

Abstract
Organophosphorus-based (OP) nerve agents represent some of the most toxic substances known to mankind. The current standard of care for exposure has changed very little in the past decades, and relies on a combination of atropine to block receptor activity and oxime-type acetylcholinesterase (AChE) reactivators to reverse the OP binding to AChE. Although these oximes can block the effects of nerve agents, their overall efficacy is reduced by their limited capacity to cross the blood-brain barrier (BBB). RS194B, a new oxime developed by Radic et al. (J. Biol. Chem., 2012) has shown promise for enhanced ability to cross the BBB. To fully assess the potential of this compound as an effective treatment for nerve agent poisoning, a comprehensive evaluation of its pharmacokinetic (PK) and biodistribution profiles was performed using both intravenous and intramuscular exposure routes. The ultra-sensitive technique of accelerator mass spectrometry was used to quantify the compound's PK profile, tissue distribution, and brain/plasma ratio at four dose concentrations in guinea pigs. PK analysis revealed a rapid distribution of the oxime with a plasma t1/2 of ∼1 hr. Kidney and liver had the highest concentrations per gram of tissue followed by lung, spleen, heart and brain for all dose concentrations tested. The Cmax in the brain ranged between 0.03-0.18% of the administered dose, and the brain-to-plasma ratio ranged from 0.04 at the 10 mg/kg dose to 0.18 at the 200 mg/kg dose demonstrating dose dependent differences in brain and plasma concentrations. In vitro studies show that both passive diffusion and active transport contribute little to RS194B traversal of the BBB. These results indicate that biodistribution is widespread, but very low quantities accumulate in the guinea pig brain, indicating this compound may not be suitable as a centrally active reactivator.
Keywords: Acetylcholinesterase, oxime, reactivator, pharmacokinetics, biodistribution
1. Introduction
The lethal effects produced by the use of chemical warfare agents (CWAs) in war- ravaged environments are well established and, in more recent times, have increased the global awareness of the destructive power of these toxic substances. Among the myriad of CWAs that are currently available, the organophosphorus (OP)-based nerve agents have been shown to be a bigger threat to the general population than other CWAs. During the Iran-Iraq war (Haines and Fox 2014) their use was tacitly confined to military conflicts, however, more recently they have also been used on civilian populations such as the Aum Shinrikyo's attack in the Tokyo subway system (Okumura et al., 1996; Okumura et al., 2005; Okumura et al., 2005; Tu 2014) and the Ghouta chemical attack in Syria (Dolgin 2013). Exposure to OP-based nerve agents (Fig.1, e.g. sarin, 1, soman, 2,) results in irreversible inhibition of cholinesterases within minutes of contact, leading to hyperactivity of the cholinergic system due to acetylcholine accumulation in both the central and peripheral nervous systems. Acetylcholine accumulation can lead to seizures, convulsions, fasciculations, secretions, respiratory distress and ultimately death if not treated properly and most importantly, immediately (O'Malley 1997; Solberg and Belkin 1997).
Figure 1.

Structures of the two OP-based nerve agents Sarin (1) and Soman (2). Their inhibition of AChE can be reversed by the use of synthetic oxime reactivators such as the water-soluble 2-PAM chloride (3) and the neutral oxime RS-194B (4).
Given the rapid onset of the symptoms associated with exposure to these chemicals, a fast acting, efficacious therapeutic treatment is needed to counteract their effects. The current standard therapeutic regimen consists of a formulation comprised of two active ingredients: atropine, a competitive muscarinic receptor agonist and pralidoxime chloride (2-PAM, 3, Fig.1), a synthetic acetylcholinesterase (AChE) oxime reactivator. These two components work together to reduce the hyperstimulation from acetylcholine accumulation, mitigating the effects of the nerve agents until the agents are degraded and active enzyme levels are restored. Although 2-PAM is an effective reactivator, one major deficiency is its inability to cross the blood-brain-barrier (BBB), thus preventing it from reactivating vital OP-inhibited AChE in the central nervous system (CNS). Over the last decade efforts have focused on screening and identifying other reactivating oximes that have a higher BBB permeability while still retaining a high reactivation profile (DeMar et al., 2010; García et al., 2010; Sit et al., 2011; Radic et al., 2012; Radic et al., 2013; Okolotowicz et al., 2014). In recent work by Radic et al., a promising centrally active oxime reactivator (RS194B, 4, Fig 1.) with enhanced BBB permeability was reported. This oxime demonstrated favorable in vitro reactivation in addition to preliminary CNS penetration and retention in rodents (Radic et al., 2012).
The current study aims to further evaluate the potential of RS194B to be a BBB-penetrating oxime reactivator by conducting a comprehensive in vivo assessment of the oxime's pharmacokinetic (PK) and biodistribution profile using accelerator mass spectrometry (AMS). AMS is an ultrasensitive measurement technique that quantifies rare, long-lived isotopes at attomolar concentrations (Vogel et al., 1995). The unmatched sensitivity of AMS enables detection of trace levels of materials using small sample sizes with high precision. Furthermore, the inherent high sensitivity of the technique allows for studies where high specific activities of radiolabeled compounds are not possible. AMS has been utilized for both rodent and human studies including: nanoparticle dosimetry (Malfatti et al., 2012) and pharmacokinetic and metabolic evaluation of therapeutics and environmental contaminants (Henderson and Pan 2010; Wagner et al., 2011; Malfatti et al., 2014; Madeen et al., 2015).
The guinea pig (Cavia porcellus) was chosen as the animal model for the current study as it more accurately reflects human susceptibility to OP-based nerve agent exposure than other rodent models. Compared to mice and rats, the guinea pig has lower carboxylesterases plasma levels, which are more similar to levels reported in humans (Bahar et al., 2012). Furthermore, the guinea pig AChE protein sequence is comparable to that of humans with a 91.7% sequence identity at the amino acid level (Cadieux et al., 2010). Using the guinea pig model, PK parameters, brain permeability and biodistribution of RS194B were quantified using AMS; four dose concentrations and two routes of administration (intravenous and intramuscular) were evaluated. The high sensitivity afforded by AMS allowed for the precise quantitation of analytes over time, especially in tissues where RS194B concentrations were low.
2. Materials and methods
2.1 Chemicals
All reagents were used as received from commercial vendors. Ethanol and hydroxylamine hydrochloride were purchased from Alfa Aesar (Ward Hill, MA). Ethyl glyoxylate and heparin sodium salt were purchased from Sigma-Aldrich (St. Louis, MO). Collagenase Type IV was obtained from Gibco, (Waltham, MA). 2-(1-azepan-1-yl)ethan-1-amine was purchased from Enamine Building Blocks (Kiev, Ukraine). 14C-labeled ethyl glyoxylate was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). All other reagents were of analytical grade or better.
