

Abstract
A rhodium-catalyzed intramolecular silylation of alkyl C–H bonds has been developed that occurs with unusual selectivity for the C–H bonds located δ to the oxygen atom of an alcohol-derived silyl ether over typically more reactive C–H bonds more proximal to the same oxygen atom. (Hydrido)silyl ethers, generated in situ by dehydrogenative coupling of tertiary alcohols with diethylsilane, undergo regioselective silylation at a primary C–H bond δ to the hydroxyl group in the presence of [(Xantphos)Rh(Cl)] as catalyst. Oxidation of the resulting 6-membered oxasilolanes generates 1,4-diols. This silylation and oxidation sequence provides an efficient method to synthesize 1,4-diols by a hydroxyl-directed, aliphatic C–H bond functionalization reaction and is distinct from the synthesis of 1,3-diols from alcohols catalyzed by iridium. Mechanistic studies show that the rhodium-catalyzed silylation of alkyl C–H bonds occurs with a resting state and relative rates for elementary steps that are significantly different from those for the rhodium-catalyzed silylation of aryl C–H bonds. The resting state of the catalyst is a (Xantphos)Rh(I)(SiR3)-(norbornene) complex, and an analogue was synthesized and characterized crystallographically. The rate-limiting step of the process is oxidative addition of the δ C–H bond to Rh. Computational studies elucidated the origin of high selectivity for silylation of the δ C–H bond when Xantphos-ligated rhodium is the catalyst. A high barrier for reductive elimination from the six-membered metalacyclic, secondary alkyl intermediate formed by cleavage of the γ C–H bond and low barrier for reductive elimination from the seven-membered metalacyclic, primary alkyl intermediate formed by cleavage of the δ C–H accounts for the selective functionalization of the δ C–H bond.
Graphical Abstract
INTRODUCTION
Catalytic functionalization of C–H bonds with main group reagents, such as boranes and silanes, has become a practical synthetic methodology because it occurs with high regioselectivity and generates intermediates that can be transformed into a variety of products.1,2 Catalytic silylations of C–H bonds occurs both intramolecularly and intermolecularly, but catalytic intramolecular silylation occurs with high regioselectivity, due to the formation of cyclic silane products.3–7 We first reported the iridium-catalyzed intramolecular silylation of primary C–H bonds of silyl ethers, that had been generated from silanes and either alcohols or ketones, to form 5-membered oxasilolanes.8 We and others have expanded the scope of this process and applied it to the derivatization of carbohydrates and additional terpene natural products.8,9 Oxidation of the oxasilolanes in one pot generates 1,3-diols (Scheme 1, top).
Scheme 1.

Intramolecular C–H Silylation Forming Cyclic Silyl Ethers and Subsequent Oxidation to Diols
C–H silylation to form a 6-membered cyclic product is also desirable because it would generate 1,4-functionalized products, instead of 1,3-functionalized products. However, formation of a 6-membered cyclic silane is less favorable because it requires the generation of a medium-sized, 7-membered metalacyclic intermediate. The few examples of C–H silylation to generate 6-membered cyclic silanes are limited to the functionalization of activated benzylic C–H bonds.10 Catalytic silylation of unactivated C–H bonds directed by a common functional group to form a 6-membered cyclic silane is unknown. Radical methods for the oxygenation of alkyl C–H bonds δ to an oxygen are known, but in most cases a cyclic ether is formed.11a A nitrate ester has been installed in certain cases at the δ position when the Barton reaction was conducted in the presence of oxygen,11b,c but this process is a stoichiometric, photochemical reaction.
The mechanism of rhodium-catalyzed silylations of C–H bonds has been investigated previously by our group.12 The resting state of the catalyst in the intermolecular silylation of arenes catalyzed by the combination of Rh and a derivative of the bisphosphine Biphep with cyclohexene as hydrogen acceptor was (bisphosphine)Rh(III)(SiR3)(H)2, and the rate-limiting step was found to be the reductive elimination of cyclohexane. The resting state of the catalyst in a subsequent enantioselective intramolecular silylation of aryl C–H bonds revealed a similar Rh(III) resting state, as well as a complex with the hydrogen acceptor, and the rate-determining step was a related reductive elimination of the reduced hydrogen acceptor.12b It is unclear whether the same type of resting state would be present in the reactions with catalysts that form larger metalacyclic intermediates, whether the rate-determining step would be the same, and, most significant for the current study, how the identity and reactivity of these intermediates would influence the regioselectivity.
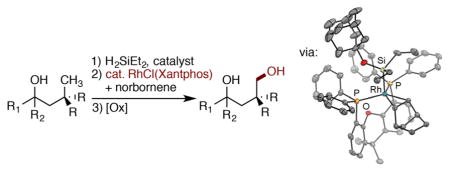
We report highly selective, silylations of unactivated, primary C–H bonds located δ to hydroxyl groups (Scheme 1, bottom) catalyzed by a rhodium complex of Xantphos (Xantphos, 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene). Oxidation of the resulting oxasilolanes generates 1,4-diols. The catalyst for this process is unusual for C–H bond functionalization; complexes of Xantphos have not been shown to catalyze the silylation of alkyl C–H bonds, and ligands like Xantphos with a wide bite-angle have rarely been used for any type of metal-catalyzed functionalization of alkyl C–H bonds.13,14 Mechanistic studies show that the rhodium-catalyzed silylation of alkyl C–H bonds occurs with a resting state and relative rates of elementary steps that are significantly different from those for the rhodium-catalyzed silylation of aryl C–H bonds and that the unusual regioselectivity results from a high barrier to reductive elimination of secondary alkyl groups from rhodium complexes ligated by Xantphos.
RESULTS AND DISCUSSION
Reaction Development
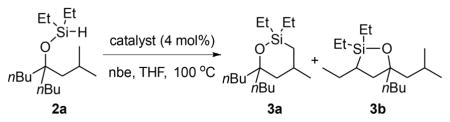
To evaluate potential silylations of alkyl C–H bonds located δ to an alcohol, we studied the reaction of silyl ether 2a in Table 1. Ether 2a is derived from the corresponding tertiary alcohol 1a by a dehydrogenative coupling with diethylsilane catalyzed by 0.1 mol % [Ir(OMe)-(COD)]2. Ether 2a contains primary, secondary, and tertiary C–H bonds that could undergo silylation. Primary C–H bonds are typically more reactive than secondary and tertiary C–H bonds toward iridium-catalyzed silylation. However, the primary C–H bond in 2a is located at the position δ to the oxygen atom of the ether, rather than the position γ to oxygen that has undergone silylation in previous work.8a Secondary C–H bonds located γ to oxygen have been shown recently to undergo silylation;8b therefore, it was unclear whether the reaction would occur at the secondary C–H bond located γ to the oxygen atom or the primary C–H bond located δ to the oxygen atom.
Table 1.
Catalytic Silylation of Primary C–H Bonda
| ||
|---|---|---|
|
| ||
| entry | catalyst | Yield% (3a: 3b) |
| 1 | [lr(OMe)(COD)]2/Me4Phen | 94 (0.18 : 1) |
| 2 | [lr(OMe)(COD)]2/BINAP (L1) | <5 (ND) |
| 3 | [RhCl(COD)]2/BINAP (L1) | 10 (0.48 : 1) |
| 4 | [RhCKCOD]2/Segphos (L2) | 15 (0.50 : 1) |
| 5 | [RhCI(COD)]2/DTBM-Segphos (L3) | 48 (0.50 : 1) |
| 6 | [RhCI(COD)]2/Xantphos (L4) | 80 (> 20 : 1) |
| 7 | [RhCI(COD)]2/MeO-Xantphos (L5) | 68 (> 20 : 1) |
| 8 | [RhCl(COD)]2/CF3-Xantphos (L6) | 71 (> 20 : 1) |
| 9 | [RhCI(COD)]2/Xantphos-NEt2 (L7) | <5 (ND) |
| 10 | [RhCI(COD)]2/Xantphos-tBu (L8) | <5 (ND) |
| 11 | RhCI(Xantphos) | 96 (>20 : 1) |
| ||
Conditions: 2a (1.0 equiv), catalyst (4 mol % monomer), nbe (1.2 equiv), THF, 100 °C, 16 h. Yields were determined by GC using n-dodecane as an internal standard. ND, not determined.
