

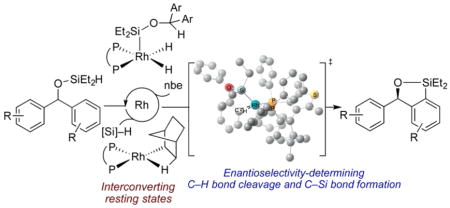
Abstract
Several classes of enantioselective silylations of C–H bonds have been reported recently, but little mechanistic data on these processes are available. We report mechanistic studies on the rhodium-catalyzed, enantioselective silylation of aryl C–H bonds. A rhodium silyl dihydride and a rhodium norbornyl complex were prepared and determined to be interconverting catalyst resting states. Kinetic isotope effects indicated that the C–H bond cleavage step is not rate-determining, but the C–H bond cleavage and C–Si bond-forming steps together influence the enantioselectivity. DFT calculations indicate that the enantioselectivity originates from unfavorable steric interactions between the substrate and the ligand in the transition state leading to the formation of the minor enantiomer.
Graphical Abstract
INTRODUCTION
The functionalization of C–H bonds with main group reagents, such as boranes and silanes, has become a synthetically valuable process because of the high regioselectivity of the reactions and the widespread utility of the products.1–3 Mechanistic studies on these reactions have provided insight into the catalyst resting states, rate-determining steps, and origins of selectivities.4–9 However, little information on the mechanism of enantioselective variants of these reactions is available.10–18 Preliminary mechanistic data on enantioselective silylation of C–H bonds have been reported,11,17,18 but systematic mechanistic studies involving the isolation of reaction intermediates and kinetic analysis of the process have not been conducted. Most relevant to the work reported here, little information on the origins of enantioselectivities has been gained.
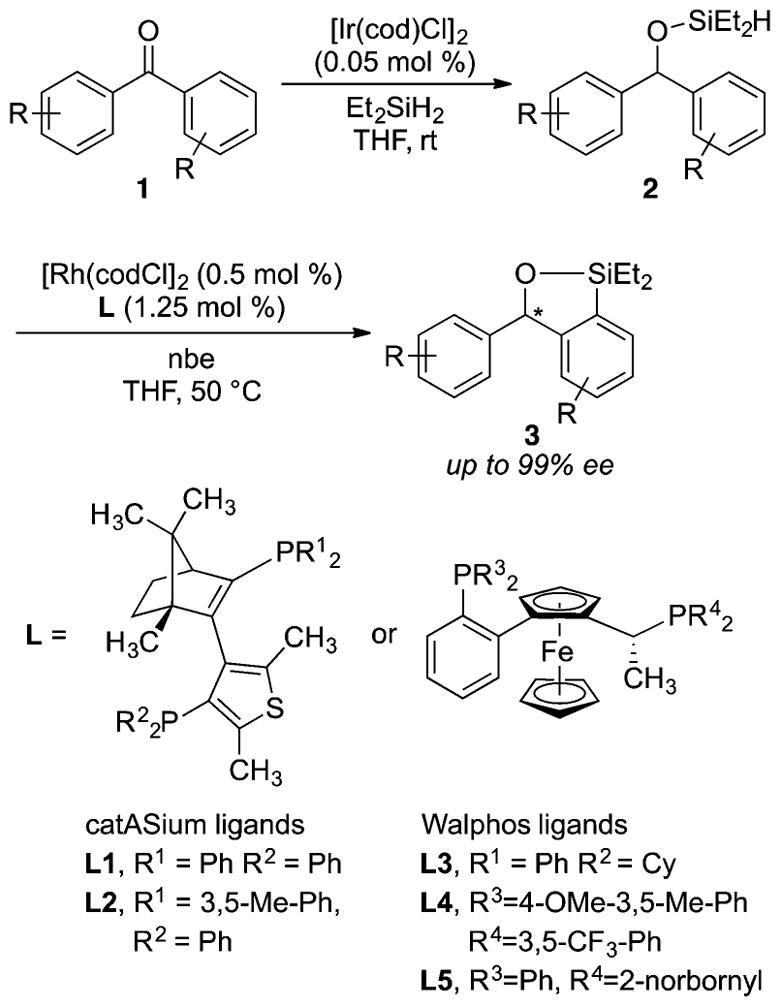
Recently, our group reported a highly enantioselective silylation of C–H bonds catalyzed by the combination of a rhodium precatalyst and a chiral bisphosphine ligand.15 In this reaction, (hydrido)silyl ethers 2, which are formed in situ by hydrosilylation of benzophenone or its derivatives 1, undergo asymmetric C–H silylation in high yield with excellent enantioselectivity to form enantioenriched benzoxasilole products 3 (Scheme 1). This reaction proceeds in the presence of [Rh(cod)Cl]2 and catASium (L1-L2) or Walphos (L3-L5) ligands with norbornene (nbe) as a hydrogen acceptor.
Scheme 1.
Rh-Catalyzed Enantioselective Silylation of Aryl C–H Bonds
Here, we describe detailed mechanistic studies on these enantioselective C–H silylation reactions. We show that the catalyst resting state is a mixture of a rhodium silyl dihydride and a rhodium norbornyl complex, the ratio of which depends on the relative concentrations of silane and norbornene. We prepared these complexes independently and showed that they are competent to be reaction intermediates. Measurements of inter- and intramolecular kinetic isotope effects (KIE) indicated that the C–H cleavage step is not rate-determining and is partially reversible. Our studies indicate that both C–H oxidative addition and C–Si reductive elimination steps influence the enantioselectivity. DFT calculations suggest that the enantioselectivity results from steric repulsion between the alkyl substituent on silicon of the substrate and the aryl group of the ligand in the transition state that forms the minor enantiomer.
