

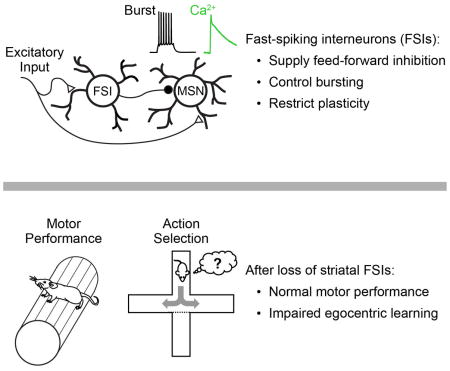
SUMMARY
Fast-spiking interneurons (FSIs) are a prominent class of forebrain GABAergic cells implicated in two seemingly independent network functions: gain control and network plasticity. Little is known, however, about how these roles interact. Here, we use a combination of cell-type-specific ablation, optogenetics, electrophysiology, imaging, and behavior to describe a unified mechanism by which striatal FSIs control burst firing, calcium influx, and synaptic plasticity in neighboring medium spiny projection neurons (MSNs). In vivo silencing of FSIs increased bursting, calcium transients, and AMPA/NMDA ratios in MSNs. In a motor sequence task, FSI silencing increased the frequency of calcium transients, but reduced the specificity with which transients aligned to individual task events. Consistent with this, ablation of FSIs disrupted the acquisition of striatum-dependent egocentric learning strategies. Together, our data support a model in which feed-forward inhibition from FSIs temporally restricts MSN bursting and calcium-dependent synaptic plasticity to facilitate striatum-dependent sequence learning.
eTOC
Fast-spiking interneurons of the striatum mediate feed-forward control of bursting, and their disruption affects learning but not performance of striatum dependent action selection.
INTRODUCTION
A complex interplay of intrinsic cellular properties, neuromodulation, and synaptic excitation and inhibition shapes neuronal activity throughout the brain (Kepecs and Fishell, 2014). Locally-projecting GABAergic interneurons are the primary source of inhibition and exhibit immense anatomical, molecular, and physiological diversity. However major questions remain unanswered regarding how this diversity translates into specialized function.
Among the most prominent interneuron types is the parvalbumin-expressing fast-spiking interneuron (FSIs), which is distributed throughout the forebrain (Kepecs and Fishell, 2014). FSIs are typically associated with feed-forward inhibition (Buzsaki and Eidelberg, 1981; Cardin et al., 2009; Cruikshank et al., 2007; Glickfeld and Scanziani, 2006; Pouille and Scanziani, 2001; Royer et al., 2012; Sohal et al., 2009), which is believed to organize local activity by sharpening spike timing (Buzsaki and Eidelberg, 1981; Cruikshank et al., 2007; Pouille and Scanziani, 2001), regulating network gain (Atallah et al., 2012; Lee et al., 2012), and expanding the dynamic range of network responses (Pouille et al., 2009). However, a separate line of research has implicated FSIs in control of network plasticity (Donato et al., 2013; Fagiolini and Hensch, 2000; Kuhlman et al., 2013; Yazaki-Sugiyama et al., 2009). The link between these apparently distinct functions of FSIs remains unclear.
The striatum, a primary input region of the basal ganglia, is critical for action selection and adaptive motor control (Yin and Knowlton, 2006). FSIs in the striatum are anatomically and physiologically similar to those in cortex, hippocampus and elsewhere, and receive potent excitatory input from primary afferent regions (Berke, 2008; Burguiere et al., 2013; Gittis et al., 2010; Koos and Tepper, 1999; Planert et al., 2010). Defects in striatal interneurons, including FSIs, have been linked to Tourette’s syndrome in human patients (Kalanithi et al., 2005; Kataoka et al., 2010), and to abnormal behaviors in animals (Gernert et al., 2000; Gittis et al., 2011; Xu et al., 2016). These findings, together with the association of FSIs with on-line network performance (Atallah et al., 2012; Burguiere et al., 2013; Cardin et al., 2009; Gage et al., 2010; Glickfeld and Scanziani, 2006; Lee et al., 2012; Pouille et al., 2009; Pouille and Scanziani, 2001; Royer et al., 2012; Sohal et al., 2009; Wilson et al., 2012), have led to a prominent hypothesis that striatal FSIs acutely shape network activity to promote appropriate neuronal ensembles and suppress undesired or conflicting motor plans (Bronfeld and Bar-Gad, 2013; Burguiere et al., 2013; Gernert et al., 2000; Gittis et al., 2011; Kalanithi et al., 2005; Kataoka et al., 2010). In this view, a loss of striatal FSIs is believed to trigger “excess” motor behavior, in the form of tics (aberrantly executed motor plans) or dyskinesias (simultaneous expression of multiple, conflicting motor plans). However, at least one recent study has called this hypothesis into question, as ablation of FSIs was not sufficient to induce dyskinesia or disrupt baseline motor behavior (Xu et al., 2016).