The combination of an iridium precatalyst and 3,4,7,8-tetramethyl-1,10-phenanthroline (Me4Phen) as ligand catalyzed the reaction of 2a to form products from C–H bond silylation in high yield. However, the reaction occurred at both secondary and primary C–H bonds to give a mixture of the five-membered and six-membered oxasilolanes (entry 1, Table 1). The reaction favored silylation at the secondary C–H bond by a factor of about 5, indicating that the ring size, rather than the degree of substitution at the C–H bond controlled the site selectivity. Silylation at the tertiary C–H bond was not observed.
To identify a catalyst that reacted with distinct selectivity to form the six-membered oxasilolane product selectively, we explored the combination of iridium and rhodium catalyst precursors with a range of bisphosphine ligands in place of bipyridine-type ligands. In the presence of [Ir(OMe)(COD)]2 as the source of iridium and BINAP as the ligand, the silylation of C–H bonds was not observed (entry 2). In contrast, the reactions conducted with a rhodium precursor and BINAP, Segphos, or DTBM-Segphos as ligand occurred to form the products of silylation of an alkyl C–H bond (entries 3–5), albeit in low to moderate yields. As observed for silylation catalyzed by the complex generated from iridium and Me4Phen, the silylation of the secondary C–H bond at the position γ to oxygen was favored over silylation of the primary C–H bond at the position δ to oxygen. However, the silylation reaction catalyzed by the combination of [RhCl(COD)]2 and Xantphos occurred at the C–H bond located δ to oxygen to give the six-membered oxasilolane exclusively (entry 6). The yield of the reaction was a high 96% when conducted with the preformed complex RhCl(Xantphos) as catalyst (entry 11).15
Reactions with a series of Xantphos derivatives showed that the electronic and steric properties of the ligand had a strong influence on the yield of the silylation process. Aromatic Xantphos derivatives generate the most active catalysts. The yields of reactions catalyzed by rhodium complexes of methoxy-substituted or trifluoromethyl-substituted Xantphos were lower than those of reactions conducted with the unsubstituted Xantphos (entries 7 and 8). The reactions catalyzed by the combination of [RhCl(COD)]2 and amino-Xantphos analogue L7 or the hindered t-butyl-Xantphos analogue L8 did not give any products from silylation of C–H bonds (entries 9 and 10).
Scope of Rh-Catalyzed Silylation of C–H Bonds
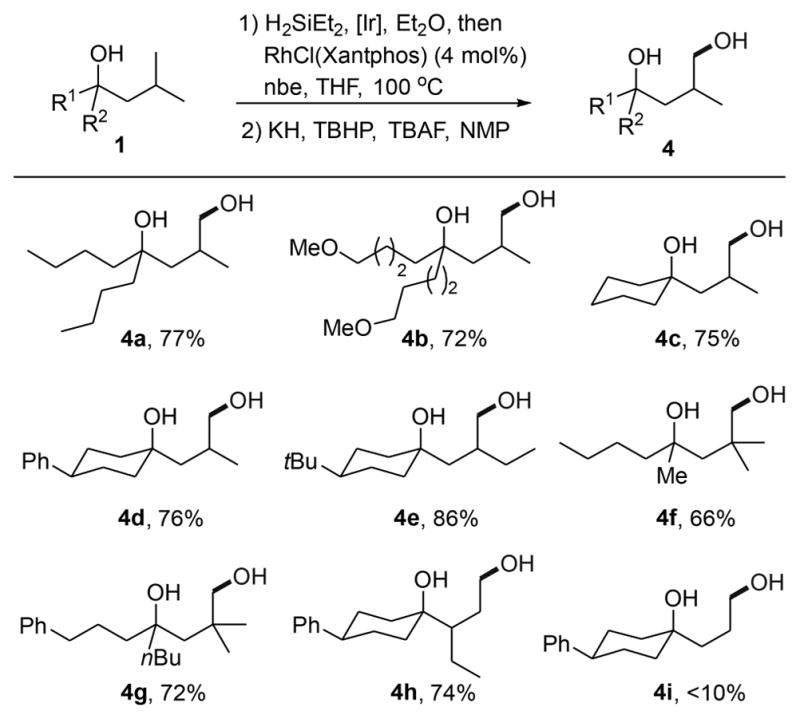
Examples of the Rh-catalyzed silylation of primary C–H bonds located δ to the oxygen of tertiary alcohols are summarized in Table 2. The yields shown correspond to the 1,4-diols formed by oxidation of the six-membered oxasilolanes. The reactions occurred in good yields with a variety of tertiary alcohols bearing primary C–H bonds δ to oxygen. Branching at the alkyl chain was important for the silylation to occur; reactions of tertiary alcohols containing an iso-butyl group occurred selectively to give the products of silylation at the primary C–H bond (4a, 4b). In the presence of primary C–H bonds at carbons located γ and δ to oxygen in the same substrate, the silylation process occurs at the γ C–H over the δ C–H bond.
Table 2.
Scope of Rh-Catalyzed C–H Silylationa
|
Conditions: 1 (1.0 equiv), Et2SiH2 (1.5 equiv), [Ir(cod)OMe]2 (0.10–0.20 mol %), Et2O, room temperature (rt); removal of volatiles, then RhCl(Xantphos) (4 mol %), nbe (1.2 equiv), THF, 100 °C. Isolated yields are reported.
In addition to acyclic alcohols, cyclic tertiary alcohols underwent silylation at a primary C–H bond δ to the hydroxyl group (4c, 4d). The reaction occurred at this primary C–H bond over the secondary C–H bond of the ring and at a primary C–H bond δ to the OH group when a primary C–H bond was present at both the δ and ε positions (e.g., to form 4a or 4e). The catalyst is not highly sensitive to steric hindrance α to the reacting C–H bond; hydroxylation occurred in 66–72% yield at the hindered methyl C–H bonds in the neopentyl groups of 4f and 4g. In addition to tertiary alcohols with branching γ to the hydroxyl group, alcohols with branching β to the hydroxyl group underwent silylation selectively at the primary C–H bond (4h). The reaction did not occur to give substantial amounts of product when the alkyl chain lacked branching altogether, as shown by the low yield of the reaction to form product 4i.
