

ABSTRACT
Staphylococcus aureus causes acute and chronic forms of infection, the latter often associated with formation of a biofilm. It has previously been demonstrated that mutation of atl, codY, rot, sarA, and sigB limits biofilm formation in the USA300 strain LAC while mutation of agr, fur, and mgrA has the opposite effect. Here we used a murine sepsis model to assess the impact of these same loci in acute infection. Mutation of agr, atl, and fur had no impact on virulence, while mutation of mgrA and rot increased virulence. In contrast, mutation of codY, sarA, and sigB significantly attenuated virulence. Mutation of sigB resulted in reduced accumulation of AgrA and SarA, while mutation of sarA resulted in reduced accumulation of AgrA, but this cannot account for the reduced virulence of sarA or sigB mutants because the isogenic agr mutant was not attenuated. Indeed, as assessed by accumulation of alpha toxin and protein A, all of the mutants we examined exhibited unique phenotypes by comparison to an agr mutant and to each other. Attenuation of the sarA, sigB and codY mutants was correlated with increased production of extracellular proteases and global changes in extracellular protein profiles. These results suggest that the inability to repress the production of extracellular proteases plays a key role in attenuating the virulence of S. aureus in acute as well as chronic, biofilm-associated infections, thus opening up the possibility that strategies aimed at the de-repression of protease production could be used to broad therapeutic advantage. They also suggest that the impact of codY, sarA, and sigB on protease production occurs via an agr-independent mechanism.
KEYWORDS: alpha toxin, AgrA, bacteremia, protein A, regulatory mutations, SarA, sepsis, Staphylococcus aureus
Introduction
The production of Staphylococcus aureus virulence factors is modulated by a complex and highly interactive regulatory circuit.1 This affords the bacterium tremendous flexibility with respect to its ability to respond to changing conditions within the host including the influence of an ongoing host immune response.2-4 This flexibility is reflected in the ability of S. aureus to cause a diverse array of infections. In general, these can be characterized as acute infections, the clinical characteristics of which are often defined by toxin production, and chronic infections, the clinical characteristics of which are often associated with formation of a biofilm.5 To some extent a general theme of the overall S. aureus regulatory circuitry is to modulate the production of specific virulence factors that contribute to these alternative forms of infection.6 A primary example is the accessory gene regulator (agr), expression of which limits biofilm formation but at the same time promotes toxin production.7,8
The treatment of all forms of S. aureus infection is complicated by the persistent emergence of antibiotic resistant strains, which accounts for its inclusion among the ESKAPE pathogens.9 The treatment of chronic biofilm-associated infections is further complicated by the presence of the biofilm itself, which confers a therapeutically-relevant level of intrinsic resistance to conventional antibiotics and host defenses.10 This has created an urgent need for new antibiotics that are both effective against the most problematic antibiotic-resistant strains and retain a therapeutically-relevant level of efficacy in the context of a biofilm. New antibiotics have been and continue to be developed,11 but given the remarkable intrinsic resistance conferred by the presence of a biofilm, accomplishing both of these goals has proven to be a formidable task.12
This has led to the suggestion that strategies targeting S. aureus regulatory circuits and/or specific virulence factors produced under the control of these circuits could be used to therapeutic benefit either alone or in combination with conventional antibiotics.13,14 Our approach has been to investigate the regulatory basis for biofilm formation itself, the goal being to identify those regulatory loci that offer the greatest opportunity for therapeutic intervention. These efforts have led us to focus on the staphylococcal accessory regulator (sarA), mutation of which limits biofilm formation to a greater extent than mutation of any other regulatory locus we have examined.15 Moreover, this limitation can be correlated with increased antibiotic susceptibility in biofilm-associated infections caused by diverse strains of S. aureus including methicillin-resistant strains.16 This suggests that inhibitors of sarA expression and/or function could be used to therapeutic advantage in the context of biofilm-associated S. aureus infections.
However, biofilms are highly dynamic structures in which changes in gene expression promote the development, maturation, and ultimately the dissemination of bacterial cells from the biofilm, at which point they can enter the systemic circulation and cause infection at secondary sites.17,18 Thus, inhibition of sarA as a means of limiting biofilm formation could have the adverse consequence of promoting acute, systemic infection. Conversely, inhibitors of agr expression and/or function may be of therapeutic benefit in the context of acute infection, but could also have the adverse consequence of promoting chronic, biofilm-associated infection. Indeed, agr-defective strains are often isolated from patients suffering from diverse forms of infection, perhaps owing at least in part to the advantage gained by the intrinsic antibiotic resistance afforded to the bacterium in the context of a biofilm.19 Moreover, one report that examined the clinical history of 814 patients with S. aureus sepsis found that agr dysfunction was associated with a statistically significant increase in mortality.20
Such results emphasize the need to consider the contribution of individual regulatory loci in diverse forms of S. aureus infection. To this end, we also examined the role of sarA in acute models of S. aureus infection, and the results confirmed that sarA mutants are attenuated in murine models of bacteremia and acute, post-traumatic osteomyelitis.21-23 This suggests that sarA may be a viable therapeutic target in diverse forms of S. aureus infection. However, many other regulatory loci have also been shown to impact various forms of infection.1 Defining the relative impact of these loci in diverse forms of S. aureus infection is difficult because most reports focused on individual regulatory loci in the context of a single form of S. aureus infection. This precludes the ability to determine which of these loci offer the greatest therapeutic promise in diverse forms of S. aureus infection. We have begun to address this by making direct comparisons between regulatory loci in the context of biofilm-associated infection,15,16 but we have not done so in the context of acute infection. Thus, in this report we extended previous experiments focusing on regulatory loci that impact biofilm-associated infection to directly assess the relative impact of these same regulatory loci on virulence in a murine bacteremia model of acute S. aureus infection.
Results
Owing to its current prominence as a cause of S. aureus infection,24-26 the experiments we report were done with a derivative of the USA300 strain LAC cured of its resident erythromycin-resistance plasmid.23 We initially focused on isogenic derivatives of this strain with mutations in codY, fur, mgrA, sarA, and sigB. These regulatory loci were chosen to allow direct comparisons with the results observed in previous biofilm studies in which mutation of these same loci was shown to either enhance (fur, mgrA) or limit (codY, sarA and sigB) biofilm formation.7,15,16 We also included a rot mutant based on a recent report concluding that mutation of rot limits biofilm formation in LAC.27 Strains were introduced into NIH-Swiss mice by tail vein injection of 5 × 107 colony-forming units (cfu) as previously described.23 Over the 7 day period of this experiment, this resulted in the death of 60% of mice infected with the LAC parent strain (Fig. 1). The mutants evaluated in this experiment were found to fall into one of three groups, with mutation of fur having no statistically significant effect on virulence, mutation of codY, sarA, and sigB significantly attenuating virulence, and mutation of mgrA and rot resulting in a significant increase in virulence relative to LAC (Fig. 1). These results were consistent with our studies examining the relative capacity of these mutants to escape the bloodstream and colonize soft tissues. Specifically, mutation of codY, sarA, and sigB attenuated virulence as assessed based on colony counts in the spleen (Fig. 2A), heart (Fig. 2B), and peripheral blood (Fig. 2C). Results observed in the kidney were less discriminatory in that the only significant difference was that between LAC and its isogenic sarA mutant (Fig. 2D).
Figure 1.
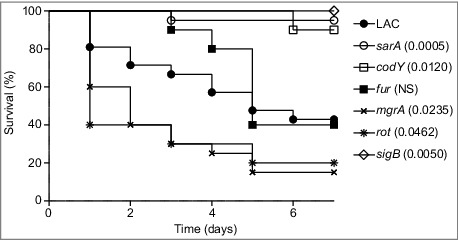
Relative virulence of S. aureus regulatory mutants in acute sepsis. Kaplan-Meier survival curves are shown for the USA300 strain LAC and the indicated isogenic mutants. Numbers in parenthesis indicate p values for each mutant by comparison to the results observed with LAC. NS = not significant.
Figure 2.
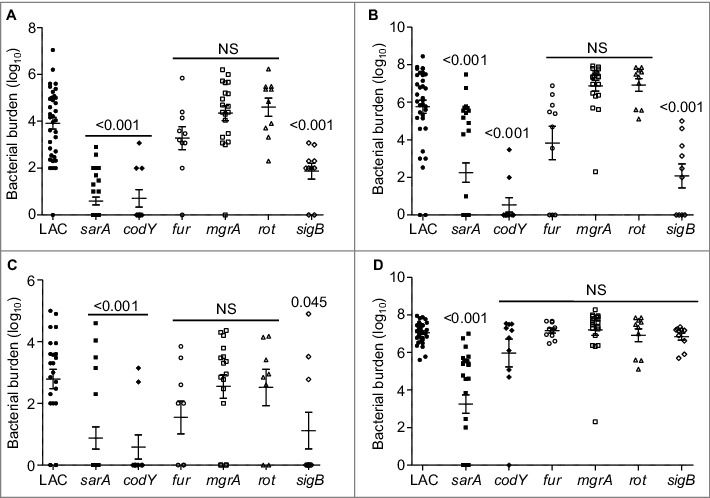
Relative virulence of S. aureus regulatory mutants as assessed by colonization. The number of colony-forming units (cfu) in the (A) spleen, (B) heart, (C) peripheral blood, and (D) kidney are shown by scatter plot. Numbers above each plot indicate p values for each mutant by comparison to the results observed with LAC. NS = not significant. Bars represent the mean ± SEM of log10 transformed values.
