

SUMMARY
Glioblastoma (GBM) is the most common primary brain cancer in adults and is notoriously difficult to treat because of its diffuse nature. We performed single-cell RNA sequencing (RNA-seq) on 3,589 cells in a cohort of four patients. We obtained cells from the tumor core as well as surrounding peripheral tissue. Our analysis revealed cellular variation in the tumor’s genome and transcriptome. We were also able to identify infiltrating neoplastic cells in regions peripheral to the core lesions. Despite the existence of significant heterogeneity among neoplastic cells, we found that infiltrating GBM cells share a consistent gene signature between patients, suggesting a common mechanism of infiltration. Additionally, in investigating the immunological response to the tumors, we found transcriptionally distinct myeloid cell populations residing in the tumor core and the surrounding peritumoral space. Our data provide a detailed dissection of GBM cell types, revealing an abundance of information about tumor formation and migration.
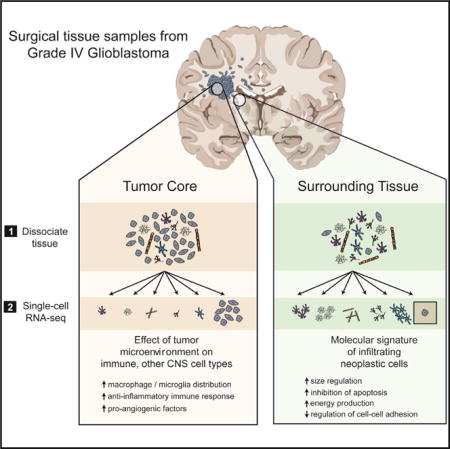
Graphical abstract
INTRODUCTION
Glioblastoma (GBM) is the most common malignant primary brain cancer in adults (Bush et al., 2016). GBMs are incurable tumors; despite aggressive treatment, including surgical resection, chemotherapy, and radiotherapy, the median overall survival remains only 12–18 months (Wen and Kesari, 2008). Unlike brain metastases, for which local control rates following surgery and radiation can reach 80%, GBMs are diffusely infiltrating (Claes et al., 2007) and invariably recur, even in distant regions of the brain. The diffuse nature of GBMs renders local therapies ineffective, as migrating cells outside of the tumor core are generally unaffected by local treatments and are responsible for the universal recurrence of GBMs in patients.
The development of novel treatment strategies is predicated upon a better understanding of the molecular features of these tumors, with a particular focus on the ability to capture and identify the infiltrating cells responsible for recurrence. Although bulk tumor sequencing approaches have been useful in generating classification schemas of GBM subtypes (Cancer Genome Atlas Research Network, 2008; Verhaak et al., 2010), they provide limited insight to the true heterogeneity of GBM tumors. Inter-patient variation and molecular diversity of neoplastic cells within individual GBMs has been previously described (Patel et al., 2014), but studies thus far have been limited in scope to the molecular complexity of cells from the tumor core; existing studies at single-cell resolution have been unable to address the nature of infiltrating GBM cells or the unexplored variety of other neuronal, glial, immune, and vascular cell types that reside within and around GBMs. The interplay between each of these cell types within the tumor microenvironment likely contributes significantly to tumor progression and resistance to therapy.
To capture and characterize infiltrating tumor cells, and to define the cellular diversity within both the tumor core and the surrounding brain, we performed high-depth single-cell RNA sequencing (RNA-seq) on a cohort of four primary GBM patients (IDH1-negative, grade IV GBMs confirmed via pathological examination). From each patient, we collected samples from two separate locations: the first residing within the tumor core and the second from peritumoral brain (Figures 1A and S1A). Additionally, from each location, we collected both unpurified cell populations and populations enriched for each of the major CNS cell types (neurons, astrocytes, myeloid cells, and endothelia) that are often overwhelmed in number by the abundance of tumor cells. This strategy allowed us to capture tumor cells that had migrated away from the primary tumor lesion into the peritumoral tissue and to specifically compare transcriptome-level effects of the tumor microenvironment on each of the various brain and immune cell types.
Figure 1. Experimental Layout.
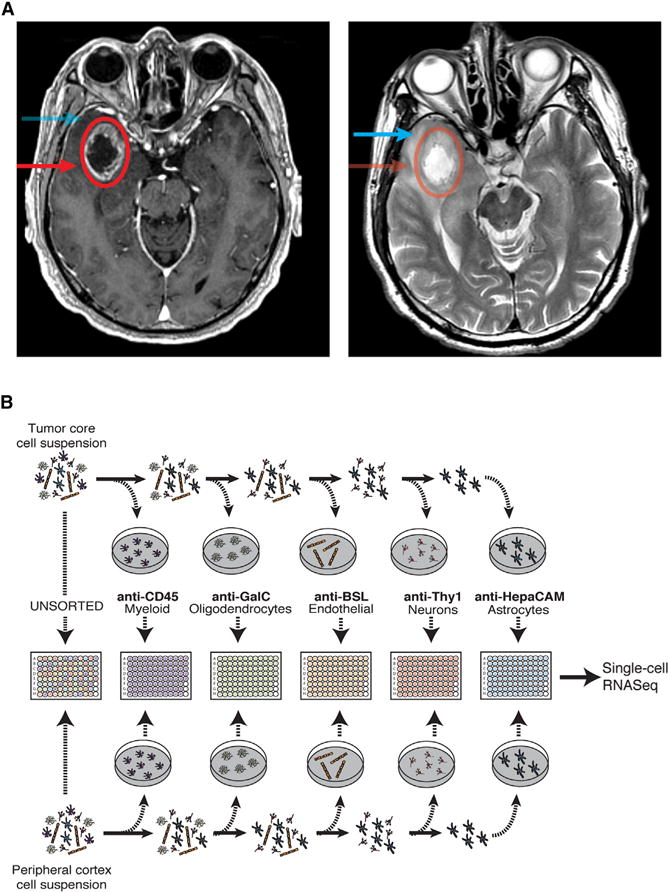
(A) Axial T1 with contrast (left side) and T2 (right side) MRI brain in a patient with a right temporal GBM. The tumor core was defined as contrast enhancing (red circle, arrow), and the peritumor was defined as enhancing yet T2 hyperintense (blue arrow).
(B) Overview of the experimental procedure.
In total, we sequenced 3,589 cells from both the tumor core and the peritumoral brain, comprising neoplastic cells and representatives from each of the major CNS cell types (vascular, immune, neuronal, and glial). Together, our data provide a large-scale dissection of GBM cell types and their respective gene expression profiles, revealing an abundance of information about tumor formation and the effects of the interaction between tumor cells and the immune system. Specifically, we managed to capture and characterize infiltrating neoplastic cells in primary tumors along the migrating front of the GBM. Additionally, we investigated the heterogeneity of GBM tumors between and within patients and characterized the effect of the tumor environment on populations of non-neoplastic cells, with particular emphasis on immune cell populations. We have deposited these transcriptomic data in a free, user-friendly website (http://www.gbmseq.org/) to ensure broad distribution and interaction with the data.
RESULTS AND DISCUSSION
Initial Clustering and Identification of Cell Types
At the onset of our efforts to sort single cells from the tumor core, we discovered that the vast majority of the cells we captured from dissociated tumor belonged to the neoplastic population, with little contribution from other glial, neuronal, vascular, or immune subtypes. Thus, to increase the relative percentage of non-neoplastic cells in our analysis, we adapted well-validated protocols for immunopanning human tissue with cell-type-specific markers (Zhang et al., 2016), with the ultimate goal of encompassing the entirety of the tumor and peri-tumor cellular landscape that is often blurred in bulk sequencing studies or insufficiently sampled in prior single cell studies. After dissociation and immuno-panning, individual cells were sorted into 96-well plates; non-immunopanned cells were also sorted to ensure that no major subpopulation of cells was excluded. This process is described in detail in the Experimental Procedures and graphically summarized in Figure 1B.