RESULT AND DISCUSSION
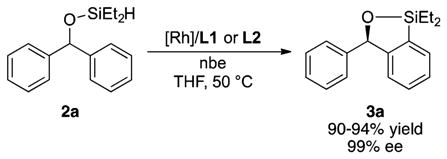
As stated in the introduction, the enantioselective silylation of diarylmethyl silyl ethers 2 occurs in the presence of [Rh(cod)Cl]2 and catASium (L1-L2) or Walphos (L3-L5) ligands with norbornene as hydrogen acceptor (Scheme 1). To gain detailed information on the mechanism of this process, we chose to study the reaction of 2a catalyzed by [Rh(cod)Cl]2 and catASium ligands L1 or L2 (eq 1). The reaction yields the silylated product 3a in 90–94% yield, as determined by 1H NMR analysis, with an enantiomeric excess of 99% with both ligands. The reaction conducted with [Rh(C2H4)2Cl]2 as the rhodium precursor occurred with the same level of reactivity and enantioselectivity.
 |
(1) |
Characterization of the Catalyst Resting States
The silylation of 2a with [Rh(C2H4)Cl]2 or [Rh(cod)Cl]2 as the source of rhodium, L1 as the ligand, and 1.2 equiv of norbornene as hydrogen acceptor was monitored in THF-d8 at 50 °C (eq 1). We observed two phosphine-ligated rhodium species 4a and 5 during the reaction (Figure 1). The 31P NMR signals corresponding to 4a consisted of two doublets of doublets at 29.4 ppm (JRh–P = 123 Hz, JP–P = 22.6 Hz) and 25.1 ppm (JRh–P = 131 Hz, JP–P = 22.6 Hz), and those corresponding to complex 5 consisted of two broad doublets at 36.6 ppm (JRh–P = 264 Hz) and 24.5 ppm (JRh–P = 178 Hz).
Figure 1.
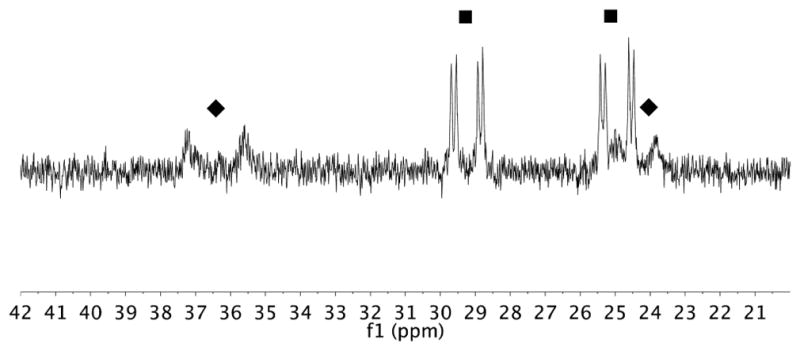
31P NMR spectrum at 50 °C of silylation reaction of 2a conducted with [Rh(C2H4)Cl]2 (2.5 mol %) and L1 (5 mol %) as the catalyst precursors and 1.2 equiv of norbornene at ca. 40% conversion of 2a. 4a (■), 5 (◆).
Two hydride species were observed in the 1H NMR spectrum. A broad singlet at −8.62 ppm and a broad doublet at −3.12 ppm were observed at 50 °C. Correlations between protons at −8.62 ppm and 31P nuclei of complex 4a and correlations between the proton at −3.12 ppm and the 31P nuclei of complex 5 were observed in the 31P–1H HMBC NMR spectrum.
The relative concentrations of 4a and 5 varied during the course of the reaction. At the beginning of the reaction, the concentration of 4a was higher than that of 5. However, the concentration of 5, relative to that of 4a, increased as the reaction progressed, and complex 4a became undetectable at greater than 90% conversion.
Monitoring of the reactions with L2 in place of L1 led to the same general observations. The 31P NMR spectrum contained two sets of signals, one of which was sharp and one of which was broad. The 1H NMR spectrum also contained two hydride signals. Finally, a similar interconversion of the two phosphine-ligated rhodium hydride complexes occurred during the course of the reaction catalyzed by the complex of L2 as occurred during the reaction catalyzed by the complex of L1, and the relative amounts of the two complexes ligated by L2 varied as did the amounts of the two complexes ligated by L1. Thus, the mechanistic data on these two catalyst systems are generally interchangeable.19
To identify complexes 4a and 5, we prepared them independently. We synthesized a complex having NMR signals that matched those of rhodium hydride species 4a. We generated this complex by the reaction of [Rh(C2H4)Cl]2 and L1 with 10 equiv of silane 2a and isolated it as a yellow solid in 77% yield (eq 2). Complex 4a is a rhodium silyl dihydride species that is analogous to the complex we observed as the resting state in the rhodium-catalyzed intermolecular silylation of aryl C–H bonds.5,20 However, 4a is distinct from the previously reported complex because 4a contains aryl C–H bonds that can react.21
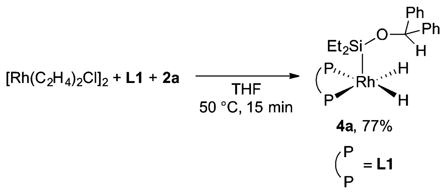 |
(2) |
The solid-state structure of 4a was determined to be a distorted square-based pyramid by single-crystal X-ray analysis (Figure 2 and Supporting Information). The phenyl groups on the silyl ether are positioned far from the rhodium center. The two hydrides are nonequivalent, but the two rhodium–hydride protons resonate as a single signal in the 1H NMR spectrum at 50 °C. Complex 4a is likely to undergo pseudorotation of the ligands in solution at that temperature, and this pseudorotation leads to the site exchange between the two rhodium hydrides. The broad hydride signal in the 1H NMR spectrum was fully resolved into two sharp signals at −20 °C. Each of the two signals is a doublet of doublets of doublets of doublets due to the J couplings with the rhodium nucleus, two phosphorus nuclei, and the other hydride proton. An energy barrier (ΔG‡310 K) for the exchange of the hydrides of 4a was calculated to be ca. 14 kcal/mol from the coalescence temperature (Tc) of 37 °C and the extrapolated peak separation of Δν = 452 Hz at the Tc in the 1H NMR spectrum.
Figure 2.