To investigate the mechanistic basis for the virulence phenotypes we observed, we performed western blots of whole cell lysates using antibodies targeting AgrA and SarA as previously described.23 Mutation of sarA and sigB resulted in a significant decrease in the accumulation of AgrA (Fig. 3A), which is the response regulator of the two component quorum-sensing system encoded by agr.28 Additionally, mutation of sigB resulted in a significant decrease in the accumulation of SarA (Fig. 3B). This suggests that the decreased virulence observed with the sarA and sigB mutants may be at least partially attributable to the impact of these mutations on expression of agr. However, comparison of agr, sarA, and sigB mutants revealed that all three had distinct phenotypes as defined by the relative accumulation of alpha toxin and protein A (Spa), which are prototype virulence factors known to be inversely regulated by agr.28 Specifically, as assessed by western blot of conditioned medium from each strain, and as expected based on previous reports,28 accumulation of Spa was increased in the LAC agr mutant while accumulation of alpha toxin was decreased (Fig. 3C and D). In contrast, accumulation of both Spa and alpha toxin was decreased in the sarA mutant, while accumulation of Spa was decreased, and accumulation of alpha toxin increased, in the sigB mutant (Fig. 3C and D).
Figure 3.
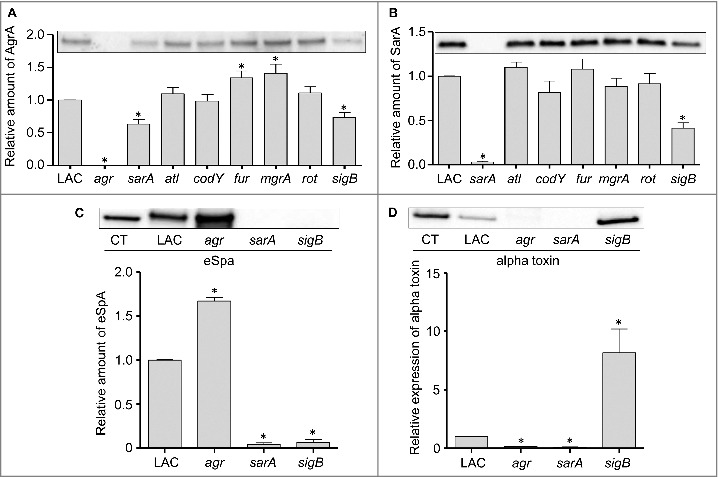
Relative accumulation of AgrA, SarA, eSpa, and alpha toxin in S. aureus regulatory mutants as assessed by immunoblot analysis. Representative western blots of cell lysates (A and B) or conditioned medium (C and D) prepared from LAC and the indicated isogenic mutants were analyzed by western blots using (A) anti-AgrA antibody, (B) anti-SarA antibody, (C) anti-Spa antibody, or (D) anti-alpha toxin antibody. Graphs indicate cumulative densitometric values obtained from all biological and experimental replicates. Asterisks indicate statistical significance (p ≤ 0.05) by comparison to values obtained with the LAC parent strain.
Additionally, if the attenuation of sarA and sigB mutants is defined by the impact of these loci on expression of agr, then it would also be anticipated that an isogenic agr mutant would be attenuated to a comparable degree by comparison to sarA and sigB mutants. To examine this issue, we used our murine bacteremia model to compare LAC with its isogenic agr mutant. Interestingly, we found that mutation of agr had little impact on virulence (Fig. 4). These results were surprising given that mutation of agr in LAC as well as other S. aureus strains has been shown to limit virulence in animal models of S. aureus infection.29-32 However, most of these models focused on some form of localized infection, primarily of the skin, and such models do not necessarily mimic the systemic infection modeled here. Indeed, Cameron et al. recently examined a number of clinical isolates of S. aureus and concluded that agr expression was not essential for virulence in a murine bacteremia model.33 However, the more important point in the context of this report is that this finding, together with the alpha toxin and Spa results discussed above, effectively rules out the possibility that the attenuation observed with the LAC sarA and sigB mutants is a function of their impact on expression of agr.
Figure 4.
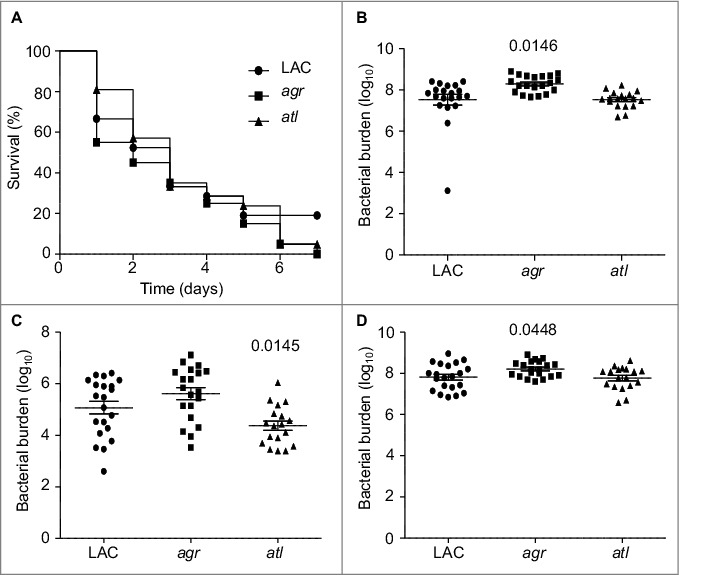
Relative virulence of S. aureus agr and atl mutants in acute sepsis. (A) Kaplan-Meier survival curves are shown for LAC and the indicated isogenic mutants. Number of cfu observed in homogenates prepared from (B) heart, (C) spleen, and (D) kidney. The number above each scatter plot cluster indicates the p value for each mutant found to significantly different by comparison to LAC. Bars represent the mean ± SEM of log10 transformed values.
While the focus of the experiments we report was on regulatory loci, we also examined the impact of mutating atl in this experiment based on the observation that mutation of atl has been shown by several laboratories, including our own, to limit biofilm formation.15,34,35 This experiment was complicated by the fact that a characteristic phenotype of atl mutants grown in vitro is the formation of large aggregates.36 To address this, we first carried out studies in which the apparent number of colony-forming units (cfu) was assessed before and after sonication. The number of detectable cfu increased in all strains after sonication, and in the case of four mutants (atl, sarA, fur, and rot) the number as assessed before sonication was significantly lower than the number observed with the LAC parent strain (Fig. 5A). However, with the exception of the atl mutant, all of the differences we observed were well within an order of magnitude (2.6 × 109 −8.9 × 109). In contrast, statistical analysis confirmed that the number of cfu as assessed before sonication was significantly higher in every strain we examined by comparison to the atl mutant (Fig. 5A). These experiments also confirmed that there was no difference between any of the strains we examined, including the atl mutant, after sonication.
Figure 5.
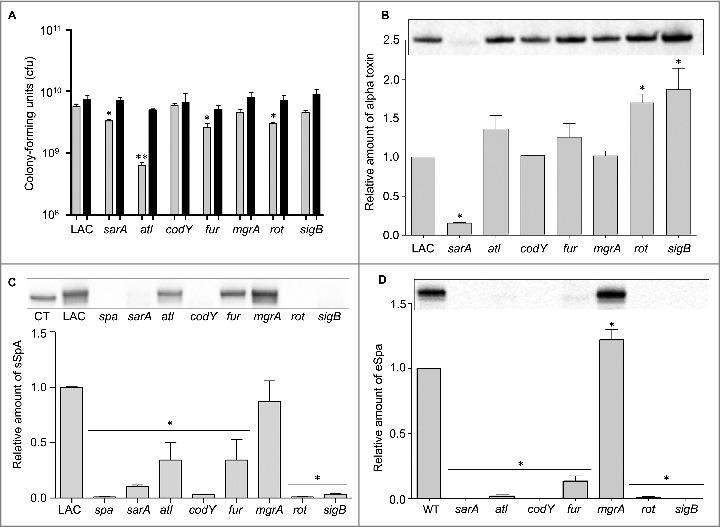
Analysis of mutant aggregative phenotypes and their respective production of alpha toxin and Spa. (A) Bars indicate the number of cfu observed before (grey) and after sonication (black). Single asterisk indicates statistical significance prior to sonication by comparison to LAC. Double asterisk indicates statistical significance of the atl mutant by comparison to all other strains prior to sonication. No significant differences were observed after sonication. (B) Representative western blot of conditioned medium from LAC and the indicated isogenic mutants using anti-alpha toxin antibody (top). Graph indicates cumulative densitometric values obtained from all biological and experimental replicates (bottom). (C) Representative western blot of surface protein preparations from LAC and the indicated isogenic mutants using anti-Spa antibody (top). CT = purified Spa control. Graph indicates cumulative densitometric values obtained from all biological and experimental replicates (bottom). (D) Representative western blot of conditioned medium from LAC and the indicated isogenic mutants using anti-Spa antibody (top). Graph indicates cumulative densitometric values obtained from all biological and experimental replicates (bottom). Asterisks in each graph indicate statistical significance (p ≤ 0.05) by comparison to values obtained with the LAC parent strain.
