


Abstract
In vivo protein ligation is of emerging interest as a means of endowing proteins with new properties in a controlled fashion. Tools to site-specifically and covalently modify proteins with small molecules, peptides, or other proteins in living cells are few and far between. Here, we describe the development of a Staphylococcus aureus sortase (SrtA)-based protein ligation approach for site-specific conjugation of fluorescent dyes and ubiquitin (Ub) to modify proteins in Caenorhabditis elegans. Hepta-mutant SrtA (SrtA7m) expressed in C. elegans is functional and supports in vitro sortase reactions in a low-Ca2+ environment. Feeding SrtA7m-expressing C. elegans with small peptide-based probes such as (Gly)3- biotin or (Gly)3-fluorophores enables in vivo target protein modification. SrtA7m also catalyzes the circularization of suitably modified linear target proteins in vivo and allows the installation of F-box domains on targets to induce their degradation in a ubiquitin-dependent manner. This is a noninvasive method to achieve in vivo protein labeling, protein circularization, and targeted degradation in C. elegans. This technique should improve our ability to monitor and alter the function of intracellular proteins in vivo.
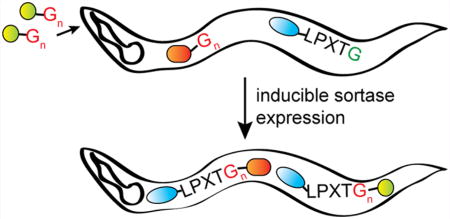
Graphical abstract
INTRODUCTION
Sortase A transpeptidases (SrtA) are a class of enzymes that covalently anchor proteins to the cell wall of Gram-positive bacteria.1–3 Staphylococcus aureus SrtA recognizes proteins that contain an LPXTG motif (where “X” indicates any amino acid) and cleaves the peptide bond between threonine and glycine; the thiol group of the catalytic cysteine serves as the nucleophile.4,5 Upon cleavage and concomitant formation of an acyl-enzyme intermediate, the substrate is subsequently linked covalently to an incoming nucleophile; typically, this occurs via the terminal amine of free glycines present in building blocks that participate in the formation of the peptidoglycan layer in Gram+ bacteria.6,7 Sortase reactions are reversible, as the reaction regenerates a nucleophile byproduct that can engage in ligation reactions that regenerate the initial, unmodified substrate. To drive a sortase reaction to completion, the incoming nucleophile must be present in molar excess over the substrate or the ligation product must be removed from the reaction environment.
Due to its ability to site-specifically link proteins or peptides through peptide bond formation, SrtA is now widely used in protein engineering applications. Recombinant SrtA enables the site-specific modification of peptides, proteins, antibodies, or polymers with a variety of ligation partners, including fluorescent dyes, oligosaccharides, biotin, nucleic acids, glycolipids, or other peptides8–13 (Figure 1). The requirements for in vitro substrate ligation are the presence of a LPXTG motif in the substrate and an excess of incoming nucleophile in the reaction, typically GGG-(G3) or GGGGG-(G5) labeled molecules, e.g., G3-biotin or G3-Alexa647. Depending on substrate, nucleophile design, and source of the sortases used, this method allows substrate ligation at the N-terminus, C-terminus, or both.9,14–16
Figure 1.

Schematic representation of sortase reactions. Protein substrates equipped with a sortase A recognition sequence (LPXTG) can participate in (A) intermolecular transpeptidation reaction with small oligoglycine nucleophiles, (B) ligation reactions with other proteins containing a terminal oligoglycine portion, or (C) intramolecular transpeptidations to yield a circular adduct if exposure to the N-terminal glycine residue is given.
S. aureus SrtA is a Ca2+-dependent enzyme that is not functional when expressed in the cytoplasm.17,18 However, Staphylococcus pyogenes SrtA, a Ca2+-independent enzyme that catalyzes the same reaction, can be used to substitute for S. aureus SrtA in reaction environments with low Ca2+ levels. Indeed, S. pyogenes SrtA enabled site-specific cell-surface and intracellular protein labeling in low Ca2+ settings, demonstrating its versatility in covalently linking substrates and nucleophiles in vivo.11,18
The means to manipulate or visualize signaling networks in vivo are critical to increase our understanding of cellular signaling and organismal development. Because the nematode Caenorhabditis elegans is transparent, many cellular and organismic processes can be monitored in vivo without the need for invasive procedures. For the visualization of proteins in vivo, worms are injected with cDNA that encodes the target protein genetically fused to either a fluorescent protein, such as GFP, or to a SNAP–CLIP tag that can be specifically labeled with synthetic probes in vivo.19–22 However, each of these modifications adds approximately 20–25 kDa to the size of the target protein and can perturb structure, stability, or activity of the fusion partner or some combination of all three. Thus, there is a need for new methods that allow site-specific in vivo labeling of proteins while minimizing interference with that protein’s function.
The ability of SrtA to catalyze intramolecular protein circularization as well as the formation of protein–protein fusions in living cells prompted us to explore in vivo applications of sortase to more complex systems, such as intact organisms. We examined the potential of in vivo sortagging in C. elegans. A Ca2+-independent mutant version of S. aureus SrtA (SrtA7m) is functional when expressed in C. elegans. It allows for sortase-dependent ligation of LPETG-tagged proteins with small molecule nucleophiles or proteins containing N-terminal oligoglycines (G3 or G5). Feeding small molecule nucleophiles to C. elegans enables sortase-dependent in vivo modification of LPETG-tagged proteins. In vivo expression of sortase in C. elegans can also catalyze the circularization of a suitably modified linear precursor of GFP and enables the rapid degradation of LPETG-tagged proteins through fusion with a G3-F-box domain. Together, we propose in vivo sortagging as a novel strategy by which to site-specifically modify LPETG-tagged proteins in C. elegans in an inducible manner.