To visualize the transcriptomic landscape across all sequenced single cells, we used dimensional reduction to generate a two-dimensional (2D) map of the 3,589 single cells that passed quality control (QC) (Figure S1B; Table 1), performing an analysis similar to that of Darmanis et al. (2015). In brief, we selected genes with the highest over-dispersion (n = 500) and used them to construct a cell-to-cell dissimilarity matrix. We then performed t-distributed stochastic neighbor embedding (tSNE) on the resulting distance matrix to create a 2D map of all cells. Finally, we used k-means clustering on the 2D tSNE map, resulting in the identification of 12 distinct cell types within separate clusters (Figure 2A).
Table 1.
Dataset Summary
| Pass QC | Fail QC | |
|---|---|---|
| Cells | 3,589 (89%) | 456 (11%) |
| Sample | ||
| BT_S1 | 489 | 11 |
| BT_S2 | 1,169 | 138 |
| BT_S4 | 1,542 | 267 |
| BT_S6 | 389 | 40 |
| Location | ||
| Tumor | 2343 | 212 |
| Periphery | 1,246 | 227 |
| Sequencing Statistics | ||
| Reads (median) | 1,706,000 | 1,019,000 |
| Uniquely mapped reads (median, %) | 75.81 | 81.93 |
| Multimapped reads (median, %) | 2.35 | 0.29 |
| Genes detected (median) | 2,129 | 33 |
Number of cells per sample, number of cells per anatomical location, and general sequencing statistics are summarized for all sequenced cells that passed or failed QC. Only cells that passed QC were further analyzed.
Figure 2. General Characteristics of Neoplastic Cells.
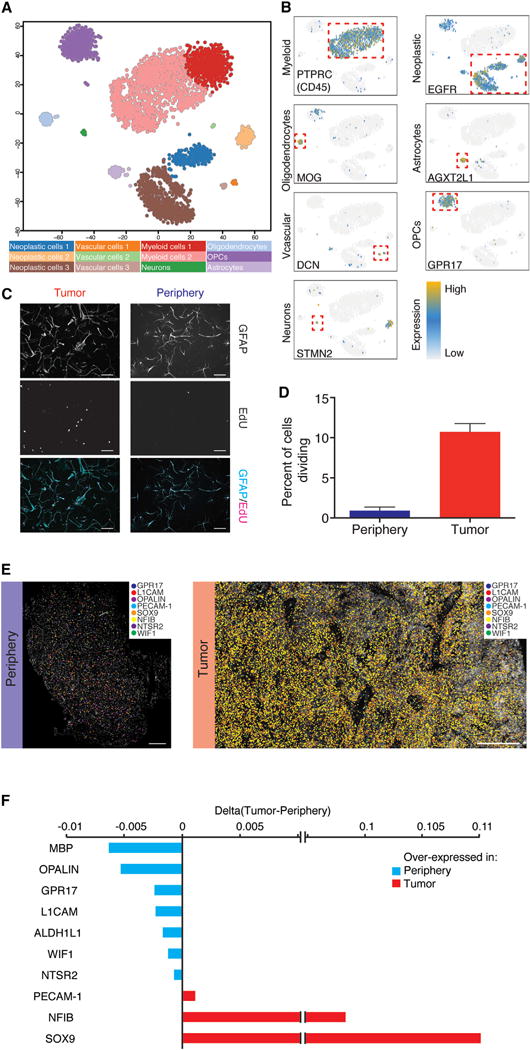
(A) 2D-tSNE representation of all single cells included in the study (n = 3,589). Cell clusters are differentially colored and identified as distinct cell classes.
(B) Expression of characteristic cell-type-specific genes overlaid on the 2D-tSNE space.
(C) GFAP and EdU staining of HepaCAM-selected cells.
(D) Quantification of EdU-positive cells as a percentage of total DAPI nuclei from the tumor core and the surrounding peritumor brain in culture for 7 days. Scale bar, 100 μm.
(E) In situ RNA staining of neoplastic and non-neoplastic specific genes in tumor tissue (right) and peritumoral brain (left). Image shown is a composite of smaller images. Scale bar, 500 μm.
(F) Quantification of in situ RNA signals shown in Figure 2E. Difference between tumor and peritumoral brain is shown.
We inferred the cellular identities of the resulting clusters from the tSNE analysis by identifying significantly overexpressed genes in each cluster. Initially, we used a Wilcoxon rank sum test to compare each gene’s expression in one cluster to its expression in all other cells, and we then cross-referenced these cluster-enriched genes (Table S1) with known cell-type-specific CNS databases (Zhang et al., 2016, 2014). We then validated the identity of the clusters by combining our dataset with published single-cell RNA-seq data from healthy human brain samples to ensure consistent cell-type classifications of each cluster (Darmanis et al., 2015). The cell-type identity of each cell cluster along with the number of cells originating from each patient and anatomical location are shown in Table S1. Genes whose expression is specific to major cell types of the brain are shown in Figure 2B. Using this method, we were able to classify all clusters into one of the major CNS neuronal, glial, or vascular subtypes found within the healthy human brain, with the exception of three clusters (1, 4, and 11). After further investigation, we preliminarily defined the cells within these three clusters as ‘‘neoplastic’’ on the basis of the following observations (as well as copy-number variant [CNV] analyses performed below). Foremost, ~94% of the cells (1,029/1,091) in neoplastic clusters originated from the tumor core. Additionally, these cells significantly overexpressed EGFR (Figure 2B), a gene upregulated in 30%–50% of all GBMs (Libermann et al., 1984, 1985a, 1985b), as well as SOX9, a transcription factor with an established oncogenic role in gliomas (Wang et al., 2012). Interestingly, the combined expression of just these two genes, EGFR and SOX9, was capable of demarcating neoplastic cells with high sensitivity and specificity (Figure S1D).
To further increase our confidence in the identity of the inferred cell types (both typical and neoplastic), we performed a direct comparison with single-cell and bulk RNA-seq data from (Darmanis et al., 2015) (healthy brain) and (Patel et al., 2014) (bulk GBM). The healthy brain dataset contains single-cell RNA-seq data from 332 cells originating from healthy adult human cortex, and the bulk GBM dataset contains RNA-seq data from bulk sequencing of five primary GBMs. The resulting tSNE map of all cells and bulk samples can be seen in Figure S1C. Using this extended dataset, we made two important observations: bulk GBM samples cluster directly in the midst of our neoplastic cell clusters, whereas single cells from the healthy brain dataset, originating from tumor-free brains, cluster together with non-neoplastic cells in each of their respective assigned cell types (Table S2). These observations provided further validation of our classification schema for both the identity of non-neoplastic cell types and the identification of GBM-specific neoplastic populations.
Because the majority of cells that were classified in the neoplastic clusters originated from the tumor core and were selected by HepaCAM immunopanning (an astrocyte-lineage marker), we maintained some of these cells in culture to compare morphological and proliferative features to HepaCAM-selected cells from the surrounding tissue. Compared with peripheral astrocytes, HepaCAM-selected cells from the tumor core exhibited a distinct morphology (Figure 2C) and were highly proliferative, as determined by EdU incorporation and expression of MKI67 (Figure 2D), further supporting the notion that they are neoplastic in origin.