Based on these results, in vivo analysis of the atl mutant was done using an inoculum prepared after sonication. As with agr, mutation of atl was also found to have no significant impact on overall virulence (Fig. 4). Mutation of atl also had no impact on the accumulation of AgrA (Fig. 3A) or SarA (Fig. 3B). Although most studies focusing on the role of atl in S. aureus pathogenesis have focused on biofilm formation, the results we observed with the atl mutant in our bacteremia model are consistent with those of Takahashi et al., who found that mutation of atl had no impact on virulence in a murine model of intraperitoneal infection.36
In contrast to sarA and sigB, mutation of fur and mgrA resulted in a modest (<2-fold) but statistically significant increase in the accumulation of AgrA (Fig. 3A). However, this was not reflected in the alpha toxin phenotype of these mutants in that neither produced alpha toxin at significantly greater levels than the isogenic LAC parent strain (Fig. 5B). In contrast, mutation of rot and sigB resulted in a significant increase in the accumulation of alpha toxin, while mutation of sarA had the opposite effect. Indeed, alpha toxin was essentially absent in conditioned medium from LAC agr and sarA mutants (Fig. 3D). The possibility that the decrease in alpha toxin observed in the sarA mutant is at least partially attributable to the impact of mutating sarA on agr cannot be completely discounted, but previous studies from our laboratory have demonstrated that the accumulation of alpha toxin can be restored to wild-type levels in a sarA mutant by eliminating the production of extracellular proteases.37,38 The relative abundance of alpha toxin in conditioned media was correlated with virulence in rot and sarA mutants, with both being increased in a rot mutant and both being decreased in a sarA mutant, but this was not the case with the sigB mutant in that the accumulation of alpha toxin was increased (Fig. 5B) while overall virulence was decreased (Fig. 1).
We also assessed the impact of each mutation on the accumulation of protein A (Spa). Because S. aureus naturally produces Spa in both extracellular and surface-associated forms, these experiments were done by western blot of both cell extracts enriched for surface-associated proteins (sSpa) and extracellular Spa (eSpa).39,40 With the exception of the mgrA mutant, the amount of both sSpa (Fig. 5C) and eSpa (Fig. 5D) was reduced relative to LAC. In contrast, accumulation of Spa was increased in a LAC mgrA mutant, particularly when assessed in its extracellular form (Fig. 5D). These results are consistent with a previous study demonstrating by RNA-seq that the amount of spa transcripts was dramatically increased in a LAC mgrA mutant.41
In western blots done with surface protein-enriched cell extracts, sSpa was essentially absent in codY, rot, sarA, and sigB mutants, and present in significantly reduced amounts atl and fur mutants (Fig. 5C). When assessed using conditioned medium, eSpa was essentially absent in every strain except LAC and its agr (Fig. 3C) and mgrA mutant (Fig. 5D). To the extent that Spa in both of these forms contributes to the virulence of S. aureus by promoting immune evasion, its virtual absence could contribute to the reduced virulence of the codY, sarA and sigB mutants.42 Conversely, its increased abundance could contribute to the increased virulence of a LAC mgrA mutant. However, it is difficult to envision how reduced accumulation of Spa would contribute to increased virulence of a LAC rot mutant.
As noted above, we focused on alpha toxin and Spa because they are prototype virulence factors in the context of the pathogenic versatility of S. aureus and are differentially regulated by agr relative to each other.6,43 A number of the loci we examined are also known to impact expression and/or function of agr. Although it was not true of the sarA or sigB mutants, it might therefore be anticipated that mutation of other regulatory loci would result in alpha toxin and Spa phenotypes comparable to those observed in the agr mutant (e.g. increased production of Spa and decreased production of alpha toxin). However, all of the mutants we examined exhibited unique alpha toxin and Spa phenotypes relative to the agr mutant and relative to each other (Table 1). This provides support for the hypothesis that the virulence changes we observed are largely agr-independent. It also emphasizes the overall complexity of S. aureus regulatory circuits and suggests that additional virulence factors that remain to be identified are involved in defining the virulence phenotypes of each of these mutants. Additional studies will be required to identify these virulence factors, but one clear correlation we did observe, and one that could potentially be exploited to help identify such virulence factors, was that attenuation of the sarA, codY, and sigB mutants was in all cases correlated with the increased production of extracellular proteases and an altered exoprotein profile (Fig. 6). Mutation of rot was also previously shown to result in an increase in protease production to an extent that could be correlated with a reduced capacity to form a biofilm, but in our comparisons we did not observe a significant increase in the accumulation of extracellular proteases in the rot mutant (Fig. 6).27 This is consistent with the observation that, unlike codY, sarA and sigB mutants, the LAC rot mutant exhibited increased rather than decreased virulence (Fig. 1).
Table 1.
Summary of phenotypes. Table summarizes whether the accumulation of AgrA, SarA, alpha (α) toxin, Spa, and virulence was increased, decreased, or unchanged (NC) in each of the indicated LAC regulatory mutants. A dash (-) indicates the indicated protein was absent. Accumulation of SarA in an agr mutant was not tested (NT).
| Mutation | AgrA | SarA | α toxin | Spa | Virulence |
|---|---|---|---|---|---|
| agr | — | NT | Down | Up | NC |
| atl | NC | NC | NC | Down | NC |
| codY | NC | NC | NC | Down | Down |
| fur | Up | NC | NC | Down | NC |
| mgrA | Up | NC | NC | Up | Up |
| rot | NC | NC | Up | Down | Up |
| sarA | Down | — | Down | Down | Down |
| sigB | Down | Down | Up | Down | Down |
Figure 6.
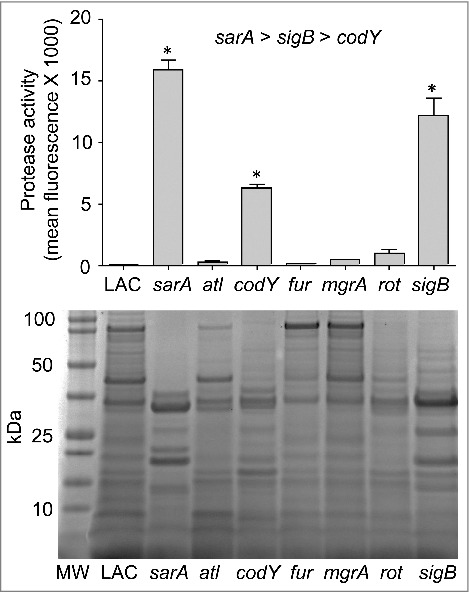
Protease activity in LAC regulatory mutants. Top: Total protease production in LAC and the indicated regulatory mutants was assessed using a commercially available FITC-casein cleavage hydrolysis assay. Results are reported as mean fluorescence values ± the standard error of the mean. Asterisks indicate statistically significant differences (p ≤ 0.05) between the indicated mutants relative to the results observed with LAC. As indicated above the graph, individual comparisons also confirmed that the amount of total protease activity observed in the sarA mutant was increased to a statistically significant extent by comparison to the results observed with both the sigB and codY mutants and that total protease activity in the sigB mutant was increased to a statistically significant extent by comparison to those observed with the codY mutant. Bottom: Extracellular protein profiles for LAC and each regulatory mutant were assessed by SDS-PAGE. MW = molecular weight markers, with the molecular weight in kilodaltons (kDa) of representative markers shown to the left.
