

Abstract
Introduction
Bronchial epithelial cell death and airway inflammation induced by cigarette smoke (CS) have been involved in the pathogenesis of COPD. GRP78, belonging to heat shock protein 70 family, has been implicated in cell death and inflammation, while little is known about its roles in COPD. Here, we demonstrate that GRP78 regulates CS-induced necroptosis and injury in bronchial epithelial cells.
Materials and methods
GRP78 and necroptosis markers were examined in human bronchial epithelial (HBE) cell line, primary mouse tracheal epithelial cells, and mouse lungs. siRNA targeting GRP78 gene and necroptosis inhibitor were used. Expression of inflammatory cytokines, mucin MUC5AC, and related signaling pathways were detected.
Results
Exposure to CS significantly increased the expression of GRP78 and necroptosis markers in HBE cell line, primary mouse tracheal epithelial cells, and mouse lungs. Inhibition of GRP78 significantly suppressed CS extract (CSE)-induced necroptosis. Furthermore, GRP78–necroptosis cooperatively regulated CSE-induced inflammatory cytokines such as interleukin 6 (IL6), IL8, and mucin MUC5AC in HBE cells, likely through the activation of nuclear factor (NF-κB) and activator protein 1 (AP-1) pathways, respectively.
Conclusion
Taken together, our results demonstrate that GRP78 promotes CSE-induced inflammatory response and mucus hyperproduction in airway epithelial cells, likely through upregulation of necroptosis and subsequent activation of NF-κB and AP-1 pathways. Thus, inhibition of GRP78 and/or inhibition of necroptosis could be the effective therapeutic approaches for the treatment of COPD.
Keywords: cigarette smoke, airway epithelium, glucose-regulated protein 78, necroptosis, airway injury
Introduction
COPD, characterized by not fully irreversible airflow limitation and abnormal lung inflammation, is the fourth leading cause of death worldwide.1,2 The pathological triad of COPD includes inflammation, protease–antiprotease imbalance, and oxidative stress. Although multiple factors increase the risk for COPD, tobacco smoking is still the main cause. It is clear that the interplay between genetic factors and environmental exposure leads to COPD, but the mechanisms by which cigarette smoke (CS) induces COPD are still poorly understood.
Glucose-regulated protein 78 (GRP78), belonging to heat shock protein 70 family, is responsible for the integrity of endoplasmic reticulum (ER) and protection from stress.3 Generally, GRP78 is regarded as an important ER chaperone, which can promote protein folding, maintain calcium homeostasis, and regulate the transmembrane ER inducers.4 GRP78 is primarily located in the ER lumen and is elevated in the early stage of ER stress.5 Interestingly, GRP78 also exists outside the ER and participates in multifaceted cellular activities. Moreover, emerging evidence indicates that ER stress can promote GRP78 to separate from the ER and to localize to multiple intracellular organelles including nucleus and mitochondria, thus regulating cell signaling and survival.6 Recently, two studies have shown that CS induces the elevation of GRP78.7,8 However, the eventual roles of GRP78 in CS-induced airway injury in COPD pathogenesis have not been illustrated.
Apoptosis and necrosis have been associated with CS-induced airway epithelial cell death. Necroptosis is a regulated form of necrosis that relies on the enzymatic activity of the receptor-interacting protein (RIP)1 and RIP3 in various diseases.9 The RIP1/3 kinases, which form a multiprotein complex termed the necrosome, are key regulators of necroptosis.10 A recent study has demonstrated that necroptosis contributes to the development of COPD.11 However, the specific mechanisms of CS-induced necroptosis in COPD are still unclear. Interestingly, Ouyang et al12 have shown that GRP78 also localizes to mitochondria in regulation of cell viability, and necroptosis has also been reported to link with the disintegration of mitochondria. Therefore, we hypothesize that GRP78 might regulate CS-induced necroptosis in airway epithelium.