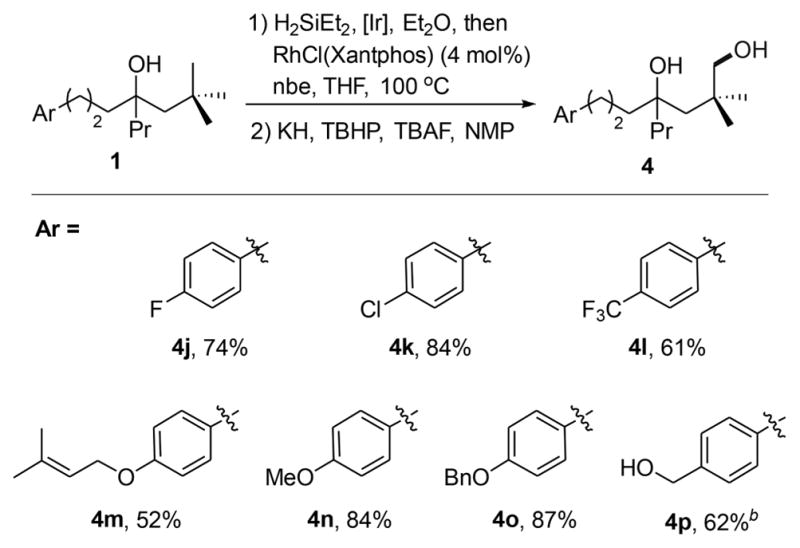
The functional group tolerance of the process leading to hydroxylation of the C–H bond located δ to the existing hydroxyl group is illustrated by the examples in Table 3. Aryl halides, an aryl trifluoromethyl group, and a trisubstituted alkene were compatible with the reaction conditions (4j–4m). A phenol and an alcohol protected as methyl, benzyl and TBS ethers were also tolerated (4n–4p). The TBS group in the silylation product cleaved during oxidation, producing a triol (4p).
Table 3.
Functional Group Tolerance of C–H Silylationa
|
Conditions: 1 (1.0 equiv), Et2SiH2 (1.5 equiv), [Ir(cod)OMe]2 (0.2 mol %), Et2O, room temperature (rt); removal of volatiles, then RhCl(Xantphos) (4 mol %), nbe (1.2 equiv), THF, 100 °C. Isolated yields are reported.
Starting from a tert-butyldimethylsilyl ether.
The compatibility of the reaction conditions with carbonyl groups was further investigated. In the presence of RuCl2(PPh3)3 as a catalyst, the reactions of diethylsilane with tertiary alcohols containing ester and carbamate groups cleanly formed the diethyl(hydrido)silyl ether precursor to the C–H bond functionalization process. The Rh-catalyzed silylation of C–H bonds then formed the six-membered oxasilolanes, demonstrating compatibility of the catalytic chemistry with the carbonyl groups in these silyl ethers. However, this functionality in products 3q–3s (eq 1) was not tolerated by
 |
(1) |
the highly basic conditions required for the oxidation of the resulting oxasilolanes under Fleming–Tamao conditions. Thus, reactions at the ester and carbamate must be conducted prior to the Fleming–Tamao oxidation. Perhaps most striking, the silylation of an alkyl C–H bond under these conditions is more tolerant of functional groups than common oxidation of a C–Si bond.
This silylation of an unactivated C–H bond also enables the functionalization of two primary C–H bonds in an iso-butyl group sequentially. Starting from alcohol 1d shown in Scheme 2, the first C–H silylation and oxidation occurred in good yield to give the hydroxylation product 4d. After protection of the primary alcohol as a silyl ether (5d), a second C–H bond was silylated. Oxidation of the resulting oxasilolane and in situ deprotection of the silyl ether under the conditions of Fleming-Tamao oxidation provided the triol product 6d.
Scheme 2.
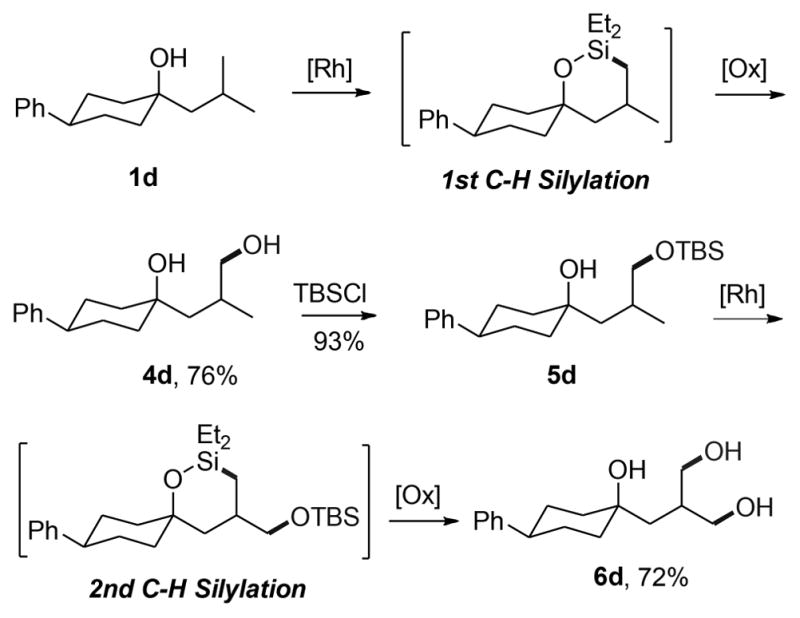
Rh-Catalyzed Sequential C–H Silylation
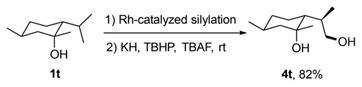
Because the Rh-catalyzed silylation is highly selective and tolerant of functional groups, this process could be applied to the direct functionalization of natural products and their derivatives bearing tertiary alcohols. In an initial example, the reaction of the silyl ether derived from 1-methylneomenthol, synthesized from menthone and MeMgBr, led to the site-selective functionalization of the primary C–H bond to give the corresponding 1,4-diol 4t. This silylation produced a single diastereomer with the relative configuration confirmed by X-ray crystallography.
 |
(2) |
Many more complex, naturally occurring compounds contain multiple hydroxyl groups. To determine the feasibility of conducting site-selective silylation directed by one of the hydroxyl groups in more complex structures, we studied the alcohol-directed oxygenation of 20(S)-hydroxycholesterol, a naturally occurring oxysterol (Scheme 3). Oxysterols function as signaling molecules that modulate a range of physiological phenomena including transportation of lipids and control over cellular states.16 Oxidation of the C–H bonds in these molecules changes their physical properties.17
Scheme 3.
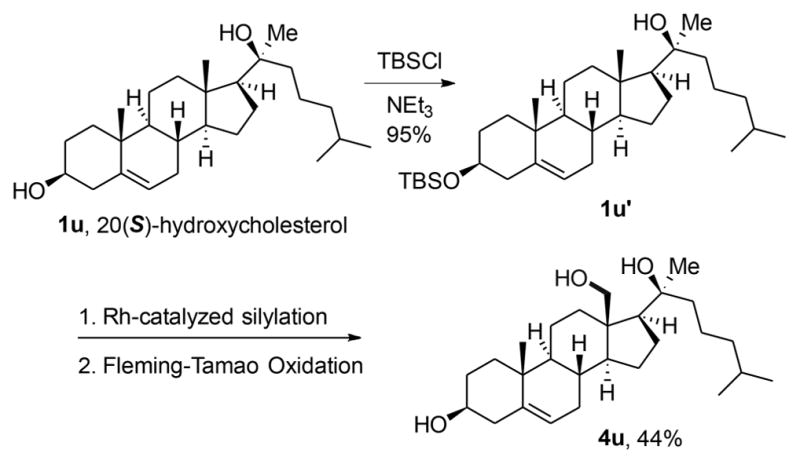
Rh-Catalyzed Silylation of Oxysterol
To enable functionalization directed by the tertiary alcohol in 20(S)-hydroxycholesterol, the secondary alcohol was selectively protected to form the corresponding silyl ether. Under the conditions we developed for alcohol-directed, site-selective silylation of C–H bonds δ to alcohols, the monoprotected diol 1u′ was functionalized at the C18 position. Subsequent Fleming–Tamao oxidation with concomitant deprotection provided triol 4u. These results show that the oxygenation of C–H bonds δ to alcohols, in addition to the previously reported oxygenation of C–H bonds γ to alcohols should allow functionalization of a range of terpenoids.