Interestingly, mutation of sarA and sigB resulted in a comparable increase in the overall production of extracellular proteases (Fig. 6) but had opposite effects on the accumulation of alpha toxin (Fig. 3D). One possible explanation that we are currently exploring is that, while sarA and sigB repress the overall production of extracellular proteases, they do not repress the production of the specific protease(s) that degrade alpha toxin to the same degree. The alternative is that mutation of sigB has a much greater impact on the production of alpha toxin than mutation of sarA, thus resulting in a net increase in the accumulation of alpha toxin in a LAC sigB mutant despite its increased production of extracellular proteases.
Finally, we did not include complementation studies in our in vivo experiments for two reasons. The first is the number of mice this would have required would have been very large given the number of mutants we examined, particularly since all of our in vivo experiments were repeated at least twice. The second is that such studies are sometimes inconclusive owing to plasmid instability in vivo. However, we did confirm that all mutations that had a significant impact on Spa and/or alpha toxin phenotypes could be complemented under in vitro conditions (Fig. S1).
Discussion
The continuing increase in antibiotic resistance has led to the suggestion that strategies targeting S. aureus virulence factors and/or the regulatory circuits that control the production of these virulence factors could be therapeutically beneficial.13,44-50 The two S. aureus regulatory loci that have been explored to the greatest extent as therapeutic targets are agr and sarA.51-53 These loci interact with each other, with SarA being required for full expression of agr.54 Although we did not assess this at the level of gene expression, we did confirm that AgrA is present in reduced amounts in a LAC sarA mutant. However, it is also clear that sarA and agr serve independent regulatory functions. This is evident in the observation that mutation of agr enhances biofilm formation while mutation of sarA has the opposite effect.7,50,55-57 Thus, the therapeutic focus on these loci has generally been targeted toward different forms of infection, with agr being proposed as a target for acute, toxin-mediated diseases and sarA as a target for chronic, biofilm-associated infections.51,52,58
Our results confirm that mutation of sarA limits the virulence of LAC in a murine sepsis model to a significant degree relative to the isogenic parent strain, but also demonstrate that mutation of codY and sigB attenuate virulence in this clinical context to a comparable degree. Mutation of these same three loci also limited the ability of LAC to form a biofilm which suggests that these loci are all viable therapeutic targets in the context of both acute and chronic forms of S. aureus infection.15 This is particularly true since mutation of all of these loci could be correlated with enhanced susceptibility to daptomycin in vivo in a murine catheter model.16 However, when assessed using ceftaroline, only mutation of sarA and sigB had a significant effect. This suggests that sarA or sigB would be preferable therapeutic targets by comparison to codY.
Further support for this hypothesis comes from the observation that, while mutation of codY has been shown to limit biofilm formation in LAC and other strains of the USA300 clonal lineage, it has also been shown to enhance biofilm formation in UAMS-1 and the clinical isolate SA564.15,59, 60 Interestingly, mutation of codY in these latter strains was also correlated with increased expression of agr,59 which would be expected to decrease rather than increase biofilm formation.7,50,55,57 At present, the mechanistic basis for these strain-dependent biofilm phenotypes is not fully understood, but it has been suggested that it may be related to the relative contribution of the polysaccharide intercellular adhesion (PIA) to biofilm formation in different strains of S. aureus.15,59, 60
There are also conflicting reports regarding the role of codY in defining the virulence of S. aureus in acute infections. For instance, our results are consistent with a report demonstrating that mutation of codY in LAC limited virulence in a neutropenic murine model of pulmonary infection,61 but they are in contrast to a report demonstrating that a codY mutant generated in the USA300 strain 923 exhibited increased virulence in murine models of skin infection and necrotizing pneumonia.62 These disparate results may simply reflect the use of different animal models and the importance of the microenvironment in vivo, or perhaps differences between strains of S. aureus. However, the more important point in the context of this report is that, when viewed collectively, such conflicting reports in the context of both biofilm-associated and acute infection further diminish enthusiasm for codY as a therapeutic target. They also emphasize the need to extend the studies we report here to include evaluation of these same loci in alternative animal models and additional strains of S. aureus.
Similarly, while mutation of sigB in LAC did have a significant impact on daptomycin susceptibility in the context of an established biofilm, it did not have a significant impact in vivo in the methicillin-sensitive, USA200 strain UAMS-1.16 This would suggest that, in the context of coverage for diverse clinical isolates of S. aureus, sarA would be the preferable target even by comparison to sigB. Moreover, much as with codY, there are conflicting reports regarding the phenotypic impact of mutating sigB. For example, Bischoff et al. concluded that mutation of sigB results in decreased levels of sarA expression.63 In contrast, Cheung et al. concluded that mutation of sigB in the 8325-4 strain RN6390 results in increased production of SarA.64 These authors also concluded that this accounts for the increased production of alpha toxin in a sigB mutant. We did not include studies assessing gene expression in the context of sarA, but we did find that the accumulation of SarA is reduced in a LAC sigB mutant. This suggests that mutation of sigB and sarA in LAC would result in similar phenotypes, and in fact we found that this was generally the case.
However, one notable exception to this is that mutation of sarA essentially abolished the accumulation of alpha toxin while mutation of sigB had the opposite effect. Despite the fact that both mutants were attenuated in our sepsis model, this provides further support for the hypothesis that sarA would be the preferred therapeutic target by comparison to sigB, particularly when combined with the observation that mutation of sarA limits biofilm formation to a greater extent than mutation of sigB.15 At the same time, it has been reported that sigB is required for intracellular persistence and development of small colony variants (SCV), both of which are considered key elements in the development of chronic S. aureus infections, and that this is not the case with sarA.65,66 Thus, the possibility that sigB would be the preferred target in the context of chronic infections cannot be ruled out without additional experimentation that includes direct comparisons between these loci in an appropriate animal model.
From a mechanistic point of view, the increased accumulation of alpha toxin in a LAC sigB mutant is consistent with a report concluding that mutation of sigB results in increased levels of agr expression.63 To the extent that rsbU is required for maximum sigB activity, this is also consistent with the observation that repair of the rsbU defect in the 8325-4 strain RN6390 resulted in increased sigB activity and reduced expression of agr.67 Repair of the rsbU defect in RN6390 also resulted in decreased hemolytic activity, increased production of Spa, and an increased capacity to form a biofilm,68 all of which are consistent with decreased levels of agr expression. Additionally, to the extent that mutation of sigB would be expected to result in the opposite phenotypes, they are also consistent with the observation that a LAC sigB mutant exhibited increased accumulation of alpha toxin, decreased production of Spa, and a decreased capacity to form a biofilm as was observed both here and in our previous reports.15,16 However, none of these phenotypes are consistent with the observation that mutation of sigB in LAC was correlated with decreased accumulation of AgrA.
One possible explanation for this apparent disparity is related to the experimental methodologies employed. Specifically, previous reports focused on agr at a transcriptional level, generally with a specific focus on RNAIII, while we focused on the accumulation of AgrA. Indeed, the mechanistic basis by which sigB impacts agr is unclear,27 and it is possible that mutation of sigB impacts the production of RNAIII differently than it does production of AgrA. An alternative possibility is that the increased production of extracellular proteases in the LAC sigB mutant results in increased degradation of AgrA, which would not be apparent in assays focusing on transcription. Indeed, we previously demonstrated that the accumulation of AgrA is limited in a sarA mutant owing to protease-mediated degradation.38 Although it remains to be determined whether this is also the case in a sigB mutant, the studies we report confirm that protease activity is increased in a LAC sigB mutant to a degree that approaches that observed in an isogenic sarA mutant.
Irrespective of the mechanism(s) involved, none of these results can explain the attenuation of a LAC sigB mutant despite the increased accumulation of alpha toxin. At the same time, while repair of rsbU in RN6390 was previously reported to result in reduced expression of agr, it was also correlated with what the authors described as a “surprising increase in mouse lethality” as assessed using a bacteremia model.68 To the extent that repair of rsbU results in increased expression of sigB, this is consistent with the observation that mutation of sigB resulted in reduced lethality in the studies we report. The fact that mutation of sigB limited the accumulation of AgrA suggests that this attenuation could be at least partially agr-dependent, although as noted above this seems unlikely given that the isogenic agr mutant was not attenuated in our model. This was surprising in light of the many reports demonstrating that agr mutants are attenuated in animal models of infection.31,69-71 However, most of these other reports focused on models other than bacteremia, and it has been shown that serum apolipoprotein B, including that from mice, binds and effectively inactivates the quorum-sensing pheromone of the agr system.72 Additionally, serum lipoproteins have been shown to inactivate phenol soluble modulins (PSMs), which have been shown in turn to be a primary determinant of the virulence of community-associated, methicillin-resistant S. aureus strains like LAC.22,26,73,74 Thus, one possible explanation for the fact that mutation of sigB limited virulence in a murine bacteremia model while mutation of agr did not is that the functionality of agr and/or PSMs is decreased owing to the presence of serum lipoproteins. Such a scenario would also suggest that the attenuation we observed in a LAC sigB is independent of its impact on agr expression.
