928812; https://doi.org/ 10.1038/nrm2275 [DOI] [PubMed] [Google Scholar]
- [16].Su B, Wang X, Zheng L, Perry G, Smith MA, Zhu X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim Biophys Acta 2010; 1802(1):135-42; PMID:19799998; https://doi.org/ 10.1016/j.bbadis.2009.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rahmani Z, Huh K-W, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol 2000; 74(6):2840-6; PMID:10684300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bouchard MJ, Wang L-H, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001; 294(5550):2376-8; PMID:11743208; https://doi.org/ 10.1126/science.294.5550.2376 [DOI] [PubMed] [Google Scholar]
- [19].Waris G, Huh K-W, Siddiqui A. Mitochondrially Associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-κB via oxidative stress. Mol Cell Biol 2001; 21(22):7721-30; PMID:11604508; https://doi.org/ 10.1128/MCB.21.22.7721-7730.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bouchard Michael J, Navas-Martin S. Hepatitis B and C virus hepatocarcinogenesis: lessons learned and future challenges. Cancer Lett 2011; 305(2):123-43; PMID:21168955; https://doi.org/ 10.1016/j.canlet.2010.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim S-J, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of parkin and subsequent mitophagy. PLoS Pathog 2013; 9(3):e1003285; PMID:23555273; https://doi.org/ 10.1371/journal.ppat.1003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim S-J, Syed GH, Khan M, Chiu W-W, Sohail MA, Gish RG, Siddiqui A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U S A 2014; 111(17):6413-8; PMID:24733894; https://doi.org/ 10.1073/pnas.1321114111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Boyapalle S, Wong T, Garay J, Teng M, San Juan-Vergara H, Mohapatra S, Mohapatra S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS One 2012; 7(2):e29386; PMID:22383950; https://doi.org/ 10.1371/journal.pone.0029386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Narayanan A, Amaya M, Voss K, Chung M, Benedict A, Sampey G, Kehn-Hall K, Luchini A, Liotta L, Bailey C, et al.. Reactive oxygen species activate NFκB (p65) and p53 and induce apoptosis in RVFV infected liver cells. Virology 2014; 449:270-86; PMID:24418562; https://doi.org/ 10.1016/j.virol.2013.11.023 [DOI] [PubMed] [Google Scholar]
- [25].Terasaki K, Won S, Makino S. The c-terminal region of rift valley fever virus NSm protein targets the protein to the mitochondrial outer membrane and exerts antiapoptotic function. J Virol 2013; 87(1):676-82; PMID:23097454; https://doi.org/ 10.1128/JVI.02192-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, Frolov I. The old world and new world alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J Virol 2007; 81(5):2472-84; PMID:17108023; https://doi.org/ 10.1128/JVI.02073-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Atasheva S, Kim Dal Y, Frolova Elena I, Frolov I. Venezuelan equine encephalitis virus variants lacking transcription inhibitory functions demonstrate highly attenuated phenotype. J Virol 2015; 89(1):71-82; PMID:25320296; https://doi.org/ 10.1128/JVI.02252-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Narayanan A, Popova T, Turell M, Kidd J, Chertow J, Popov SG, Bailey C, Kashanchi F, Kehn-Hall K. Alteration in superoxide dismutase 1 causes oxidative stress and p38 MAPK activation following RVFV infection. PloS One 2011; 6(5):e20354; PMID:21655261; https://doi.org/ 10.1371/journal.pone.0020354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kehn-Hall K, Narayanan A, Lundberg L, Sampey G, Pinkham C, Guendel I, Van Duyne R, Senina S, Schultz KL, Stavale E, et al.. Modulation of GSK-3β activity in venezuelan equine encephalitis virus infection. PLoS One 2012; 7(4):e34761; PMID:22496857; https://doi.org/ 10.1371/journal.pone.0034761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lundberg L, Pinkham C, Baer A, Amaya M, Narayanan A, Wagstaff KM, Jans DA, Kehn-Hall K. Nuclear import and export inhibitors alter capsid protein distribution in mammalian cells and reduce venezuelan equine encephalitis virus replication. Antiviral Res 2013; 100(3):662-72; PMID:24161512; https://doi.org/ 10.1016/j.antiviral.2013.10.004 [DOI] [PubMed] [Google Scholar]
- [31].Chen TH, Lo YP, Yang CF, Chen WJ. Additive protection by antioxidant and apoptosis-inhibiting effects on mosquito cells with dengue 2 virus infection. PLoS Negl Trop Dis 2012; 6(4):e1613; PMID:22530071; https://doi.org/ 10.1371/journal.pntd.0001613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen T-H, Tang P, Yang C-F, Kao LH, Lo Y-P, Chuang C-K, Shih YT, Chen WJ. Antioxidant defense is one of the mechanisms by which mosquito cells survive dengue 2 viral infection. Virology 2011; 410(2):410-7; PMID:21216424; https://doi.org/ 10.1016/j.virol.2010.12.013 [DOI] [PubMed] [Google Scholar]