RESULTS AND DISCUSSION
Lysates of C. elegans Expressing SrtA7m Showing Sortase Activity in Vitro
Is it possible to use sortase-mediated protein labeling in vivo to site-specifically modify proteins in C. elegans? By germline injection, we generated a transgenic C. elegans strain that contains an extrachromosomal array encoding HA-tagged S. aureus hepta-mutant SrtA (SrtA7m) under the control of a heat shock promoter (phsp-16.2::srtA7m; pmyo-3::mCherry). This particular mutant version of sortase was engineered to enhance its catalytic activity while simultaneously rendering the enzyme independent of Ca2+.23–25 To obtain maximal sortase expression upon induction, we heat-shocked approximately 1000 adult animals per sample for 5–180 min at 37 °C and tested intracellular SrtA7m levels 5–1080 min post-induction (Figure S1A,B). We observed robust SrtA7m expression following a short heat pulse of 15 min, and SrtA7m expression levels plateaued 240 min after induction. To visualize sortase expression within the worm, we first heat-shocked SrtA7m as well as N2 animals for 30 min at 37 °C and fixed the animals 30, 90, or 150 min after induction. Microscopic evaluation of these animals confirmed a time-dependent increase in sortase levels and showed that SrtA7m is expressed throughout the animal body expect for the germline (Figure S2A). We also observed a strong signal from unhatched eggs, suggesting embryonic expression of sortase upon induction. In contrast, mCherry levels of the pmyo-3::mCherry co-injection marker stayed constant (Figure S2B). Based on these results, we set our standard SrtA7m induction condition as 30 min of heat-shock at 37 °C followed by 2 h of recovery at RT. Using these conditions, we first tested if sortase expression alters animal physiology or fitness. We heat-shocked L1, L3, and day 1 adult animals and tested their lifespan or ability to develop into fertile adults (Figures 2A,B and S2C). Our results showed that sortase expression did not affect lifespan, nor did srtA-expressing animals show any changes in body anatomy or experienced delays in development. We also probed if sortase expression would cause acute toxicity that might impair animals for only a few hours prior to recovery. We repeated our tests with day 1 adults and analyzed their motility 4 or 8 h after heat-shock treatment (Figure 2C,D). We did not observe any changes in C. elegans motility. SrtA expression in C. elegans thus has no obvious negative consequences on animal health. Having established that sortase expression is well-tolerated by C. elegans, we evaluated the ability of C. elegans-expressed SrtA7m to catalyze a sortase reaction in vitro and ascertained the enzymatic activity of in vivo expressed SrtA7m. Total worm lysates from SrtA7m-expressing animals were mixed with a purified protein substrate, VHH-LPETG-His6 and an excess of a triglycine tetramethylrhodamine nucleophile (G3-TMR) was added. To prevent detergents from interfering with putative SrtA7m activity, worms were lysed using a detergent-free lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl). We analyzed the reaction products by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and fluorescent in situ gel scanning and detected the formation of the specific conjugate (VHH–TMR) in the reaction mixture, whereas controls from which either precursor substrate proteins or nematode lysate were omitted failed to yield VHH–TMR (Figure S3). Next, we tested whether our labeling method could achieve site-specific biotinylation of a target protein expressed in C. elegans. We analyzed in vitro reactions combining C. elegans (phsp-16.2::srtA7m;peft-3::GFP-LPETG) lysate with G3-biotin. This showed successful biotinylation of the GFP-LPETG substrate in vitro, confirming that sortase can label substrates coexpressed with the enzyme in C. elegans (Figure 3A). We conclude that inducibly expressed SrtA7m in C. elegans catalyzes in vitro sortase reactions.
Figure 2.

SrtA7m expression and lack of toxicity in C. elegans. (A,B) Longevity assays of animals expressing SrtA7m as L3s or day 1 adults. L3 larvae (A) or day 1 adults (B) were heat-shocked (HS) for 30 min at 37 °C and thereafter incubated at 20 °C. Animals were scored in 2 day intervals. The p-values are >0.05 (Gehan–Breslow–Wilcoxon test). (C,D) motility assays of animals expressing SrtA7m. Day 1 adults were heat-shocked for 30 min at 37 °C and thereafter incubated at 20 °C for 4 h (C) or 8 h (D) prior to testing. Number of body bends per 30 s was assessed. At least 30 animals were scored per condition.
Figure 3.

Sortase-mediated protein labeling. (A) A lysate of transgenic animals expressing GFP-LPETG and SrtA7m following heat-shock was supplemented with G3-biotin (1 mM) and 100 µL of sortagging reaction buffer (50 mM Tris, pH 7.5, 150 mM NaCl). (B) In vivo sortagging in C. elegans: animals expressing SrtA7m upon heat-shocking and GFP-LPETG substrate were fed with G3-biotin. The mixing control consists of a 1:1 mixture of lysates from animals either expressing GFP-LPETG substrate or SrtA7m enzyme, previously fed with G3-biotin (1 mM) and lysed in buffer supplemented with 1% SDS and 2% NP40. (C,D) Phsp-16.2::srtA7m; Peft-3::GFP-LPETG strain was fed with G3-Alexa 647 for 5 h at RT. Worms were transferred to a fresh plate for a further 24 h and (C) collected for imaging by confocal fluorescence microscopy or (D) lysed and resolved by SDS-polyacrylamide gel electrophoresis (PAGE). Samples were visualized by immnoblotting (upper panel) or Typhoon in gel scanning (lower panel) at λem = 647 nm. The scale bar represents 10 µm. GFP-LPETG and SrtA7m signal distributions shown in (C) are representative for the entire animal. Pearson colocalization coefficient for images shown in panel 4 is 0.754. All images were acquired with the same laser and exposure settings.