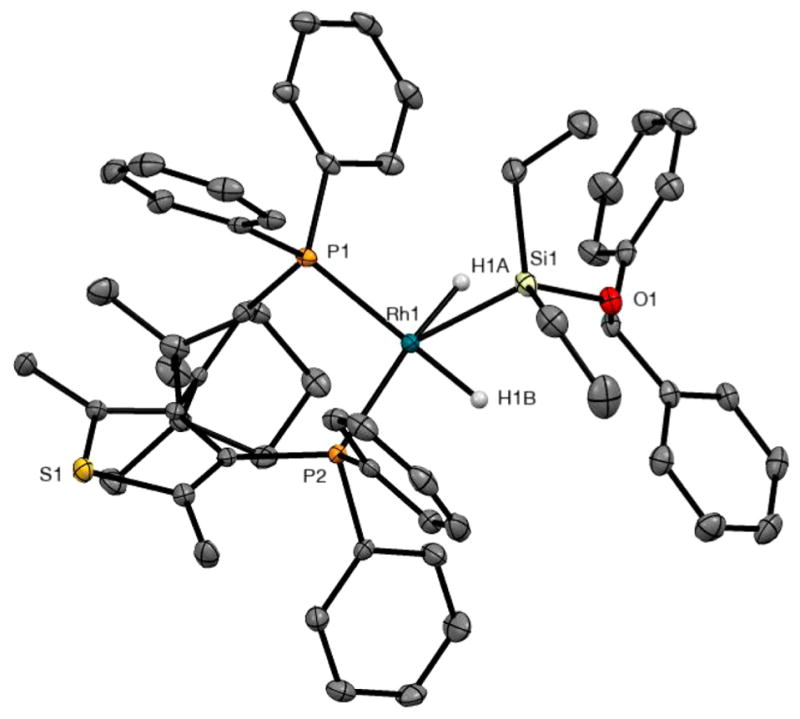
ORTEP diagram of 4a (thermal ellipsoids at the 50% probability level). Hydrogen atoms have been omitted except for the hydrides bound to rhodium. Selected bond lengths (Å): Rh1–P1 = 2.2763(8), Rh1–P2 = 2.3456(8), Rh1–H1A = 1.48(3), Rh1–H1B = 1.49(3), and Rh1–Si1 = 2.3126(8). Selected bond angles (deg): P1–Rh1–Si1 = 115.52(3), P2–Rh1–Si1 = 129.64(3), H1A–Rh1–Si1 = 53(1), and H1B–Rh1–Si1 = 63(1).
In addition, we independently prepared a complex with NMR signals that matched those of rhodium complex 5, which is the major species observed during the catalytic reaction at high conversion of silane 2a. This complex was generated by the addition of lithium triethylborohydride to a solution of [Rh(C2H4)Cl]2, L1 and norbornene (eq 3), followed by heating at 50 °C for 2 h.22 Complex 5′, containing L2 in place of L1, was prepared in the same manner. We were unable to isolate complexes 5 and 5′ in pure form because of their high solubility, but they were generated in approximately 70% yields, as determined by 1H NMR spectroscopy with an internal standard.
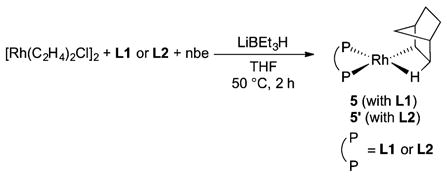 |
(3) |
We assigned 5 and 5′ to be norbornyl–rhodium complexes containing β-agostic interactions between the rhodium and a C–H bond of the norbornyl ligand. The signals at the chemical shift of −3.12 ppm (5) and −3.24 ppm (5′) were assigned to the agostic protons. These chemical shifts are more downfield than those of terminal rhodium hydride complexes23–25 and are similar to those of other metal complexes that contain norbornyl ligands with agostic interactions.26–31 The 1JC–H coupling constant of the agostic C–H bond of 5′ was ~60 Hz at −80 °C, as determined by 2D J-resolved 13C NMR spectroscopic analysis. This value is significantly lower than those of the analogous free C–H bonds and agrees with the previously reported values for C–H bonds engaged in agostic interactions.32 In addition, the infrared spectrum of 5′ did not contain any bands attributable to Rh–H stretches, which further supports the assertion that 5′ is not a rhodium species containing a terminal metal hydride.33
Considering the structures of 4a and 5, we propose that the resting state of the catalytic reaction changes from 4a to 5 during the catalytic process because the relative concentration of norbornene to the substrate 2a increases. The change in the relative concentrations occurs because norbornene was used in slight excess relative to 2a and because a pathway for the catalytic conversion of 2a to 3a that does not require norbornene is followed in parallel with a pathway that requires norbornene. Consistent with this proposal, complex 5 was the major species at all times when the reaction was conducted with a larger excess (2.2 equiv) of norbornene.
Evaluation of the Kinetic Competency of 4a
The reactivity of the isolated complex 4a was investigated to assess the relevance of the complex to the catalytic system (Scheme 2). The reaction of 4a with norbornene in THF at 50 °C formed 3a in 72% yield with 98% ee. The enantiomeric excess of this stoichiometric reaction matched that of the catalytic reaction (99% ee). In addition, complex 5 formed as a product of this reaction. This result suggests that 4a and 5 are potential intermediates in the catalytic reaction. The intermediacy of 4a was further demonstrated by the reaction catalyzed by 4a. The reaction of 2a catalyzed by complex 4a resulted in the formation of 3a in quantitative yield and 97% ee.
Scheme 2.
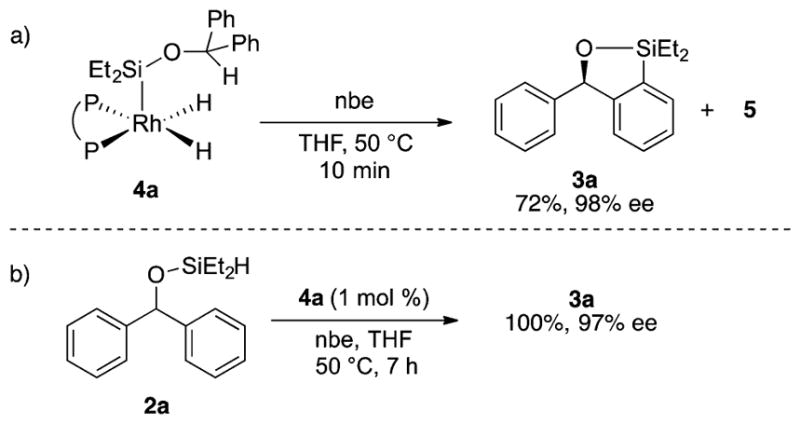
Reactivity of Complex 4a
Determination of the Experimental Rate Law
We determined the rate law of the silylation process by the method of initial rates with [Rh(cod)Cl]2 and L1 (Rh:L1=1:1.25) as the catalyst. The reaction was first order in catalyst and first order in silane 2a when the concentration of 2a was below ~0.33 M (0.83 equiv to nbe) and was zeroth order in 2a when the concentration of 2a was higher than ~0.33 M (Figure 3a). The dependence of the rate on the concentration of norbornene was more complex (Figure 3b). The reaction proceeded in the absence of norbornene,34 but the reaction in the presence of norbornene was faster than that in the absence of norbornene. The rate was first order in norbornene up to ~0.40 M of norbornene (1.2 equiv to 2a). However, the rate decreased with increasing concentrations of norbornene when the concentration of norbornene was higher than ~0.40 M.