2.2.Radiolabeling of RS194B
14C-RS194B (specific activity = 2.5 μCi/mmol) was synthesized in two steps using 14C-labeled ethyl glyoxylate as a precursor. The radiolabeled product was synthesized as follows: ethyl glyoxylate (50% in toluene, 9.2 g, 45.1 mmol, 102.1 g/mol) and 0.5 mL of 14C-labeled glyoxylate (∼1.7 mg, 0.017 mmol, 0.250 mCi/mL, specific activity = 7.5 mCi/mmol) were dissolved in a 9:1 acetonitrile:water (50 mL) solution in a 100 mL round bottom flask equipped with a stir bar treated with hydroxylamine hydrochloride (3.13 g, 45.1 mmol, 69.49 g/mol). The resulting suspension was treated with triethylamine (6.35 mL, 45.1 mmol, 101.2 g/mol, 0.726 g/mL) dropwise over 10-20 minutes using an addition funnel and then stirred at room temperature overnight. The solvents were removed in vacuo at 70°C for 1-2 hours to yield a semisolid oily residue. This semisolid was dissolved with 10 mL water and the aqueous phase was washed with diethyl ether (3 × 15 mL). The organic phase was dried over anhydrous Na2SO4, suction filtered, and evaporated in vacuo to yield the crude oxime-ethyl glyoxylate. The crude oxime-ethyl glyoxylate (1.2 g, 10.24 mmol) was dissolved in ethanol (3 mL) in a 50 mL glass vial equipped with a stir bar. N-2-aminoethylhomopiperidine (1.6 g, 11.26 mmol, 1.1 equiv. in ethanol) was added to the solution and the resulting mixture was stirred at 55°C overnight. The mixture was then cooled to room temperature and the solid was suspended in 10 mL of cold ethanol. The fine white solid was collected after filtration, washed with cold ethanol (3 × 10 mL), and dried in vacuo for 1-2 h. Radiopurity was assessed by high performance liquid chromatography and liquid scintillation counting. The product was determined to be 98% pure.
2.3. Animals
Animal experiments were conducted at the Lawrence Livermore National Laboratory (LLNL) AAALAC accredited animal care facility. The protocol for the animal experiments was reviewed and approved by the LLNL Institutional Animal Care and Use Committee prior to the initiation of the study. Male Hartley guinea pigs weighing 250-300 g with a surgically implanted jugular vein catheter were obtained from Charles River Laboratories (Wilmington, MA). Guinea pigs were housed individually in polystyrene cages containing hardwood bedding and kept on a 12 h light/dark cycle in a ventilated room maintained at 24°C. Food and water were provided ad libitum.
2.4. RS194B Plasma Pharmacokinetics
To evaluate the pharmacokinetics of RS194B at several dose levels guinea pigs (n=6 per dose concentration) were administered a single intravenous (IV)dose of 10, 50, 100, or 200 mg/kg 14C-RS194B dissolved in sterile saline (1.6 ml/kg), pH 5.6, through an implanted jugular vein catheter. The 14C-RS194B injection was immediately followed by an injection of 50 μL of sterile saline into the catheter to ensure complete infusion of 14C-RS194B into the blood stream. Following dose administration, whole blood samples (approximately 0.3 mL) were collected from each animal via the jugular vein at 0.08, 0.25, 0.5, 1, 2, 4, 8 and 24 h post dose, placed into Microtainer® tubes coated with lithium heparin (Becton Dickinson, Franklin Lakes, NJ) and placed on ice. Within one hour of collection the plasma was separated from the whole blood by centrifugation following the manufacturers recommendations (8,000 × g for 2 min). The volume of plasma obtained was recorded and the sample was stored at -80°C until analysis by AMS. To compare dose routes and to obtain absolute bioavailability, experiments were repeated using a single intramuscular (IM) dose of 50 mg/kg 14C-RS194B dissolved in sterile saline (0.83 ml/kg), pH 5.6, delivered in the right hind thigh.
The plasma PK parameters of RS194B were calculated by non-compartmental analysis using PK Solutions software (Summit Research Services, Montrose, CO). A two-stage approach was used to independently fit the plasma concentration data from each guinea pig, and then determine the means ± standard errors. The half-life (t1/2) and the initial concentration observed in plasma (C0) were determined from the concentration-versus-time data. The area under the concentration vs. time curve (AUC) was calculated for the intervals from time zero to time t (AUC0 –t), where t is the time of the last measurable concentration (24 h), and for time zero to infinity (AUC0 –inf), using the linear trapezoidal method. The volume of distribution (Vd) was determined on the basis of the AUC determination and reflects the Vd during the elimination phase. The clearance (CL) calculation is based on the AUC0-inf.
2.5. RS194B Biodistribution
To assess biodistribution of RS194B, guinea pigs (n=6 per time point) were administered a single IV dose of 10, 50, 100, or 200 mg/kg 14C-RS194B as described above. Following dose administration animals were euthanized by CO2 asphyxiation at 0.08, 0.25, 0.5, 1, 2, 4, 8 and 24 h post dose. The 24 h dose group of animals was placed in metabolism cages and urine was collected for 24 h after 14C-RS194B administration. Immediately following euthanasia, whole animal perfusion was performed to ensure all the blood was removed from the tissues prior to collection. Animals were perfused with heparinized phosphate buffered saline (PBS, 50,000 U/L sodium heparin) through the posterior end of the heart through the left ventricle into the aorta using a 20 gauge × 1.5 inch gavage needle (VWR, Radnor, PA). The needle was clamped into place and connected to a 50 mL syringe via a 14” minibore extension tube (Vygon, Lansdale PA). An incision was made in the right atrium to serve as an outlet; 150 mL of perfusion buffer was then pumped through the animal at a rate of 40 mL/min with a syringe pump (Cole Parmer, Vernon Hills, IL). After perfusion, tissues (brain, liver, kidney, heart, spleen and lung) were excised from the carcass and rinsed twice in PBS. All tissues were then stored in glass vials (28 × 60mm) with the exception of livers which were stored in 50 mL conical polypropylene tubes at -80°C until analysis. Clean, unused surgical tools were used for each animal to avoid cross contamination. To compare dose routes experiments were repeated using a single IM dose of 50 mg/kg 14C-RS194B delivered in the right hind thigh.