SrtA7m Mediation of in Vivo Sortase Reactions in C. elegans
The successful ligation of small molecule nucleophiles (G3-TMR or G3-biotin) to target proteins in complex C. elegans lysates prompted us to ask whether sortase could catalyze such reactions also in living nematodes. To exclude the possibility that the nucleophiles intended for use in in vivo ligation reactions might intoxicate the nematodes, we first cultured day 1 adult N2 animals for 10 h in liquid nematode growth medium (NGM) supplemented with G3-biotin (1 mM), G3-TMR (1 mM), or G3-Alexa647 (1 mM) and scored their survival after 24 h. Prolonged exposure to high concentrations of nucleophile did not affect survival (Figure S3B). Visual inspection of the animals confirmed that exposed animals were indistinguishable from untreated controls. To then test if sortase could catalyze in vivo ligation reactions, we cultured L4 larvae inducibly expressing SrtA7m and GFP-LPETG (phsp-16.2::srtA7m;pef t-3::GFP-LPETG) for 5 h in liquid nematode growth medium (NGM) supplemented with G3-biotin (1 mM) and analyzed total worm lysates for the presence of the ligation product. GFP-biotin was readily detected in lysates from animals induced for sortase expression, while uninduced controls did not contain the transpeptidation product (Figure 3B). Control samples (mixing control) consisting of a mixture of lysates from strains individually expressing either SrtA7m or GFP-LPETG and fed with G3-biotin (1 mM) for 5 h in liquid nematode growth medium (NGM). The worm lysis buffer used contained 1% SDS and 2% NP-40 to instantly denature all proteins and prevent ongoing sortase reactions from proceeding. The presence of GFP-biotin was seen only in lysates from animals that coexpress SrtA7m and GFP-LPETG and that were fed with G3-biotin (1 mM). We therefore conclude that the biotinylation reaction occurred in vivo rather than post-lysis in vitro. To test if in vivo sortase expression might result in off-target ligation events, we first conducted a bioinformatics analysis to identify all proteins with LPXTG motifs present in the C. elegans proteome (Table S1). We identified 92 proteins containing a LPXTG motif; however, none of the motifs were close to either the N- or C-terminus, nor were they embedded within a favorable amino acid stretch that might allow more distant LPXTG motifs to engage in sortase ligations. To address the possibility that endogenous proteins are modified in off-target reactions, we constructed animals that express the substrate protein G5-GFP-LPETG in a SrtA7m -positive background (phsp-16.2::srtA7m;pef t-3::G5-GFP-LPETG). Using mass spectrometry, we analyzed total lysates form induced animals 4 h after heat-shocking for the presence of unique peptides that would only emerge upon modification of endogenous proteins (Table S2). We did not identify any such peptide, suggesting that sortase expression in C. elegans does not result in measurable off-target ligation events. This finding was supported by the absence of any nonspecific bands in our immunoblot-based tests for in vivo sortase reactions (Figure S3C).
Next, we tested in vivo protein labeling using a different nucleophile, G3-Alexa647. Successful labeling would allow the detection of sortase reaction products by immunofluorescence microscopy of live animals. A total of 30 adult worms per condition were incubated for 5 h in liquid NGM supplemented with G3-Alexa647 and then transferred to OP50 plates. After 24 h of incubation, worms were washed off of the plates and analyzed by fluorescence microscopy. In the absence of SrtA7m, G3-Alexa647 was found to be enriched in the pharynx in 30 out of 30 animals (Figure 3C, panel 2). In contrast, animals that coexpress SrtA7m and GFP-LPETG showed abundant Alexa647 signal in all body parts. The signal partially overlapped with GFP throughout the body (Figure 3C, panel 4) in 28 out of 30 animals, indicating the formation of GFP-Alexa647 conjugates. Biochemical analysis of similarly treated animals confirmed in vivo ligation of GFP-LPETG to G3-Alexa647 (Figure 3D). These results demonstrated the possibility of executing in vivo sortase-mediated protein labeling using different probes.
Together, our results represent the first account of successful in vivo protein labeling using sortase in C. elegans. Feeding worms with synthetically readily accessible small molecules constitutes a simple means of nucleophile administration for sortase reactions in vivo.
SrtA7m Enabling of in Vivo Protein Circularization in C. elegans
Intramolecular circularization confers several advantages to proteins in comparison to the linear form, such as enhanced thermostability and increased resistance to exoproteolytic attack.26,27 To evaluate the ability of SrtA7m to promote protein circularization in C. elegans, we generated transgenic animals that coexpress SrtA7m and a circularizable GFP variant, G5-GFP-LPETG (phsp-16.2::srtA7m;pef t-3::G5-GFP-LPETG). Circular GFP species show accelerated mobility on SDS-polyacrylamide gels, while their fluorescence properties are not affected.26 The N-terminal pentaglycine motif of G5-GFP-LPETG can function as a nucleophile to resolve acyl-enzyme intermediates (G5-GFP-LPET-SrtA7m), thus enabling sortase-mediated GFP circularization.26 We analyzed total worm lysates of induced and uninduced phsp-16.2::srtA7m;pef t-3::G5-GFP-LPETG animals by immunoblotting and observed accumulation of faster migrating (circular) GFP species as well as GFP oligomers upon induction of sortase expression (Figure 4A, lanes 1 and 2). Because protein circularization regenerates a LPETG motif at the site of covalent closure, SrtA7m should be able to cleave the circularized species, too. We supplemented C. elegans lysates from induced phsp-16.2::srtA7m;pef t-3::G5-GFP-LPETG animals with a large molar excess of purified SrtA7m and G3-Biotin and observed the conversion of low-molecular-weight cyclic GFP as well as GFP oligomers to linear monomeric GFP species in the presence of both exogenous enzyme and nucleophile (Figure 4A, lanes 3, 4). Quantification of the different reaction products in Figure 4A showed that the sortase reaction products accounted for approximately 30% (29% ± 3%) of the total GFP pool, highlighting the reversibility of the reaction. This confirms that the products sortase-mediated protein circularization can be resolved by sortase in the presence of excess nucleophile. We conclude that SrtA7m can mediate reversible protein cyclization in C. elegans.