Figure 3.
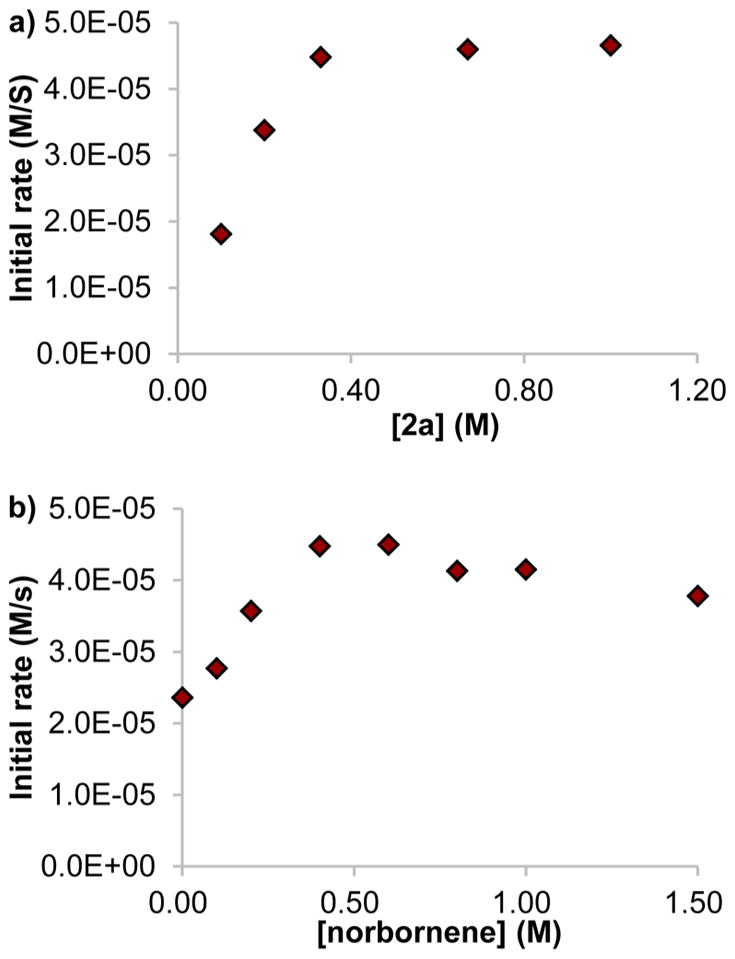
Dependence of the rate of C–H silylation on the concentration of (a) 2a and (b) norbornene. Reactions were conducted with 3.3 mM of catalyst, (a) 0.40 M of nbe, and (b) 0.33 M of 2a.
These findings are consistent with our proposal that the resting state of the reaction depends on the ratio of 2a to norbornene. When [2a] was higher than [nbe], silyl complex 4a was the major species, and this resting state led to a zeroth-order dependence of the rate on [2a] and positive-order dependence of the rate on [nbe].35 On the other hand, when [2a] was lower than [nbe], norbornyl complex 5 was the major rhodium species, and this change in the resting state led to a first-order dependence on [2a] and a partial inverse-order dependence on [nbe].
The partial inverse-order dependence on [nbe] when complex 5 was the major rhodium species is likely to result from competitive pathways with different rate dependences on the concentration of norbornene (Figure 4). For the pathway that is inversely dependent on the concentration of norbornene, complex 5 would react by reversible dissociation of norbornene to generate a 14-electron Rh(I) hydride 6. For the pathway that is independent of the concentration of norbornene, complex 5 could react directly with silane 2a to form silyl hydride complex 7a, or 5 could react in a multistep sequence involving release of norbornene to form 6, addition of silane 2a to 6 to form dihydride silyl complex 4a, and insertion of norbornene into the hydride of 4a to generate 7a. This multistep process would be zero-order in norbornene because norbornene dissociates and adds prior to the rate-limiting transition state. If pathways with inverse and zero-order dependences on norbornene occur competitively, then the complex kinetic behavior in Figure 3b would be observed.
Figure 4.
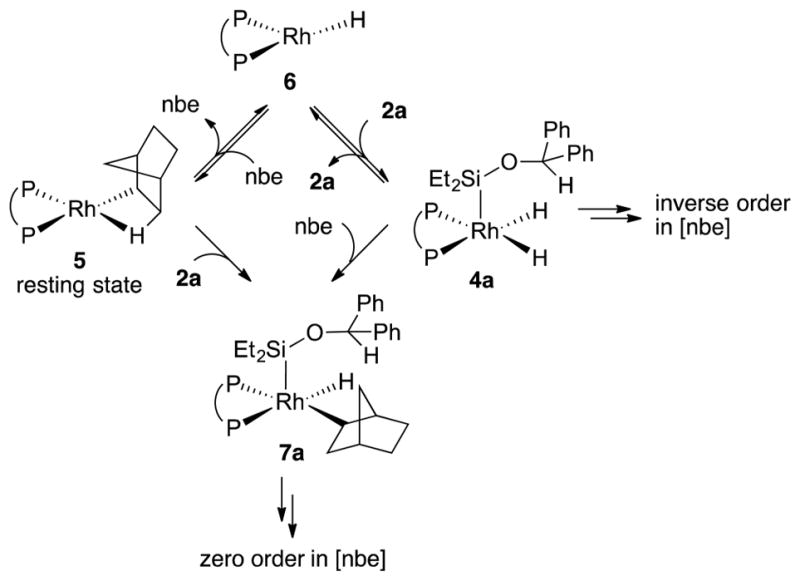
Competitive pathways responsible for the partial inverse-order rate dependence on [nbe] when complex 5 is the resting state.