Although the tumor core and surrounding samples were separated by magnetic resonance imaging (MRI)-guided surgical excision, we also looked for a molecular signature to help confirm the identity of the tumor core cells from the surrounding cells. We hypothesized that expression of hypoxic genes would correlate with the relatively oxygen-poor tumor core where cells are forced to adapt to the low-oxygen environment. Thus, we calculated a ‘‘hypoxic score’’ for each cell on the basis of the combined expression of classic hypoxic genes (PGK1, CA9, VEGFA, SPP1, and HIF1A) (Figure S1E). As expected, we observed that these hypoxic genes were expressed significantly more within the tumor core than in the surrounding regions.
It should be noted that the tumors themselves are heterogeneous and contain both neoplastic cells and non-neoplastic cells. Out of 2,343 tumor-core-originating cells, only 1,029 were members of the aforementioned neoplastic cell clusters (~44%). The vast majority of the remaining cells (n = 1,182, ~50%) belonged to immune cell clusters, with the residual cells assigned to the oligodendrocyte precursor cell (OPC) cluster (n = 50, 2.13%), one of the endothelial cell clusters (n = 47, 2%), the oligodendrocyte cluster (n = 34, ~1.5%), or the neuronal cluster (n = 1, ~0.05%). Interestingly, the only cell population without a cell originating from the tumor tissue sample is the mature astrocyte cluster.
Neoplastic Cell Characteristics
Neoplastic cells share common characteristics that distinguish them from other cells of the brain. Differential expression analysis (DESeq2) between neoplastic and non-neoplastic cells revealed genes enriched within all neoplastic cells (Figure S2A; Supplemental Experimental Procedures). Unsurprisingly, EGFR showed the highest enrichment of any gene. In addition, among the highest enriched genes, we found CHL1, a member of the L1 family of neural cell adhesion molecules that is involved in migration and positioning of neurons in the developing neocortex. Furthermore, we found upregulation of the transcription factors SOX2 and SOX9, which are also involved in brain development and lineage specification. Notably, SOX2 has been reported as a marker of glioma stem cells, along with the genes POU3F2, OLIG2, and SALL2 (Suvà et al., 2014), whereas SOX9 has been shown to correlate with poor clinical outcome (Wang et al., 2012). NFIB was another neoplastic-specific transcription factor that has been implicated in brain development (Campbell et al., 2008; Steele-Perkins et al., 2005) with an important role in the induction of quiescence in neural stem cells (Martynoga et al., 2013) and promotion of metastasis (Denny et al., 2016). We verified the upregulation of both NFIB and SOX9 by using multiplexed in situ RNA staining, with padlock probes and rolling circle amplification (Ke et al., 2013), on sections of tumor core as well as peripheral tissue (Figures 2D and S2C). We also looked for gene expression that was specifically limited to non-neoplastic cell populations and verified their expression patterns via in situ RNA staining (Figures 2D and S2C). Unsurprisingly, many of these markers were genes known to define mature, differentiated CNS cell types: MBP and OPALIN (oligodendrocytes), GPR17 (OPCs), and L1CAM (neurons), as well as ALDH1L1, WIF1, and NTSR2 (astrocytes).
In light of recent advances in our understanding of how tumors affect immune cells within their microenvironment, as well as a number of clinical studies pursuing the use of immune checkpoint inhibitors for the treatment of GBM, we next looked at the expression of major histocompatibility complex (MHC) class I genes (Figure S3D) and genes coding for ligands of PD1 (CD274 and PDCD1LG2) and CTLA4 (CD80 and CD86) on neoplastic cells (Figure S3E). We found that most neoplastic cells do not express the transcripts for ligands of PD1 and CTLA4, whereas the opposite is true of MHC class I genes. Nonetheless, we did notice a high degree of heterogeneity both within and between patients. Specifically, fewer than 25% of neoplastic cells of BT_S1 express HLA-A, HLA-B, and HLA-C, whereas more than 75% of neoplastic cells from patients BT_S2, BT_S4, and BT_S6 express these genes (Table S3). We observed the same heterogeneity in the expression of ligands of PD1 and CTLA4 as well. Interestingly, most of the neoplastic cells do not express CD274, PDCD1LG2, CD80, or CD86, with the exception of a small subset cells from BT_S2 expressing both ligands of PD1 (Figure S3F), which suggests that therapeutics directed against these targets (so-called checkpoint inhibitors) could have limited efficacy in this tumor type. We also compared the expression of immune checkpoint receptor ligands in myeloid cells from the tumor core and the peritumoral tissue, as discussed below.
Within the greater neoplastic cell cluster, we found a high degree of both inter- and intra-tumor variation. From an inter-tumor perspective, we observed that neoplastic cells largely separated on the basis of patient of origin, with each patient’s neoplastic cells clustering preferentially with other neoplastic cells from the same patient (Figure S3A); this enabled identification of genes that differentiated between each patient’s tumor (Figure S3B). Inter-patient heterogeneity was even more apparent at the level of unique CNV abnormalities between patients, as discussed below. From an intra-tumor perspective, we wondered whether heterogeneity within individual patient tumors might reflect differences in the Verhaak classification (Verhaak et al., 2010) of these tumors. However, like previous findings (Patel et al., 2014), we found that each patient’s tumor represents an ensemble of cells belonging to each of the different Verhaak molecular subtypes (see Supplemental Experimental Procedures). We therefore quantified the degree of intratumor heterogeneity, and used the endogenous heterogeneity as other CNS cell types as a benchmark for comparison (i.e., oligodendrocytes, astrocytes, and OPCs). In Figure S3C, we plotted histograms of the distributions of all pairwise distances (self-to-self distances were removed) between neoplastic cells of each patient and cells belonging to each the non-neoplastic clusters. We observed a clear and statistically significant (p < 10−16) difference between the distribution of distances of neoplastic cells originating from each of the patients and those found within the healthy cell clusters, suggesting that the neoplastic cell clusters had a higher level of internal transcriptomic heterogeneity than non-neoplastic CNS cell types.
Neoplastic Cell CNV Analysis Using RNA-Seq
Genomic CNVs are known to be among the triggers of tumor formation (Shlien and Malkin, 2009), and tumor progression is commonly associated with further variations in copy number. Using the RNA-seq data, we calculated CNV vectors for each individual cell and then clustered cells on the basis of their respective profile CNV vector (as opposed to the typical gene expression profile used for tSNE analysis). The resulting dendrogram was composed of three primary branches (Figure 3A): one (CNV 1) consisted exclusively of neoplastic cells, whereas the remaining two contained the majority of non-neoplastic cells. More specifically, one of the two non-neoplastic clusters contained all CD45-positive antigen-presenting cells (APCs) (CNV 3), and the remaining branch consisted of all other non-neoplastic cells (CNV 2). The most likely reason for the separate clustering of normal cells and APCs is the overexpression of the MHC class II genes, which cluster together on chromosome 6 (data not shown). To evaluate the degree of correlation between the CNV analysis and the initial tSNE clustering, we calculated the agreement between classifications in the two platforms. Of the cells identified as neoplastic in the initial tSNE plot, 1,047 out of 1,091 were similarly identified as neoplastic in CNV1 (4% misclassification). Additionally, 2,483 out of 2,498 cells that were initially identified as non-neoplastic belonged to either of the non-neoplastic clusters (CNV 2 or 3) (0.6% misclassification).
Figure 3. Single-Cell CNV and Variant Analysis.