Experimental Mechanistic Studies
To understand the origin of the regioselectivity of this silylation reaction and the origin of the effect of the ligand on this selectivity, we conducted detailed studies of the mechanism by a combination of experiment and computation. To determine the rate limiting step of this reaction, the resting state of the catalyst was characterized, the reaction orders for each reagent were determined, and the rate of silylation of C–H and C–D bonds was measured. In addition, several rhodium complexes were synthesized that elucidate the path by which the precatalyst enters the catalytic cycle. Finally, to gain information on the energetics that control selectivity, computations were conducted on the Segphos-ligated catalyst, which generates products from silylation of secondary C–H bonds γ to the original hydroxyl group, and on the Xantphos ligated catalyst, which generates products from silylation of primary C–H bonds δ to the original hydroxyl group.
Mechanism for the Generation of the Active Catalyst and Identification of the Resting State
To elucidate the pathway by which the precatalyst enters the catalytic reaction and the structure of the resting state of the catalyst, the reaction of (Xantphos)Rh(Cl) with silyl ether 2a and norbornene was conducted. Although (Xantphos)Rh(Cl) is insoluble, the reaction occurred quickly (<10 m) at room temperature to form a clear homogeneous solution containing complex 7 shown in eq 3. The 31P NMR spectrum of the solution
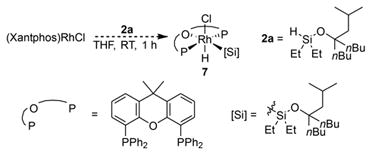 |
(3) |
consisted of a doublet (due to coupling to 103Rh) at 36.6 ppm, indicating two equivalent phosphines, and the 1H NMR spectrum contained a doublet of triplets at −14.1 ppm, indicating that the Rh complex contains a metal hydride coupled to rhodium and two equivalent phosphorus atoms. The same species formed from (Xantphos)Rh(Cl) and the silyl ether 2a in the absence of norbornene, but it was not formed from (Xantphos)Rh(Cl) and norbornene in the absence of the silyl ether.
Heating of the catalytic silylation reaction at 100 °C for 1 h led to a loss of the 31P NMR signal at 36.6 ppm. The major resonance observed in the 31P NMR spectrum after heating was a doublet at 22.6 ppm with a 31P-103Rh coupling constant of 142 Hz. No signal in the hydride region of the 1H NMR spectra was observed. Based on these data, we hypothesized that the resting state of the Rh catalyst is a Xantphos-ligated Rh(I) silyl complex.
We were unable to isolate complex 7 formed from (Xantphos)Rh(Cl) and the silyl ether 2a in pure form, due to its high solubility in organic solvents, but we isolated an analogous complex containing the 1-adamantyl silyl ether 2v.18 The reaction of 2v with (Xantphos)Rh(Cl) formed silyl hydride complex 8 (Figure 1). The solution-phase NMR spectral data of complex 8 were analogous to those of the rhodium complex formed in the catalytic system before heating. A single doublet for the phosphine was observed in the 31P NMR spectrum at 35.7 ppm, and a doublet of triplets at −14.0 ppm for a hydride ligand was observed in the 1H NMR spectrum. These data are consistent with the formula (Xantphos)Rh(Cl)(H)(SiEt2OAd), resulting from oxidative addition of the Si–H bond of the silane to (Xantphos)Rh(Cl).
Figure 1.

Synthesis and solid-state structure of (Xantphos)Rh(H)-(Cl)(SiEt2OAd) (8).
The identity of Rh complex 8 was confirmed by single-crystal X-ray diffraction. The solid-state molecular structure of Rh silyl hydride 8 consists of an octahedral Rh(III) center containing a meridional, tridentate Xantphos ligand. The silyl group is oriented trans to the Xantphos oxygen, and the chloride occupies a position axial to the plane of the Xantphos ligand and the silyl group. The hydride ligand, shown by 1H NMR spectroscopy to be present, would be located at the site trans to the chloride.
The reaction of (Xantphos)Rh(Cl) with silane in the presence of additional Xantphos produced a complex that was different from that formed by the reaction of (Xantphos)-Rh(Cl) with silane in the absence of added Xanphos. The resulting complex (9) is not soluble in common laboratory solvents, but was analyzed in the solid state by single-crystal X-ray crystallography. The structure, shown in Figure 2, is a rhodium(I) hydride complex containing two Xantphos ligands with phosphorus atoms in a nearly tetrahedral array. The P–Rh–P bite angle for each ligand is 106°. The hydride was not located by X-ray diffraction, but the infrared spectrum contains a stretching frequency at 2156 cm–1, which is consistent with the presence of a rhodium hydride. The mild conditions under which the complex forms imply that the barrier to reductive elimination of the silyl chloride from complex 8 is low. We propose that complex 8 forms initially in the catalytic reaction, and that it undergoes reductive elimination of the silyl chloride, producing a Rh(I) hydride complex, which enters the catalytic cycle. In the presence of excess Xantphos, this hydride complex forms the crystalline, insoluble 9.
Figure 2.

Synthesis and solid-state structure of (Xantphos)2Rh(H) (9). Selected bond lengths (Å) and angles (deg): Rh–P1, 2.35; Rh–P2, 2.34; P1–Rh–P2 (same ligand), 106.4; P1–Rh–P2 (between ligands), 109.8; P1–Rh–P1, 105.9; P2–Rh–P2, 118.0.
The catalytic competencies of complexes 8 and 9 were determined. The rate of the reaction conducted with isolated 8 as catalyst was the same as that for the reaction initiated with (Xantphos)Rh(Cl) as catalyst (see the Supporting Information for details). Thus, complex 8 is catalytically competent to be an intermediate in the silylation of C–H bonds located δ to a silyl ether. In contrast, no product was observed when the reaction was conducted with complex 9 as catalyst. Thus, complex 9 is not competent to be part of the catalytic cycle, likely due to the insolubility of this species in THF at 100 °C.
We also sought to identify the resting state of the Rh complex in the catalytic silylation of C–H bonds. We hypothesized that this species was a Xantphos-ligated Rh(I) silyl complex because no signal was observed in the hydride region of the 1H NMR spectra of the reaction obtained after heating. To test this hypothesis, we independently prepared Rh(I) silyl complex 10 (Figure 3) from the reaction of Rh hydride complex 8 with LiHMDS (Figure 3).19 The Rh(I) silyl complex [Rh(Xantphos)(SiEt2OAd)] (10) was characterized by 1H, and 31P NMR spectroscopy of the crude reaction mixture. The 1H and 31P NMR spectra of the complex are consistent with a Rh(I) silyl complex; the 31P NMR spectra contain a resonance at 32.6 ppm with a strong 103Rh coupling of 193 Hz, indicating that Xantphos remained bound to the metal. Resonances corresponding to the adamantyl group were also observed, showing that the silyl group is still connected to rhodium. NMR spectroscopy does not reveal whether the oxygen of Xantphos is bound to Rh in complex 10. Silyl complex 10 formed in this manner decomposed during attempts to isolate the complex in pure form.
Figure 3.

Synthesis and solid-state structure of (Xantphos)Rh(H)-(nbe)(SiEt2OAd) (10).
However, a norbornene adduct of Rh(I) silyl complex 10 was isolated and fully characterized. The deprotonation of Rh-silyl complex 8 containing hydride and chloride ligands in the presence of norbornene formed the isolable norbornene silyl Rh(I) species 11 (Figure 3). The 31P NMR spectrum of this complex contains a single resonance, which is a doublet at 21.4 PPM with a 31P-103Rh coupling of 140 Hz. The 1H NMR spectrum of isolated 11 contains alkyl resonances for the silyl unit and a resonance at 2.6 ppm for the coordinated alkene unit.