As shown in Figure 4, the insertion of norbornene into rhodium silyl dihydride 4a or the addition of silane 2a into 5 leads to formation of complex 7a. The distinction between these two pathways is subtle because they are indistinguishable by the kinetic analysis of the catalytic reaction. Therefore, we conducted DFT calculations to determine which pathway is preferred (Figure 5).
Figure 5.
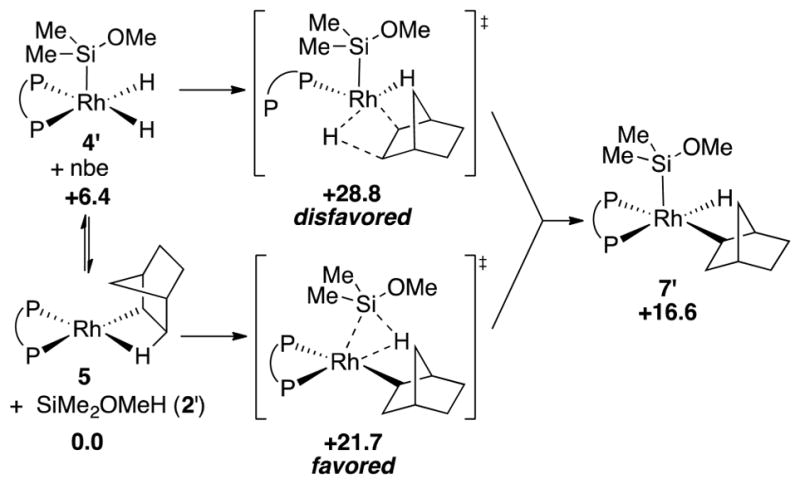
Computed pathways for the formation of 7′. Values given for Gibbs free energy (kcal/mol).
This calculation was performed on model compounds 2′, 4′, and 7′ to simplify the calculation while maintaining the electronic environment on the silicon. The structures of intermediates and transition states were optimized using the B3LYP functional with the lanl2dz basis set for rhodium and the 6-31G** basis set for all other atoms. Single-point energy calculations with solvent correction (THF, SMD) were conducted using the M06 functional with the lanl2tz basis set for rhodium and the 6-311++G** basis set for all other atoms.36
As shown in Figure 5, the computed transition state for the insertion of norbornene into 4′ to form 7′ was 7.1 kcal higher than that for the oxidative addition of the Si–H of 2′ into 5. These calculations imply that complex 7a is likely to be generated from the reaction between 5 and 2a, not from the reaction between 4a and norbornene.
Measurement of the Kinetic Isotope Effect
We determined the kinetic isotope effect (KIE) by measuring the initial rates of the silylation of 2a and 2a-d10 in separate vessels (Figure 3a). The KIE from this set of experiments was 1.1 ± 0.1. This value indicates that C–H bond cleavage is not the rate-determining step.37 To assess the reversibility of the C–H bond cleavage step, we measured an intramolecular KIE. The reaction of 2a-d2 (Scheme 3b) gave a mixture of 3a-d2 and 3a-d1 in a 3.0 ± 0.1:1 ratio. This KIE is primary, but the modest value suggests that the C–H bond oxidative addition step could be partially reversible38 and that both the oxidative addition of the C–H bond and reductive elimination to form the C–Si bond could affect the enantioselectivity.
Scheme 3.
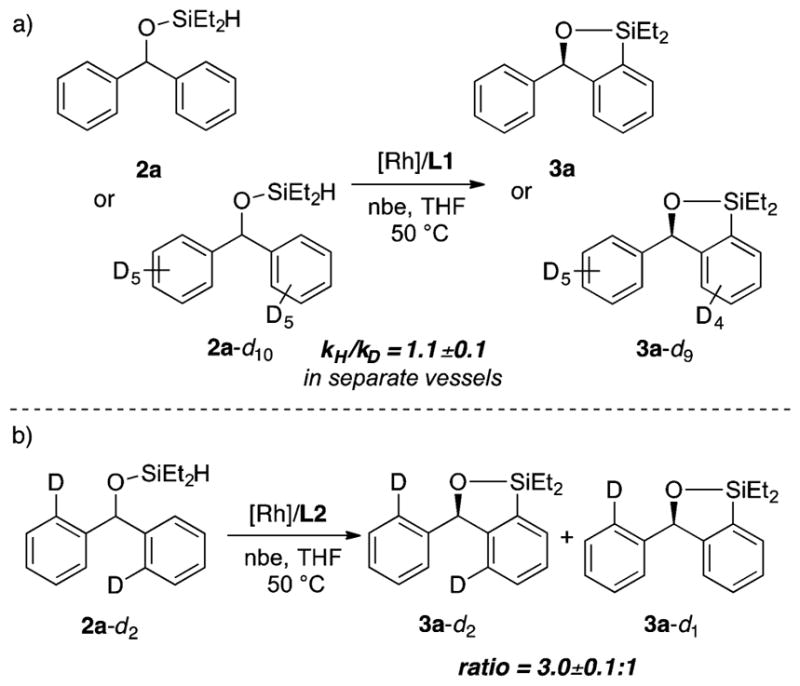
Kinetic Isotope Effects
Deuterium Labeling Study
We studied reactions of a substrate 2b-d1 to gain further insight into the reaction mechanism (eq 4). When the silylation of 2b-d1 was conducted with the catalyst containing L2 and norbornene, deuterium was incorporated only into norbornane. The absence of deuterium in aryl C–H bonds of the remaining 2b-d1 at partial conversion or in 3b at partial or full conversion indicates that the rhodium deuteride species does not cleave the C–H bond to generate an intermediate containing two equivalent or interconverting hydrides. Because the C–H cleavage step is likely to be partially reversible, according to the moderate intramolecular KIE value and computational studies described in the next section, incorporation of deuterium into the aryl C–H bonds in 2b-d1 or 3b would be observed if a rhodium deuteride species were to undergo oxidative addition of the C–H bond.