Figure 4.

GFP circularization by SrtA7m in living nematodes. (A) Lanes 1 and 2: GFP circularization upon SrtA7m expression: Phsp-16.2::srtA;pef t-3::GFP strains expressing circularizable G5-GFP-LPETG substrate under the control of the eft-3 promoter and SrtA7m-HA controlled by a hsp-16 promoter were lysed. The migration pattern of GFP was analyzed by SDS-PAGE, followed by immunoblotting. Lanes 3 and 4: Reversibility of GFP circularization in the presence of excess nucleophile was tested by adding an excess nucleophile (G3-biotin) at 4 °C overnight to detergent-free lysates of induced (heat-shocked) or uninduced Phsp-16.2::srtA7m; Peft-3::GFP animals. Reversion of GFP circularization is characterized by the disappearance of the circular GFP signal observed in the immunoblot.
SrtA7m Allowance of Targeted Degradation of Suitably Modified Substrates in C. elegans
To expand the range of applications for in vivo sortagging in C. elegans, we engineered strains that would allow specific engagement of the Ub-proteasome pathway. We hypothesized that sortase-dependent ligation of an F-box domain onto a target protein of interest should recruit the target to SKP1-CUL1-F-box complexes and to E2 ubiquitin ligases and thus tag the target protein for degradation (Figure 5B). We constructed transgenic strains that either expressed Ced-10-LPETG-MYC driven by an eft-3 promoter or encoded a nucleophile G3-F-box-FLAG construct under the control of a heat-inducible promoter. Expression of both constructs was verified by immunoblotting (Figure 5A). Through crossing, we obtained strains that encoded all three components required for F-box installation onto the target protein (srtA7m, ced-10-LPETG-MYC, G3-F-box-flag). We observed robust expression of the Ced-10-LPETG-MYC substrate at 20 °C. However, Ced-10-LPETG-MYC levels markedly decreased when these transgenic nematodes were heat-shocked to induce coexpression of the nucleophile G3-F-box-FLAG and sortase, together with Ced-10-LPETG-MYC (Figure 5C). We conclude that Ced-10-LPETG-MYC is proteolytically degraded upon sortase-dependent F-box attachment. Control samples (Ced10::SrtA7m or Ced10:Fbox) did not show a significant reduction in total Ced-10-LPETG-MYC levels (Figure 5A,D). We also tested the kinetics of SrtA7m -mediated Ced-10-LPETG-MYC degradation and observed the initiation of degradation as soon as 30 min after heat shock. At 5 h post-induction, the Ced-10-LPETG-MYC protein signal was almost undetectable, indicating near-complete degradation of the substrate (Figure 5E,F). The addition of the proteasome inhibitor (MG-132) to the plates 30 min after heat-shock onset largely inhibited degradation of Ced-10-LPETG-MYC (Figure S4A). The slight decrease in total Ced-10-LPETG-MYC levels can be attributed to early proteolytic events that occurred before intracellular MG-132 levels reached inhibitory concentrations. SrtA7m can therefore catalyze the in vivo ligation of a G3-F-box domain to its designated, LPETG-tagged target in less than 30 min, mediating substrate depletion within 5 h through engagement of the Ub proteasome pathway.
Figure 5.

SrtA7m mediation of protein degradation in C. elegans. (A) Schematic illustration of ubiquitin-dependent degradation. SKP1-CUL1-Fbox protein ligase complexes (SCFs) mediate ubiquitin transfer from E2 enzymes to target substrates. The resulting ubiquitin-tagged substrates are degraded by the proteasome. Target specificity of the SCFs depends on substrate binding to F-box proteins (left panel). SrtA7m-dependent covalent binding of substrates to F-box proteins enables degradation of an LPETG-tagged protein (right panel). (B) Immunoblot analysis of Ced10-LPETG-MYC substrate and G3-Fbox-Flag nucleophile levels in Pef t-3::Ced10; Phsp-16.2::Fbox strain lysates. N2 strains were used as the control. (C) Specific depletion of Ced10-LPETG-MYC following sortagging of the F-box to Ced10 in Phsp-16.2::srtA7m; Pef t-3::Ced10; phsp-16.2::Fbox strains. N2 and Phsp-16.2::srtA7m strains were used as controls. (D) Immunoblot analysis of different transgenic strains expressing combinations of Ced10-LPETG-MYC substrate, G3-Fbox-flag nucleophile, and SrtA7m. (E) Immnoblot analysis of Ced10-LPETG-MYC substrate degradation following SrtA7m-mediated F-box ligation. Phsp-16.2::srtA7m; Pef t-3::Ced10 strains were heat-shocked for 90 min and thereafter kept at 20 °C. Samples were taken at the indicated time points. (F) Quantification of Ced10-LPETG-MYC substrate degradation shown in (E).
Conclusions
The ability to introduce site-specific covalent protein modifications in a living organism, and to do so at will, poses an experimental challenge. Here, we show that in vivo expression of SrtA7m in C. elegans enables reliable site-specific protein modification. We demonstrate the versatility of this approach through the labeling of protein targets with small molecules, by showing intramolecular protein circularization as well as protein–protein ligation. We apply this method to attenuate intracellular protein levels in a sortase-dependent manner through engagement of the proteasome-degradation pathway.