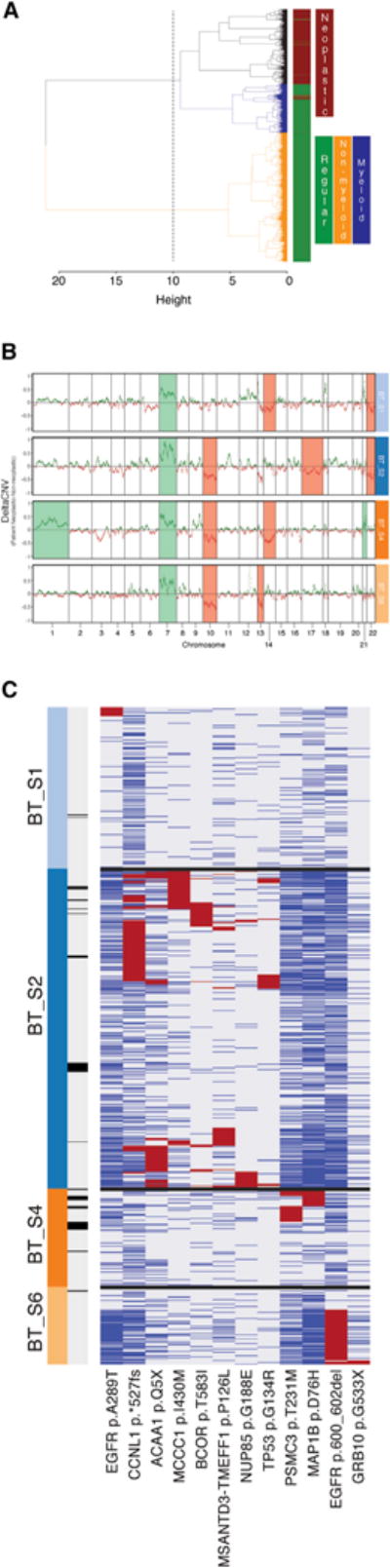
(A) Hierarchical clustering of all cells on the basis of their RNA-seq-derived CNV profile. Dendrogram branches are colored to denote different clusters of cells. Color bar demarcates neoplastic (brown) versus non-neoplastic (green) cells.
(B) DeltaCNV profiles of each patient’s neoplastic cells and non-neoplastic non-myeloid cells. Specific chromosomes that were found to be over- or underrepresented in each of the patients are highlighted.
(C) Exonic non-synonymous variants occurring in greater than 5% of any patient’s neoplastic cells. Cells (columns) might contain a variant (red), not contain the wild-type (blue), or display insufficient read coverage at that position to make a determination (gray). Cells are labeled by patient of origin (top color bar) and infiltrating status (bottom color bar; dark is infiltrating).
To determine which chromosomal regions were most affected in neoplastic cells, we subtracted the median CNV profile of the non-neoplastic, non-myeloid populations (CNV 2) from that of the neoplastic cells (CNV 1). The individual median CNV profiles along with their differences (DeltaCNV) can be seen in Figure S4A. If the differences between RNA-seq-derived CNV profiles across different cell populations were merely a result of gene-expression fluctuations with no underlying structural chromosomal variation, we would expect the DeltaCNV in Figure S4A to randomly fluctuate around zero. Instead, we specifically looked for the presence of entire chromosomes that were over-or under-represented in the neoplastic cells by counting the number of positive (higher in neoplastic cells) and negative (higher in non-neoplastic cells) occurrences (Figure S4A) for each chromosome. The RNA-derived CNV profiles of neoplastic cells revealed two well-described chromosomal alterations in gliomas (Reifenberger and Collins, 2004): the amplification of chromosome 7 (including EGFR) and a putative deletion of chromosome 10 (including PTEN and MGMT). In addition, we noticed a similar pattern for chromosome 22 as we did with chromosome 10, suggesting another GBM-specific putative deletion. Although the amplification of chromosome 7 was observed in all four patients, inter-patient heterogeneity was largely reflected in the neoplastic CNV profiles for each of the patients’ tumors, as can be seen in Figure 3B.
To validate the utility of the RNA-derived CNV profiles, we performed DNA sequencing (DNA-seq) on DNA isolated from the tumor and the peritumoral tissue of one of the patients (BT_S4). We found that RNA-derived CNV profiles closely represented those derived from genomic data. As predicted, bulk DNA-seq revealed a richer landscape of chromosomal aberrations in the tumor not evident in single-cell RNA-seq-derived CNV profiles. Nonetheless, all identified chromosomal aberrations (over-representation of chromosomes 1, 7, and 21 and under-representation of chromosomes 10 and 14) in that patient’s tumor found by RNA-seq were similarly classified by DNA-seq (Figure S4B).
Single-Cell Genomic Variant Analysis Using RNA-Seq Data
Single-cell RNA-seq data also have the capacity to reveal clinically relevant tumor variants, such as single-nucleotide variants (SNVs) as well small indels, in heterogeneous tumors. We called variants by using the GATK pipeline (see “Variant analysis” in the Supplemental Experimental Procedures) for single-cell RNA-seq data and subsequently limited the variant set to non-synonymous exonic variants occurring in at least 5% of a patient’s neoplastic cells. To exclude likely germline mutations, we removed variants observed in the 1000 Genomes Project, those found in greater than three non-neoplastic cells and those where greater than 2% of non-neoplastic cells with reads covering the variant position expressed the variant allele. Cross-referencing the remaining putative tumor-specific variants with previously identified somatic tumor variants by using the Catalogue of Somatic Mutations in Cancer led to the identification of neoplastic and patient specific subpopulations harboring both new and well-established mutations on a number of tumor-related genes, absent in non-neoplastic cells (Figure S4C). Specifically, we found two EGFR variants: a previously reported missense substitution (c.G865A:p.A289T, COSM21686) and a non-frameshift deletion of nucleotides 1,799–1,804 on exon 15. Interestingly, the first variant is found on a subset of BT_S1 neoplastic cells and the second in a subset of neoplastic cells of BT_S6 (Figure 3C). Neoplastic cells of patient BT_S4 harbor a missense mutation (c.G226C:p.D76H) on MAP1B, which is involved in regulation of cytoskeletal changes occurring during neurogenesis. We were able to confirm this variant as tumor specific by using whole-genome sequencing of BT_S4 tumor and peripheral bulk DNA. Of note, the same position of MAP1B has been previously found mutated in skin carcinomas but with a different substitution (c.G226A:p.D76N, COSM5908518). In a sub-population of BT_S2 neoplastic cells, we observed a potential cancer driver mutation in the form of a missense TP53 mutation (c.G226A:p.D76N, COSM5908518), previously found in multiple cancer types, including gliomas as well as lung and breast carcinomas. In addition, a subset of cells from the same patient carry a CCNL1 frameshift mutation in exon 11 resulting in deletion of the stop codon as well as a missense mutation (c.C1748T:p.T583I) in BCOR, a gene that has previously been shown to be somatically mutated in GBM (Frattini et al., 2013). Additional tumor-specific somatic tumor variants are illustrated in Figure 3C, which further highlights intra-tumor heterogeneity and inter-patient heterogeneity.