- [33].Yang T-C, Lai C-C, Shiu S-L, Chuang P-H, Tzou B-C, Lin YY, Tsai FJ, Lin CW. Japanese encephalitis virus down-regulates thioredoxin and induces ROS-mediated ASK1-ERK/p38 MAPK activation in human promonocyte cells. Microbes Infect 2010; 12(8–9):643-51; PMID:20430109; https://doi.org/ 10.1016/j.micinf.2010.04.007 [DOI] [PubMed] [Google Scholar]
- [34].Murata T, Goshima F, Daikoku T, Inagaki-Ohara K, Takakuwa H, Kato K, Nishiyama Y. Mitochondrial distribution and function in herpes simplex virus-infected cells. J Gen Virol 2000; 81(2):401-6; PMID:10644838; https://doi.org/ 10.1099/0022-1317-81-2-401 [DOI] [PubMed] [Google Scholar]
- [35].Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic alzheimer's disease patients. Am J Pathol 2008; 173(2):470-82; PMID:18599615; https://doi.org/ 10.2353/ajpath.2008.071208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gegg Matthew E, Cooper JM, Chau K-Y, Rojo M, Schapira Anthony HV, Taanman J-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010; 19(24):4861-70; PMID:20871098; https://doi.org/ 10.1093/hmg/ddq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Narendra Derek P, Youle Richard J. Targeting mitochondrial dysfunction: role for PINK1 and parkin in mitochondrial quality control. Antioxid Redox Signal 2011; 14(10):1929-38; PMID:21194381; https://doi.org/ 10.1089/ars.2010.3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al.. PINK1 autophosphorylation upon membrane potential dissipation is essential for parkin recruitment to damaged mitochondria. Nat Commun 2012; 3:1016; PMID:22910362; https://doi.org/ 10.1038/ncomms2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Barry G, Fragkoudis R, Ferguson MC, Lulla A, Merits A, Kohl A, Fazakerley JK. Semliki forest virus-induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J Virol 2010; 84:7369-77; PMID:20427528; https://doi.org/ 10.1128/JVI.02310-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sharma A, Bhattacharya B, Puri RK, Maheshwari RK. Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genomics 2008; 9:289; PMID:18558011; https://doi.org/ 10.1186/1471-2164-9-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jackson AC, Rossiter JP. Apoptotic cell death is an important cause of neuronal injury in experimental venezuelan equine encephalitis virus infection of mice. Acta Neuropathol (Berl) 1997; 93:349-53; https://doi.org/ 10.1007/s004010050626 [DOI] [PubMed] [Google Scholar]
- [42].Baer A, Lundberg L, Swales D, Waybright N, Pinkham C, Dinman JD, Jacobs JL, Kehn-Hall K. Venezuelan equine encephalitis virus induces apoptosis through the unfolded protein response activation of EGR1. J Virol 2016; 90(7):3558-72; PMID:26792742; https://doi.org/ 10.1128/JVI.02827-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Atkins GJ. The pathogenesis of alphaviruses. Int Sch Res Not 2012; 2013:e861912; https://doi.org/ 10.5402/2013/861912 [DOI] [Google Scholar]
- [44].Coleman Mathew L, Sahai Erik A, Yeo M, Bosch M, Dewar A, Olson Michael F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 2001; 3(4):339-45; PMID:11283606; https://doi.org/ 10.1038/35070009 [DOI] [PubMed] [Google Scholar]
- [45].Lazarou M, Sliter Danielle A, Kane Lesley A, Sarraf Shireen A, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015; 524(7565):309-14; PMID:26266977; https://doi.org/ 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Williams R, Yao H, Peng F, Yang Y, Bethel-Brown C, Buch S. Co-operative induction of CXCL10 involves NADPH oxidase: implications for HIV dementia. Glia 2010; 58(5):611-21; PMID:19941336; https://doi.org/ 10.1002/glia.20949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Santana S, Sastre I, Recuero M, Bullido Maria J, Aldudo J. Oxidative stress enhances neurodegeneration markers induced by herpes simplex virus type 1 infection in human neuroblastoma cells. PLoS One 2013; 8(10):e75842; PMID:24124518; https://doi.org/ 10.1371/journal.pone.0075842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen Eunice C, Yagi S, Kelly Kristi R, Mendoza Sally P, Maninger N, Rosenthal A, Spinner A, Bales KL, Schnurr DP, Lerche NW, et al.. Cross-species transmission of a novel adenovirus associated with a fulminant pneumonia outbreak in a new world monkey colony. PLoS Pathog 2011; 7(7):e1002155; PMID:21779173; https://doi.org/ 10.1371/journal.ppat.1002155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kramer T, Enquist Lynn W. Alphaherpesvirus infection disrupts mitochondrial transport in neurons. Cell Host Microbe 2012; 11(5):504-14; PMID:22607803; https://doi.org/ 10.1016/j.chom.