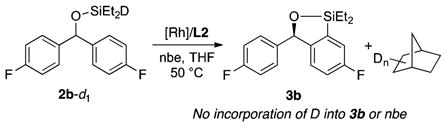 |
(4) |
We sought to investigate the reversibility of insertion of the hydrogen acceptor into the rhodium hydride by testing for the incorporation of deuterium into the remaining hydrogen acceptor. Although we used norbornene in our synthetic studies, norbornene is not a suitable hydrogen acceptor for this study because deuterium would not be incorporated into norbornene, even if the insertion process were reversible. The insertion occurs by syn-insertion, and the reverse reaction occurs by syn-β-H elimination. Thus, deuterium would not be present in nobornene after the β-H elimination (eq 5).
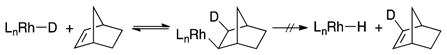 |
(5) |
Instead, we conducted the silylation of 2b-d1 with cyclo-hexene as a hydrogen acceptor (eq 6). In this experiment, we observed the incorporation of deuterium into both cyclohexene and cyclohexane.39 This result suggests that the insertion of the rhodium hydride into the hydrogen acceptor is reversible.
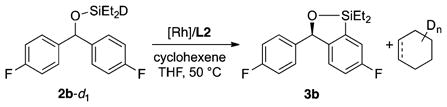 |
(6) |
Computational Study of the Origin of Enantioselec-tivity
We conducted DFT calculations to investigate the origin of enantioselectivity. We calculated the energies of intermediates and transition states involved in the oxidative addition of the C–H bond and reductive elimination to form the C–Si bond (Figure 6).40 The computational method described previously was used in this calculation.
Figure 6.

Computed pathways for the C–H oxidative addition and C–Si reductive elimination.
As shown in Figure 6, the computed transition state for the oxidative addition of the C–H bond in the pathway to form the major enantiomer was 2.8 kcal higher than that for the reductive elimination of the C–Si bond in the same pathway. This result suggests that oxidative addition is the enantioselectivity-determining step. However, the intramolecular KIE was calculated to be 4.2, and this value is higher than the experimental value of 3.0. This difference implies that the difference in energy between the two steps could be overestimated by the DFT calculation. If the barrier for the C–H oxidative addition step were reduced by 3.0 kcal or the barrier for the C–Si reductive elimination were increased by 3.0 kcal without changing the KIE’s of each elementary step, the computed KIE would be 3.5 (see the Supporting Information for details). On the basis of this analysis, we suggest that the energy barriers for the oxidative addition of the C–H bond and reductive elimination to form the C–Si bond are likely to be similar to each other, and both steps are likely to affect the enantioselectivity of the silylation process.
The difference in free energy between the transition states to form the major and minor enantiomers was calculated to be 6.3 kcal for the C–H oxidative addition step and 4.2 kcal for the C–Si reductive elimination step. These values are in line with the observed enantiomeric excess of 99%.
Comparison of the diastereomeric transition states leading to the formation of two enantiomers revealed the origin of the enantioselectivity (Figure 7). Three phenyl groups of L1 project toward the coordination sites of the bound substrate, rendering quadrants II, III, and IV less accessible than quadrant I (Figure 7a). In TS-IR and TS-IS, an ethyl group on the silicon of the substrate points toward L1 (Figure 7, panels b and c). In TS-IR, the silyl group is placed in quadrant I, minimizing the steric repulsion between the ethyl group of the substrate and the phenyl group of the ligand. A similar arrangement exists in the experimental solid-state structure of the silyl complex 4a. In this structure, the silyl group resides in quadrant I, presumably to avoid unfavorable interaction with the ligand (Figure 2). In TS-IS, however, the silyl group lies in quadrant II to avoid the steric interaction between L1 and the phenyl group of the substrate that is not reacting. This conformation places the ethyl group proximal to the phenyl group in quadrant II, causing an unfavorable steric interaction. Similar analysis of the transition states TS-IIR and TS-IIS indicates that an analogous interaction causes the energy of TS-IIS to be higher than that of TS-IIR (see the Supporting Information). Thus, these calculations indicate that the difference in orientations of the substrate in the diastereomeric transition states for oxidative addition of the C–H bond, as well as reductive elimination to form the C–Si bond, gives rise to the high level of enantioselectivity.
Figure 7.

Computed structures of the transition states of the C–H oxidative addition step: (a) Rhodium ligated by L1 in TS-IR, (b) TS-IR, and (c) TS-IS. Hydrogen atoms have been omitted except for the ones in C–H bonds being cleaved.
Proposed Mechanism
On the basis of the results described above, we propose that the rhodium-catalyzed enantioselective silylation of aryl C–H bonds occurs by the mechanism shown in Scheme 4.
Scheme 4.