GBM Infiltrating Tumor Cells
We observed that although the vast majority of cells (n = 1,029) within the neoplastic clusters were collected from the tumor core, a small number of cells with a neoplastic signature originated from the peripheral tissue (n = 62) (Figure 4A). We hypothesized that these might represent neoplastic cells that migrated from the tumor core to the surrounding peritumoral space and hereafter refer to them as infiltrating cells. To further verify that these cells are neoplastic in origin, we found that 57 of 62 were found in cluster 1 (the neoplastic cluster) of the CNV analysis. Furthermore, it is evident from the map of tumor-specific somatic variants in Figure 3C that infiltrating cells are indeed neoplastic as they share a number of SNV variants with other core-derived neoplastic cells of each patient. A support vector machine (SVM) classification analysis using genomic variants, trained on non-infiltrating neoplastic and all non-neoplastic cells, also identified 57 out of 62 cells as neoplastic in origin.
Figure 4. Analysis of Infiltrating Tumor Cells.
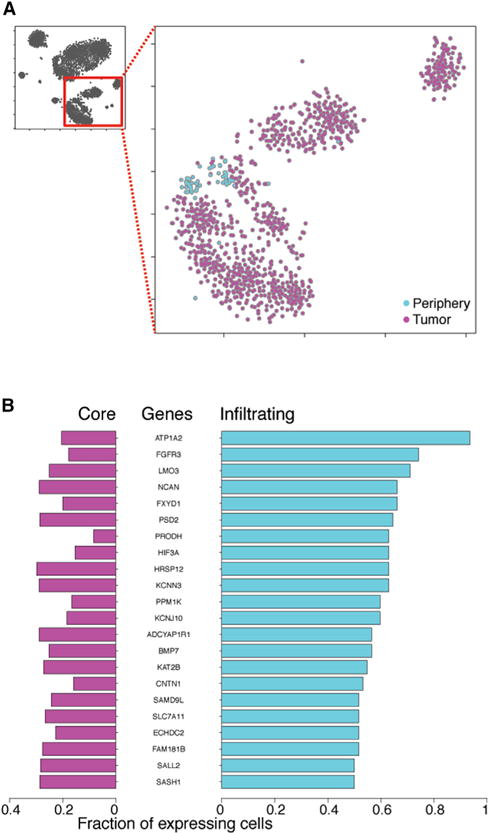
(A) 2D-tSNE representation of all neoplastic cells colored by location (tumor versus periphery).
(B) Differentially expressed genes between neoplastic cells originating from the tumor (core) or the periphery (infiltrating). The fraction of tumor core and infiltrating cells expressing any given gene is plotted.
The infiltrating cells are unlikely to represent contamination from the tumor core, given that the peripheral tissue was the first sample to be removed during the surgery under MRI guidance. Furthermore, despite the inter-patient heterogeneity across neoplastic cells (each patient’s tumor cells form their own distinct clusters on the tSNE plot), the infiltrating cells from each patient clustered closely to each other irrespective of sample of origin. To better quantify the relationship between core and infiltrating cells, we used nearest-neighbor classification to predict the site of origin (tumor or peripheral) for each neoplastic cell on the basis of the tissue of origin of three of its closest neighbors. Despite the fact that our dataset contains ~15 times more neoplastic cells from the tumor core than from the periphery, we were able to correctly classify peripheral neoplastic cells for more than 85% of the cells. Unlike the cells residing in the tumor core, infiltrating cells seem to have downregulated genes involved in adaptation to hypoxic environments as they are migrating through the (relative) oxygen-rich proximal brain tissue (p < 10−5 lower hypoxic scores as determined by a Wilcoxon rank sum test) (Figure S1E).
To understand the molecular features that distinguish infiltrating cells, we performed DESeq2 and found that ~1,000 and ~250 genes were down- or upregulated, respectively, in infiltrating cells in comparison with tumor core cells. Further selection of differentially expressed genes expressed in greater than 50% of infiltrating cells and less than 30% of core neoplastic cells narrowed down the list of upregulated genes to 22 (Figure 4B). Among the top ten genes enriched in the infiltrating cell population, we found genes with functions involving the invasion of the interstitial matrix. For example, size regulation might be achieved via the overexpression of Na+/K+ ATPases such as ATP1A2 (also further regulated by FXYD1), whereas the over-expression of enzymes such as PRODH involved in proline catabolism might contribute to increased ATP energy demands of migrating cells. We also noticed increased infiltrating cell expression of cell survival signaling via the FGFR3 receptor as well as LMO3 via inhibition of TP53-mediated apoptosis. Fibroblast growth factor (FGF)-signaling-mediated survival has been previously linked to chemotherapy resistance. Interestingly, FGF signaling is important for cell migration during embryogenesis, whereas re-activation of the pathway has been linked with cell migration and invasion in prostate tumors (Turner and Grose, 2010).
Gene Ontology (GO) analysis (Figure S5A) of genes upregulated in infiltrating cells revealed significant enrichment of GO categories (query space: Biological Processes) that are highly relevant to tumor cell migration (Demuth and Berens, 2004) such as cell-cell adhesion (ECM2, ANGPT1, and TSPAN7), anion-transport (TTYH1/2 and AQP1), nervous system development (BCAN, HES6, and GLI3), and a number of metabolic processes. A similar analysis using PAGODA (Fan et al., 2016) gave consistent results (Figure S5B).
It has remained a long-standing question whether infiltrating GBM cells actively proliferate while disseminating throughout the brain. When we looked specifically at the proportion of proliferating cells within the tumor core or infiltrating neoplastic cells, we found that 7.7% (80/1,029) of neoplastic tumor core cells were actively proliferating in comparison to only 1.6% (1/62) of infiltrating tumor cells, as determined by MKI67 expression. It should be also noted that within the cells included in this dataset, we noted the strong resemblance of neoplastic cells to OPCs (Figure S2B), suggesting functional similarity or a possibly alternate cell of origin to astrocytes for GBM (Liu et al., 2011). Of note, we also observed a strong resemblance of neoplastic cells to fetal human astrocyte gene signatures from a separate dataset (Zhang et al., 2016).
Analysis of Immune Cells
Two of the largest tSNE clusters that we observed were composed of immune cells, defined by the specific expression of numerous myeloid-specific genes, including PTPRC (CD45) and MHC class II genes (Figure 2B). Further analysis, summarized in the Supplemental Experimental Procedures, demonstrated that the vast majority of cells within these populations could be classified as either macrophages or microglia (>95%), with the remaining population comprised primarily of dendritic cells (~4.5%). Of the two primary myeloid clusters, we noticed that each contained cells almost exclusively from either the peritumoral space or tumor core, respectively (Table S1; Figure 5A), suggesting pronounced gene expression differences between the intra- and extra-tumor core myeloid subpopulations.
Figure 5. Analysis of Immune Cells.
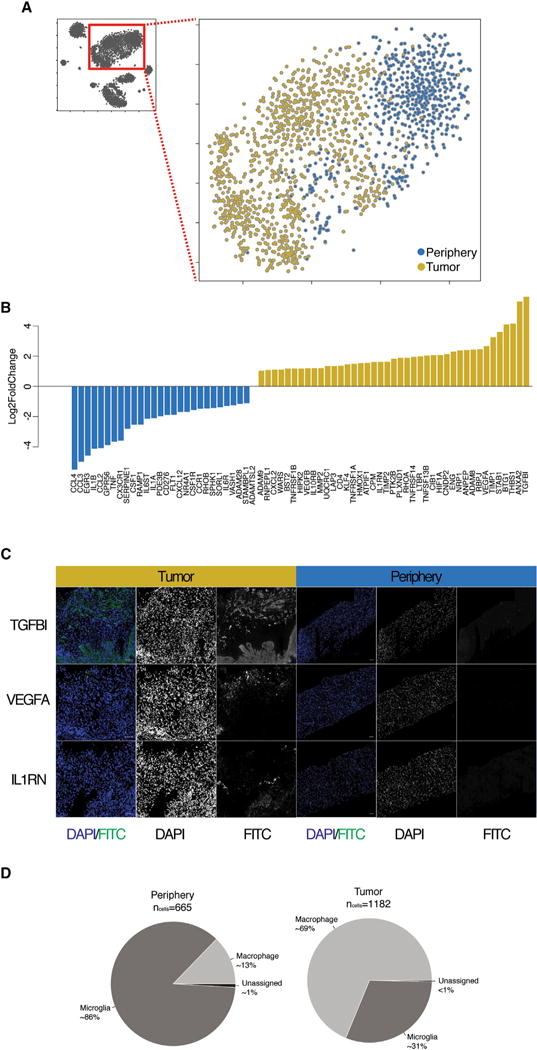
(A) 2D-tSNE representation of all immune cells colored by location (tumor versus periphery).