Mutation of rot was previously reported to result in increased virulence and increased production of both alpha toxin and extracellular proteases.75 We also observed increased virulence and increased accumulation of alpha toxin. However, we did not observe a significant increase in the production of extracellular proteases in our LAC rot mutant. This suggests that the increased virulence we observed with a rot mutant is likely due to changes in the production of important virulence factors relative to the rate of their protease-mediated degradation. This is consistent with the observation that rot was originally identified as a repressor of S. aureus toxin production.75 This was subsequently shown to involve an interaction between the agr-encoded RNAIII and rot mRNA that limits translation of the latter.76,77 Thus, it would be anticipated that mutation of agr and mutation of rot would have opposite effects on virulence. This is consistent with the observation that mutation of rot enhanced the virulence of LAC while mutation of agr did not.
In contrast, our results demonstrating that mutation of mgrA enhances virulence are not consistent with reports demonstrating that mutation of mgrA attenuates virulence in murine models of sepsis and septic arthritis as well as rabbit models of sepsis and endocarditis.41,78 We have no explanation for this disparity, but would note that these collective studies were also done using different strains of S. aureus. Indeed, none of the mgrA mutants employed in these earlier virulence studies were generated in LAC. Rather, they were generated in the commonly-studied S. aureus strain Newman, which has a recognized mutation in the saeRS regulatory system,68 or in MW2 and 502A.41 Even aside from recognized regulatory defects like those present in Newman, different strains of S. aureus exhibit a great deal of genetic and phenotypic diversity. In fact, a recent report confirmed that there is as much phenotypic diversity within different clonal lineages of S. aureus as there is between these clonal lineages.79 It is also virtually certain that all strains of S. aureus carry mutations,68 thus making it essentially impossible to define a definitive wild-type strain. Although no specific mutations have been identified in LAC that we are aware of, this is presumably true of USA300 strains. Nevertheless, the clinical predominance of such strains makes them worthy of investigation, and this is the primary reason we chose LAC for our studies. Thus, we believe the possibility that the impact of mutating mgrA on virulence in acute infection is strain-dependent diminishes enthusiasm for mgrA as a therapeutic target, particularly when viewed in light of the fact that mutation of mgrA enhances biofilm formation.15,41
While much remains to be explored regarding the virulence phenotypes of the mutants we examined, one common phenotype observed with the attenuated codY, sarA and sigB mutants is that they all produced extracellular proteases at significantly increased levels relative to LAC. However, the impact of mutating these loci was not equivalent. Specifically, protease production was highest in the sarA mutant and decreased progressively in the sigB and codY mutants, respectively. As discussed above, mutation of sigB results in increased accumulation of alpha toxin, and to the extent that mutation of sarA resulted in a greater increase in protease production than mutation of sigB, one possible explanation for the disparate alpha toxin phenotypes we observed in LAC sigB and sarA mutants may be due to the relative impact of these loci on the production vs. protease-mediated degradation of alpha toxin. Alternatively, our protease assays did not allow us to distinguish between the activity of different proteases, and it is also possible that mutation of sigB vs. sarA has a differential impact on the production of specific proteases that contribute to the degradation of alpha toxin. Nevertheless, the correlation between the increased production of extracellular proteases and decreased virulence suggests that the inability to repress the production of extracellular proteases may play a key role in attenuating the virulence of S. aureus in acute as well as chronic, biofilm-associated infections. The observations that mutation of sarA results in a greater increase in protease production than any other mutant we have examined, and that this can be correlated with a reduced capacity to cause both acute and biofilm-associated infections suggests that sarA may be the best target by which this observation can be exploited to therapeutic advantage.21,23
Materials and methods
Bacterial strains and growth conditions
The strains used in these experiments are summarized in Table S1. With the exception of the spa mutant and the complemented rot mutant, the methods used to generate and confirm all mutants and the corresponding complemented strains were described in previous reports.15,34,80-82 The Nebraska Transposon Mutant Library (NTML) was utilized to generate the spa mutant by phage transduction from the original JE2 mutant into our strain of LAC. The rot complementation strain was constructed similarly by transducing a previously described rot complementation plasmid into the rot mutant.15,83 All strains were maintained at −80°C in a suspension containing tryptic soy broth (TSB) and 25% (v/v) glycerol. For each experiment, strains under study were retrieved from cold storage by plating on tryptic soy agar (TSA) with appropriate antibiotic selection. Antibiotics were incorporated into the culture media as appropriate at the following concentrations: erythromycin, 10 μg ml−1; chloramphenicol 10 μg ml−1; tetracycline, 5 μg ml−1; kanamycin, 50 μg ml−1; and neomycin, 50 μg ml−1; spectinomycin, 1 mg ml−1. Kanamycin and neomycin were always used together to avoid selection of spontaneously resistant strains.
Murine bacteremia model
Bacterial strains were retrieved from cold storage and grown at 37˚C to stationary phase (16-17 hrs) in TSB with appropriate antibiotic selection. Cultures were standardized to an OD560 of 0.05 in TSB without antibiotics and grown to an OD560 of 1.0. Bacterial cells were harvested by centrifugation and separate aliquots were resuspended in an equal volume of sterile phosphate-buffered saline (PBS) containing 10% DMSO and 5% bovine serum albumin (BSA) which were stored at −80°C. The number of colony-forming units (cfu) in aliquots prepared from each strain was confirmed by plate count after 20 hrs incubation at 37oC.
The bacteremia model used in this study was previously described by Zielinska et al. (2012).23 Briefly, the strains under study were removed from cold storage, washed with PBS, and standardized in PBS to a cell density of 5 × 108 cfu per ml. For each experiment, groups of ten 5–8 week-old female NIH-Swiss mice were infected via tail vein injection with 5 × 107 cfu of LAC or one of its isogenic mutants. Organs and tissues were harvested from any mice found dead or which required compassionate euthanasia; otherwise, tissues were harvested at 7 days post-infection. Organs were removed aseptically and homogenized. Serial dilutions of each homogenate were then plated on TSA without selection, and the number of cfus per organ determined following overnight incubation at 37°C. To rule out the possibility of contaminants skewing the results, replicate samples were also plated on CHROMagar (BBL™, Cat. # 254102/215081). All experiments were repeated at least twice, with the total number of mice infected with each strain indicated in the scatter plots.
Western blotting
Samples for western blots and the primary and secondary antibodies used were all prepared and used as previously described.23,38 Western blots included at least two biological replicates with at least two experimental replicates of each. Densitometric values were obtained with a Bio-Rad ChemiDocMP Imaging System and Image Lab Software (Bio-Rad Laboratories, Inc., Irvine, CA).
Sonication assay
To ensure that the results were not skewed by the impact of any given mutation on cellular clumping, all strains were grown in overnight cultures with appropriate selection and standardized to an OD560 of 10.0 in a volume of 5 ml. Serial dilutions of a 100 μl aliquot were performed on ice and plated on TSA without selection. The remaining cultures were kept on ice and sonicated (QSonica S4000, Newtown, CT) for a period of 4 minutes at 6 watts. Serial dilutions were then prepared post-sonication and plated on TSA. The number of cfu before and after sonication was determined by plate count after 20 hrs incubation at 37oC.
Characterization of exoprotein profiles
Exoprotein profiles were examined as described by Zielinksa et al. (2012).23 Briefly, overnight (16-17 hrs) cultures were standardized to an OD560 of 10.0. Conditioned medium from each culture was then harvested by centrifugation and the supernatant filter sterilized. Samples were resolved by SDS-PAGE using 4–12% gradient Novex Bis-Tris Plus gels (Life Technologies, cat. # NW04125BOX). Proteins were visualized by staining with SimplyBlue™ SafeStain (Life Technologies, cat. # LC6060) and imaging using Bio-Rad ChemiDocMP Imaging System (Bio-Rad Laboratories, Inc., Irvine, CA).
Total protease activity
Total protease activity was assessed using conditioned media prepared as described above and the Protease Fluorescent Detection Kit (Sigma Chemical Co., cat # PF0100). MFI values for conditioned medium from LAC were set to a value of 100% activity, with the activity observed in each mutant shown relative to this value. All assays included two biological replicates with at least three experimental replicates of each.
Cell lysis procedure
Bacterial cells from overnight cultures standardized as described above were harvested by centrifugation and resuspended in 750 μl of ice-cold TEG buffer (25 mM Tris at a pH of 8 and 25 mM EGTA). Cell suspensions were then transferred to 2 ml RNase/DNase free Fastprep Lysing Matrix B tubes (MP Biomedicals, cat. # 116911050). Cell suspensions were then lysed in a FastPrep®-24 benchtop homogenizer (MP Biomedicals, Solon, OH) for two separate 40 second intervals at a rate of 6.0 m/sec (interrupted by a 5 minute interval in which the homogenates were chilled on ice). After centrifugation at 15,000 X g at 4oC for 10 minutes, supernatants were aliquoted and stored at -20oC until use.