Here we use a combination of ex vivo and in vivo physiological recording and calcium imaging, together with extensive behavioral characterization, to develop a unified model for the role of striatal FSIs in regulating MSN activity, synaptic plasticity, and learning. We show that FSIs are the primary source of feed-forward inhibition in the striatum, and that acute optogenetic silencing of FSIs in vivo not only disinhibits MSN spiking, but also alters the pattern of MSN spiking to enhance bursting and calcium signaling throughout the network. This change in MSN spiking activity is not sufficient to disrupt motor coordination or performance of well-trained behavioral tasks: a finding that is surprising in the context of several established papers (Bronfeld and Bar-Gad, 2013; Gernert et al., 2000; Gittis et al., 2011; Kalanithi et al., 2005; Kataoka et al., 2010), yet in agreement with at least one recent report (Xu et al., 2016). Instead, we observe a specific deficit in striatum-dependent action selection learning. These results support a model in which defects in FSI function result in elevated calcium signaling and non-specific synaptic plasticity across the MSN network, effectively adding noise to striatal encoding and decreasing the efficiency of learning. Consistent with the functional importance of this physiological mechanism, we show that ablating striatal FSIs impairs the acquisition of striatum-dependent sequence learning strategies.
RESULTS
To develop a mechanistic understanding of the role of FSIs in the striatal network, we first sought to establish the contribution of FSIs to feed-forward inhibition. We stimulated cortico-striatal afferents in acute, para-horizontal slices (Smeal et al., 2007), which activated local striatal interneurons and elicited di-synaptic feed-forward inhibition onto MSNs (Fig S1a–f). To gain optogenetic control over FSIs, we injected an adeno-associated virus to drive Cre-dependent expression of the light-activated chloride pump halorhodopsin (AAV-DIO-eNpHR3.0) in dorsal striatum of PV-2A-Cre BAC transgenic mice that express Cre-recombinase in striatal FSIs under the parvalbumin promoter with high penetrance (Madisen et al., 2010) (Fig 1a, S1g). Optogenetic silencing of FSIs with eNpHR3.0 (Fig 1a–e), or pharmacological blockade of excitatory inputs onto FSIs with IEM-1460 (Gittis et al., 2011) (Fig S2a–d), selectively suppressed 70–80% of feed-forward inhibition onto MSNs of both the direct and indirect pathways (Fig S2e–l) without affecting monosynaptic excitatory transmission. In simultaneous paired recordings, we found that FSIs received stronger excitatory cortical input than neighboring MSNs (Fig S1i–l), providing a physiological mechanism for the rapid recruitment of FSIs in feed-forward inhibition. These results are consistent with the prevailing hypothesis that striatal FSIs are the predominant source of feed-forward inhibition onto MSNs and provide a mechanistic basis for further investigation into how FSIs influence neighboring MSNs and impact overall striatal network function.
Figure 1. Striatal FSIs deliver feed-forward inhibition and modulate burst firing in MSNs.

(A) Immunostaining of parvalbumin-expressing interneurons in striatum of PV-2A-cre mice injected with AAV-DIO-eNpHR3.0. (B) Exemplar current clamp recording from eNpHR3.0-expressing FSI confirms fast-spiking physiology in response to current step injections. Light-activation of eNpHR3.0 (amber) confirms silencing of spiking. (C) Recording configuration for optogenetic suppression of FSIs in feed-forward inhibition. (D,E) Voltage-clamp recording from MSNs isolates monosynaptic EPSC (black) and di-synaptic feed-forward IPSC (red) at different holding potentials. Optogenetically silencing FSIs defines key role in feed-forward inhibition. (N=6 mice, n=14 MSNs; paired two-tailed t-test. P>0.1 EPSC. P<0.001 IPSC). (F) Recording configuration for freely-moving in vivo electrophysiology. (G) Average spike waveforms from exemplar FSI and MSN. (H) Classification of cell types based on waveform and firing rate (for panels H-M, N=4 PV-eNpHR3.0 mice, n=64 MSNs, n=16 FSIs; N=2 control mice, n=25 MSNs, n=6 FSIs). (I) Raster and PSTH for exemplar FSI suppressed by eNPHR3.0. (J) Average firing rates of all putative FSIs and MSNs. Gray shading indicates mean +/− s.e.m. Statistical tests for group data summarized in Fig 2F. (K) Spike rate variability (Fano Factor, σ2/μ), is increased by FSI silencing (sign rank test P>0.8 pre-vs-light and P>0.7 light-vs-post for control; P<10−3 pre-vs-light and P<10−4 light-vs-post for PV-eNpHR3.0). (L) Raster and PSTH for individual spikes from exemplar MSN disinhibited by optogenetic suppression of FSIs. (M) Raster and PSTH for bursts detected in same MSN depicted in Panel L reveals greater disinhibition of spike bursts than individual spike rates. Bursts are ≥8 spikes at 20 Hz. (N) Comparison of spiking and bursting (N=5 PV-eNpHR3.0 mice and n=34 MSNs; N=2 mCherry control mice and n=10 MSNs; sign rank test P>0.05 for control; P<10−3 for PV-eNpHR3.0 mice). All error bars are s.e.m. and center values are group means.