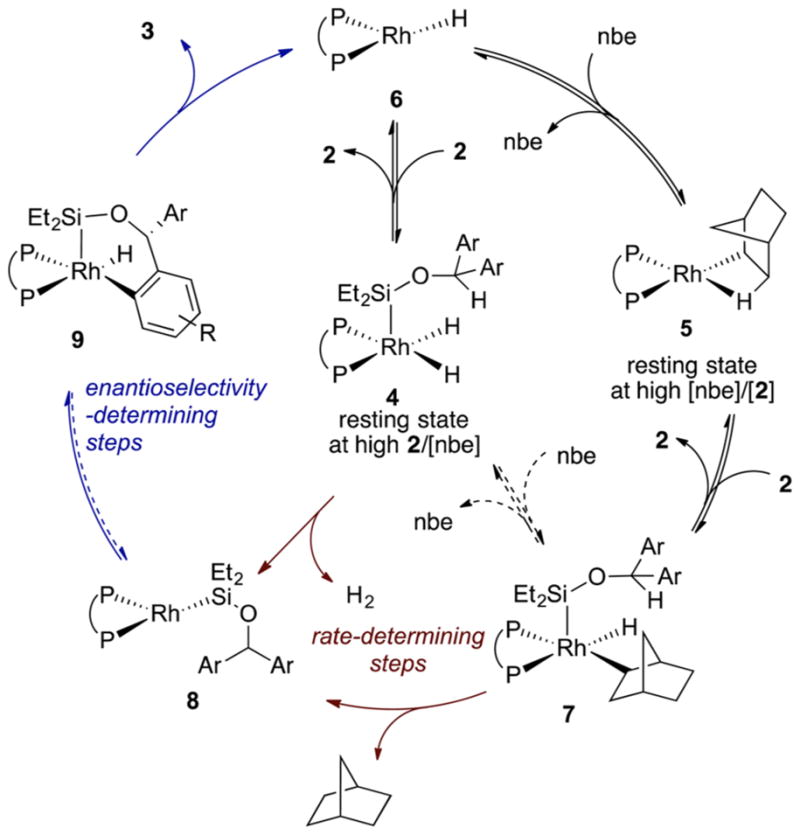
Proposed Mechanism of Enantioselective Silylation of Aryl C–H bonds
In this mechanism, the catalyst resting state is either 4 or 5, and the identity of the resting state depends on the relative concentrations of the silane 2 and norbornene. The addition of silane 2 to 5 generates 7. Although intermediate 7 can be formed by the insertion of norbornene into rhodium silyl dihydride 4, we disfavor this alternative mechanism based on the higher barrier computed by DFT for this pathway than that computed for the addition of 2 to 5. Either process that forms 7 is likely to be reversible because we observed the incorporation of deuterium into a hydrogen acceptor, cyclohexene, when a silane containing a Si–D bond was used.
Intermediate 7 can then undergo reductive elimination to generate intermediate 8 and norbornane. Alternatively, 8 can be formed by the reductive elimination of H2 from 4. Intermediate 8 would then undergo oxidative addition of the C–H bond. Oxidative addition of the C–H bond by complex 8 is consistent with the lack of deuterium incorporation into the aryl C–H bonds from reactions with the substrate containing an Si–D bond. If other intermediates, such as 4 or 7, cleaved the C–H bond, incorporation of deuterium into the aryl C–H bonds in 2 or 3 would occur because the oxidative addition of the C–H bond is partially reversible.
Reductive elimination occurs to form the C–Si bond of silylation product 3 and rhodium hydride species 6. The transition-state energy for this step is likely to be similar to that of the previous step (oxidative addition of the C–H bond) and to influence the enantioselectivity together with the preceding C–H bond cleavage step, according to the results from the measurement of the KIE from an intramolecular competition between the silylations of C–H and C–D bonds and DFT calculations. Intermediate 6 then can be converted to the catalyst resting states 4 or 5 by addition of the Si–H bond of 2 or by insertion of norbornene.
Because the C–H bond cleavage step is not rate-determining, according to the KIE value of 1.1 for side-by-side reactions, and the energies of the transition states for oxidative addition of the C–H bond and reductive elimination to form the C–Si bond are likely to be similar to each other, for the reasons discussed above, we propose that reductive elimination of norbornane or dihydrogen to generate a Rh(I) silyl complex 8 is the rate-determining step for this silylation process. The generation of a Rh(I) silyl complex is the same process we proposed to be rate-determining in the mechanism of intermolecular silylation of aryl C–H bonds.5
CONCLUSION
The identity of the resting states of the catalyst, the kinetic data, and the results of DFT calculations provide detailed insights into the mechanism of intramolecular enantioselective silylation of aryl C–H bonds and the origin of enantioselectivity. We showed that the rhodium silyl dihydride and the rhodium norbornyl complex are the catalyst resting states. The kinetic isotope effect measured in separate vessels was close to 1, implying that oxidative addition of the C–H bond is not the rate-determining step. However, the KIE from an intra-molecular competition between the silylations of C–H and C–D bonds was 3.0. This value and the results from the DFT calculations suggest that the transition-state energies of the C–H bond cleavage step and the C–Si bond forming step are close to each other and, therefore, affect the enantioselectivity of this silylation process together. In addition, the examination of calculated diastereomeric transition states indicates that the steric interaction between the alkyl substituent of the silicon on the substrate and the aryl group on the bisphosphine ligand lead to the high level of enantioselectivity.
This work constitutes a rare systematic mechanistic study on enantioselective C–H bond functionalization41–44 and the first on the enantioselective functionalization of C–H bonds with main group reagents. By conducting a combination of experimental and theoretical studies, we elucidated the enantioselectivity-determining steps and the origin of the high enantioselectivity. We envision that insights obtained from this study may serve as a guideline for the design of catalysts for additional enantioselective C–H functionalizations.
Supplementary Material
Acknowledgments
We thank the NIH (GM-115812 to J.F.H. and S10-RR027172 to the X-ray facility) and the NSF (CHE-0840505 to the Molecular Graphics and Computation Facility at UC Berkeley) for support of this work. T.L. thanks the Samsung Scholarship for a graduate fellowship, Dr. Antonio G. DiPasquale for help wtih X-ray crystallography, Dr. Qian Li, Dr. Kathleen Durkin, and Dr. Olayinka Olatunji-ojo for help with DFT calculations, Dr. Hasan Celik for help with NMR spectroscopy, and Dr. Per Ryberg, Chen Cheng, Matthew A. Larsen, and Dr. Sarah Y. Lee for insightful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b00737.