(B) Barplots of log2 fold changes between immune cells from the tumor and periphery for a curated list of genes involved in ECM remodeling, angiogenesis, and immune regulation.
(C) IFC staining of TGFBI, VEGFA, and IL1RN in tumor (left) and peripheral tissue (right). Scale bars, 500 μm.
(D) Percentage of immune cells from the tumor or periphery classified as macrophages or microglia.
To better specify the identity of each myeloid cell within and surrounding the tumor core, we correlated gene expression of each cell with a panel of established macrophage and microglia specific genes (TMEM119, P2RY12, GPR34, OLFML3, SLC2A5, SALL1, and ADORA3 for microglia and CRIP1, S100A8, S100A9, ANXA1, and CD14 for macrophages) (Bennett et al., 2016). We then classified each cell as macrophage or microglia on the basis of the combined expression of those genes and found that the majority of cells within the tumor core tended to express genes characteristic of macrophages (nmacrophage = 813, nmicroglia = 365), whereas cells from the surrounding space expressed genes characteristic of microglia (nmacrophage = 85, nmicroglia = 574) (Figure 5D). These data suggest that tumor-infiltrating macrophages and resident brain microglia preferentially occupy the tumor and peritumoral spaces, respectively.
We then examined the differential expression of selected gene sets within and surrounding the tumor core. These genes included cytokines, chemokines, chemokine receptors, matrix metalloproteinases (MMPs), and genes involved in angiogenesis. For a broader view of the role of tumor residing myeloid cells, we examined any gene of our curated gene list that was significantly (padj < 0.01, as determined by DESeq2) up- or down-regulated in tumor or surrounding myeloid cells in at least one of the patients. We observed discrete gene expression differences between tumor and surrounding myeloid cells, such that more pro-inflammatory markers were expressed in the tumor periphery and more anti-inflammatory and pro-angiogenic factors were expressed in the tumor core (Figure 5B). We subsequently validated the tumor-core-specific presence of several biologically relevant examples of these molecules via in situ hybridization staining. For example, whereas the inflammatory markers IL1A/B were upregulated in the peritumor brain, IL1RN, an inhibitor of IL1A/B, was upregulated in the tumor core (Figure 5C). IL1RN is an important anti-inflammatory regulator that acts as a dominant-negative interactor with IL1R1 to actively suppress immune activation. Another, highly enriched gene in immune cells within the tumor core was TGFBI (Figure 5C), a molecule with a yet unclear role in tumor formation (Han et al., 2015) that was previously thought to be expressed by neoplastic cells. TGFBI, which is induced by TGF-beta, is part of the non-Smad-mediated TGF-beta signaling pathway. The expression of TGFBI by immune cells of the tumor microenvironment could be potentially beneficial for tumor growth and dissemination, given that TGFBI has been shown to inhibit cell adhesion (dissemination) and promote survival of cells with DNA damage (survival or resistance to treatment) and is a potential angiogenic factor (growth). Immune cells within the tumor also seem to further promote angiogenesis via the expression of VEGFA (Figure 5C), a hypoxia-induced (via HIF1A) angiogenic factor that promotes both vascular permeability and endothelial cell growth.
We also compared the expression of immune checkpoint receptor ligands in myeloid cells from the tumor core versus the peritumoral tissue. We found a small but statistically significant expression difference (padj < 0.01, as determined by DESeq2) between tumor and peritumoral myeloid cells for both ligands of the receptors PD1 (CD274 [PDL1], PDCD1LG2 [PDL2]) and CTLA4 (CD80 and CD86), as well as the genes ICOSLG (ligand of ICOS receptor), CD276 (B7-H3), TNFRSF14 (ligand of BTLA), and LGALS9 (ligand of TIM3). All genes were found to be upregulated in the peritumoral compartment, with the exception of LGALS9 and CD80, which were upregulated in the tumor (Figure S5C).
Conclusions
GBM is the most frequent and deadly primary brain cancer in adults (Kleihues and Sobin, 2000). Current treatment strategies combining surgery with radiotherapy and chemotherapy prolong survival, but tumor recurrence usually occurs within 2 years. Recurrence stems from infiltrating neoplastic cells originating from the tumor core and spreading quickly across long distances within the brain. Thus, there is great hope in new treatment strategies that specifically target neoplastic cells in general as well as the infiltrating populations of cells migrating away from the primary tumor. In addition, immunotherapies are also being developed (Binder et al., 2015); these include immune checkpoint blockade, adoptive T cell transfer, and combinatorial immunotherapies.
This work represents the identification and characterization of individual infiltrating tumor cells in the tissue surrounding the GBM tumor core. Furthermore, our high-depth and full-length RNA-seq data allowed us to use gene expression information to infer genomic variation on the level of large chromosomal aberrations as well as smaller genomic variants such as insertions or deletions and single-nucleotide somatic mutations. Our analysis revealed a large degree of heterogeneity between and within different tumors. We found that different patients’ tumors harbor different chromosomal aberrations while sharing hallmark CNVs such as the amplification of chromosome 7. Furthermore, each patient’s neoplastic cells have a unique set of smaller-scale mutations that also demarcate different populations of neoplastic cells within each tumor and potentially derive from different intra-tumoral lineages.
We defined infiltrating tumor cells as a population of neoplastic cells originating from the peripheral tissue whose transcriptional and genomic variant profiles resembled tumor core cells. Despite the heterogeneity of neoplastic cells originating from each of the individual patients, infiltrating cells share common characteristics regardless of patient of origin. The homogeneous gene signature of infiltrating GBM cells presents the potential of a convergent strategy for the mechanism of infiltration between highly variable tumors, which could provide new therapeutic avenues. When we examined the genes specifically upregulated in infiltrating neoplastic cells, we found groups of genes involved in size regulation, energy production, inhibition of apoptosis, regulation of cell-cell adhesion, and CNS development. In addition, infiltrating cells are likely hijacking machinery used during CNS development by cells migrating over long distances. It should be noted that the specific gene expression changes observed in infiltrating cells could either be intrinsic to the cell type or be a result of migrating through a non-tumor microenvironment.