Wild-type S. aureus SrtA is a Ca2+-dependent enzyme that does not support sortase reactions in the low intracellular Ca2+ milieu of metazoan cells. Although previous efforts used S. pyrogenes SrtA, a Ca2+-independent transpeptidase, to substitute for S. aureus SrtA in in vivo applications, we now expressed a hepta-mutant S. aureus, SrtA7m, in C. elegans. A pair of mutations (E105 K/E108A) confer Ca2+ independence, and the remaining five mutations (P94R/D160N/D165A/K190E/K196T) improve the catalytic properties of the enzyme.23–25 We demonstrate the in vivo activity of SrtA7m, not only to site-specifically label LPETG-tagged target proteins with distinct G3-containing nucleophiles but also to promote intramolecular circularization as a second example.28 Feeding sortase-expressing C. elegans with G3-labeled fluorescent dyes, such as G3-TMR or G3-Alexa647, resulted in modification of the intended target proteins. Our results indicate that after 5 h of incubation with G3-Alexa647, the dye is distributed throughout the animal. Although tissue penetration of the G3 nucleophiles presumably depends largely on active and passive diffusion, the expression of sortase using tissue-specific promoters may create new possibilities for localizing and tracking proteins of interest in different tissues or organelles. Covalent ligation of a biotin handle to LPETG-tagged proteins may offer an alternative to the use of BirA biotin ligase to enrich for targets in immunoprecipitation assays.
As expected, in vivo protein labeling reactions using our sortase-based ligation setup in C. elegans did not run to completion with the exception of the F-box-target fusions. Sortase reactions using a G3- or G5-nucleophile are reversible because upon completion of the reaction, the LPXTG motif is restored. Only the immediate removal of the ligation product from the reaction environment, such as the case for the FBox-target fusion that was readily degraded by the ubiquitin–proteasome pathway,29,30 allows the reaction to reach completion. Alternatively, the presence of a large molar excess of the intact nucleophile can drive the equilibrium in favor of the desired reaction product. In vivo reactions with both substrate and nucleophile at approximately equimolar concentrations are expected to yield significantly less than 50% ligation product, even after prolonged incubation. Dealing with the reaction’s reversibility, for example by using depsipeptides, β-hairpin linkers or a nickel-peptide complex may prove useful to enhance the efficiency of in vivo sortase reactions and shift the equilibrium in favor of the intended ligation product.13,31–33
We show that we can specifically deplete a target protein in vivo in a sortase-dependent, inducible manner. The proteolytic removal of F-box-target fusions, a reaction product required for reversing the sortase reaction, is expected to drive the reaction to complete substrate turnover, even at low local nucleophile concentrations.
Methods to target a tagged protein for degradation and to do so inducibly in intact cells or organisms are scarce. Armenti and co-workers showed that expression of the E3 ubiquitin ligase substrate recognition unit Zif-1 is sufficient to enable the rapid degradation of ZF-1-tagged proteins in C. elegans,34 and Zhang et al. introduced the plant auxin-inducible degradation system (AID) into C. elegans and demonstrated reliably depletion of degron-tagged proteins within 30 min in an auxin-dependent manner.35 Compared to these two very efficient proteolytic systems, sortase-dependent target depletion exhibits slower overall reaction kinetics. Considering that it takes approximately 2 h for srtA to be expressed at maximal levels post induction (Figure S1A), however, the actual ligation-based depletion reaction is only slightly slower than the ZF-1 or auxin-dependent setups. Importantly, the implementation of our sortase-based target depletion system requires the installment of only a five amino acid LPXTG tag on the target protein’s N- or C-terminus, while the ZF-1 tag (44 amino acids) and the degron tag (36 amino acids) are fairly large. The relatively small size of the LPXTG tag minimizes the chance for tag-related off-target effects, translational limitations, or changes in overall target protein structure. Unlike the ZF-1 and auxin systems that are specifically designed to decrease target protein levels, sortase mediated protein depletion is only one of numerous applications for sortase-dependent in vivo protein ligation, rendering this new method more versatile than other approaches.
In conclusion, we provide a proof of concept for a SrtA7mbased protein modification strategy in living C. elegans. The method is specific, robust, and complements existing protein modification and tracking approaches to study protein function in C. elegans.
MATERIALS AND METHODS
C. elegans Strains and Growth Conditions
Wild-type N2 Bristol was maintained at 20 °C on nematode growth medium (NGM) plates containing Escherichia coli OP50 bacteria as a food source.36 Synchronized worm populations were obtained by bleaching gravid adults washed off NGM plates and culturing the recovered eggs on fresh NGM plates. All strains containing phsp16.2-based arrays were maintained at 15 °C to suppress basal activation of the hsp-16.2 promoter.
C. elegans Strains Used in This Study
N2, MT23063 (nEx2249[Pmyo-3::mCherry, Phsp16.2::G5-GFP-LPETG]), MT23311 (nIs699[Pmyo-3::mCherry, Phsp16.2::srtA7m]), MT23699 (nIs699; nEx2431) MT23698 (nEx2431[Pef t-3::GFP-LPETG]), MT23700 (nIs699; nEx2432[Peft-3::Ced-10-LPETG-MYC, Phsp16.2::G3-SCFbox-FLAG, pRF4[rol-6(su1006dm)]]), MT23701 (nIs699, nEx2433[Peft-3::Ced-10-LPETG-MYC, pRF4]), and MT23702 (nEx2432) were used.