2.6. Renal Clearance
Urine was collected at time intervals of 0-4 h, 4-8 h and 8-24 h post-administration of the 14C-RS194B dose, and immediately frozen and stored at −80°C. For analysis, each sample was thawed and the volume recorded. A 0.1 mL aliquot from each fraction was then analyzed by liquid scintillation counting (Packard, Perkin Elmer, Waltham, MA) to measure the 14-carbon content. The 0-4 h urine samples were analyzed by reversed-phase HPLC to determine the presence of RS194B metabolites or breakdown products. Samples consisting of approximately 1000 disintegrations per minute (dpm) of 14-carbon from each urine sample were dried in vacuo and subsequently rehydrated in 100 μL 10% acetonitrile. Each sample was then directly injected into an Alliance HPLC system (Waters, Milford, MA) equipped with a 4 μm, 4.6 × 250 mm Synergi Max-RP 80A column (Phenomenex, Torrance, CA), and monitored at λ = 245 nm. Analytes were eluted at 0.5 mL/min using a solvent of 54% 100 mM ammonium acetate and 46% acetonitrile. The column eluent was collected at 1 min intervals and radioactivity of each fraction was quantified by scintillation counting. Approximately 90% of the radioactivity was recovered after sample preparation and analysis. A RS194B authentic standard had a retention time of 4.5 min.
2.7. AMS sample processing and analysis
To ensure a uniform sampling distribution of 14C-RS194B within each tissue, tissues were digested to create a homogenous solution that is compatible with AMS analysis. All tissues were homogenized using a digestion buffer of 0.11 M KCl, 3.4 mM NaCl, 0.4 mM MgSO4, 40 mM CaCl2, 4mg/mL Collagenase Type IV, and 1.1 U/mL DNase 1 using previously established methods (Malfatti et al., 2012). Samples were incubated overnight at 37°C in a shaking water bath with gentle agitation. After the incubation time, samples were vortexed vigorously for 1-3 min to break up any solid particles to ensure complete digestion of the tissues. Plasma samples were analyzed neat without any treatment. A small aliquot of each sample (10-100 μl, depending on tissue carbon content) was used for AMS analysis. Carbon content for each tissue was measured using an Elementar Vario Isotope Cube (Elementar Analysensysteme, Hanau, Germany). Tissue homogenates were determined to have a carbon content ranging between 2-7%. Plasma carbon content was determined to be 3.2%
Preparation of the samples for radiocarbon analysis by AMS requires conversion of the samples to graphite. This procedure has been described previously (Ognibene et al., 2003). All the tissues and reagents were handled carefully to avoid radiocarbon cross-contamination. This required using disposable materials for any item that might come into contact with the samples. A 10-100 μL aliquot of each tissue homogenate or plasma sample was pipetted into 6 × 55 mm quartz tubes using aerosol resistant tips. All samples were subsequently dried under vacuum centrifugation. The dried samples were then converted to graphite by a two-step process using reported methods (Ognibene et al., 2003). Briefly, the dried samples were oxidized to CO2 by heating at 900° C for 4 h in the presence of copper oxide. The CO2 was then cryogenically transferred to a septa-sealed vial under vacuum and reduced to filamentous graphite in the presence of cobalt, titanium hydride, and zinc powder.
The total radiocarbon content of the samples was quantified by AMS as described previously (Ognibene et al., 2003). The 14C/12C ratios from the graphitized samples obtained by AMS were converted to either μg RS194B per mL of plasma or μg RS194B per gram of tissue after subtraction of the background carbon contribution and correction for the specific activity of the 14C-labeled RS194B dosing material. The precision of the AMS measurements is based on the standard deviation of several measurements for equivalent samples and is a nominal 3%. Accuracy is determined by normalization to well-defined standards and with high-accuracy counting measurements and is ∼1%(Vogel et al., 1995).
2.8 Parallel artificial membrane permeability assay (PAMPA)
Passive diffusion of RS194B was assessed using the Gentest Precoated PAMPA Plate System (Corning Discovery Labware). This system is composed of a 96-well plate with an insert containing a phospholipid-oil-phospholipid trilayer. Compound was dissolved at 100 μM in Hanks Balanced Salt Solution (HBSS) and added to the lower (donor) well of the plate, followed by addition of the insert to the plate. The system was incubated for five hours at 25°C, after which buffer was removed from the upper (acceptor) well. Five replicate wells were employed in each plate. All compounds were quantified by UPLC separation and UV detection, and effective permeability (cm/s) was calculated. All UPLC detection protocols and calculation parameters have been previously described (Bennion et al, 2017).
2.9. Brain endothelial cell traversal
Traversal of RS194B across brain endothelial cells was assessed using Human cerebral microvascular cells (HCMEC/D3), obtained from Cornell University. Cells were grown in EndoGRO-MV Complete media (EMD Millipore). For transcellular assays, cells were seeded at approximately 5 × 104 cells/cm2 in 12-well transwell tissue culture inserts (0.4 μm pore size, polycarbonate membrane) (Corning Life Sciences). All inserts were collagen coated prior to use. Cells were seeded in media containing 10 mM LiCl, and media was exchanged every 2-3 days. Monolayer integrity was monitored continuously via transendothelial electrical resistance using the EVOM2 voltohmmeter (World Precision Instruments). Inserts were used in assays 6-7 days following seeding. Compound was dissolved at 100 μM in HBSS for testing and added to the upper (apical) well of the insert. Buffer samples were removed from the lower (basolateral) well at 0, 15, 30, 60, 90, and 120 minutes after dosing. Each compound was examined in at least three replicates. As above, compound in buffer was quantified by UPLC as previously described (Bennion et al, 2017). Permeability of each compound across inserts not containing cells was examined as a control in each assay plate. Effective permeability (Peff [cm/s]) was calculated using the permeability surface area product method, as described by Deli et al.
2.10. MDR1 efflux assessment
Efflux capacity of RS194B was assessed in MDR1-MDCK and MDCK parent cells, obtained from the National Institutes of Health (Bethesda, MD). Both cell types were grown in Dulbecco's modified Eagle's medium (DMEM) containing glucose (4.5 g/L) and sodium pyruvate (110 mg/L), supplemented with 10% fetal bovine serum (FBS), L-glutamine (5 mM), and penicillin/streptomycin (50 U/mL; 50 μg/mL). MDR1 expression was maintained in MDR1-MDCK cells through culture in the presence of colchicine (80 ng/mL). For traversal studies, cells were seeded at approximately 5 × 105 cells/cm2 in 24-well HTS Transwell supports (0.4 μm pore size, polycarbonate membrane) (Corning Life Sciences). Media was exchanged every 2-3 days, and cells were used 5-6 days after seeding. For testing, compounds were dissolved at 100 μM in HBSS and added to the donor well. Transport was examined in both the A > B (apical to basolateral) and B > A (basolateral to apical) directions. This experiment was performed in both MDR1-MDCK cells and control MDCK parent cells. The efflux ratio was calculated as the ratio of permeability in the B > A direction to permeability in the A > B direction. As above, quantity of compound in buffer was quantified by UPLC as previously described (Bennion et al, 2017). Results are represented as the net flux ratio (NFR), calculated as the efflux ratio in MDR-MDCK cells to efflux ratio in MDCK cells. NFR value greater than one indicates that the compound is a possible substrate for efflux by p-gp (MDR1). The calculations outlined above were performed as described by Feng et al.