Preparation of samples for analysis of surface-associated Spa (sSpa)
To examine relative amounts of sSpa, samples enriched for surface-associated proteins were prepared as previously described.23 Here, bacterial cells from overnight cultures standardized as described above were harvested by centrifugation. Cell pellets were then resuspended to a density 1 × 109 cells per ml. Cells were then washed in distilled water before resuspending in 200 μl of filter-sterilized digestion buffer consisting of 100 μl of 1M Tris-HCl (pH 7.5), 50 μl of 5M NaCl, 675 mg of sucrose, 50 μl of 1M MgCl2, 25 μl of lysostaphin (10 mg/ml), 100 μl mutanolysin (1.25 mg/ml), 5 units of DNase I, 50 μl of 100 mM PMSF, 2.5 μl of 1M benzamidine, 50 μl of 100 mM Nα-p-Tosyl-L-arginine methyl ester hydrochloride (TAME), 25 μl leupeptin (1 mg/ml), and 12.5 μl pepstatin (1 mg/ml). Samples were then brought to a final volume of 2.5 ml using distilled water. Cell suspensions were then incubated at 37oC for 4 hours. The lysis reactions were then centrifuged at 6000 X g for 20 minutes at 4oC and the supernatants aliquoted and stored at -80oC until used for western blot.
Ethics statement
All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences and performed according to NIH guidelines, the Animal Welfare Act, and US Federal law.
Statistical analysis
To allow for statistical comparison across biological and experimental replicates from all in vitro assays, the results obtained for each mutant were averaged across all replicates. This average was then plotted relative to the results observed with LAC after setting the value observed with LAC either to 1.0 (western blots) or 100% (protease assay). Analysis of variance (ANOVA) models were then used to assess the statistical significance of the results observed with each mutant relative to LAC (Bonferroni correction). ANOVA methods were also used to analyze cfu data. Specifically, Dunnett's procedure was used to compare each mutant mean to the mean of LAC. The cfu data were log10-transformed prior to analysis, and P-values were calculated using permutation methods. P-values less than or equal to 0.05 were considered to be statistically significant. Statistical analyses were performed using the statistical programming language R version 3.3.3 (Vienna, Austria), SAS 9.4 (Cary, NC) and GraphPad Prism 5.0 (La Jolla, CA).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH grant R01-AI119380 to MSS. Additional support was provided by core facilities supported by the Center for Microbial Pathogenesis and Host Inflammatory Responses (P20-GM103450) and the Translational Research Institute (UL1TR000039).
References
- [1].Priest NK, Rudkin JK, Feil EJ, van den Elsen JM, Cheung A, Peacock SJ, Laabei M, Lucks DA, Recker M, Massey RC. From genotype to phenotype: Can systems biology be used to predict Staphylococcus aureus virulence? Nature Rev Microbiol. 2012;10:791-97. https://doi.org/ 10.1038/nrmicro2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bröker BM, Mrochen D, Péton V. The T cell response to Staphylococcus aureus. Pathogens. 2016;5:31. https://doi.org/ 10.3390/pathogens5010031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Flannagan RS, Heit B, Heinrichs DE. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens. 2015;4:826-68. https://doi.org/ 10.3390/pathogens4040826. PMID:26633519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wilde AD, Snyder DJ, Putnam NE, Valentino MD, Hammer ND, Lonergan ZR, Hinger SA, Aysanoa EE, Blanchard C, Dunman PM, et al.. Bacterial hypoxic responses revealed as critical determinants of the host-pathogen outcome by TnSeq analysis of Staphylococcus aureus invasive infection. PLoS Pathog. 2015;11:e1005341. https://doi.org/ 10.1371/journal.ppat.1005341. PMID:26684646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brady RA, Leid JG, Calhoun JH, Costerton JW, Shirtliff ME. Osteomyelitis and the role of biofilms in chronic infection. FEMS Immunol Med Microbiol. 2008;52:13-22. https://doi.org/ 10.1111/j.1574-695X.2007.00357.x. PMID:18081847 [DOI] [PubMed] [Google Scholar]
- [6].Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520-32. https://doi.org/ 10.1056/NEJM199808203390806. PMID:9709046 [DOI] [PubMed] [Google Scholar]
- [7].Beenken KE, Mrak LN, Griffin LM, Zielinska AK, Shaw LN, Rice KC, Horswill AR, Bayles KW, Smeltzer MS. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS ONE. 2010;5:e10790.5. https://doi.org/ 10.1371/journal.pone.0010790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yarwood JM, Bartels DJ, Volper EM, Greenberg PE. Quorum sensing in Staphylococcus aureus biofilms. J Bacteriol. 2004;186:1838-50. https://doi.org/ 10.1128/JB.186.6.1838-1850.2004. PMID:14996815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:11-12. https://doi.org/ 10.1086/595011 [DOI] [PubMed] [Google Scholar]
- [10].Lewis K. Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol. 2008;322:107-31. PMID:18453274 [DOI] [PubMed] [Google Scholar]
- [11].Fernandes P, Martens E. Antibiotics in late clinical development. Biochem Pharmacol. 2017;133:152-63. https://doi.org/ 10.1016/j.bcp.2016.09.025. PMID:27687641 [DOI] [PubMed] [Google Scholar]
- [12].Meeker DG, Beenken KE, Mills WB, Loughran AJ, Spencer HJ, Lynn WB, Smeltzer MS. Evaluation of antibiotics active against methicillin-resistant Staphylococcus aureus based on activity in an established biofilm. Antimicrob Agents Chemother. 2016;60:5688-94. https://doi.org/ 10.1128/AAC.01251-16. PMID:27401574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Maura D, Ballok AE, Rahme LG. Considerations and caveats in anti-virulence drug development. Curr Opin Microbiol. 2016;33:41-46. https://doi.org/ 10.1016/j.mib.2016.06.001. PMID:27318551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503:365-70. https://doi.org/ 10.1038/nature12790. PMID:24226776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Atwood DN, Loughran AJ, Courtney AP, Anthony AC, Meeker DG, Spencer HJ, Gupta RK, Lee CY, Beenken KE, Smeltzer MS. Comparative impact of diverse regulatory loci on Staphylococcus aureus biofilm formation. MicrobiologyOpen. 2015;4:436-51. https://doi.org/ 10.1002/mbo3.250. PMID:25810138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Atwood DN, Beenken KE, Lantz TL, Meeker DG, Lynn WB, Mills WB, Spencer HJ, Smeltzer MS. Regulatory mutations impacting antibiotic susceptibility in an established Staphylococcus aureus biofilm. Antimicrob Agents Chemother. 2016;60:1826-29. https://doi.org/ 10.1128/AAC.02750-15. PMID:26824954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boles BR, Horswill AR. Staphylococcal biofilm disassembly. Trends Microbiol. 2011;19:449-55. https://doi.org/ 10.1016/j.tim.2011.06.004. PMID:21784640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moormeier DE, Bayles KW. Staphylococcus aureus biofilm: A complex developmental organism. Mol Microbiol. 2017;104:365-76. https://doi.org/ 10.1111/mmi.13634. PMID:28142193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Painter KL, Krishna A, Wigneshweraraj S, Edwards AM. What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia? Trends Microbiol. 2014;22:676-85. https://doi.org/ 10.1016/j.tim.2014.09.002. PMID:25300477 [DOI] [PubMed] [Google Scholar]
- [20].Schweizer ML, Furuno JP, Sakoulas G, Johnson JK, Harris AD, Shardell MD, McGregor JC, Thom KA, Perencevich EN. Increased mortality with accessory gene regulator (agr) dysfunction in Staphylococcus aureus among bacteremic patients. Antimicrob Agents Chemother. 2011;55:1082-87. https://doi.org/ 10.1128/AAC.00918-10. PMID:21173172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Loughran AJ, Atwood DN, Anthony AC, Harik NS, Spencer HJ, Beenken KE, Smeltzer MS. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. MicrobiologyOpen. 2014;3:897-909. https://doi.org/ 10.1002/mbo3.214. PMID:25257373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Loughran AJ, Gaddy D, Beenken KE, Meeker DG, Morello R, Zhao H, Byrum SD, Tackett AJ, Cassat JE, Smeltzer MS. Impact of sarA and phenol-soluble modulins on the pathogenesis of osteomyelitis in diverse clinical isolates of Staphylococcus aureus. Infect Immun. 2016;84:2586-94. https://doi.org/ 10.1128/IAI.00152-16. PMID:27354444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zielinska AK, Beenken KE, Mrak LN, Spencer HJ, Post GR, Skinner RA, Tackett AJ, Horswill AR, Smeltzer MS. sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol Microbiol. 2012;86:1183-96. https://doi.org/ 10.1111/mmi.12048. PMID:23075270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mediavilla JR, Chen L, Mathema B, Kreiswirth BN. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA). Curr Opin Microbiol. 2012;15:588-95. https://doi.org/ 10.1016/j.mib.2012.08.003. PMID:23044073 [DOI] [PubMed] [Google Scholar]