We also characterized the effect of the tumor microenvironment on non-neoplastic cells. Among the non-neoplastic cell populations, OPCs, neurons, mature oligodendrocytes, and vascular cells originating from both the tumor core and surrounding regions are essentially indistinguishable from each other. In contrast, myeloid cells, consisting primarily of macrophages and microglia, are greatly affected by the tumor microenvironment. Despite belonging to the same cell class, myeloid cells within the tumor display unique gene expression profiles, demonstrating the direct effect of the tumor milieu on these important immune mediators. Specifically, we found that immune cells within the tumor mass play a crucial role in enhancing tumor growth, survival, and dissemination via suppression of inflammation, promotion of angiogenesis and extracellular matrix (ECM) remodeling. Additionally, a subset of the changes induced by the tumor are shared between macrophages and microglia. These data, in addition to the full transcriptomes of immune cells within and surrounding the tumor, could help refine the development of novel therapies targeting cells that enhance tumor growth but are not themselves neoplastic. In particular, the findings from our study have implications for therapeutic immunotherapy efforts in GBM. For example, although we did find robust expression of MHC class I gene expression, which would allow for T-cell-mediated immune responses, the expression of these genes varied greatly between patients. We also observed similar expression patterns for the ligands of PD1, which play a role in abrogating immune responses in some tumors. These data suggest that targeted immune therapies could vary vastly between patients and that phenotyping patient tumors either via tumor resection or using technologies that capture tumor cells non-invasively in the cerebrospinal fluid (CSF) could be essential for screening patients who might be particularly responsive to therapy.
EXPERIMENTAL PROCEDURES
Further information and requests for resources and reagents should be directed to (and will be fulfilled by) the Lead Contact, Stephen R. Quake (quake@stanford.edu). Further details and an outline of resources used in this work can be found in the Supplemental Experimental Procedures.
Tissue Dissociation, Immunopanning, and Single-Cell Sorting
The experimental layout is outlined in Figure 1B. We analyzed samples from four patients with confirmed cases of primary GBM. Clinical information for each patient included in the study can be found in Table S3. Informed consent was obtained from all subjects.
An additional sample from each patient was sent to pathology to confirm the diagnosis of GBM and identify IDH1 status (negative). From each patient, we collected two separate tissue samples: one originating from the tumor core and another from the peritumoral space (cortex) immediately adjacent to the tumor core. The tumor core was demarcated on MRI as strongly enhancing (with gadolinium contrast) (Figure 1A), unlike the non-contrast-enhancing peritumor space. Peritumor cortex was always removed prior to resecting tumor core in order to prevent potential cross-contamination. For each sample, tumor and peritumor tissue were processed separately.
Tissue samples were transported to the laboratory immediately from the operating room in order to begin dissociating samples within 1 hr of resection. The samples were processed similarly to existing protocols for human brain dissection. In brief, the tissue was first chopped into small pieces (<1 mm3) with a #10 scalpel blade and then incubated in 30 U/mL papain at 34°C for 100 min. After digestion, the tissue was washed with a protease inhibitor stock solution. The tissue was then gently triturated in order to yield a single-cell suspension. The single-cell suspension was then added to a series of plastic petri dishes pre-coated with cell-type-specific antibodies (see below) and incubated for 10–30 min at room temperature. Unbound cells were transferred to the subsequent Petri dish, and the dish with bound cells was rinsed eight times with PBS to wash away loosely bound contaminating cell types. The antibodies used included anti-CD45 to capture microglia and macrophages, anti-O4 hybridoma to harvest oligodendrocytes lineage cells, anti-Thy1 (CD90) to harvest neurons, anti-HepaCAM to harvest astrocytes, and Banderiaea simplicifolia lectin 1 (BSL-1) to harvest endothelial cells. Once bound to the Petri dish and rinsed, adherent cells were removed by incubating in a trypsin solution at 37°C for 5–10 min before gently squirting the cells off the plate. These single-cell suspensions were then transferred to a fluorescence-activated cell sorting (FACS) buffer before proceeding with single cell sorting.
Single-cell suspensions were sorted by a SONY SH800 Cell Sorter. All events were gated with four consecutive gates: (1) forward scatter-area (FCS-A)/side scatter-area (SSC-A), (2) FCS-height (FCS-H)/FCS-width (FCS-W), (3) propidium iodide (PI) negative, and (4) Hoechst positive. Single cells were sorted in 96-well plates containing 4 μL lysis buffer (4U Recombinant RNase Inhibitor, Takara Bio), 0.05% Triton X-100 (Thermo Fisher), 2.5 mM dNTP mix (Thermo Fisher), 2.5 μM Oligo-dT30VN (5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′), spun down for 2 min at 1,000 × g, and snap frozen. Plates containing sorted cells were stored at −80°C until processing.
cDNA Synthesis and Library Preparation
Reverse transcription and PCR amplification were performed according to the Smart-seq2 protocol described previously (Picelli et al., 2014). In brief, 96-well plates containing single-cell lysates were thawed on ice followed by incubation at 72°C for 3 min and placed immediately on ice. Reverse transcription was carried out after adding 6 μL of reverse transcription-mix (100 U SMARTScribe Reverse Transcriptase (Takara Bio), 10 U Recombinant RNase Inhibitor (Takara Bio), 1× First-Strand Buffer (Takara Bio), 8.5 mM DTT (Invitrogen), 0.4 mM betaine (Sigma), 10 mM MgCl2 (Sigma), and 1.6 μM TSO (5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′) for 90 min at 42°C, followed by 5 min at 70°C.
Reverse transcription was followed by PCR amplification. PCR was performed with 15 μL PCR mix (1× KAPA HiFi HotStart ReadyMix, Kapa Bio-systems), 0.16 μM ISPCR oligo (5′-AAGCAGTGGTATCAACGCAGAGT-3′), and 0.56 U Lambda exonuclease (NEB) according to the following thermal-cycling protocol: (1) 37°C for 30 min; (2) 95°C for 3 min; (3) 21 cycles of 98°C for 20 s, 67°C for 15 s, and 72°C for 4 min; and (4) 72°C for 5 min.
PCR was followed by bead purification using 0.7× AMPure beads (Beckman Coulter), capillary electrophoresis, and smear analysis using a Fragment Analyzer (AATI). Calculated smear concentrations within the size range of 500 and 5,000 bp for each single cell were used to dilute samples for Nextera library preparation as described previously (Darmanis et al., 2015).
Sequencing and QC
In total, we sequenced 4,028 single cells by using 75-bp-long paired-end reads on a NextSeq instrument (Illumina) and High-Output v2 kits (Illumina). Raw reads were preprocessed and aligned to the human genome (hg19) according to the exact pipeline described previously (Darmanis et al., 2015).
As a quality metric, we first performed hierarchical clustering on all cells by using a list of housekeeping genes (Figure S1B) and removed any cells with uniformly low expression across all genes (likely a result of low quality RNA or cDNA synthesis). We separated the resulting dendrogram into two clusters containing cells that passed (n = 3,589) or failed (n = 456) this QC. All downstream analyses were performed with only the cells that passed QC. A summary of the number of cells by QC cluster (pass or fail), with respect to patient and tissue of origin along with sequencing statistics, can be found in Table 1.
Preparation of Libraries from Genomic DNA
Human genomic DNA was sheared to an average size of 500 bp with a Covaris S220 according to the manufacturer’s protocol in snap-cap microtubes. Sheared DNA (500 ng) was ligated into indexed Illumina sequencing adapters (made by IDT) with the Kapa Hyper Prep Kit for Illumina. Libraries were quantitated according to average DNA fragment length (AATI Fragment Analyzer) and concentration determined by qPCR (Kapa Library Quantification Kit for NGS). Pooled libraries were sequenced as paired-end 150-bp reads on an Illumina NextSeq500.