2.11. Statistics
Values are expressed as the mean ± SE (n=6), unless otherwise noted. Dose linearity was assessed using the power regression model on log-transformed data for AUC0–t, and C0 (Balani et al., 2006). Coefficients of determination (R2 values), and 95% confidence limits were determined from the power regression analysis. Results were considered linear when R2 was ∼1, and lower confidence limits were ≥0.8 and upper confidence limits were ≤1.25 (Smith et al., 2000). For the cellular assays, triplicate wells were run for each experiment.
3. Results
3.1. Plasma pharmacokinetics
Pharmacokinetic parameters of RS194B were evaluated over an intravenous administered 20-fold dose range that was designed to capture the potential therapeutic doses. Mean plasma concentrations over time of RS194B are illustrated in Figure 2 and the mean PK parameters are presented in Table 1. The plasma concentration vs. time curve for all four dose concentrations were similar following first order kinetics. The C0 of RS194B was back extrapolated to time zero from the plasma-concentration curve and had mean values ranging from 6.23 to 157.66 μg/mL. Across the four doses studied, the mean apparent distribution half-life (t1/2α) ranged from 0.78-1.5 h, and the terminal half-life (t1/2 β)spanned from 9.75 to 12.87 h. Total clearance of RS194B from plasma (CL) ranged from 400.51 to 590.51 mL/h/kg and the apparent volume of distribution (Vd) was 5802.08 to 8675.15 mL/kg suggesting rapid and extensive distribution beyond the plasma compartment. The lowest Vd and CL values (5802.08 mL/kg, 400.51 mL/h/kg, respectively) and the longest t1/2α were observed in the highest dose concentration (200 mg/kg) indicating a higher proportion of the compound remained in the plasma, compared to the lower dose exposures.
Figure 2.

Mean concentration-time profiles of RS194B in plasma (solid line) and brain (dotted line) following single intravenous administrations of 10 (◆), 50 (■), 100 (▲) or 200 (●) mg/kg 14C-RS194 to male guinea pigs. Plasma data is depicted as μg RS194B/ml plasma; brain data is depicted as μg RS194B/mg brain tissue. Data are expressed as the mean of 6 animals ± the standard error.
Table 1. Mean pharmacokinetic parameters of RS194B following a single administration of 10, 50, 100 or 200 mg/kg 14C-RS194B to male guinea pigs.
| Dose | C0 | t1/2α | T1/2;β | AUC0-t | AUC0-inf | Vd | CL |
|---|---|---|---|---|---|---|---|
| (mg/kg) | (μg/ml) | (h) | (h) | (μg.h/mL) | (μg.h/mL) | (mL/kg) | (mL/h/kg) |
| 10a | 6.23 ± 0.15 | 0.79 ± 0.07 | 10.41 ± 1.54 | 15.27 ± 1.23 | 17.53 ± 1.35 | 8675.15 ± 1999.77 | 590.51 ± 51.87 |
| 50a | 42.77 ± 2.5 | 1.03 ± 0.28 | 10.68 ± 1.94 | 83.80 ± 4.05 | 92.60 + 4.38 | 8378.18 ± 1448.68 | 546.65 ± 27.44 |
| 100a | 93.18 ± 3.4 | 0.78 ± 0.05 | 12.87 ± 2.16 | 188.05 ± 11.10 | 223.60 ±14.56 | 8322.03 ± 1442.65 | 455.89 ± 26.85 |
| 200a | 157.66 ± 4.34 | 1.50 ± 0.20 | 9.75 ± 1.83 | 488.52 ± 63.16 | 530.83 ± 57.86 | 5802.08 ± 1329.87 | 400.51 ± 44.01 |
| 50b | 24.90 ± 1.44 | 0.87 ± 0.13 | 8.53 ± 1.49 | 73.50 ± 2.41 | 78.74 ± 3.75 | 7729 ± 1088.99 | 641.80 ± 31.32 |
Expoure was through IV route. Data expressed as the mean of six animals ± the standard error
Exposure was through IM route. Data expressed as the mean of six animals ± the standard error
The mean C0 and AUC0-t values of RS194B for all four doses are summarized in Table 1. Following an increase in dose from 10 to 200 mg/kg (20-fold), the increases in mean C0 (25-fold) and AUC0-t (32-fold) values were greater than expected for a dose proportional response. Although the AUC0-t and C0 did not reflect precise dose proportionality, power regression analysis of both AUC0-t and C0 across the entire dose range (10-200 mg/kg) revealed a linear relationship between exposure vs. dose with R2 values of 0.9756 (slope 1.13) and 0.9885 (slope 1.09) for AUC0-t and C0, respectively.
A comparison of PK parameters from IV administration vs. IM administration of a 50 mg/kg dose of RS194B showed a statistically significant (p < 0.01) lower maximum observed concentration (Cmax), and a later tmax following the IM exposure (Figure 3). The observed tmax of 0.5 h indicates rapid absorption from the injection site into the plasma compartment. The mean AUC0-t was 83.80 ± 4.05 and 73.50 ± 2.41 μg•hr/mL for the IV and IM dose route, respectively (Table 1). Based on the difference in AUC0-t between the two dose routes the absolute bioavailability (F) was determined to be 0.88, indicating abundant absorption into the systemic circulation after an IM exposure.
Figure 3.

Mean plasma concentration-time profiles of RS194B following single intravenous (◆) or intramuscular (■) administration of 50mg/kg 14C-RS194 to male guinea pigs. Data are expressed as the mean of 6 animals ± the standard error.
3.2. BBB penetration
The Cmax of RS194B in the brain occurred at the first sampling time-point of 5 min post dose with mean values ranging from 0.22 – 25.99 ng/mg tissue over the four doses (Figure 2)indicating rapid distribution to the brain. These levels, however, accounted for only 0.03% - 0.180% of the administered dose signifying that very little of the dose permeated the BBB and trans-located to the brain. The difference in RS194B percentage in the brain was dose dependent with the lowest percentage (0.03 %) observed from the lowest dose (10 mg/kg) and the highest percentage (0.18%) observed from the highest dose (200 mg/kg) (Table 2). A dose–dependent brain/plasma ratio was also observed. At the Cmax of RS194B in the brain (0.08 h), the brain/plasma ratios were 0.04, 0.09, 0.12, 0.18 for the doses of 10 mg/kg, 50 mg/kg, 100 mg/kg and 200 mg/kg, respectively (Table 3), indicating dose dependent differences in brain and plasma RS194B concentrations. There were also observed differences in the brain/plasma ratio over time. Although not statistically significant, at 24 h post dose the brain-to-plasma ratio was increased over the values obtained at the brain Cmax at the 10, 50 and 200 mg/kg doses (Table 3) suggesting differences in brain and plasma clearance rates. When comparing the IV and IM dose routes at 50 mg/kg, there was no significant difference in the brain/plasma ratio between the two dose routes.