- [25].Nimmo GR. USA300 abroad: Global spread of a virulent strain of community-associated methicillin-resistant Staphylococcus aureus. Clin Microbiol Infect. 2012;18:725-34. https://doi.org/ 10.1111/j.1469-0691.2012.03822.x. PMID:22448902 [DOI] [PubMed] [Google Scholar]
- [26].Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Ann Rev Microbiol. 2010;64:143-62. https://doi.org/ 10.1146/annurev.micro.112408.134309 [DOI] [PubMed] [Google Scholar]
- [27].Mootz JM, Benson MA, Heim CE, Crosby HA, Kavanaugh JS, Dunman PM, Kielian T, Torres VJ, Horswill AR. Rot is a key regulator of Staphylococcus aureus biofilm formation. Mol Microbiol. 2015;96:388-404. https://doi.org/ 10.1111/mmi.12943. PMID:25612137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet. 2008;42:541-64. https://doi.org/ 10.1146/annurev.genet.42.110807.091640. PMID:18713030 [DOI] [PubMed] [Google Scholar]
- [29].Abdelnour A, Arvidson S, Bremell T, Rydén C, Tarkowski A. The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect Immun. 1993;61:3879-85. PMID:8359909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cheung GYC, Wang R, Khan BA, Sturdevant DE, Otto M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect Immun. 2011;79:1927-35. https://doi.org/ 10.1128/IAI.00046-11. PMID:21402769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun. 1995;63:3373-80. PMID:7642265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Montgomery CP, Boyle-Vavra S, Daum RS. Importance of the global regulators agr and saeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS ONE. 2010;5:e15177. https://doi.org/ 10.1371/journal.pone.0015177. PMID:21151999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cameron DR, Lin YH, Trouillet-Assant S, Tafani V, Kostoulias X, Mouhtouris E, Skinner N, Visvanathan K, Baines SL, Howden B, et al.. Vancomycin-intermediate Staphylococcus aureus isolates are attenuated for virulence when compared to susceptible progenitors. Clin Microbiol Infect. 2017;pii: S1198-743X(17)30199-4. https://doi.org/ 10.1016/j.cmi.2017.03.027. PMID:28396035 [DOI] [PubMed] [Google Scholar]
- [34].Bose JL, Lehman MK, Fey PD, Bayles KW. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS ONE. 2012;7:e42244. https://doi.org/ 10.1371/journal.pone.0042244. PMID:22860095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Houston P, Rowe SE, Pozzi C, Waters EM, O'Gara JP. Essential role for the major autolysin in the fibronectin-binding protein-mediated Staphylococcus aureus biofilm phenotype. Infect Immun. 2011;79:1153-65. https://doi.org/ 10.1128/IAI.00364-10. PMID:21189325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Takahashi J, Komatsuzawa H, Yamada S, Nishida T, Labischinski H, Fujiwara T, Ohara M, Yamagishi J, Sugai M. Molecular characterization of an atl null mutant of Staphylococcus aureus. Microbiol Immunol. 2002;46:601-12. https://doi.org/ 10.1111/j.1348-0421.2002.tb02741.x. PMID:12437027 [DOI] [PubMed] [Google Scholar]
- [37].Zielinska AK, Beenken KE, Joo H-S, Mrak LN, Griffin LM, Luong TT, Lee CY, Otto M, Shaw LN, Smeltzer MS. Defining the strain-dependent impact of the staphylococcal accessory regulator (sarA) on the alpha-toxin phenotype of Staphylococcus aureus. J Bacteriol. 2011;193:2948-58. https://doi.org/ 10.1128/JB.01517-10. PMID:21478342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Beenken KE, Mrak LN, Zielinska AK, Atwood DN, Loughran AJ, Griffin LM, Matthews KA, Anthony AM, Spencer HJ, Post GR, et al.. Impact of the functional status of saeRS on in vivo phenotypes of Staphylococcus aureus sarA mutants. Mol Microbiol. 2014;92:1299-312. https://doi.org/ 10.1111/mmi.12629. PMID:24779437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Movitz J. Formation of extracellular protein A by Staphylococcus aureus. Eur J Biochem. 1976;68:291-99. https://doi.org/ 10.1111/j.1432-1033.1976.tb10788.x. PMID:964266 [DOI] [PubMed] [Google Scholar]
- [40].O'Hallorana DP, Wynneb K, Geoghegana JA. Protein A Is released into the Staphylococcus aureus culture supernatant with an unprocessed sorting signal. Infect Immun. 2015;83:1598-609. https://doi.org/ 10.1128/IAI.03122-14. PMID:25644005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Crosby HA, Schlievert PM, Merriman JA, King JM, Salgado-Pabón W, Horswill AR. The Staphylococcus aureus global regulator MgrA modulates clumping and virulence by controlling surface protein expression. PLoS Pathog. 2016;12:e1005604. https://doi.org/ 10.1371/journal.ppat.1005604. PMID:27144398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Foster TJ. Immune evasion by staphylococci. Nature Rev Microbiol. 2005;3:948-58. https://doi.org/ 10.1038/nrmicro1289 [DOI] [PubMed] [Google Scholar]
- [43].Li S, Arvidson S, Möllby R. Variation in the agr-dependent expression of alpha-toxin and protein A among clinical isolates of Staphylococcus aureus from patients with septicaemia. FEMS Microbiol Lett. 1997;152:155-61. https://doi.org/ 10.1016/S0378-1097(97)00195-X. PMID:9228782 [DOI] [PubMed] [Google Scholar]
- [44].Moellering RC. MRSA: The first half century. J Antimicrob Chemother. 2012;67:4-11. https://doi.org/ 10.1093/jac/dkr437. PMID:22010206 [DOI] [PubMed] [Google Scholar]
- [45].Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother. 1997;40:135-36. https://doi.org/ 10.1093/jac/40.1.135. PMID:9249217 [DOI] [PubMed] [Google Scholar]
- [46].Sievert DM, Boulton ML, Stoltman G, Johnson D, Stobierski MG, Downes FP, Somsel PA, Rudrik JT, Brown W, Hafeez W, et al.. Staphylococcus aureus resistant to vancomycin — United States. Atlanta: (GA: ): CDC MMWR; 2002. [Google Scholar]
- [47].Kumar K, Chopra S. New drugs for methicillin-resistant Staphylococcus aureus: An update. J Antimicrob Chemother. 2013;68:1465-70. https://doi.org/ 10.1093/jac/dkt045. PMID:23429643 [DOI] [PubMed] [Google Scholar]
- [48].Rodvold KA, McConeghy KW. Methicillin-resistant Staphylococcus aureus therapy: Past, present, and future. Clin Infect Dis. 2014;58:S20-S27. https://doi.org/ 10.1093/cid/cit614. PMID:24343828 [DOI] [PubMed] [Google Scholar]
- [49].Jansen KU, Girgenti DQ, Scully IL, Anderson AS. Vaccine review: "Staphyloccocus aureus vaccines: Problems and prospects.” Vaccine. 2013;31:2723-30. https://doi.org/ 10.1016/j.vaccine.2013.04.002. PMID:23624095 [DOI] [PubMed] [Google Scholar]
- [50].Boles BR, Horswill AR. agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 2008;4:e1000052. https://doi.org/ 10.1371/journal.ppat.1000052. PMID:18437240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Arya R, Ravikumar R, Santhosh RS, Princy SA. SarA based novel therapeutic candidate against Staphylococcus aureus associated with vascular graft infections. Front Microbiol. 2015;6:416. https://doi.org/ 10.3389/fmicb.2015.00416. PMID:26074884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gray B, Hall P, Gresham H. Targeting agr- and agr-like quorum sensing systems for development of common therapeutics to treat multiple gram-positive bacterial infections. Sensors. 2013;13:5130-66. https://doi.org/ 10.3390/s130405130. PMID:23598501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Muhs A, Lyles JT, Parlet CP, Nelson K, Kavanaugh JS, Horswill AR, Quave CL. Virulence inhibitors from Brazilian Peppertree block quorum sensing and abate dermonecrosis in skin infection models. Sci Rep. 2017;7:42275. https://doi.org/ 10.1038/srep42275. PMID:28186134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chien Y, Cheung AL. Molecular interactions between two global regulators, sar and agr, in Staphylococcus aureus. J Biol Chem. 1998;273:2645-52. https://doi.org/ 10.1074/jbc.273.5.2645. PMID:9446568 [DOI] [PubMed] [Google Scholar]