Also, it is well established that airway inflammation and mucus hypersecretion are important cardinal COPD features. However, can the GRP78 or necroptosis regulate airway inflammation and/or mucus secretion? The current study aims to explore the roles and mechanisms of GRP78 in regulation of CS-induced necroptosis and subsequent inflammatory response and mucus hyperproduction in cultured human bronchial epithelial (HBE) cells.
Materials and methods
Cell culture, CS extract (CSE) preparation, and cell viability
HBE cell lines were obtained from a cell bank (CRL-2741; American Type Culture Collection [ATCC], Manassas, VA, USA) and cultured in Roswell Park Memorial Institute (RPMI) 1640 containing 10% fetal bovine serum (FBS) at 37°C in the presence of 5% CO2. CSE was prepared and treated as described previously.13,14 Primary mouse tracheal epithelial cells (MTECs) were isolated from the tracheas of wild-type (WT) mice. MTECs were cultured and differentiated according to the previously described methods.15,16 Cell viability was evaluated by cell counting kit 8 (CCK8) assay (LiankeBio, Hangzhou, China) according to the manufacturer’s instructions.
Chemicals and reagents
Necrostatin-1 (NEC-1) was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). The following antibodies were used: ACTIN, GRP78, phosphorylated (p)-P65, c-FOS, c-JUN, MUC5AC, and RIP1 obtained from Cell Signaling Technology (Danvers, MA, USA); phosphorylated mixed lineage kinase domain-like protein (p-MLKL) obtained from EMD Millipore (Billerica, MA, USA); and RIP3 obtained from Abcam (Cambridge, MA, USA).
Transfection
siRNAs for control and GRP78 were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The siRNA transfection reagent (GenMute) was purchased from SignaGen Laboratories (Rockville, MD, USA). All the transfections were accomplished following the manufacturer’s protocol as described previously.17 After 24 h, HBE cells were used for CSE exposure experiments.
RNA isolation and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was isolated with Trizol (Takara Bio Inc., Beijing, China) following the manufacturer’s protocol. Reverse transcription reagents (Takara Bio Inc.) were applied to reverse transcribe RNA. qRT-PCR was carried out to quantify the target genes’ expression with SYBR Green system (Takara Bio Inc.).
ELISA
Cellular supernatants were collected from treated cells. Human IL6 and IL8 enzyme-linked immunosorbnent assay (ELISA) kits (R&D Systems, Inc., Minneapolis, MN, USA) were used to measure IL6 and IL8 levels in cellular supernatants according to the manufacturer’s protocols.
Mice
WT mice from C57BL/6 background were purchased from the Animal Center of Slaccas (Shanghai, China). All the mice were exposed to room air or whole-body CS (2 h per day, 5 days per week) for 3 months, generated from research-grade cigarettes (3R4F; University of Kentucky, Lexington, KY, USA) in 5-l smoking chambers as previously described.17 The total particulate matter concentrations measured in the chamber were 160–180 mg/m3. At 24 h after the last CS exposure, mice were euthanized and lung tissues were removed for Western blotting. All the animal experimental protocols were approved by the ethical committee for Animal Studies at Zhejiang University and were in accordance with the “Guide for the care and use of laboratory animals” approved by the committee.
Western blotting
Total proteins from HBE cells, MTECs, or mouse lung tissues were extracted with ice-cold radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors. Protein samples were separated on 6%–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto the polyvinylidene fluoride (PVDF) membrane. After being blocked with 5% non-fat milk for 1 h, the membranes were immunoblotted using the above-mentioned antibodies by following the standard methods. Afterward, the membranes were washed with tris-buffered saline with tween 20 (TBST) for 5 min (three times) and then incubated with secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h. Target bands were visualized by a two-color infrared imaging system (Odyssey; LI-COR, Lincoln, NE, USA). Band densities were quantified using densitometry (Odyssey; LI-COR).
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Statistical tests were analyzed using GraphPad Prism software (version 6; GraphPad software, Inc., La Jolla, CA, USA). Differences between experimental and control groups were identified with an unpaired, two-tailed Student’s t-test. P-value <0.05 was considered significant.