Experimental procedures and computational details (PDF)
X-ray crystallographic data of 4a (CIF)
References
- 1.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 2.Hartwig JF. Acc Chem Res. 2012;45:864. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 3.Cheng C, Hartwig JF. Chem Rev. 2015;115:8946. doi: 10.1021/cr5006414. [DOI] [PubMed] [Google Scholar]
- 4.Choi G, Tsurugi H, Mashima K. J Am Chem Soc. 2013;135:13149. doi: 10.1021/ja406519u. [DOI] [PubMed] [Google Scholar]
- 5.Cheng C, Hartwig JF. J Am Chem Soc. 2014;136:12064. doi: 10.1021/ja505844k. [DOI] [PubMed] [Google Scholar]
- 6.Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. J Am Chem Soc. 2005;127:14263. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 7.Li Q, Liskey CW, Hartwig JF. J Am Chem Soc. 2014;136:8755. doi: 10.1021/ja503676d. [DOI] [PubMed] [Google Scholar]
- 8.Larsen MA, Wilson CV, Hartwig JF. J Am Chem Soc. 2015;137:8633. doi: 10.1021/jacs.5b04899. [DOI] [PubMed] [Google Scholar]
- 9.Obligacion JV, Semproni SP, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:10645. doi: 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]
- 10.Kuninobu Y, Yamauchi K, Tamura N, Seiki T, Takai K. Angew Chem, Int Ed. 2013;52:1520. doi: 10.1002/anie.201207723. [DOI] [PubMed] [Google Scholar]
- 11.Murai M, Takeuchi Y, Yamauchi K, Kuninobu Y, Takai K. Chem - Eur J. 2016;22:6048. doi: 10.1002/chem.201504718. [DOI] [PubMed] [Google Scholar]
- 12.Shibata T, Shizuno T, Sasaki T. Chem Commun. 2015;51:7802. doi: 10.1039/c5cc00723b. [DOI] [PubMed] [Google Scholar]
- 13.Zhang QW, An K, Liu LC, Yue Y, He W. Angew Chem, Int Ed. 2015;54:6918. doi: 10.1002/anie.201502548. [DOI] [PubMed] [Google Scholar]
- 14.Murai M, Matsumoto K, Takeuchi Y, Takai K. Org Lett. 2015;17:3102. doi: 10.1021/acs.orglett.5b01373. [DOI] [PubMed] [Google Scholar]
- 15.Lee T, Wilson TW, Berg R, Ryberg P, Hartwig JF. J Am Chem Soc. 2015;137:6742. doi: 10.1021/jacs.5b03091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murai M, Takeshima H, Morita H, Kuninobu Y, Takai K. J Org Chem. 2015;80:5407. doi: 10.1021/acs.joc.5b00920. [DOI] [PubMed] [Google Scholar]
- 17.Lee T, Hartwig JF. Angew Chem, Int Ed. 2016;55:8723. doi: 10.1002/anie.201603153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su B, Zhou TG, Li XW, Shao XR, Xu PL, Wu WL, Hartwig JF, Shi ZJ. Angew Chem, Int Ed. 2017;56:1092. doi: 10.1002/anie.201609939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The parallels between the mechanistic observations with the two ligands were valuable because the commercial availability of L1 ended during the course of this study.
- 20.Cheng C, Hartwig JF. Science. 2014;343:853. doi: 10.1126/science.1248042. [DOI] [PubMed] [Google Scholar]
- 21.In this reaction mixture, we observed the formation of 3a.
- 22.Swartz BD, Ateşin TA, Grochowski MR, Oster SS, Brennessel WW, Jones WD. Inorg Chim Acta. 2010;363:517. [Google Scholar]
- 23.Green MLH, Joyner DS, Wallis JM. J Chem Soc, Dalton Trans. 1987:2823. [Google Scholar]
- 24.Strauss SH, Shriver DF. Inorg Chem. 1978;17:3069. [Google Scholar]
- 25.Pregosin PS. NMR in Organometallic Chemistry. Wiley-VCH; Weinham: 2012. pp. 67–73. [Google Scholar]
- 26.Carr N, Dunne BJ, Orpen AG, Spencer JL. J Chem Soc, Chem Commun. 1988:926. [Google Scholar]
- 27.Carr N, Dunne BJ, Mole L, Orpen AG, Spencer JL. J Chem Soc, Dalton Trans. 1991:863. [Google Scholar]
- 28.Zayya AI, Spencer JL. Organometallics. 2012;31:2841. [Google Scholar]
- 29.Urtel H, Meier C, Eisenträger F, Rominger F, Joschek JP, Hofmann P. Angew Chem, Int Ed. 2001;40:781. doi: 10.1002/1521-3773(20010216)40:4<781::aid-anie7810>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 30.Speckman DM, Knobler CB, Hawthorne MF. Organometallics. 1985;4:426. [Google Scholar]
- 31.Pisareva IV, Dolgushin FM, Godovikov IA, Chizhevsky IT. Inorg Chem Commun. 2008;11:1202. [Google Scholar]
- 32.Brookhart M, Green MLH, Wong L-L. Carbon-Hydrogen-Transition Metal Bonds. Vol. 36. John Wiley & Sons, Inc; Hoboken, NJ: 1988. pp. 1–124. [Google Scholar]
- 33.We were unable to unambiguously identify the stretching vibrations of the agostic C–H bond in the IR spectrum of 5′.
- 34.The reaction of 2a catalyzed by [Rh(cod)Cl]2 (0.5 mol %) and L1 (1.25 mol %) in the absence of norbornene formed 3a in 87% yield and 97% ee.
- 35.The reaction rate is zero-order in [2a] in the absence of nbe.
- 36.See the Supporting Information for the citations on the calculation methods.
- 37.Simmons EM, Hartwig JF. Angew Chem, Int Ed. 2012;51:3066. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- 38.This intramolecular KIE is similar to the KIE of 2.9 measured by a competition between the reactions of 1,3-bis(trifluoromethyl) benzene and 5-D-1,3-bis(trifluoromethyl)benzene during our previous study on Rh-catalyzed intermolecular silylation of arenes (ref 5). In this prior study, we suggested that the C–H bond cleavage step is partially reversible. In the previous work, the partial reversibility was further supported by the H–D exchange between the arene and the silane when the reaction was conducted with a silane reagent containing Si–D.
- 39.Trace amount of deuterium (less than 1%) was incorporated into 3b.
- 40.Figure 6 does not indicate that the reaction is endothermic because Figure 6 only includes part of the catalytic cycle.
- 41.Musaev DG, Kaledin A, Shi BF, Yu JQ. J Am Chem Soc. 2012;134:1690. doi: 10.1021/ja208661v. [DOI] [PubMed] [Google Scholar]
- 42.Larionov E, Nakanishi M, Katayev D, Besnard C, Kündig EP. Chem Sci. 2013;4:1995. [Google Scholar]
- 43.Cheng GJ, Chen P, Sun TY, Zhang X, Yu JQ, Wu YD. Chem - Eur J. 2015;21:11180. doi: 10.1002/chem.201501123. [DOI] [PubMed] [Google Scholar]
- 44.Gwon D, Park S, Chang S. Tetrahedron. 2015;71:4504. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.