The composition of complex 11, deduced in solution, was confirmed by X-ray crystallography (Figure 3). The structure consists of a Rh(III) center in which the two phosphorus atoms, but not the oxygen atom, of Xantphos are bound to rhodium, along with one silyl group, one hydride, and one alkene unit. The geometry at rhodium is trigonal pyramidal or square pyramidal, depending on how one considers the bonding of the alkene. The C–C double bond bound to rhodium is significantly longer (1.45 Å) than that of the free alkene (1.34 Å), suggesting that the alkene is bound in a metallacyclopropane structure and that the overall geometry is a square pyramid. The two norbornene carbons bonded to rhodium, the two phosphorus atoms, and the rhodium center lie in a plane; the axial position of the square pyramidal structure is occupied by the silyl group. This 16-electron complex could undergo oxidative addition of a C–H bond. However, the large size of the bound norbornene would hinder the approach of a C–H bond to the metal center.
The full characterization of 11 allows clear assignment of the resting state of the rhodium in the catalytic reaction. As noted earlier in this paper, the 31P NMR signal for the resting state is a doublet at 22.6 ppm with a coupling constant of 142 Hz. Likewise, the 31P NMR signal for complex 11 is a doublet at 21.4 ppm with a coupling constant of 140 Hz. For comparison, the 31P NMR signal for complex 10 lacking norbornene is a doublet at 32.6 ppm with a coupling constant of 193 Hz. Therefore, the resting state of the catalyst is a Rh complex ligated by one Xantphos, one norbornene, and one alkoxy diethylsilyl ligand.
Kinetic Analysis of the Catalytic Process
The dependence of reaction rate on the concentration of each reagent was measured to reveal the rate-determining step of the catalytic process. The reaction orders in silane, catalyst, and hydrogen acceptor were determined by measuring initial rates of the silylation at 100–120 °C with varied concentrations of each reagent. The reactions were conducted with chloride complex 7 as the precatalyst. Reactions with concentrations of catalyst ranging from 2 to 16 mM showed that the reaction is first order in catalyst. Reactions with concentrations of silane varying from 0.04 to 0.3 M showed that the reaction is zero order in silane. Finally, reactions conducted with concentrations of norbornene varying from 0.4 to 3 M showed that the reaction is inverse first order in norbornene. With the resting state shown to be a silyl norbornene complex, the zero-order behavior in silane indicates that the silane bound to the resting state undergoes C–H bond functionalization, and the inverse reaction order in norbornene indicates that norbornene dissociates reversibly from this resting state to form a 14-electron compound prior to the functionalization process.
The kinetic isotope effect on the catalytic process was measured to determine the potential reversibility of the C–H bond cleavage process (Scheme 4). The rates of the reaction of the protio substrate (2a) and deuterio substrate (2a–d7) containing deuterium at the position δ to the alcohol were measured separately. A kinetic isotope effect of 4.6 was obtained from these rates (5.7 and 1.2 × 10−5 M/s), indicating that C–H bond cleavage is irreversible and rate limiting. This result is consistent with the reaction orders and the observed resting state and also implies that the reversible dissociation of norbornene occurs before irreversible cleavage of the C–H bond.
Scheme 4.
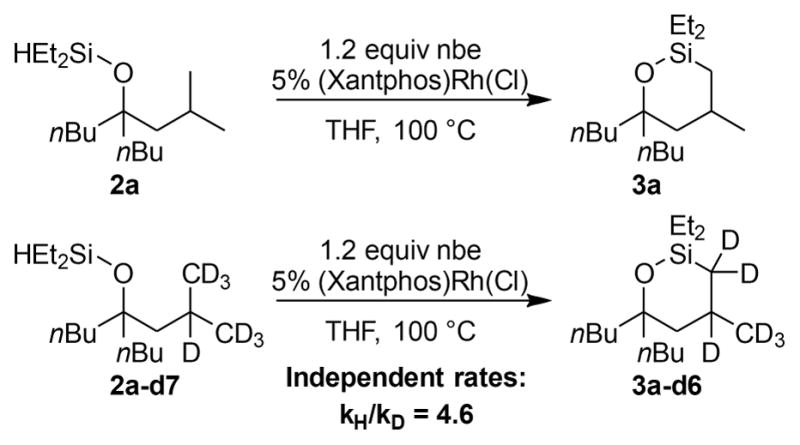
Kinetic Isotope Effect
Based on the orders in substrate, hydrogen acceptor, and catalyst, the kinetic isotope effect, and the isolated model of the resting state, we conclude that the reaction occurs by the catalytic cycle in Scheme 5. The active catalyst is generated by oxidative addition of a silyl hydride to the RhCl precatalyst to form complex A. Complex A forms the Rh(I) hydride compound B, which lies on the catalytic cycle. Norbornene undergoes migratory insertion into the Rh–H bond to form norbornyl complex C, which undergoes oxidative addition of a silyl hydride and reductive elimination of norbornane to produce the 14-electron Rh(I) silyl complex D. Complex D binds norbornene reversibly to form the resting-state E, which lies off the catalytic cycle. Intramolecular oxidative addition of the δ C–H bond to the rhodium center in D forms the seven-membered rhodacycle F. Reductive elimination from F to form the C–Si bond produces a six-membered cyclic silyl ether and regenerates Rh(I) hydride complex B.
Scheme 5.

Proposed Catalytic Cycle for the Silylation of the C–H Bond γ to a Silyl Ether
Computational Study and Origin of Selectivity
DFT computations were performed to gain insight into the origin of the selectivity of the Xantphos catalyst for formation of the six-membered oxasilolane product. To do so, we computed the energies and geometries of Rh(I) silyl complexes that are potential catalytic intermediates, transition states for intra-molecular cleavage of C–H bonds in those complexes, rhodacycles that result from C–H cleavage, and transition states for reductive eliminations to form C–Si bonds. Geometry optimizations were conducted with the Gaussian 09 software package, B3LYP functional (with gd3 dispersion correction), and LANL2DZ basis set for Rh and 6-31g(d,p) basis set for all other atoms. Single-point energy calculations were conducted with the M06 functional and LANL2TZ basis set for Rh and the 6-31++g** basis set for all other atoms, along with the SMD THF solvent correction. Thermal corrections were applied to the optimized geometries to provide Gibbs free energies. Each Rh(I) hydride complex, Rh(I) silyl complex, transition state, and Rh(III) metallacycle was computed with Segphos (experimental selectivity for silylation of C–H bonds located γ to a silyl ether) and Xantphos (experimental selectivity for silylation of C–H bonds located δ to a silyl ether) as the ligand to reveal the origin of selectivity. To reduce the computational requirements, the calculations were conducted on a model substrate containing methyl groups, instead of ethyl groups, on silicon.
The minimization of the energy of octahedral Rh-Xantphos complexes that result from oxidative addition of a C–H bond showed that many reasonable isomeric products can form. Xantphos can coordinate in a facial or meridonal manner. Any of the three X-type ligands could be trans to the weakly coordinating oxygen of the Xantphos ligand in either coordination mode. The length of the alkyl chain of the substrate is too short to form a complex with the alkyl ligand and the silyl ligand trans to each other. Thus, the five 6-coordinate isomers in which the alkyl and silyl ligands are cis to each other were modeled. The square pyramidal complex lacking coordination by the oxygen atom in Xantphos and containing the silyl group in the axial position was also computed. The isomer with the silyl group in this position was computed because the two methyl substituents on silicon in this isomer lie over the backbone of Xantphos, whereas these two methyl substituents clash with the phenyl substituents on phosphorus in the isomer containing the silyl group in an equatorial position.