Plasmids
To construct heat-inducible plasmids, we used a pPD49.78 plasmid backbone containing a hsp-16.2 promoter linked to a multiple cloning site. The Fire Lab C. elegans Vector Kit was a gift from Andrew Fire. Target genes were cloned into NheI–SacI digested plasmid backbones using the Invitrogen Gibson Assembly kit following the manufacturer’s instructions. ORFs encoding for SrtA7m, GFP-LPETG, GGG-GFP-LPETG, Ced-10-LPETG-MYC, and GGG-F-Box-FLAG were designed and, if required, the codons optimized in silico. To optimize gene expression, 51 base pairs (bps) long synthetic introns (gtaagtttaaacatgattttactaactaactaatctgatttaaattttcag) were introduced in one or two copies into all open reading frames (ORFs). ORFs were synthesized (idtDNA) and amplified by PCR to introduce the required flanking restriction sites followed by 20 bps homology overlap using the primers listed in Table 1. Purifed PCR products were ligated into NheI–SacI-digested pPD49.78 using Gibson assembly. For constitutive expression, we exchanged phsp-16.2 in pPD49.78 with a synthetic DNA fragment that encodes pef t-3. Table 1 lists plasmids and primers used in this study.
Table 1.
Sortase, Substrate, and Nucleophile Constructs Used in This Study
| protein | plasmid | promoter | forward primer | reverse primer |
|---|---|---|---|---|
| SrtA7m | pPD 49–100-HA-SrtA | hsp promoter | GAAGCTAGCATGCGAAGCTCGTATCCTTACGATGTGCCTGA CTAC | GAACCATGGTTATTCGAGTTTAACCTCAGTGGCG |
| eGFP-LPETG | pPD 49–101-GFP-LPETG | eft-3 promoter | ATG AGT AAA GGA GAA GAA CTT TTC ACT GGA | TTATCCGGTCTCCGGGAGTTTGTAT |
| G5-GFP-LPETG | pPD 49–102-G5-GFP-LPETG | eft-3 promoter | CATTTTCAGGAGGACCCTTGGCTAGC | TGATGACAGCGGCCGATGCGGAGCTCTTATTTGTATAGTTCATCCATGCC |
| Ced10-LPETG-MYC | pPD 49–103-Ced10-LPETG-MYC | eft-3 promoter | CATTTTCAGGAGGACCCTTGGCTAGCATGCAAGCGATCAAATGTG | GATGACAGCGGCCGATGCGGAGCTCTTAGAGATCCTCTTCGCTGA |
| G3-Fbox-FLAG | pPD 49–104-G3-Fbox-FLAG | hsp promoter | ATGTCTTCAC CGCACCGAGC | GATTTTTGGATATAATTTACTGTAGAAGACAT |
Generation of Transgenic C. elegans Strains
Transgenic animals were obtained by germline injection of N2 worms. Plasmids pmyo-3::mCherry as well as PRF4 (rol-6(su1006dm)) were used as co-injection markers. Array-positive animals were selected, cultured as individual strains, and evaluated. For array integration, 30 L4 animals were exposed to γ irradiation and afterward grown for 3 weeks at 20 °C to allow Dauer larvae to emerge. A total of 50 array-positive Dauer animals were singled out, and resultant strains were tested for genomic array integration and homozygosity by microscopy and Western blotting post heat-shocking.
Heat-Shocking Protocol
To ensure reproducibility, we performed all experiments that required heat shock using a standardized protocol: animals on NGM plates were placed on a preheated metal tray (37 °C) and immediately transferred into a 37 °C incubator. Following 30 min of incubation, plates were removed and immediately spread out on a lab bench in a 20 °C room without stacking. After 3 h of initial recovery, plates were stacked and stored at 20 °C for the indicated times.
Protein Expression in C. elegans
For inducible protein expression, synchronized L1 populations were distributed on fresh NGM plates and grown for 24–80 h at 20 °C prior to heat shocking. Worms containing p49.78-based arrays were heat-shocked at indicated larval or adult stages at 37 °C for the indicated times and transferred to room temperature to allow for protein expression. The number of animals per biochemical experiment was set such that the resulting worm pellet available for analysis would have a volume of ~50 µL, which corresponds to ~1000 adult animals.
Generation of C. elegans Lysate
Synchronized C. elegans populations were washed off of NGM plates, resuspended in appropriate buffer (lysis buffer: 50 mM Tris, 100 mM NaCl, pH 7.5, 1% SDS, 2% NP40; sortagging buffer: 50 mM Tris–HCl, pH 7.5, 150 mM NaCl) and sonicated for 30 s (intervals of 3 s on and 3 s off at 10% duty cycle control). The lysate was then centrifuged for 10 min at 15000g at 4 °C. Protein concentration was measured by Nanodrop.
In Vitro Sortase Reaction
Synchronized phsp-16.2::srtA7m; pmyo-3::mCherry worms were collected, resuspended in approximately 100 µL of sortagging buffer, and lysed as described above. Total protein concentration in worm lysates was determined by Nanodrop. Transpeptidation reactions were performed by combining worm lysates normalized for protein concentration in the presence or absence of G3-TMR nucleophile or G3-biotin (1 mM) followed by incubation at 4 °C overnight. Reactions were terminated by the addition of SDS-PAGE loading buffer, boiled for 5 min, and analyzed by SDS-PAGE.
Immunoblotting
Samples were supplemented with an equal volume of 2× SDS-PAGE sample buffer and boiled for 5 min at 95 °C. A total of 20 µL of each sample was loaded on 15% SDS-PAGE gels (Mini-Gels, Bio-Rad). Gels were run at 100 mV for 20 min and then 150 mV for 40 min. Proteins were transferred to PVDF membranes using a semidry transfer system. Membranes were blocked overnight in PBS–0.1% TritonX–5% milk and then probed with the appropriate antibody (Table 2). Membranes were developed with ECL reagent (PerkinELmer Life Science), imaged on XAR-5 film (Kodak) and processed.
Table 2.