Table 2.
Tissue levels of RS194B at Cmax expressed as percent of administered dose based on total tissue weight ± the standard error.
| Dose (mg/kg) | Percent of administered dose | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Liver | Kidney | Heart | Lung | Spleen | Brain | |
| 10 | 8.76 ± 0.32 | 5.19 ± 0.23 | 0.05 ± 0.01 | 0.45 ± 0.04 | 0.04 ± 0.01 | 0.03 ± 0.002 |
| 50 | 8.60 ± 0.48 | 5.16 ± 0.52 | 0.06 ± 0.01 | 0.95 ± 0.12* | 0.08 ± 0.01* | 0.09 ± 0.01* |
| 100 | 8.32 ± 0.42 | 3.81 ± 0.42 | 0.05 ± 0.01 | 0.77 ± 0.04* | 0.07 ± 0.01* | 0.11 ± 0.01* |
| 200 | 5.25 ± 0.32 | 3.65 ± 0.48 | 0.08 ± 0.01* | 0.85 ± 0.04* | 0.12 ± 0.01* | 0.18 ± 0.01* |
| 50a | 7.04 ± 0.61 | 2.87 ± 0.48 | 0.07 ± 0.02 | 0.67 ± 0.16 | 0.03 ± 0.01 | 0.02 ± 0.005 |
Exposure was through IM route
Significantly higher than 10 mg/kg dose (p< 0.01)
Table 3. Brain plasma ratio over time of RS194B following a single IV or IM administration of 14C-RS194B to male guineapigsa.
| Time (hr) | 10 mg/kg | 50 mg/kg | 100 mg/kg | 200 mg/kg | 50 mg/kgb |
|---|---|---|---|---|---|
|
| |||||
| brain/ plasma | brain/ plasma | brain/ plasma | brain/ plasma | brain/ plasma | |
| 0.08 | 0.04 ± 0.001 | 0.09 ± 0.006 | 0.12 ± 0.006 | 0.18 ± 0.013 | 0.06 ± 0.002 |
| 0.25 | 0.040 ±0.002 | 0.08 ± 0.005 | 0.10 ± 0.008 | 0.19 ± 0.012 | 0.04 ± 0.006 |
| 0.5 | 0.03 ± 0.002 | 0.05 ± 0.003 | 0.12 ± 0.007 | 0.27 ± 0.080 | 0.03 ± 0.002 |
| 1 | 0.02 ± 0.001 | 0.03 ± 0.014 | 0.05 ± 0.001 | 0.15 ± 0.028 | 0.02 ± 0.010 |
| 2 | 0.03 ± 0.003 | 0.03 ± 0.002 | 0.05 ± 0.005 | 0.08 ± 0.009 | 0.02 ± 0.001 |
| 4 | 0.05 ± 0.003 | 0.07 ± 0.004 | 0.08 ± 0.005 | 0.08 ± 0.005 | 0.06 ± 0.005 |
| 8 | 0.09 ± 0.011 | 0.07 ± 0.009 | 0.08 ± 0.010 | 0.12 ± 0.010 | 0.08 ± 0.007 |
| 24 | 0.11 ± 0.035 | 0.18 ± 0.062 | 0.06 ± 0.008 | 0.22 ± 0.071 | 0.19 ± 0.045 |
Data is the mean of 6 animals per time point ± the standard error.
Exposure was through IM administration
3.3. Tissue distribution
Analysis of 14C-RS194B in tissue revealed time and dose dependent concentrations in all tissue examined (Figure 4). After IV administration, the initial biodistribution of RS194B was rapid with the tissue Cmax occurring between 5-15 min post dose for all tissues except for the liver, which occurred at 30 min post dose. The concentrations of RS194B in the tissues varied with the highest concentrations per gram of tissue occurring in the kidney and liver, followed by lung, spleen, heart and brain for all dose concentrations tested.
Figure 4.

Mean tissue concentration of RS194B over a 24 h sampling time following a single administration of 14C-RS194B to male guinea pigs. A) 10 mg/kg IV administration; B) 50 mg/kg IV administration; C) 100 mg/kg IV administration; D) 200 mg/kg IV administration; E) 50 mg/kg IM administration. Data are expressed as the mean of 6 animals ± the standard error.
When adjusted to total tissue weight, at the Cmax, the liver contained the highest proportion of the dose with a mean of 7.7% of the administered IV dose over the 20-fold dose range followed by the kidney at 4.4%. All other tissues contained less than 1% of the administered dose with the heart having the least (0.06%) (Table 2). These values were relatively consistent across the dose range tested. In the IV dosed animals, lung, spleen, and brain showed statistically significant concentration dependent increases in total tissue concentration of RS194B when expressed as percent of administered dose. As expected the levels of RS194B in the tissues from the IM exposed animals were lower compared to the IV dosed animals.
3.4. Renal clearance
Clearance of RS194B occurred primarily through the renal elimination route with the urine accounting for 49-77%, 46-68%, and 59-79% of the administered dose within the first 24 hr post dose for the animals receiving a 14C-RS194B dose of 50 mg/kg, 100 mg/kg and 200 mg/kg, respectively (Figure 5). The animals that received 10 mg/kg of RS194B excreted only 11-28% of the administered dose in the first 24 h. The low concentration of compound in the urine could be attributed to low urinary output from some of the study animals. Renal clearance was rapid with over 60% of the radioactivity being recovered in the urine in the first 4 h post dose at the three highest dose concentrations. These results indicate clearance through the urine is the primary route of elimination RS194B.
Figure 5.

Percent of administered RS194B recovered in urine over 24 h post dose following a single intravenous administration of 14C-RS194B in male guinea pigs. Data are expressed as the mean of 6 animals ± the standard error.
HPLC separation of the urine samples revealed one radioactive peak at a retention time of 4.5 min for all samples analysed. This retention time corresponded to the retention time of an authentic RS194B standard indicating that RS194B is metabolically stable over the course of the experiment
3.5 In vitro assessment of BBB permeability mechanism
In an effort to elucidate the mechanism of RS194B behavior at the BBB, a panel of in vitro assays was used to evaluate the compound. A range of factors may contribute to the relative capacity of a compound to traverse the BBB. The luminal portion of this barrier is composed of tightly apposed microvascular endothelial cells that severely restrict trans- and intercellular traffic. A given compound may employ a variety of routes to achieve traversal, for instance the compound may passively diffuse across the cellular membrane or pass via an active transport mechanism. Permeability may also be limited by efflux pumps such as p-glycoprotein (p-gp), also known as multidrug resistance protein 1 (MDR1). This transmembrane protein exhibits broad substrate specificity for transporting compounds out of the CNS.