Although the energy of six-coordinate, 18-electron structures in which the oxygen atom in Xantphos binds to rhodium might be expected to be lower than that of square pyramidal 16-electron complexes lacking coordination of the oxygen atom, our calculations showed that the energy of the square pyramidal complex in which this oxygen does not interact with rhodium is lower than that of the complexes in which this oxygen does interact with rhodium (Figure 4). The isolated resting state, complex 11, also lacked a bonding interaction between the oxygen atom of Xantphos and the rhodium center in the solid-state structure. These two results suggest that the ability of Xantphos to bind as a tridentate ligand does not account for the difference in selectivity between the catalysts containing Xantphos and Segphos as ligand.
Figure 4.

Modeling of rhodacycles in various geometries and coordination modes.
Computation of the Pathways for Silylation of C–H Bonds δ and γ to a Hydroxyl Group
The computed rate-determining step of the Rh-catalyzed silylation of primary C–H bonds δ to a silyl ether unit is different from the that for the competing silylation of secondary C–H bonds γ to a silyl ether (Figure 5). The highest calculated transition-state energy for functionalization of the δ C–H bond by the rhodium complexes ligated by both Xantphos (21.7 kcal/mol, shown in red in the left energy diagram) and Segphos (24.1 kcal/mol, shown in red in the right energy diagram) is that for oxidative addition of the C–H bond (Figure 6). The highest calculated transition-state energy for functionalization of the γ C–H bond by complexes of both ligands, shown in blue in both diagrams, is that for reductive elimination to form the C–Si bond. The barrier to reductive elimination from the secondary alkylrhodium complex to functionalize the γ C–H bond with the Xantphos-ligated system is almost 19 kcal/mol, and the transition-state free energy is 25.8 kcal/mol higher than that of the catalyst and reactants, whereas the analogous barrier for the complex ligated by Segphos is only 8.3 kcal/mol and the transition state free energy is 20.4 kcal/mol higher than that of the catalyst and reactants.
Figure 5.

Computed energies for silylation of C–H bonds δ to a silyl ether (red) and γ to a silyl ether (blue). Silylation with the Rh-Xantphos catalyst (left). Silylation with the Rh-Segphos catalyst (right).
Figure 6.
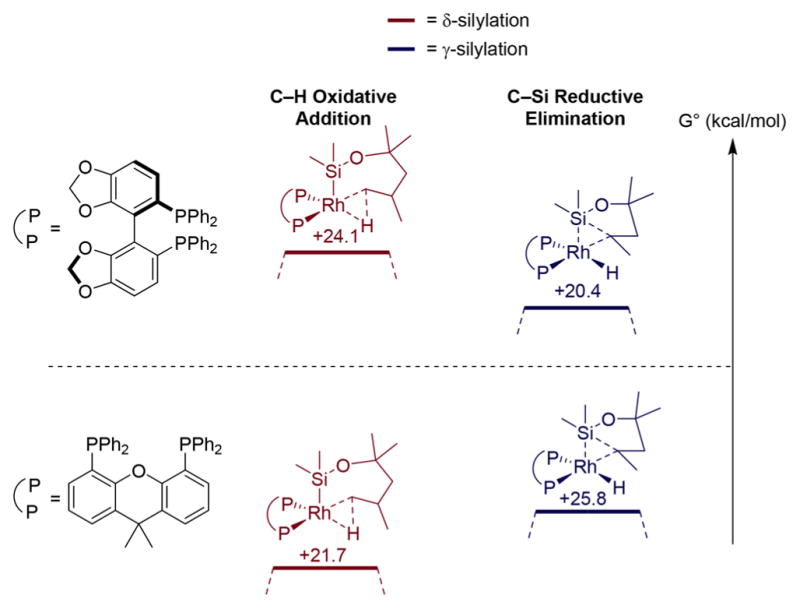
Computed energies for oxidative addition of C–H Bonds δ to a silyl ether (red) and reductive elimination of C–Si bond γ to a silyl ether (blue). Silylation with Rh/Segphos as catalyst (top). Silylation with Rh/Xantphos as catalyst (bottom).
These calculations show that the unusual selectivity for formation of the six-membered oxasilolane with the Rh-Xantphos catalyst results from a high barrier for reductive elimination from a 6-membered metallacycle containing a secondary alkyl and a transition-state energy that is higher than that for oxidative addition of a δ C–H bond to the same Rh-Xantphos catalyst to form a seven-membered metallacycle. The selectivity for formation of the more common 5-membered oxasilolane with the Rh-Segphos catalysts results from a transition-state energy for oxidative addition of the δ C–H bond to form the seven-membered metallacycle that is higher than that for reductive elimination from a 6-membered metallacycle containing a secondary alkyl.
Computational Model of the Silylation of Primary C–H Bonds γ to the Silyl Ether
The calculations just discussed show that the rate-determining steps for functionalization of C–H bonds δ to a silyl ether by both catalysts are different from those for functionalization of C–H bonds γ to a silyl ether, but they do not reveal why the oxidative addition of a primary C–H bond δ to the silyl ether is slower than the oxidative addition of a secondary C–H bond γ to the silyl ether for complexes of both ligands. Oxidative addition of a primary C–H bond is usually faster than oxidative addition of a secondary C–H bond. Thus, the relative rates for reactions of primary and secondary C–H bonds are, most likely, influenced by the size of the ring formed in the intramolecular processes. Further, these calculations do not reveal the extent to which formation of a five-membered cyclic silyl ether by reductive elimination from a secondary alkyl intermediate is slow because of the size of the ring that is formed versus the extent to which it is slow because reductive elimination from a secondary alkyl complex to form an Si–C bond is slower than reductive elimination to form a Si–C bond from a primary alkyl complex.
To address these questions, we computed the reaction coordinate for the silylation of a primary C–H bond γ to a silyl ether by the two catalysts and compared the energies in these reaction coordinates to those discussed in the prior section on the silylation of the substrate containing primary C–H bonds δ to a silyl ether and the substrate containing secondary C–H bonds γ to a silyl ether.
The reaction coordinates for silylations of primary C–H bonds γ to a silyl ether in the model compound catalyzed by the two rhodium-ligand combinations are shown in green color in Figure 7, along with the reaction coordinate (in blue color) presented earlier for the silylation of a secondary C–H bond γ to a silyl ether catalyzed by the same two systems. The left side shows the computed reaction coordinates for the silylation of primary and secondary C–H bonds γ to a silyl ether catalyzed by Xantphos-ligated system and the right side shows the computed reaction coordinate for the silylation of the same silyl ethers catalyzed by the Segphos-ligated system. For silylation of a primary C–H bond γ to the oxygen catalyzed by complexes of either ligand, the rate-determining step was computed to be oxidative addition of the C–H bond because reductive elimination from the primary alkyl intermediate has a low barrier. In contrast, for silylation of the secondary C–H bond γ to the silyl ether catalyzed by complexes of either ligand, the rate-determining step was computed to be reductive elimination to form the C–Si bond (vide supra). Thus, the rate-determining step for reaction at the primary C–H bond is computed to be different from that for reaction at a secondary C–H bond located the same distance from the ether oxygen as the primary C–H bond.
Figure 7.

Computational model of silylation of a primary and secondary C–H bonds γ to a Silyl ether with Rh/Segphos and Rh/Xantphos.