Results
CS exposure induces GRP78 expression in HBE cells, in MTECs, and in mouse lungs
In vitro, HBE cells were treated with CSE, and the expression of GRP78 was measured. As expected, we observed that CSE exposure significantly induced the mRNA levels of GRP78 in dose- and time-dependent manners (Figure 1A), and this induction was further confirmed by Western blotting analysis (Figure 1B). In MTECs, GRP78 was induced significantly by CSE in protein levels (Figure 1C). To further confirm the expression of GRP78 in response to CS in vivo, WT mice were exposed to air or CS for 3 months. Western blotting analysis demonstrated that the levels of GRP78 were significantly increased in lung tissues of CS-exposed mice (Figure 1D).
Figure 1.

CSE induces GRP78 expression in HBE cells, MTECs, and mouse lungs.
Notes: HBE cells were exposed to various concentrations of CSE for 24 h or 2% CSE at indicated times. Expression of GRP78 was measured by qRT-PCR (A) or Western blotting (B). MTECs were treated with 2% CSE at indicated times, and the levels of GRP78 were assessed by Western blotting (C). Wild-type mice were exposed to room air or whole-body CS, and protein levels of GRP78 from mouse lung homogenate samples were measured by Western blotting analysis (D). Data (A–D) were mean ± SEM of three independent experiments. Western blots (B–D) were representative of three independent experiments. *P<0.05 and **P<0.01 (Student’s t-test).
Abbreviations: CS, cigarette smoke; CSE, cigarette smoke extract; GRP78, glucose-regulated protein 78; HBE, human bronchial epithelial; MTECs, mouse tracheal epithelial cells; qRT-PCR, quantitative reverse transcription polymerase chain reaction; SEM, standard error of the mean.
GRP78 regulates CS-induced necroptosis
It is clear that multiple patterns of cell death, eg, apoptosis, autophagy, and necroptosis, contribute to the development of COPD.11,13 We next assessed the role of GRP78 in regulation of CS-induced cell death. We utilized siRNA targeting GRP78 gene, and the knockdown effects of GRP78-siRNA are shown in Figures 2 and 3. Apparently, inhibition of GRP78 by siRNA attenuated CSE-induced cell death in HBE cells (Figure 2A). We further explored the functional significance of necroptosis in relationship to cell death caused by CS. Not surprisingly, we observed that CSE-induced cell death was markedly decreased by necroptosis inhibitor NEC-1, targeting RIP1 kinase activity (Figure 2B). Also, CS exposure induced an elevation in RIP1, RIP3, and p-MLKL protein levels in HBE cells, in MTECs, and in mouse lungs (Figure 2C–E), indicating an increased necroptotic cell death by CS in airway epithelial cells.
Figure 2.


GRP78 regulates CSE-induced necroptosis.
Notes: (A) HBE cells were transfected with control or GRP78-siRNA for 24 h and then were treated with 4% CSE for an additional 24 h. Cell death was determined by CCK8 assay. (B) HBE cells were cultured with NEC-1 (50 μM) or vehicle (DMSO) together with 4% CSE for 24 h. Cell death was determined by CCK8 assay. (C) HBE cells were exposed to 4% CSE at indicated times. Expressions of RIP1, RIP3, and p-MLKL were measured by Western blotting. MTECs were treated with 4% CSE at indicated times, and the protein levels of RIP1 and RIP3 were assessed by Western blotting (D). Wild-type mice were exposed to room air or CS, and protein levels of RIP1 and RIP3 from mouse lung homogenate samples were measured by Western blotting analysis (E). HBE cells were pretreated with control or GRP78-siRNA for 24 h. After that, cells were incubated with 4% CSE for an additional 12 h, and cell lysates were then subjected to Western blotting for RIP1, RIP3, p-MLKL, and GRP78 (F). Data (A–F) were mean ± SEM of three independent experiments. Western blots (C–F) were representative of three independent experiments. *P<0.05 and **P<0.01 (Student’s t-test).