To examine the role of such factors with respect to RS194B, three in vitro assays were performed (Figure 6). The parallel artificial membrane permeability assay (PAMPA) was employed to examine traversal across a continuous phospholipid bilayer, and the effective permeability (Peff [cm/s]) was calculated. Though RS194B demonstrated increased diffusion relative to the 2-PAM standard of care, rate of permeation was dramatically lower than the diazepam positive control and short of defined thresholds for “moderate” (2.0 × 10-6 cm/s) or “high” (4.0 × 10-6 cm/s) permeability at the BBB (Di et al.) (Figure 6A). Experiments were also performed to examine traversal across an in vitro monolayer of human cerebral microvascular endothelial cells (HCMEC/D3). As observed in the PAMPA assessment, permeability was improved over that of 2-PAM, but was lower than that of a known permeable compound (Figure 6B).
Figure 6.
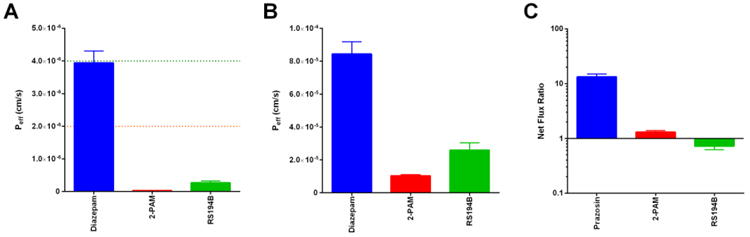
In vitro assessment of RS194B permeability. (A) The parallel artificial membrane permeability assay (PAMPA) was used to examine passive diffusion across a lipid membrane. Diazepam was employed as a positive control for permeability. A threshold for moderate permeability is shown as an orange dotted line, while high permeability is indicated by a green dotted line. (B) A monolayer of human microvascular endothelial cells (HCMEC/D3) was used to examine transport across the brain endothelium. Diazepam is again shown as a positive control for permeability. (C) MDR1-MDCK cells were used to determine the potential of a compound to be effluxed from the brain by p-gp (MDR1). Net flux ratio (NFR) greater than one indicates a potential substrate for efflux. Prazosin is shown as a positive control for efflux.
In addition to diffusion-mediated limitations, permeability may also be restricted when a compound exhibits substrate specificity for the p-gp (MDR1) efflux pump. An in vitro model of p-gp activity was used to examine RS194B using the MDR1-MDCK transgenic cell line, where a net flux ratio (NFR) greater than one (indicating disproportionately high basolateral to apical transport) suggests substrate specificity. Testing of RS194B did not result in NFR > 1, suggesting that any limitations in permeability are not due to p-gp efflux (Figure 6C).
4. Discussion
The guinea pig was used as a model to determine the PK profile, tissue distribution, metabolism, and the BBB penetrability of RS194B over a 20-fold dose range. The results showed that the PK parameters were proportional with dose and was confirmed using a power regression model (R2 = 0.9756 and 0.9885 for AUC0-t and C0, respectively). The PK parameters at all dose concentrations exhibited a multi-compartment PK profile, initially showing a rapid decline in plasma concentration post-dose followed by a slower terminal phase. The short plasma t1/2α and a large apparent volume of distribution (Vd) in all dose groups indicated rapid and extensive distribution beyond the plasma compartment. This is in agreement for what has been reported for this compound in the mouse and for an amide-oxime in the rat (Radic et al., 2012; Okolotowicz et al., 2014). The relatively lower Vd and longer t1/2α observed in the 200 mg/kg dose group from the current study indicates a higher proportion of the compound remaining in the plasma suggesting saturation of the distribution mechanisms. The mean t1/2α of 62 min from all IV dose concentrations, and 52 min for the IM dose route, was significantly longer when compared to the t1/2α of 12 min reported by Radic et al. for an IM exposure of 80 mg/kg in the mouse (Radic et al., 2012). These differences could be attributed to the difference in animal models used between the two studies, since plasma half-life of xenobiotics has been correlated to species, animal body weight, and route of administration (Ioannides et al.,1982).
RS194B was detected in all tissues examined confirming that the compound is widely distributed throughout the body. The lower levels of RS194B observed in the tissue after exposure via the IM route is consistent with the lower levels observed in the plasma, confirming the decreased bioavailability (F) after IM exposure compared to IV exposure (F=0.88). The relatively high levels found in the kidney support the conclusion that most of RS194B is eliminated through renal excretion. Analysis of the urine revealed unchanged RS194B with no metabolites or breakdown products detected indicating that it is metabolically stable.
Analysis of homogenized brain tissue revealed low RS194B levels, with a reported Cmax concentration range of 0.216 ng/mg - 26.22 ng/mg across the dose range, which amounts values of 0.03-0.18% of the administered dose. These levelsare lower than the 10% that was reported for the current standard of care, 2-PAM, from a study that used a microdialysis technique to determine BBB penetration in rats after IV exposures of 10, 50, and 100 mg/kg (Sakurada et al., 2003). The observed low levels of RS194B in the brain correlated with the in vitro results that showed a low rate of diffusion across a phospholipid bilayer (PAMPA assay) and limited traversal across a mono layer of human cerebral microvessel endothelial cells suggesting that the mechanism of entry into the brain of RS194B is limited by both passive diffusion and active transport. Even though overall penetration into the brain was quite low, a dose dependent increase in RS194B was observed. The higher brain concentration observed at the higher doses could be due to a rapid equilibrium mechanism. The Cmax for RS194B was attained within the first 5 min after dosing. During this time, the concentration gradient over the BBB could have been sufficiently large enough to drive passage into the CNS especially at the high dose levels (Ligtenstein and Kossen, 1983). The presence of a rapidly equilibrating compartment in the brain behind the BBB has been previously reported (Bradbury, 1979; Davson and Welch.1971; Sisson and Oldendorf, 1971; Reed and Woodbury, 1963). Additionally, the build-up in the brain could be the result of the saturation of efflux transporters. Evidence has suggested that certain oximes may transverse the BBB by active processes involving influx and efflux transporters (Sakurada et al., 2003). The lack of substrate specificity for the P-gp efflux pump for RS194B suggests that this mechanism is not involved, however, other transporters, cannot be discounted.