Antibodies Used in This Study
| provider | specificity | dilution used |
|---|---|---|
| Santa Cruz Biotechnology | HA | 1:20 000 |
| Abcam | GFP | 1:10 000 |
| Cell Signaling | biotin | 1:20 000 |
| Santa Cruz Biotechnology | MYC | 1:10 000 |
| Abcam | FLAG | 1:10 000 |
Nucleophile Synthesis and Feeding
G3-Biotin, G3-TMR, or G3-Alexa647 were synthesized, as described in refs 14, 37, and 38. For feeding, nucleophiles in water were diluted to a final concentration of 1 mM in NGM containing living OP50 E. coli bacteria as food-source L4 stage larvae or young adult nematodes were added, and animals were incubated for 5 h at RT with gentle shaking. Control worms were grown in plain NGM. Worms were then transferred to the fresh OP50 plates and incubated for at least 12 h before the preparation of lysates.
Microscopy
For fluorescence microscopy, worms at the L3–L4 stage were paralyzed with levamisole on an agar pad. All images were collected on a PerkinElmer Ultraview multispectral spinning disk confocal microscope equipped with a Zeiss 1.4 numerical aperture oil immersion ×63 objective lens and a Prior piezoelectric objective focusing device. Images were acquired with a Hamamatsu ORCA ER cooled CCD camera controlled with Volocity software. Post-acquisition image manipulations as well as Pearson correlation coefficient calculations were performed using Fiji software.39 Images shown in Figure 3C represent projections of acquired z-stacks.
Typhoon Scanning
To analyze reaction products of in vitro sortagging reactions using G3-TMR or G3-Alexa647 as nucleophiles, samples were separated by 10% SDS-PAGE, and gels were scanned using a Typhoon scanner (GE healthcare). Excitation and emission filters were set to 555 and 580 nm for TMR, respectively, and 633 and 670 nm for Alexa647, respectively. Gels were stained afterward with InstantBlue to visualize total sample content.
Longevity, Survival, and Motility Assays
For longevity assays, animals were grown on NGM plates and transferred to fresh NGM plates every 2 days for the first 10 days. At least 100 animals per condition were scored. For survival assays, animals exposed to indicated nucleophiles were transferred to NGM plates and incubated for 24 h at 20 °C before assessing. Animals were considered dead if they did not react to 10 pinches with a platinum loop. Motility assays were performed as described in ref 40. In brief, animals were transferred to a 15 µL drop of M9 and rested for 3 min. Afterward, body bending was scored for 30 s.
Supplementary Material
Acknowledgments
We are grateful to the members of the Ploegh and Horvitz laboratories for technical support, comments, and discussions. We thank G. Bell and the Whitehead bioinformatics group for their help with C. elegans proteome in silico analysis as well as E. Spooner and the Whitehead Mass Spec facility for their service. This work was supported by an NIH Award (grant no. 2R01AI087879-06 to H.L.P.).
Footnotes
ASSOCIATED CONTENT
The authors declare no competing financial interest.
References
- 1.Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- 2.Ton-That H, Mazmanian SK, Faull KF, Schneewind O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH(2)-Gly(3) substrates. J. Biol. Chem. 2000;275:9876–9881. doi: 10.1074/jbc.275.13.9876. [DOI] [PubMed] [Google Scholar]
- 3.Mazmanian SK, Ton-That H, Schneewind O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001;40:1049–1057. doi: 10.1046/j.1365-2958.2001.02411.x. [DOI] [PubMed] [Google Scholar]
- 4.Ilangovan U, Ton-That H, Iwahara J, Schneewind O, Clubb RT. Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6056–6061. doi: 10.1073/pnas.101064198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruger RG, Otvos B, Frankel BA, Bentley M, Dostal P, McCafferty DG. Analysis of the substrate specificity of the Staphylococcus aureus sortase transpeptidase SrtA. Biochemistry. 2004;43:1541–1551. doi: 10.1021/bi035920j. [DOI] [PubMed] [Google Scholar]
- 6.Popp MW, Ploegh HL. Making and breaking peptide bonds: protein engineering using sortase. Angew. Chem., Int. Ed. 2011;50:5024–5032. doi: 10.1002/anie.201008267. [DOI] [PubMed] [Google Scholar]
- 7.Clancy KW, Melvin JA, McCafferty DG. Sortase transpeptidases: insights into mechanism, substrate specificity, and inhibition. Biopolymers. 2010;94:385–396. doi: 10.1002/bip.21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ritzefeld M. Sortagging: a robust and efficient chemoenzymatic ligation strategy. Chem. - Eur. J. 2014;20:8516–8529. doi: 10.1002/chem.201402072. [DOI] [PubMed] [Google Scholar]
- 9.Schmohl L, Schwarzer D. Sortase-mediated ligations for the site-specific modification of proteins. Curr. Opin. Chem. Biol. 2014;22:122–128. doi: 10.1016/j.cbpa.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 10.Proft T. Sortase-mediated protein ligation: an emerging biotechnology tool for protein modification and immobilisation. Biotechnol. Lett. 2010;32:1–10. doi: 10.1007/s10529-009-0116-0. [DOI] [PubMed] [Google Scholar]