Conclusion on the Origin of Selectivity for Five vs Six-Membered Cyclic Silyl Ethers with Segphos and Xantphos as Ligand
Insight into the origin of the selectivity for the formation of the various-sized cyclic silyl ethers can be gained by comparing the transition-state energies for the oxidative addition of the primary C–H bond γ to a silyl ether to that for oxidative addition of the primary C–H bond δ to a silyl ether. The comparison of the computed transition-state energies for oxidative addition to form six-membered rhodacycles with primary alkyl groups to that to form the seven-membered rhodacycles with primary alkyls ligated by Xantphos or Segphos shows that oxidative additions that form seven-membered metallacycles are slower than those forming six-membered metallacycles for complexes of both ligands. The difference in computed free energies between the two transition states for reaction of the Xantphos-ligated catalyst is smaller (2.6 kcal/mol) than that for reaction of the Segphos-ligated catalyst (8 kcal/mol). However, this comparison of the transition-state energies for oxidative addition of primary C–H bonds to form six or seven-membered rhodacycles shows that formation of the six-membered rhodacycle is faster for complexes of both ligands. These data show quantitatively the extent to which formation of seven-membered metallacycles is slower than formation of six-membered metallacycles.
The comparison of the computed barriers to reductive elimination that form five-membered cyclic silyl ethers from primary alkyl complexes to those that form the same size ring from secondary alkyl complexes ligated by Xantphos or Segphos shows that reductive eliminations to form C–Si bonds involving secondary alkyl groups are much slower than those involving primary alkyl groups for complexes of both ligands. However, the difference between the transition-state energies of reductive elimination from the primary and secondary alkyl groups to form the C–Si bond in 5-membered rings is much greater for the catalyst containing the Xantphos ligand (10.5 kcal/mol) than it is for the catalyst containing the Segphos ligand (4.9 kcal/mol), and it is the difference in rates for reductive elimination from primary and secondary alkyl groups that lead to the unusual selectivity observed for the silylation of alkyl C–H bonds located γ to the silyl ether over C–H bonds located δ to the silyl ether.
This effect of the two ligands on these relative rates can be ascribed to the greater steric demand of Xantphos ligand. During the reductive elimination of secondary alkyl groups, the substituents on that carbon bound to rhodium tilt away from the silicon, causing these substituents to be closer to the phenyl groups of the ligand in the transition state than in the ground state. The catalyst formed from the Segphos ligand is more able to accommodate this increased steric demand than that formed from the Xantphos ligand possessing a wider bite angle. Thus, the difference in rates of reductive elimination from the primary or secondary alkyl complex containing Segphos as ligand is smaller than the differences in rates of reductive elimination from the analogous complexes containing Xantphos as ligand.
CONCLUSION
In summary, we have developed a rhodium-catalyzed site-selective silylation of primary C–H bonds to form six-membered oxasilolanes in the presence of [Rh(Xantphos)Cl] and conducted studies that reveal the origin of the new regioselectivity. After oxidation of the silylation products, 1,4-diols are obtained. This C–H oxidation tolerates many functional groups and has the potential for applications in the synthesis and functionalization of complex molecules.
The mechanism of this process is distinct from that for silylation of arenes. In contrast to the rate-determining reductive elimination to form the C–H bond of the reduced hydrogen acceptor that occurs during the silylation of arenes, the rate-limiting step of the rhodium-catalyzed silylation of alkyl C–H bonds δ to a silyl ether is cleavage of the C–H bond undergoing functionalization. The resting state of the catalyst for this process catalyzed by Xantphos-ligated rhodium is (Xantphos)Rh(SiEt2OR)(nbe). Consistent with this structure, the reaction is zero-order in substrate and inverse first-order in norbornene because norbornene dissociates prior to rate-determining intramolecular cleavage of the alkyl C–H bond. The resting states of Rh-catalyzed C–H silylation reactions published previously undergo reductive elimination of dihydrogen or reduction of the hydrogen acceptor before C–H bond cleavage.
Computational studies have revealed the origins of the so-far unique selectivity for silylation of a δ C–H bond over a γ C–H bond with the Rh-Xantphos catalyst. This selectivity results from a higher barrier to reductive elimination from the secondary alkyl intermediate with Xantphos as ligand than with Segphos (and presumably related phosphines) as ligand. This high barrier results from increased steric repulsion between the ligand and the substrate during reductive elimination to form a secondary C–Si bond. These mechanistic insights lay the groundwork for the development of new regioselective C–H silylation methodologies and provide a framework to predict the site of C–H silylation in complex molecules with multiple accessible C–H bonds.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support by the Einstein Foundation Berlin (J.F.H.) and the Cluster of Excellence UniCat (financed by the DFG and administered by the TU Berlin to M.D.) for initial data and the NIH (GM5R01-GM115812) (J.F.H.) for support. We thank Dr. Antonio DiPasquale for assistance with crystallographic data. We thank Dr. Kathleen Durkin, of the Molecular Graphics and Computation Facility (supported by NIH Grant S10OD023532), for assistance with DFT calculations. B.L. thanks Dr. Qian Li and Dr. Qingwei Zhang for assistance with some experiments. C.K. thanks Dr. Ala Bunescu for helpful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11964.
Experimental procedures, characterization of new compounds, and spectroscopic data (PDF)
Crystallographic data for compound 5t (CIF)
Crystallographic data for compound 8 (CIF)
Crystallographic data for compound 9 (CIF)
Crystallographic data for compound 11 (CIF)
References
- 1.(a) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (b) Hartwig JF. Acc Chem Res. 2012;45:864. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 2.Cheng C, Hartwig JF. Chem Rev. 2015;115:8946. doi: 10.1021/cr5006414. [DOI] [PubMed] [Google Scholar]
- 3.selected early examples of C-H silylation Gustavson WA, Epstein PS, Curtis MD. Organometallics. 1982;1:884.Sakakura T, Tokunaga Y, Sodeyama T, Tanaka M. Chem Lett. 1987;16:2375.
- 4.selected examples of intramolecular silylation: Tsukada N, Hartwig JF. J Am Chem Soc. 2005;127:5022. doi: 10.1021/ja050612p.Furukawa S, Kobayashi J, Kawashima T. J Am Chem Soc. 2009;131:14192. doi: 10.1021/ja906566r.Ureshino T, Yoshida T, Kuninobu Y, Takai K. J Am Chem Soc. 2010;132:14324. doi: 10.1021/ja107698p.Simmons EM, Hartwig JF. J Am Chem Soc. 2010;132:17092. doi: 10.1021/ja1086547.Kuznetsov A, Gevorgyan V. Org Lett. 2012;14:914. doi: 10.1021/ol203428c.Kuznetsov A, Onishi Y, Inamoto Y, Gevorgyan V. Org Lett. 2013;15:2498. doi: 10.1021/ol400977r.Kuninobu Y, Yamauchi K, Tamura N, Seiki T, Takai K. Angew Chem, Int Ed. 2013;52:1520. doi: 10.1002/anie.201207723.Zhang QW, An K, Liu LC, Yue Y, He W. Angew Chem, Int Ed. 2015;54:6918. doi: 10.1002/anie.201502548.Murai M, Matsumoto K, Takeuchi Y, Takai K. Org Lett. 2015;17:3102. doi: 10.1021/acs.orglett.5b01373.Lee T, Wilson TW, Berg R, Ryberg P, Hartwig JF. J Am Chem Soc. 2015;137:6742. doi: 10.1021/jacs.5b03091.Murai M, Takeshima H, Morita H, Kuninobu Y, Takai K. J Org Chem. 2015;80:5407. doi: 10.1021/acs.joc.5b00920.Ghavtadze N, Melkonyan FS, Gulevich AV, Huang C, Gevorgyan V. Nat Chem. 2014;6:122. doi: 10.1038/nchem.1841.