When comparing the brain/plasma ratios, for the IV exposed animals, at Cmax the ratio ranged from 0.04 to 0.18 across the dose range. This contrasts with the mouse model that reported a brain/plasma ratio of 0.3 from mice dosed with 80 mg/kg RS194B (Radic et al., 2012). The higher ratio in the mouse could be due to residual blood remaining in the brain capillaries, whereas, in the guinea pig, the tissues were perfused prior to collection to remove any blood that might interfere with the analysis. The brain/plasma ratio at Cmax of 0.058 in the current study for the IM exposure was similar to what has been reported for 2-PAM (0.056) from brain homogenates of rats after a 50 μmol IM exposure (Petroianu et al., 2007). Taken together, these results indicate that brain penetration for both RS194B and 2-PAM is poor.
The change in the brain/plasma ratio over time indicates that elimination from the brain lagged behind plasma clearance. At the later time points, the brain-to-plasma ratios were up to 2.5 times higher over the ratios at Tmax in three of the four IV doses, and 3.2 times higher in the IM exposure. These observations are similar to what was reported in the mouse (Radic et al., 2012), and for the oxime reactivator HI-6 (Asoxime chloride) (Ligtenstein and Kossen, 1983). The slower clearance of RS194B in the brain compared to the plasma suggest that a longer residence time in the brain could positively affect the therapeutic outcome of the oximes. However, given the poor BBB penetrating ability of RS194B, it remains to be established if these levels are therapeutically significant.
Overall, the results from the current study have shown that RS194B plasma PK parameters are linear with dose and follow first order kinetics with a rapid distribution phase followed by a slower elimination phase. Tissue distribution is widespread with the majority of the administered dose confined to the liver and kidneys, with no evidence of metabolic degradation over the course of the experiments. Concentrations of RS194B in the brain tissue at Cmax was found to be less than 0.2% of the administered dose demonstrating that RS194B is poor at permeating the blood brain barrier, and does not significantly accumulate in the brain in the guinea pig model as indicated by the low brain/plasma ratios. Based on these results, RS194B warrants further evaluation as a potential candidate for peripheral nervous system reactivation. However, its poor blood brain barrier permeability and low retention in the brain may limit its efficacy as a central nervous system reactivator.
Acknowledgments
This work was performed under the auspices of the U.S. DOE by LLNL under Contract DE-AC52-07NA27344 and supported by DTRA [CBS-03-2-004] and the National Institute of Health General Medical Sciences [2P41GM103483-16].
Abbreviations
- AChE
acetylcholinesterase
- AMS
accelerator mass spectrometry
- BBB
blood-brain barrier
- CNS
central nervous system
- OP
organophosphorus
References
- Bahar FG, Ohura K, Ogihara T, Imai T. Species Difference of Esterase Expression and Hydrolase Activity in Plasma. J Pharm Sci. 2012;101:3979–3988. doi: 10.1002/jps.23258. [DOI] [PubMed] [Google Scholar]
- Balani SK, Nagaraja NV, Qian MG, Costa AO, Daniels JS, Yang H, Shimoga PR, Wu JT, Gan LS, Lee FW, Miwa GT. Evaluation of microdosing to assess pharmacokinetic linearity in rats using liquid chromatography-tandem mass spectrometry. Drug Metab Dispos. 2006;34:384–388. doi: 10.1124/dmd.105.007195. [DOI] [PubMed] [Google Scholar]
- Bennion BJ, Be NA, McNerney MW, Lao V, Carlson EM, Valdez CA, Malfatti MA, Enright HA, Nguyen TH, Lightstone FC, Carpenter TS. Predicting a Drug's Membrane Permeability: A Computational Model Validated With In Vitro Permeability Assay Data. J Phys Chem B. 2017 doi: 10.1021/acs.jpcb.7b02914. Just Accepted Manuscript • Publication Date (Web): 28 Apr 2017. [DOI] [PubMed] [Google Scholar]
- Bradbury MW. The concept of a blood-brain barrier. Wiley, Chichester; England: 1979. pp. 107–108. [Google Scholar]
- Cadieux CL, Broomfield CA, Kirkpatrick MG, Kazanski ME, Lenz DE, Cerasoli DM. Comparison of human and guinea pig acetylcholinesterase sequences and rates of oxime-assisted reactivation. Chem Biol Interact. 2010;187:229–233. doi: 10.1016/j.cbi.2010.04.020. [DOI] [PubMed] [Google Scholar]
- Davson H, Welch K. The permeation of several materials into the fluids of rabbit brain. J Physiol. 1971;18:337–351. doi: 10.1113/jphysiol.1971.sp009621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability Studies on In Vitro Blood–Brain Barrier Models: Physiology, Pathology, and Pharmacology. Cellular and Molecular Neurobiology. 2005;25:59–127. doi: 10.1007/s10571-004-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMar JC, Clarkson ED, Ratcliffe RH, Campbell AJ, Thangavelu SG, Herdman CA, Leader H, Schulz SM, Marek E, Medynets MA, Ku TC, Evans SA, Khan FA, Owens RR, Nambiar MP, Gordon RK. Pro-2-PAM therapy for central and peripheral cholinesterases. Chem Biol Interact. 2010;187:191–198. doi: 10.1016/j.cbi.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood-brain barrier. Euro J Med Chem. 2003;38:223–232. doi: 10.1016/s0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- Dolgin E. Syrian gas attack reinforces need for better anti-sarin drugs. Nat Med. 2013;19:1194–1195. doi: 10.1038/nm1013-1194. [DOI] [PubMed] [Google Scholar]
- García GE, Campbell AJ, Olson J, Moorad-Doctor D, Morthole VI. Novel oximes as blood–brain barrier penetrating cholinesterase reactivators. Chem Biol Interact. 2010;187:199–206. doi: 10.1016/j.cbi.2010.02.033. [DOI] [PubMed] [Google Scholar]
- Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, de Morais SM. In Vitro P-glycoprotein Assays to Predict the in Vivo Interactions of P-glycoprotein with Drugs in the Central Nervous System. Drug Metab Dispos. 2008;36:268–275. doi: 10.1124/dmd.107.017434. [DOI] [PubMed] [Google Scholar]
- Haines DD, Fox SC. Acute and Long-Term Impact of Chemical Weapons: Lessons from the Iran-Iraq War. Forensic Sci Rev. 2014;26:97–114. [PubMed] [Google Scholar]