Abbreviations: CS, cigarette smoke; CSE, cigarette smoke extract; GRP78, glucose-regulated protein 78; HBE, human bronchial epithelial; MTECs, mouse tracheal epithelial cells; CCK8, cell counting kit 8; NEC-1, necrostatin-1; DMSO, dimethyl sulfoxide; RIP, receptor-interacting protein; SEM, standard error of the mean.
Figure 3.


GRP78 regulates CSE-induced inflammatory response and mucus production in HBE cells.
Notes: (A and B) HBE cells were exposed to 2% CSE at indicated times, and qRT-PCR was performed to assess the relative level of IL6 and IL8 mRNA transcripts. HBE cells were transfected with control or GRP78-siRNA for 24 h, the relative mRNA levels of GRP78 were measured (C) and then were treated with 2% CSE for an additional 24 h. The relative levels of IL6 (D), IL8 (E), and MUC5AC (H) mRNA transcripts were measured by qRT-PCR, and the protein levels of IL6 (F) and IL8 (G) in the culture supernatants were measured by ELISA. (I) Expression of MUC5AC was measured by Western blotting. Data (A–I) were mean ± SEM of three independent experiments. Western blots (I) were representative of three independent experiments. *P<0.05 and **P<0.01 (Student’s t-test).
Abbreviations: CSE, cigarette smoke extract; GRP78, glucose-regulated protein 78; HBE, human bronchial epithelial; IL, interleukin; qRT-PCR, quantitative reverse transcription polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean.
GRP78 has been reported to regulate CSE-induced apoptosis in alveolar epithelial cells,18 and we next examined whether GRP78 could regulate necroptosis in HBE cells. Interestingly, knockdown of GRP78 by siRNA significantly attenuated necroptosis, as revealed by the downregulation of RIP1, RIP3, and p-MLKL (Figure 2F).
GRP78 and necroptosis regulate CSE-induced inflammatory response and mucus production in HBE cells
Inflammatory cytokines interleukin 6 (IL6) and IL8 have been involved in the pathogenesis of COPD. In HBE cells, CSE exposure time dependently increased the mRNA levels of IL6 and IL8 (Figure 3A and B). We next sought to assess the role of GRP78 and necroptosis in CS-induced inflammation. Not surprisingly, CSE exposure elicited the mRNA expression and protein secretion of IL6 and IL8, which were markedly attenuated in GRP78-deficient cells (Figure 3D–G). MUC5AC, as the predominant mucin, is highly inducible in pathological conditions and plays a pivotal role in COPD pathogenesis.19 Again, we observed that CSE induced a significant increase in MUC5AC mRNA transcripts, as well as protein levels in HBE cells, which were also alleviated by knockdown of GRP78 (Figure 3H and I). Moreover, inhibition of necroptosis by NEC-1 significantly attenuated CSE-induced production of IL6, IL8, and MUC5AC (Figure 4A–E). These data suggest that the GRP78 together with necroptosis cooperatively regulate the CS-induced inflammatory response and mucin hyperproduction in airway epithelium. GRP78 promotes CSE-induced inflammatory response and mucus hyperproduction by CSE in airway epithelial cells, likely through the induction of necroptosis.
Figure 4.


Necroptosis regulates CSE-induced inflammatory response and mucus production in HBE cells.
Notes: HBE cells were cultured with NEC-1 (50 μM) or vehicle (DMSO) together with 2% CSE for 24 h. The relative levels of IL6 (A) and IL8 (B) mRNA transcripts were measured by qRT-PCR, the protein levels of IL6 (C) and IL8 (D) in the culture supernatants were measured by ELISA, and the expression of MUC5AC was measured by Western blotting (E). Data (A–E) were mean ± SEM of three independent experiments. Western blots (E) were representative of three independent experiments. *P<0.05 and **P<0.01 (Student’s t-test).