- [55].Beenken KE, Blevins JS, Smeltzer MS. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun. 2003;71:4206-11. https://doi.org/ 10.1128/IAI.71.7.4206-4211.2003. PMID:12819120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Valle J, Toledo-Arana A, Berasain C, Ghigo J-M, Amorena B, Penadés JR, Lasa I. SarA and not σB is essential for biofilm development by Staphylococcus aureus. Mol Microbiol. 2003;48:1075-87. https://doi.org/ 10.1046/j.1365-2958.2003.03493.x. PMID:12753197 [DOI] [PubMed] [Google Scholar]
- [57].Vuong C, Saenz HL, Götz F, Otto M. Impact of the agr quorum-sensing system on adherence to polystyrene in Staphylococcus aureus. J Infect Dis. 2000;182:1688-93. https://doi.org/ 10.1086/317606. PMID:11069241 [DOI] [PubMed] [Google Scholar]
- [58].Balamurugan P, Hema M, Kaur G, Sridharan V, Prabu PC, Sumana MN, Princy SA. Development of a biofilm inhibitor molecule against multidrug resistant Staphylococcus aureus associated with gestational urinary tract infections. Front Microbiol. 2015;6:832. https://doi.org/ 10.3389/fmicb.2015.00832. PMID:26322037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol. 2008;190:2257-65. https://doi.org/ 10.1128/JB.01545-07. PMID:18156263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tu Quoc PH, Genevaux P, Pajunen M, Savilahti H, Georgopoulos C, Schrenzel J, Kelley WL. Isolation and characterization of biofilm formation-defective mutants of Staphylococcus aureus. Infect Immun. 2007;75:1079-88. https://doi.org/ 10.1128/IAI.01143-06. PMID:17158901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ibberson CB, Jones CL, Singh S, Wise MC, Hart ME, Zurawski DV, Horswill AR. Staphylococcus aureus hyaluronidase is a CodY-regulated virulence factor. Infect Immun. 2014;82:4253-64. https://doi.org/ 10.1128/IAI.01710-14. PMID:25069977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, Daum RS. CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect Immun. 2012;80:2382-89. https://doi.org/ 10.1128/IAI.06172-11. PMID:22526672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bischoff M, Entenza JM, Giachino P. Influence of a functional sigB operon on the global regulators sar and agr in Staphylococcus aureus. J Bacteriol. 2001;183:5171-79. https://doi.org/ 10.1128/JB.183.17.5171-5179.2001. PMID:11489871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cheung AL, Chien Y, Bayer AS. Hyperproduction of alpha-hemolysin in a sigB mutant is associated with elevated SarA expression in Staphylococcus aureus. Infect Immun. 1999;67:1331-37. PMID:10024579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mitchell G, Fugère A, Gaudreau KP, Brouillette E, Frost EH, Cantin AM, Malouin F. SigB Is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS ONE. 2013;8:e65018. https://doi.org/ 10.1371/journal.pone.0065018. PMID:23705029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Tuchscherr L, Bischoff M, Lattar SM, Noto Llana M, Pförtner H, Niemann S, Geraci J, Van de Vyver H, Fraunholz MJ, Cheung AL, et al.. Sigma factor SigB is crucial to mediate Staphylococcus aureus adaptation during chronic infections. PLoS Pathog. 2015;11:e1004870. https://doi.org/ 10.1371/journal.ppat.1004870. PMID:25923704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shaw LN, Aish J, Davenport JE, Brown MC, Lithgow JK, Simmonite K, Crossley H, Travis J, Potempa J, Foster SJ. Investigations into σB-modulated regulatory pathways governing extracellular virulence determinant production in Staphylococcus aureus. J Bacteriol. 2006;188:6070-80. https://doi.org/ 10.1128/JB.00551-06. PMID:16923874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Herbert S, Ziebandt A-K, Ohlsen K, Schäfer T, Hecker M, Albrecht D, Novick R, Götz F. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect Immun. 2010;78:2877-89. https://doi.org/ 10.1128/IAI.00088-10. PMID:20212089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cheung AL, Eberhardt KJ, Chung E, Yeaman MR, Sullam PM, Ramos M, Bayer AS. Diminished virulence of a sar−/agr− mutant of Staphylococcus aureus in the rabbit model of endocarditis. J Clin Invest. 1994;94:1815-22. https://doi.org/ 10.1172/JCI117530. PMID:7962526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Heyer G, Saba S, Adamo R, Rush W, Soong G, Cheung A, Prince A. Staphylococcus aureus agr and sarA functions are required for invasive infection but not inflammatory responses in the lung. Infect Immun. 2002;70:127-33. https://doi.org/ 10.1128/IAI.70.1.127-133.2002. PMID:11748173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Smeltzer MS, Hart ME, Iandolo JJ. Phenotypic characterization of xpr, a global regulator of extracellular virulence factors in Staphylococcus aureus. Infect Immun. 1993;61:919-25. PMID:8432612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Peterson MM, Mack JL, Hall PR, Alsup AA, Alexander SM, Sully EK, Sawires YS, Cheung AL, Otto M, Gresham HD. Apolipoprotein B is an innate barrier against invasive Staphylococcus aureus infection. Cell Host Microbe. 2008;4:555-66. https://doi.org/ 10.1016/j.chom.2008.10.001. PMID:19064256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Surewaard BGJ, Nijland R, Spaan AN, Kruijtzer JAW, de Haas CJC, van Strijp JAG. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog. 2012;8:e1002606. https://doi.org/ 10.1371/journal.ppat.1002606. PMID:22457627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, Smeltzer MS, Torres VJ, Skaar EP. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe. 2013;13:759-72. https://doi.org/ 10.1016/j.chom.2013.05.003. PMID:23768499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].McNamara PJ, Milligan-Monroe KC, Khalili S, Proctor RA. Identification, cloning, and initial characterization of rot, a locus encoding a regulator of virulence factor expression in Staphylococcus aureus. J Bacteriol. 2000;182:3197-203. https://doi.org/ 10.1128/JB.182.11.3197-3203.2000. PMID:10809700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al.. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 2007;21:1353-66. https://doi.org/ 10.1101/gad.423507. PMID:17545468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Geisinger E, Adhikari RP, Jin R, Ross HF, Novick RP. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol Microbiol. 2006;61:1038-48. https://doi.org/ 10.1111/j.1365-2958.2006.05292.x. PMID:16879652 [DOI] [PubMed] [Google Scholar]
- [78].Jonsson IM, Lindholm C, Luong TT, Lee CY, Tarkowski A. mgrA regulates staphylococcal virulence important for induction and progression of septic arthritis and sepsis. Microbes Infect. 2008;10:1229-35. https://doi.org/ 10.1016/j.micinf.2008.07.026. PMID:18692591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].King JM, Kulhankova K, Stach CS, Vu BG, Salgado-Pabón W. Phenotypes and virulence among Staphylococcus aureus USA100, USA200, USA300, USA400, and USA600 clonal lineages. mSphere. 2016;1:e00071-16. https://doi.org/ 10.1128/mSphere.00071-16. PMID:27303750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Blevins JS, Gillaspy AF, Rechtin TM, Hurlburt BK, Smeltzer MS. The staphylococcal accessory regulator (sar) represses transcription of the Staphylococcus aureus collagen adhesin gene (cna) in an agr-independent manner. Mol Microbiol. 1999;33:317-26. https://doi.org/ 10.1046/j.1365-2958.1999.01475.x. PMID:10411748 [DOI] [PubMed] [Google Scholar]
- [81].Torres VJ, Attia AS, Mason WJ, Hood MI, Corbin BD, Beasley FC, Anderson KL, Stauff DL, McDonald WH, Zimmerman LJ, et al.. Staphylococcus aureus fur regulates the expression of virulence factors that contribute to the pathogenesis of pneumonia. Infect Immun. 2010;78:1618-28. https://doi.org/ 10.1128/IAI.01423-09. PMID:20100857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Luong TT, Sau K, Roux C, Sau S, Dunman PM, Lee CY. Staphylococcus aureus ClpC divergently regulates capsule via sae and codY in strain Newman but activates capsule via codY in strain UAMS-1 and in strain Newman with repaired saeS. J Bacteriol. 2011;193:686-94. https://doi.org/ 10.1128/JB.00987-10. PMID:21131496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Benson MA, Lilo S, Nygaard T, Voyich JM, Torres VJ. Rot and SaeRS cooperate to activate expression of the staphylococcal superantigen-like exoproteins. J Bacteriol. 2012;194:4355-65. https://doi.org/ 10.1128/JB.00706-12. PMID:22685286 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.