- 5.Selected examples of intermolecular sp2 C–H silylation: Ishikawa M, Okazaki S, Naka A, Sakamoto H. Organometallics. 1992;11:4135.Uchimaru Y, El Sayed AMM, Tanaka M. Organometallics. 1993;12:2065.Ishikawa M, Naka A, Ohshita J. Organometallics. 1993;12:4987.Ezbiansky K, Djurovich PI, LaForest M, Sinning DJ, Zayes R, Berry DH. Organometallics. 1998;17:1455.Ishiyama T, Sato K, Nishio Y, Miyaura N. Angew Chem, Int Ed. 2003;42:5346. doi: 10.1002/anie.200352399.Ishiyama T, Sato K, Nishio Y, Saiki T, Miyaura N. Chem Commun. 2005:5065. doi: 10.1039/b511171d.Sadow AD, Tilley TD. J Am Chem Soc. 2005;127:643. doi: 10.1021/ja040141r.Saiki T, Nishio Y, Ishiyama T, Miyaura N. Organometallics. 2006;25:6068.Murata M, Fukuyama N, Wada J-i, Watanabe S, Masuda Y. Chem Lett. 2007;36:910.Lu B, Falck JR. Angew Chem, Int Ed. 2008;47:7508. doi: 10.1002/anie.200802456.Klare HF, Oestreich M, Ito J-i, Nishiyama H, Ohki Y, Tatsumi K. J Am Chem Soc. 2011;133:3312. doi: 10.1021/ja111483r.
- 6.Selected examples of directed sp2 C–H silylation: Williams NA, Uchimaru Y, Tanaka MJ. Chem Soc, Chem Commun. 1995:1129.Kakiuchi F, Matsumoto M, Sonoda M, Fukuyama T, Chatani N, Murai S, Furukawa N, Seki Y. Chem Lett. 2000;29:750.Kakiuchi F, Igi K, Matsumoto M, Chatani N, Murai S. Chem Lett. 2001;30:422.Kakiuchi F, Igi K, Matsumoto M, Hayamizu T, Chatani N, Murai S. Chem Lett. 2002;31:396.Kakiuchi F, Matsumoto M, Tsuchiya K, Igi K, Hayamizu T, Chatani N, Murai S. J Organomet Chem. 2003;686:134.Tobisu M, Ano Y, Chatani N. Chem - Asian J. 2008;3:1585. doi: 10.1002/asia.200800090.Ihara H, Suginome M. J Am Chem Soc. 2009;131:7502. doi: 10.1021/ja902314v.Oyamada J, Nishiura M, Hou Z. Angew Chem, Int Ed. 2011;50:10720. doi: 10.1002/anie.201105636.Sakurai T, Matsuoka Y, Hanataka T, Fukuyama N, Namikoshi T, Watanabe S, Murata M. Chem Lett. 2012;41:374.Chen C, Guan M, Zhang J, Wen Z, Zhao Y. Org Lett. 2015;17:3646. doi: 10.1021/acs.orglett.5b01393.
- 7.Selected examples of directed sp3 C–H silylation: Kakiuchi F, Tsuchiya K, Matsumoto M, Mizushima E, Chatani N. J Am Chem Soc. 2004;126:12792. doi: 10.1021/ja047040d.Mita T, Michigami K, Sato Y. Org Lett. 2012;14:3462. doi: 10.1021/ol301431d.Mita T, Michigami K, Sato Y. Chem -Asian J. 2013;8:2970. doi: 10.1002/asia.201300930.Ihara H, Ueda A, Suginome M. Chem Lett. 2011;40:916.
- 8.(a) Simmons EM, Hartwig JF. Nature. 2012;483:70. doi: 10.1038/nature10785. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li B, Driess M, Hartwig JF. J Am Chem Soc. 2014;136:6586. doi: 10.1021/ja5026479. [DOI] [PubMed] [Google Scholar]
- 9.(a) Frihed TG, Heuckendorff M, Pedersen CM, Bols M. Angew Chem, Int Ed. 2012;51:12285. doi: 10.1002/anie.201206880. [DOI] [PubMed] [Google Scholar]; (b) Frihed TG, Pedersen CM, Bols M. Angew Chem, Int Ed. 2014;53:13889. doi: 10.1002/anie.201408209. [DOI] [PubMed] [Google Scholar]
- 10.Hua Y, Jung S, Roh J, Jeon J. J Org Chem. 2015;80:4661. doi: 10.1021/acs.joc.5b00564. [DOI] [PubMed] [Google Scholar]
- 11.(a) Čeković Z. Tetrahedron. 2003;59:8073. [Google Scholar]; (b) Allen J, Boar RB, McGhie JF, Barton DHR. J Chem Soc, Perkin Trans. 1973;1:2402. [Google Scholar]; (c) Boar RB, Copsey DBJ. J Chem Soc, Perkin Trans. 1979;1:563. [Google Scholar]
- 12.(a) Cheng C, Hartwig JF. J Am Chem Soc. 2014;136:12064. doi: 10.1021/ja505844k. [DOI] [PubMed] [Google Scholar]; (b) Lee T, Hartwig JF. J Am Chem Soc. 2017;139:4879. doi: 10.1021/jacs.7b00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Catalysts bound by ligands with wide bite angles have been used for specific types of hydroacylation reactions: Moxham GL, Randell-Sly HE, Brayshaw SK, Woodward RL, Weller AS, Willis MC. Angew Chem. 2006;118:7780. doi: 10.1002/anie.200603133.Chaplin AB, Hooper JF, Weller AS, Willis MC. J Am Chem Soc. 2012;134:4885. doi: 10.1021/ja211649a.
- 14.(a) Mita T, Hanagata S, Michigami K, Sato Y. Org Lett. 2017;19:5876. doi: 10.1021/acs.orglett.7b02871. [DOI] [PubMed] [Google Scholar]; (b) Michigami K, Mita T, Sato Y. J Am Chem Soc. 2017;139:6094. doi: 10.1021/jacs.7b02775. [DOI] [PubMed] [Google Scholar]; (c) Cabrera-Pardo JR, Trowbridge A, Nappi M, Ozaki K, Gaunt MJ. Angew Chem, Int Ed. 2017;56:11958. doi: 10.1002/anie.201706303. [DOI] [PubMed] [Google Scholar]; (d) Probst N, Grelier G, Ghermani N, Gandon V, Alami M, Messaoudi S. Org Lett. 2017;19:5038. doi: 10.1021/acs.orglett.7b02170. [DOI] [PubMed] [Google Scholar]
- 15.The nuclearity of this species is unknown, in part due to its lack of solubility. Although many L2RhCl complexes are dimeric with bridging chlorides, the complex of the Xantphos analogue with iPr groups in place of Ph groups is monomeric with Xantphos coordinated in a tridentate fashion through two phosphorus and one oxygen atom.
- 16.Stappenbeck F, Xiao W, Epperson M, Riley M, Priest A, Huang D, Nguyen K, Jung ME, Thies RS, Farouz F. Bioorg Med Chem Lett. 2012;22:5893. doi: 10.1016/j.bmcl.2012.07.073. [DOI] [PubMed] [Google Scholar]
- 17.Michaudel Q, Journot G, Regueiro-Ren A, Goswami A, Guo Z, Tully TP, Zou L, Ramabhadran RO, Houk KN, Baran PS. Angew Chem, Int Ed. 2014;53:12091. doi: 10.1002/anie.201407016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.For the synthesis of related Rh(III) silyl complexes, see: Esteruelas MA, Oliván M, Vélez A. Inorg Chem. 2013;52:12108–12119. doi: 10.1021/ic401931y.
- 19.Prepared in a manner similar to that used to prepare (Xantphos-iPr)Rh(H): Haibach MC, Wang DY, Emge TJ, Krogh-Jespersen K, Goldman AS. Chem Sci. 2013;4:3683–3692.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.