- 11.Swee LK, Guimaraes CP, Sehrawat S, Spooner E, Barrasa MI, Ploegh HL. Sortase-mediated modification of alphaDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proc. Natl. Acad. Sci. U. S. A. 2013;110:1428–1433. doi: 10.1073/pnas.1214994110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat. Chem. Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- 13.Antos JM, Truttmann MC, Ploegh HL. Recent advances in sortase-catalyzed ligation methodology. Curr. Opin. Struct. Biol. 2016;38:111–118. doi: 10.1016/j.sbi.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Theile CS, Witte MD, Blom AE, Kundrat L, Ploegh HL, Guimaraes CP. Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013;8:1800–1807. doi: 10.1038/nprot.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antos JM, Chew GL, Guimaraes CP, Yoder NC, Grotenbreg GM, Popp MW, Ploegh HL. Site-specific N- and C-terminal labeling of a single polypeptide using sortases of different specificity. J. Am. Chem. Soc. 2009;131:10800–10801. doi: 10.1021/ja902681k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorr BM, Ham HO, An C, Chaikof EL, Liu DR. Reprogramming the specificity of sortase enzymes. Proc. Natl. Acad. Sci. U. S. A. 2014;111:13343–13348. doi: 10.1073/pnas.1411179111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Race PR, Bentley ML, Melvin JA, Crow A, Hughes RK, Smith WD, Sessions RB, Kehoe MA, McCafferty DG, Banfield MJ. Crystal structure of Streptococcus pyogenes sortase A: implications for sortase mechanism. J. Biol. Chem. 2009;284:6924–6933. doi: 10.1074/jbc.M805406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strijbis K, Spooner E, Ploegh HL. Protein ligation in living cells using sortase. Traffic. 2012;13:780–789. doi: 10.1111/j.1600-0854.2012.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautier A, Juillerat A, Heinis C, Correa IR, Jr, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Gautier A, Nakata E, Lukinavicius G, Tan KT, Johnsson K. Selective cross-linking of interacting proteins using self-labeling tags. J. Am. Chem. Soc. 2009;131:17954–17962. doi: 10.1021/ja907818q. [DOI] [PubMed] [Google Scholar]
- 21.Ullrich M, Liang V, Chew YL, Banister S, Song X, Zaw T, Lam H, Berber S, Kassiou M, Nicholas HR, Gotz J. Bio-orthogonal labeling as a tool to visualize and identify newly synthesized proteins in Caenorhabditis elegans. Nat. Protoc. 2014;9:2237–2255. doi: 10.1038/nprot.2014.150. [DOI] [PubMed] [Google Scholar]
- 22.Machleidt T, Robers M, Hanson GT. Protein labeling with FlAsH and ReAsH. Methods in molecular biology. 2007;356:209–220. doi: 10.1385/1-59745-217-3:209. [DOI] [PubMed] [Google Scholar]
- 23.Hirakawa H, Ishikawa S, Nagamune T. Design of Ca2+-independent Staphylococcus aureus sortase A mutants. Biotechnol. Bioeng. 2012;109:2955–2961. doi: 10.1002/bit.24585. [DOI] [PubMed] [Google Scholar]
- 24.Swee LK, Lourido S, Bell GW, Ingram JR, Ploegh HL. One-step enzymatic modification of the cell surface redirects cellular cytotoxicity and parasite tropism. ACS Chem. Biol. 2015;10:460–465. doi: 10.1021/cb500462t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirakawa H, Ishikawa S, Nagamune T. Ca2+-independent sortase-A exhibits high selective protein ligation activity in the cytoplasm of Escherichia coli. Biotechnol. J. 2015;10:1487–1492. doi: 10.1002/biot.201500012. [DOI] [PubMed] [Google Scholar]
- 26.Antos JM, Popp MW, Ernst R, Chew GL, Spooner E, Ploegh HL. A straight path to circular proteins. J. Biol. Chem. 2009;284:16028–16036. doi: 10.1074/jbc.M901752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popp MW, Dougan SK, Chuang TY, Spooner E, Ploegh HL. Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. U. S. A. 2011;108:3169–3174. doi: 10.1073/pnas.1016863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirakawa H, Ishikawa S, Nagamune T. Ca2+-independent sortase-A exhibits high selective protein ligation activity in the cytoplasm of Escherichia coli. Biotechnol. J. 2015;10:1487–1492. doi: 10.1002/biot.201500012. [DOI] [PubMed] [Google Scholar]
- 29.Caussinus E, Kanca O, Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 2012;19:117–121. doi: 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- 30.Shin YJ, Park SK, Jung YJ, Kim YN, Kim KS, Park OK, Kwon SH, Jeon SH, Trinh le A, Fraser SE, Kee Y, Hwang BJ. Nanobody-targeted E3-ubiquitin ligase complex degrades nuclear proteins. Sci. Rep. 2015;5:14269. doi: 10.1038/srep14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamura Y, Hirakawa H, Yamaguchi S, Nagamune T. Enhancement of sortase A-mediated protein ligation by inducing a beta-hairpin structure around the ligation site. Chem. Commun. 2011;47:4742–4744. doi: 10.1039/c0cc05334a. [DOI] [PubMed] [Google Scholar]
- 32.Yazawa K, Numata K. Recent advances in chemoenzymatic peptide syntheses. Molecules. 2014;19:13755–13774. doi: 10.3390/molecules190913755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.David Row R, Roark TJ, Philip MC, Perkins LL, Antos JM. Enhancing the efficiency of sortase-mediated ligations through nickel-peptide complex formation. Chem. Commun. 2015;51:12548–12551. doi: 10.1039/c5cc04657b. [DOI] [PubMed] [Google Scholar]
- 34.Armenti ST, Lohmer LL, Sherwood DR, Nance J. Repurposing an endogenous degradation system for rapid and targeted depletion of C. elegans proteins. Development. 2014;141:4640–4647. doi: 10.1242/dev.115048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Ward JD, Cheng Z, Dernburg AF. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development. 2015;142:4374–4384. doi: 10.1242/dev.129635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Theile CS, Chen GY, Bilate AM, Duarte JN, Avalos AM, Fang T, Barberena R, Sato S, Ploegh HL. Fluorophore-Conjugated Holliday Junctions for Generating Super-Bright Antibodies and Antibody Fragments. Angew. Chem., Int. Ed. 2015;54:11706–11710. doi: 10.1002/anie.201505277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AE, Ploegh HL. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013;8:1787–1799. doi: 10.1038/nprot.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brignull HR, Moore FE, Tang SJ, Morimoto RI. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J. Neurosci. 2006;26:7597–7606. doi: 10.1523/JNEUROSCI.0990-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.