Abbreviations: CSE, cigarette smoke extract; HBE, human bronchial epithelial; IL, interleukin; qRT-PCR, quantitative reverse transcription polymerase chain reaction; NEC-1, necrostatin-1; ELISA, enzyme-linked immunosorbent assay; DMSO, dimethyl sulfoxide; SEM, standard error of the mean.
GRP78 and necroptosis regulate CSE-induced inflammation and mucus production through the activation of nuclear factor (NF)-κB and activator protein 1 (AP-1) pathway, respectively
It has been well recognized that NF-κB is closely associated with the regulation of COPD-related inflammatory cytokines.20 We also observed that knockdown of P65 by siRNA significantly attenuated the mRNA transcripts of IL6 and IL8 induced by CSE (Figure 5A and B). Interestingly, inhibition of GRP78 by siRNA significantly decreased the activation of p-P65 (Figure 5D). Similar results were also observed in the NEC-1-treated cells (Figure 5E). It has been reported that AP-1 regulates MUC5AC production through two binding sites on the −3700/−3337 area of MUC5AC promoter.21 Our previous study reveals that the AP-1 is required for MUC5AC expression in HBE cells.22 Again, we confirmed that inhibition of c-JUN by siRNA obviously suppressed CSE-induced MUC5AC expression (Figure 5C). Next, we sought to clarify whether GRP78 along with necroptosis regulates CSE-induced production of MUC5AC through AP-1 pathways. As expected, we observed that CSE induced elevated protein levels of c-FOS and c-JUN, two subunits of AP-1, in HBE cells, which were effectively suppressed by GRP78 siRNA or NEC-1 (Figure 5D and E). These data suggest that GRP78 together with necroptosis upregulate CSE-induced inflammation and mucin through activation of NF-κB and AP-1 pathways, respectively.
Figure 5.


GRP78 and necroptosis regulate CSE-induced inflammation and mucus production through the activation of NF-κB and AP-1 pathways, respectively.
Notes: HBE cells were treated with indicated siRNA for 24 h, following treatment with 2% CSE for an additional 24 h. The relative mRNA levels of IL6 (A), IL8 (B), and MUC5AC (C) were measured by qRT-PCR. After siRNA transfection for 24 h, HBE cells were cultured with 2% CSE for 12 h, and the expressions of p-P65, c-FOS, and c-JUN were detected by Western blotting (D). HBE cells were cultured with NEC-1 (50 μM) or vehicle (DMSO) together with 2% CSE for 12 h, and the expressions of p-P65, c-FOS, and c-JUN were detected by Western blotting (E). Data (A–E) were mean ± SEM of three independent experiments. Western blots (D and E) were representative of three independent experiments. *P<0.05 and **P<0.01 (Student’s t-test).
Abbreviations: CSE, cigarette smoke extract; GRP78, glucose-regulated protein 78; HBE, human bronchial epithelial; NEC-1, necrostatin-1; p-P65, phosphorylated-P65; qRT-PCR, quantitative reverse transcription polymerase chain reaction; SEM, standard error of the mean; NF-κB, nuclear factor-κB; AP-1, activator protein 1.
Discussion
The major findings of this study can be summarized as follows (Figure 6): 1) CS increases the expression of GRP78, which orchestrates the epithelial necroptosis; 2) GRP78 and necroptosis are required for CS-induced pulmonary inflammation and mucus hyperproduction in COPD pathogenesis; 3) GRP78 and necroptosis modulate CS-induced pulmonary inflammation and mucus hyper-production possibly through NF-κB and AP-1 pathways, respectively. Collectively, these data provide new insights into the deleterious role of GRP78 and necroptosis in COPD pathogenesis.
Figure 6.