- Henderson PT, Pan CX. Human microdosing for the prediction of patient response. Bioanalysis. 2010;2:373–376. doi: 10.4155/bio.10.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannides C, Parke DV, Taylor IW. Elimination of glycerol trinitrate: Effects of sex, age, species and route of administration. Br J Pharmac. 1982;77:83–88. doi: 10.1111/j.1476-5381.1982.tb09272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligtenstein DA, Kossen SP. Kinetic Profile in Blood and Brain of the Cholinesterase Reactivating Oxime HI-6 after Intravenous Administration to the Rat. Toxicol Appl Parmacol. 1983;71:177–183. doi: 10.1016/0041-008x(83)90334-4. [DOI] [PubMed] [Google Scholar]
- Madeen E, Corley RA, Crowell S, Turteltaub K, Ognibene T, Malfatti M, McQuistan TJ, Garrard M, Sudakin D, Williams DE. Human in Vivo Pharmacokinetics of [(14)C]Dibenzo[def,p]chrysene by Accelerator Mass Spectrometry Following Oral Microdosing. Chem Res Toxicol. 2015;28:126–134. doi: 10.1021/tx5003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfatti MA, Lao V, Ramos CL, Ong VS, Turteltaub KW. Use of microdosing and accelerator mass spectrometry to evaluate the pharmacokinetic linearity of a novel tricyclic GyrB/ParE inhibitor in rats. Antimicrob Agents Chemother. 2014;58:6477–6483. doi: 10.1128/AAC.03300-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfatti MA, Palko HA, Kuhn EA, Turteltaub KW. Determining the Pharmacokinetics and Long-Term Biodistribution of SiO2 Nanoparticles In Vivo Using Accelerator Mass Spectrometry. Nano Lett. 2012;12:5532–5538. doi: 10.1021/nl302412f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley M. Clinical evaluation of pesticide exposure and poisonings. Lancet. 1997;349:1161–1166. doi: 10.1016/S0140-6736(96)07222-4. [DOI] [PubMed] [Google Scholar]
- Ognibene TJ, Bench G, Vogel JS, Peaslee GF, Murov S. A High-Throughput Method for the Conversion of CO2 Obtained from Biochemical Samples to Graphite in Septa-Sealed Vials for Quantification of 14C via Accelerator Mass Spectrometry. Anal Chem. 2003;75:2192–2196. doi: 10.1021/ac026334j. [DOI] [PubMed] [Google Scholar]
- Okolotowicz KJ, Dwyer M, Smith E, Cashman JR. Preclinical Studies of Noncharged Oxime Reactivators for Organophophate Exposure. J Biochem Mol Toxcicol. 2014;28:23–31. doi: 10.1002/jbt.21519. [DOI] [PubMed] [Google Scholar]
- Okumura T, Hisaoka T, Naito T, Isonuma H, Okumura S, Miura K, Maekawa H, Ishimatsu S, Takasu N, Suzuki K. Acute and chronic effects of sarin exposure from the Tokyo subway incident. Environ Toxicol Pharmacol. 2005;19:447–450. doi: 10.1016/j.etap.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Okumura T, Hisaoka T, Yamada A, Naito T, Isonuma H, Okumura S, Miura K, Sakurada M, Maekawa H, Ishimatsu S, Takasu N, Suzuki K. The Tokyo subway sarin attack—lessons learned. Toxicol Appl Pharmacol. 2005;207:471–476. doi: 10.1016/j.taap.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Okumura T, Takasu N, Ishimatsu S, Miyanoki S, Mitsuhashi A, Kumada K, Tanaka K, Hinohara S. Report on 640 victims of the Tokyo subway sarin attack. Ann Emerg Med. 1996;28:129–135. doi: 10.1016/s0196-0644(96)70052-5. [DOI] [PubMed] [Google Scholar]
- Petroianu GA, Lorke DE, Hasan MY, Adem A, Sheen R, Nurulain SM, Kalasz H. Paraoxon has only a minimal effect on pralidoxime brain concentration in rats. J Appl Toxicol. 2007;27:350–357. doi: 10.1002/jat.1213. [DOI] [PubMed] [Google Scholar]
- Radic Z, Sit RK, Garcia E, Zhang L, Berend S, Kovarik Z, Amitai G, Fokin VV, Barry Sharpless K, Taylor P. Mechanism of interaction of novel uncharged, centrally active reactivators with OP-hAChE conjugates. Chem Biol Interact. 2013;203:67–71. doi: 10.1016/j.cbi.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic Z, Sit RK, Kovarik Z, Berend S, Garcia E, Zhang L, Amitai G, Green C, Radic B, Fokin VV, Sharpless KB, Taylor P. Refinement of structural leads for centrally acting oxime reactivators of phosphylated cholinesterases. J Biol Chem. 2012;287:11798–11809. doi: 10.1074/jbc.M111.333732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed DJ, Woodbury DM. Kinetics of movement of iodide, sucrose, inuline and radio-iodinated serum albumin in the central nervous system and cerebrospinal fluid of the rat. J Physiol. 1963;169:816–850. doi: 10.1113/jphysiol.1963.sp007298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurada K, Matsubara K, Shimizu K, Shiono H, Seto Y, Tsuge K, Yoshino M, Sakai I, Mukoyama H, Takatori T. Pralidoxime Iodide (2-PAM) Penetrates across the Blood-Brain Barrier. Neurochem Res. 2003;28:1401–1407. doi: 10.1023/a:1024960819430. [DOI] [PubMed] [Google Scholar]
- Sisson WB, Oldendorf W. Brain distribution spaces of 3H-mannitol, 14C-inulin and 14C-dextrane in the rat. Amer J Physiol. 1971;221:214–217. doi: 10.1152/ajplegacy.1971.221.1.214. [DOI] [PubMed] [Google Scholar]
- Sit RK, Radić Z, Gerardi V, Zhang L, Garcia E, Katalinić M, Amitai G, Kovarik Z, Fokin VV, Sharpless KB, Taylor P. New Structural Scaffolds for Centrally Acting Oxime Reactivators of Phosphylated Cholinesterases. J Biol Chem. 2011;286:19422–19430. doi: 10.1074/jbc.M111.230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, Forgue ST. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278–1283. doi: 10.1023/a:1026451721686. [DOI] [PubMed] [Google Scholar]
- Solberg Y, Belkin M. The role of excitotoxicity in organophosphorous nerve agents central poisoning. Trends Pharmacol Sci. 1997;18:183–185. doi: 10.1016/s0165-6147(97)89540-5. [DOI] [PubMed] [Google Scholar]
- Tu AT. Aum Shinrikyo's Chemical and Biological Weapons: More Than Sarin. Forensic Sci Rev. 2014;26:115–120. [PubMed] [Google Scholar]
- Vogel JS, Turteltaub KW, Finkel R, Nelson DE. Accelerator Mass Spectrometry. Anal Chem. 1995;67:353A–359A. doi: 10.1021/ac00107a001. [DOI] [PubMed] [Google Scholar]
- Wagner CC, Simpson M, Zeitlinger M, Bauer M, Karch R, Abrahim A, Feurstein T, Schutz M, Kletter K, Muller M, Lappin G, Langer O. A combined accelerator mass spectrometry-positron emission tomography human microdose study with 14C- and 11C-labelled verapamil. Clin Pharmacokinet. 2011;50:111–120. doi: 10.2165/11537250-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]