Antibody Staining
Tissue sections were removed from −80°C and allowed to dry at room temperature for 30 min followed by fixation with 4% paraformaldehyde (PFA) (in PBS). Sections were washed three times with PBS (5-min washes at room temperature) and blocked with blocking buffer (1× PBS, 10% donkey serum, and 0.1% Triton) for 1 hr at room temperature. After blocking, sections were probed with primary antibodies (for antibodies used and working concentrations, see Table S4) and incubated overnight at 4°C. Sections were washed, and secondary antibodies (Table S4) were added and incubated with the tissue for 1 hr at 37°C followed by three 5-min washes with PBS. Sections were stained with DAPI (1× in PBS) for 30 min at room temperature, washed another two times as before, and then dried and mounted with SlowFade Gold antifade reagent (Life Technologies) prior to imaging.
In Situ RNA
In situ RNA staining was performed on 10-μm-thick fresh-frozen tissue sections with padlock probes and rolling-circle amplification (RCA) as described before (Ke et al., 2013). In situ reverse transcription was carried out with primers containing 2-O-Me modified bases. The sequences of the primers, padlock probes, and fluorescently labeled detection oligos are listed in Table S5. In order to reduce the high-fluorescence background due to the presence of lipofuscins in the biopsy specimen from tumor periphery, the sections were incubated in 1% Sudan black in 70% ethanol for 30 min at room temperature in the dark prior to fluorescence labeling of RCA products.
Multiplexed detection was achieved via combinatorial labeling of RCA products by sequential hybridization four distinct fluorescence detection oligos. Three cycles of hybridization, imaging, and detection oligo removal allow identification of 64 gene-specific fluorescence barcodes (43), which is in principle enough to distinguish 54 probes used in this study. To further reduce the complexity of the barcodes and facilitate the identification of false positives (errors), we split the padlock probes in two distinct pools of 27 and 27 probes and performed the in situ RNA staining on consecutive sections. To distinguish true signals from tissue autofluorescence (as described below), only two colors are used to label RCA products at third cycle.
Statistical Methods
All data analysis was performed with R. Specific packages and functions used are described in greater detail below. All codes are available upon request.
Dimensionality Reduction and Clustering
Dimensionality reduction was performed in three steps. First, we calculated the over-dispersion of each gene as described previously (Fan et al., 2016). We then selected the top 500 over-dispersed genes and constructed a cell-to-cell distance matrix (1 − absolute correlation) of all cells. The distance matrix was reduced to two dimensions with tSNE as previously described (Darmanis et al., 2015) and as implemented in package ‘‘tsne’’ for R (perplexity = 50). Clustering of groups of similar cells was performed on the 2D tSNE space with k-means as implemented in package ‘‘stats’’ for R.
Differential Expression Analysis
Differential expression analysis was performed with package DESeq2 (Love et al., 2014) for R. All functions in the DESeq2 pipeline were used with default settings.
DATA AND SOFTWARE AVAILABILITY
The accession number for the data reported in this paper is GEO: GSE84465. In addition, all processed data are available for exploration and download at http://gbmseq.org/.
Supplementary Material
Highlights.
Vast genomic and transcriptomic heterogeneity of GBM tumor cells
Characterization of individual GBM infiltrating cells in peritumoral tissue
Possible convergent strategy of infiltration between highly variable tumors
Myeloid cells participate in enhancing tumor growth, survival, and dissemination
Acknowledgments
The authors thank Jennifer Okamoto and Gary Mantalas for assistance with sequencing and library preparation. This study was supported by the Hearst Foundation, the Curci Foundation, the NIH (grants K08-NS901527, R01 CA216054-01, and R21-CA193046-01 to M.H.G. and grants R01NS081703 and MH099555-03 to B.A.B.), the California Institute for Regenerative Medicine (grant GC1R-06673-A), and the Center of Excellence for Stem Cell Genomics (S.R.Q.). S.D. was supported by a fellowship from Svenska Sällskapet för Medicinsk Forskning. S.A.S. was supported by the National Institute of Mental Health (T32GM007365 and F30MH106261) and Bio-X Bowes predoctoral fellowships.
Footnotes
Data and Software Availability
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.10.030.
AUTHOR CONTRIBUTIONS
S.D., S.A.S., M.H.G., and S.R.Q. designed the research. S.D., S.A.S., C.C., M.H.G., N.N., M.M., and M.K. performed the research. M.H.G., G.L., S.C., I.D.C., P.S., Y.Z., S.D.C., and Y.L. contributed new reagents or analytic tools. S.D., D.C., S.A.S., and S.R.Q. analyzed the data. S.D., S.A.S., B.A.B., and S.R.Q. wrote the paper.
References
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. 2016;113:E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DC, Davis AA, Wainwright DA. Immunotherapy for cancer in the central nervous system: current and future directions. Oncolmmunology. 2015;5:e1082027. doi: 10.1080/2162402X.2015.1082027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush NAO, Chang SM, Berger MS. Current and future strategies for treatment of glioma. Neurosurg Rev. 2016;40:1–14. doi: 10.1007/s10143-016-0709-8. [DOI] [PubMed] [Google Scholar]
- Campbell CE, Piper M, Plachez C, Yeh YT, Baizer JS, Osinski JM, Litwack ED, Richards LJ, Gronostajski RM. The transcription factor Nfix is essential for normal brain development. BMC Dev Biol. 2008;8:52. doi: 10.1186/1471-213X-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes A, Idema AJ, Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol. 2007;114:443–458. doi: 10.1007/s00401-007-0293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Hayden Gephart MG, Barres BA, Quake SR. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA. 2015;112:7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70:217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Grüner BM, Chiou SH, Schep AN, Baral J, Hamard C, et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. 2016;166:328–342. doi: 10.1016/j.cell.2016.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan JB, Zhang K, Chun J, Kharchenko PV. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods. 2016;13:241–244. doi: 10.1038/nmeth.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Cai H, Chen Y, Hu B, Luo H, Wu Y, Wu J. The role of TGFBI (βig-H3) in gastrointestinal tract tumorigenesis. Mol Cancer. 2015;14:64. doi: 10.1186/s12943-015-0335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wählby C, Nilsson M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013;10:857–860. doi: 10.1038/nmeth.2563. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Sobin LH. World Health Organization classification of tumors. Cancer. 2000;88:2887. doi: 10.1002/1097-0142(20000615)88:12<2887::aid-cncr32>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Razon N, Bartal AD, Yarden Y, Schlessinger J, Soreq H. Expression of epidermal growth factor receptors in human brain tumors. Cancer Res. 1984;44:753–760. [PubMed] [Google Scholar]
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J Cell Sci Suppl. 1985a;3:161–172. doi: 10.1242/jcs.1985.supplement_3.16. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985b;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RGW, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B, Mateo JL, Zhou B, Andersen J, Achimastou A, Urbán N, van den Berg D, Georgopoulou D, Hadjur S, Wittbrodt J, et al. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 2013;27:1769–1786. doi: 10.1101/gad.216804.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Reifenberger G, Collins VP. Pathology and molecular genetics of astrocytic gliomas. J Mol Med (Berl) 2004;82:656–670. doi: 10.1007/s00109-004-0564-x. [DOI] [PubMed] [Google Scholar]
- Shlien A, Malkin D. Copy number variations and cancer. Genome Med. 2009;1:62. doi: 10.1186/gm62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL, Litwack ED, Richards LJ, Gronostajski RM. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol Cell Biol. 2005;25:685–698. doi: 10.1128/MCB.25.2.685-698.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvà ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, He S, Yuan J, Mao X, Cao Y, Zong J, Tu Y, Zhang Y. Oncogenic role of SOX9 expression in human malignant glioma. Med Oncol. 2012;29:3484–3490. doi: 10.1007/s12032-012-0267-z. [DOI] [PubMed] [Google Scholar]
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.