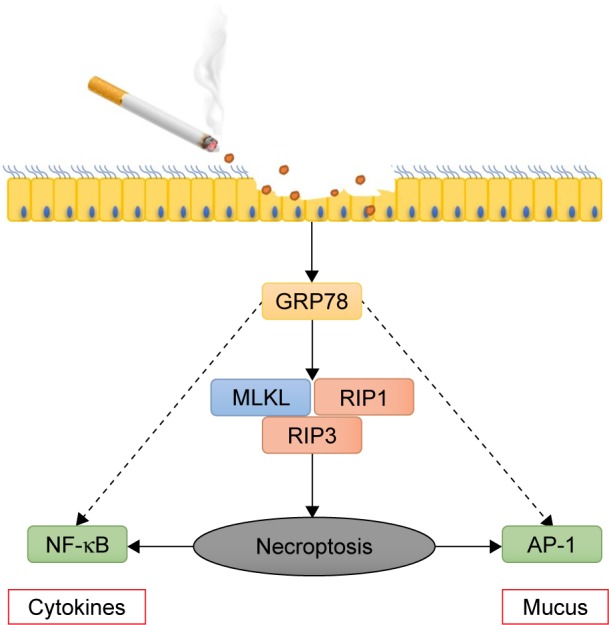
Schematic graphical summary of the deleterious role of GRP78 in CS-induced necroptosis and injury in bronchial epithelium.
Notes: CS exposure induces GRP78 and subsequently elicits necroptosis in bronchial epithelium. GRP78 promotes CSE-induced pulmonary inflammation and mucus hyperproduction, likely through upregulation of necroptosis and subsequent activation of NF-κB and AP-1 pathways, eventually contributing to COPD development.
Abbreviations: CS, cigarette smoke; CSE, cigarette smoke extract; GRP78, glucose-regulated protein 78; NF-κB, nuclear factor-κB; AP-1, activator protein 1.
The eventual functions of GRP78 in human diseases are likely cell- and pathogen-dependent, being either deleterious or protective. For example, liver-specific deletion of GRP78 increases the susceptibility to a variety of acute and chronic hepatic disorders,23 whereas mice with endometrium-specific deletion of GRP78 block endometrial cancer development.24 Accumulated studies have shown that the level of GRP78 is elevated in pulmonary diseases, including neutrophilic asthma, idiopathic pulmonary fibrosis, and acute lung injury.25–27 Consistent with these studies, our current data demonstrate that CS exposure induces the GRP78 expression in airway epithelial cells and in mouse lungs. We observe that knockdown of GRP78 significantly attenuates CSE-induced inflammation and mucus hyperproduction in HBE cells, indicating that GRP78 plays a deleterious role in the pathogenesis of COPD. In terms of mechanisms, we also provide evidence that the deleterious role of GRP78 in COPD is linked with epithelial necroptosis and subsequent activation of the NF-κB and AP-1 pathways. To the best of our knowledge, this is the first study to illuminate the role and mechanisms of GRP78 in CS-induced epithelial injury.
GRP78 is conventionally regarded as a critical ER chaperone, promoting protein folding and regulating ER stress signaling. Fan et al28 have reported that 4-phenyl butyric acid, an ER stress inhibitor, blocks oxygen–glucose deprivation-induced necroptosis in microglia. We also found that knockdown of GRP78 in HBE cells significantly alleviated CSE-induced necroptosis, suggesting GRP78 as a key positive regulator of necroptosis. Of note, the specific mechanisms of GRP78 in the regulation of necroptosis remain poorly known, and further studies are needed. Interestingly, Mizumura et al11 showed that RIP3 is elevated in mouse lungs with chronic CS exposure, which is consistent with our current results. The detailed mechanisms of how necroptosis regulates cellular inflammation remain largely unclear. In general, cells undergoing necroptosis are unlikely to produce massive cytokines or other proteins, but they could release certain toxic molecules such as high mobility group box-1 protein (HMGB1),29 which then damages the neighbor cells to produce inflammatory cytokines.