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, et al.. Parkinson's disease-associated kinase PINK1 regulates miro protein level and axonal transport of mitochondria. PLoS Genet 2012; 8(3):e1002537; PMID:22396657; https://doi.org/ 10.1371/journal.pgen.1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Willows S, Ilkow Carolina S, Hobman Tom C. Phosphorylation and membrane association of the rubella virus capsid protein is important for its anti-apoptotic function. Cell Microbiol 2014; 16(8):1201-10; PMID:24456140; https://doi.org/ 10.1111/cmi.12272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shiba-Fukushima K, Inoshita T, Hattori N, Imai Y. PINK1-mediated phosphorylation of parkin boosts parkin activity in drosophila. PLOS Genet 2014; 10(6):e1004391; PMID:24901221; https://doi.org/ 10.1371/journal.pgen.1004391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang X, Winter D, Ashrafi G, Schlehe J, Wong Yao L, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011; 147(4):893-906; PMID:22078885; https://doi.org/ 10.1016/j.cell.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ordureau A, Heo J-M, Duda David M, Paulo Joao A, Olszewski Jennifer L, Yanishevski D, Rinehart J, Schulman BA, Harper JW. Defining roles of parkin and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A 2015; 112(21):6637-42; PMID:25969509; https://doi.org/ 10.1073/pnas.1506593112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Beatch Martin D, Hobman Tom C. Rubella virus capsid associates with host cell protein p32 and localizes to mitochondria. J Virol 2000; 74(12):5569-76; PMID:10823864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gottlieb Robert A, Bernstein D. Mitochondrial remodeling: rearranging, recycling, and reprogramming. Cell Calcium 2016; 60(2):88-101; PMID:27130902; https://doi.org/ 10.1016/j.ceca.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Morciano G, Pedriali G, Sbano L, Iannitti T, Giorgi C, Pinton P. Intersection of mitochondrial fission and fusion machinery with apoptotic pathways: role of Mcl-1. Biol Cell 2016; 108:279-293; PMID:27234233; https://doi.org/ 10.1111/boc.201600019 [DOI] [PubMed] [Google Scholar]
- [58].Nykky J, Vuento M, Gilbert L. Role of mitochondria in parvovirus pathology. PLoS One 2014; 9(1):e86124; PMID:24465910; https://doi.org/ 10.1371/journal.pone.0086124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chu Victor C, Bhattacharya S, Nomoto A, Lin J, Zaidi Syed K, Oberley TD, Weinman SA, Azhar S, Huang TT. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS One 2011; 6(12):e28551; PMID:22164304; https://doi.org/ 10.1371/journal.pone.0028551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sasaki M, Miyazaki K, Koga Y, Kimura G, Nomoto K, Yoshida H. Calcineurin-dependent mitochondrial disturbances in calcium-induced apoptosis of human immunodeficiency virus gp160-expressing CD4+ cells. J Virol 2002; 76(1):416-20; PMID:11739707; https://doi.org/ 10.1128/JVI.76.1.416-420.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gullberg RC, Jordan SJ, Moon SL, Soltani E, Geiss Brian J. Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology 2015; 475:219-29; PMID:25514423; https://doi.org/ 10.1016/j.virol.2014.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lopez Sanchez MI, Crowston JG, Mackey DA, Trounce IA. Emerging mitochondrial therapeutic targets in optic neuropathies. Pharmacol Ther 2016; 165:132-152; PMID:27288727; https://doi.org/ 10.1016/j.pharmthera.2016.06.004 [DOI] [PubMed] [Google Scholar]
- [63].Hall Andrew M, Schuh Claus D. Mitochondria as therapeutic targets in acute kidney injury. Curr Opin Nephrol Hypertens 2016; 25(4):355-62; PMID:27166518; https://doi.org/ 10.1097/MNH.0000000000000228 [DOI] [PubMed] [Google Scholar]
- [64].Kinney RM, Chang GJ, Tsuchiya KR, Sneider JM, Roehrig JT, Woodward TM, Trent DW. Attenuation of venezuelan equine encephalitis virus strain TC-83 is encoded by the 5′-noncoding region and the E2 envelope glycoprotein. J Virol 1993; 67(3):1269-77; PMID:7679745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vialat P, Muller R, Vu Thuy H, Prehaud C, Bouloy M. Mapping of the mutations present in the genome of the rift valley fever virus attenuated MP12 strain and their putative role in attenuation. Virus Res 1997; 52(1):43-50; PMID:9453143; https://doi.org/ 10.1016/S0168-1702(97)00097-X [DOI] [PubMed] [Google Scholar]
- [66].Caplen H, Peters CJ, Bishop David HL. Mutagen-directed attenuation of rift valley fever virus as a method for vaccine development. J Gen Virol 1985; 66(10):2271-7; PMID:4045430; https://doi.org/ 10.1099/0022-1317-66-10-2271 [DOI] [PubMed] [Google Scholar]
- [67].Kim S-J, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog 2013; 9(12):e1003722; PMID:24339771; https://doi.org/ 10.1371/journal.ppat.1003722 [DOI] [PMC free article] [PubMed] [Google Scholar]