Neutrophilic inflammation is a cardinal feature for CS-induced lung injury, which induces the release of substantial proteases, thus ultimately contributing to airway injury and emphysematous lung destruction.30 In rheumatoid arthritis, pro-inflammatory cytokines can activate GRP78 that in turn induce more inflammatory cytokines, establishing a vicious inflammation cycle.31 Similarly, the present study shows that knockdown of GRP78 apparently attenuates CSE-induced inflammation in airway epithelial cells. We also provide evidence that inhibition of necroptosis by NEC-1 effectively relieves CSE-induced inflammation in cultured HBE cells. Moreover, it is well known that mucus hyperproduction can lead to airway obstruction, indicative of poor prognosis. Mucus hyperproduction is another important feature of COPD, which can contribute to morbidity and mortality especially in advanced COPD patients. This study clearly indicates that GRP78 and necroptosis are required for CSE-induced mucus hyperproduction in HBE cells. Thus, these data altogether suggest that GRP78 exerts a crucial role in CS-induced lung inflammation and mucus hyperproduction, likely through the activation of necroptosis. However, GRP78 acts in multiple cellular processes, and NEC-1 also blocks other cellular processes such as RIPK1-dependent apoptosis and indoleamine 2,3-dioxygenase, and thus, it is likely that GRP78 regulates CS-induced inflammation and mucus production through different pathways other than necroptosis, which requires further investigation.
NF-κB and AP-1 pathways have been involved in the pathogenesis of COPD.32 Here, we provide clear evidence that activation of NF-κB and AP-1 pathways is responsible for the regulation of CSE-induced inflammation and mucus hyperproduction, respectively. We also demonstrate that GRP78 together with necroptosis modulates the NF-κB and AP-1 activation in HBE cells. Recent research focuses on the relation between necroptosis and NF-κB pathway,33,34 while little is known about necroptosis’s role in the AP-1 pathway. Interestingly, we found that necroptosis also regulates the AP-1 pathway in HBE cells. Furthermore, CSE also induced c-JUN or c-FOS transcription levels, which might be the result of the phosphorylation of c-JUN or c-FOS.35 But our study showed that inhibition of GRP78 or necroptosis only downregulated the protein levels of c-JUN and c-FOS, but failed to affect their phosphorylated status.
Conclusion
We show for the first time that GRP78 and subsequent activation of necroptosis are essential for CS-induced inflammation and mucus hyperproduction in airway epithelial cells, likely through modulation of NF-κB and AP-1 pathways. Thus, inhibition of GRP78 and/or inhibition of necroptosis may represent novel therapeutic strategies for CS-induced airway injury.
Acknowledgments
This work was supported by the General Projects (81370142 and 81670031 to Z-H Chen) from the National Natural Science Foundation of China, the Key Science-Technology Innovation Team of Zhejiang Province (2011R50016), the National key research and development program (2016YFA0501802 to Z-H Chen), and project from the National Clinical Research Center of China for Respiratory Disease.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 2.Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD) Lancet. 2004;364(9434):613–620. doi: 10.1016/S0140-6736(04)16855-4. [DOI] [PubMed] [Google Scholar]
- 3.Tavaria M, Gabriele T, Kola I, Anderson R. A Hitchhiker’s guide to the human Hsp70 family. Cell Stress Chaperones. 1996;1(1):23–28. doi: 10.1379/1466-1268(1996)001<0023:ahsgtt>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behnke J, Feige MJ, Hendershot LM. BiP and its nucleotide exchange factors Grp170 and Sil1: mechanisms of action and biological functions. J Mol Biol. 2015;427(7):1589–1608. doi: 10.1016/j.jmb.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197(7):857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434(2):181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tagawa Y, Hiramatsu N, Kasai A, et al. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP) Free Radic Biol Med. 2008;45(1):50–59. doi: 10.1016/j.freeradbiomed.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Kelsen SG, Duan X, Ji R, Perez O, Liu C, Merali S. Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am J Respir Cell Mol Biol. 2008;38(5):541–550. doi: 10.1165/rcmb.2007-0221OC. [DOI] [PubMed] [Google Scholar]
- 9.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 11.Mizumura K, Cloonan S, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouyang YB, Xu LJ, Emery JF, Lee AS, Giffard RG. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion. 2011;11(2):279–286. doi: 10.1016/j.mito.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10):e3316. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–18885. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam HC, Choi AM, Ryter SW. Isolation of mouse respiratory epithelial cells and exposure to experimental cigarette smoke at air liquid interface. J Vis Exp. 2011;48:2513. doi: 10.3791/2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu YP, Wu YF, Zhang C, et al. Activating transcription factor 3 is essential for cigarette smoke-induced mucin expression via interaction with activator protein-1. Am J Pathol. 2017;187(2):280–291. doi: 10.1016/j.ajpath.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Zhou J, Zhao Y, Zhou H, et al. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1042–L1052. doi: 10.1152/ajplung.00418.2015. [DOI] [PubMed] [Google Scholar]
- 18.He B, Luo B, Chen Q, Zhang L. Cigarette smoke extract induces the expression of GRP78 in A549 cells via the p38/MAPK pathway. Mol Med Rep. 2013;8(6):1683–1688. doi: 10.3892/mmr.2013.1724. [DOI] [PubMed] [Google Scholar]
- 19.Cohn L. Mucus in chronic airway diseases: sorting out the sticky details. J Clin Invest. 2006;116(2):306–308. doi: 10.1172/JCI27690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee IT, Yang CM. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013;2013:791231. doi: 10.1155/2013/791231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gensch E, Gallup M, Sucher A, et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J Biol Chem. 2004;279(37):39085–39093. doi: 10.1074/jbc.M406866200. [DOI] [PubMed] [Google Scholar]
- 22.Chen ZH, Wu YF, Wang PL, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12(2):297–311. doi: 10.1080/15548627.2015.1124224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology. 2011;54(1):229–239. doi: 10.1002/hep.24368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin YG, Shen J, Yoo E, et al. Targeting the glucose-regulated protein-78 abrogates Pten-null driven AKT activation and endometrioid tumorigenesis. Oncogene. 2015;34(43):5418–5426. doi: 10.1038/onc.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim SR, Kim DI, Kang MR, et al. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor kappaB activation. J Allergy Clin Immunol. 2013;132(6):1397–1408. doi: 10.1016/j.jaci.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 26.Zeng M, Sang W, Chen S, et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett. 2017;271:26–37. doi: 10.1016/j.toxlet.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 27.Ayaub EA, Kolb PS, Mohammed-Ali Z, et al. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. Am J Pathol. 2016;239(4):411–425. doi: 10.1002/path.4738. [DOI] [PubMed] [Google Scholar]
- 28.Fan H, Tang HB, Kang J, et al. Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience. 2015;311:362–373. doi: 10.1016/j.neuroscience.2015.10.049. [DOI] [PubMed] [Google Scholar]
- 29.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Sharafkhaneh A, Hanania NA, Kim V. Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc. 2008;5(4):475–477. doi: 10.1513/pats.200708-126ET. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoo SA, You S, Yoon HJ, et al. A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. J Exp Med. 2012;209(4):871–886. doi: 10.1084/jem.20111783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Stefano A, Caramori G, Oates T, et al. Increased expression of nuclear factor-B in bronchial biopsies from smokers and patients with COPD. Eur Respir J. 2002;20(3):556–563. doi: 10.1183/09031936.02.00272002. [DOI] [PubMed] [Google Scholar]
- 33.Yatim N, Jusforgues-Saklani H, Orozco S, et al. RIPK1 and NF-B signaling in dying cells determines cross-priming of CD8+ T cells. Science. 2015;350(6258):328–334. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moriwaki K, Chan FK-M. Necroptosis-independent signaling by the RIP kinases in inflammation. Cell Mol Life Sci. 2016;73(11–12):2325–2334. doi: 10.1007/s00018-016-2203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitani A, Ito K, Vuppusetty C, Barnes PJ, Mercado N. Restoration of corticosteroid sensitivity in chronic obstructive pulmonary disease by inhibition of mammalian target of rapamycin. Am J Respir Crit Care Med. 2016;193(2):143–153. doi: 10.1164/rccm.201503-0593OC. [DOI] [PMC free article] [PubMed] [Google Scholar]