To examine the effect of FSIs on MSN spiking in vivo, we injected PV-2A-Cre mice with AAV-DIO-eNpHR3.0 and implanted chronic, 32-channel microwire recording arrays with optical fibers into dorsal striatum (Fig 1f). Whole-cell recordings in acute slices have previously established that striatal FSIs form large-conductance GABAergic connections onto neighboring MSNs with a high connection probability (Gittis et al., 2010; Koos and Tepper, 1999; Straub et al., 2016), but several detailed recording studies have failed to find evidence for this inhibition in vivo (Bakhurin et al., 2016; Burguiere et al., 2013; Gage et al., 2010; Lee et al., 2017). Using standard waveform and firing rate criteria (Berke, 2008; Burguiere et al., 2013; Gage et al., 2010; Gittis et al., 2011), we were able to differentiate putative FSIs from MSNs (Fig 1g,h). In response to light delivery (3 mW, 532nm, 1 sec pulses), the firing rate of most putative FSIs was rapidly suppressed in PV-eNpHR3.0 mice, but unaffected in mice that received mCherry control virus (Fig 1i, S3a–c). In contrast, neighboring MSNs significantly increased their firing rate, consistent with a net disinhibition of the striatal network during FSI silencing (Fig 1j,l, S3a–c). These results, obtained from mice expressing eNpHR3.0 with high selectivity and penetrance in parvalbumin-expressing striatal neurons (Fig S1g,h), help clarify a fundamental controversy in the field, by clearly indicating that FSIs inhibit MSNs under normal physiological conditions in vivo as well as in acute slice preparations.
In addition to its canonical role in controlling spike timing (Pouille and Scanziani, 2001), feed-forward inhibition can regulate gain (Atallah et al., 2012; Pouille et al., 2009; Royer et al., 2012; Wilson et al., 2012), bursting (Blitz and Regehr, 2005; Pouille et al., 2009; Pouille and Scanziani, 2001; Verret et al., 2012), and dynamic range (Atallah et al., 2012; Pouille et al., 2009) of network responses to afferent input. We therefore hypothesized that loss of FSIs may affect the pattern as well as the frequency of MSN spiking activity. To examine spike rate variability, we calculated the Fano Factor (σ2/μ – a measure of variability in spiking activity that is independent of overall changes in firing rate). This analysis revealed that the disinhibition of MSNs was accompanied by an increase in spike rate variability (Fig 1k). We next tested whether this variability was associated with an increase in bursting. We detected and analyzed bursts in single unit in vivo recordings from MSNs using frequency and duration criteria that, according to our own measurements (Fig S4m) and previous published reports (Kerr and Plenz, 2002), are predicted to drive somatic and dendritic calcium transients (at least 8 spikes at a firing rate of at least 20 Hz). We found that these bursts were more strongly modulated than individual MSN spikes (Fig 1l–n).
Bursts of action potentials are tightly correlated with calcium influx in MSNs (Kerr and Plenz, 2002). The increase in MSN bursting during optogenetic silencing of FSIs therefore suggested that FSIs might regulate calcium signaling in neighboring MSNs. We therefore used a head-mounted microendoscope-based miniature microscope capable of simultaneous calcium imaging and optogenetic control in deep brain structures of freely-moving mice (Fig 2a,b). In agreement with prior studies in striatum (Barbera et al., 2016) and other brain regions (Jennings et al., 2015; Ziv et al., 2013), we observed sparsely-occurring calcium transients in striatal MSNs (e.g. Fig 2c). Optogenetic silencing of FSIs with eNpHR3.0 elevated the rate of MSN calcium transients nearly 7-fold in the open field (Fig 2c–g, S4c). This increase was not observed in mice that received the control virus AAV-DIO-mCherry (Fig 2e,g, S3c). Notably, the increase in calcium transients during FSI silencing (Modulation Index = 0.7647) was significantly larger than the increase in overall spiking rate (Modulation Index = 0.6292) (Fig 2f, S3c, rank sum test P<10−3), suggesting that control over MSN calcium signaling may be a primary role for FSIs. In addition to this substantial increase in calcium transient frequency, we also observed a modest increase in calcium transient amplitude (Fig 2h, sign rank test P<10−9).
Figure 2. Striatal FSIs control MSN calcium transient rates.

(A) Recording configuration and (B) schematic of light path for head-mounted microscope enabling imaging with simultaneous optogenetic suppression of FSIs in vivo. (C) Calcium signals recorded in an open field. Sweeps represent successive light pulses from one exemplar MSN. (D) Raster and PSTH of calcium transients detected in the MSN recording depicted in Panel C. (E) Cell fields for control (left) and PV-eNpHR3.0 (right) mice, color-coded to reveal optogenetically driven change in calcium transient rates. (F) Electrophysiological spikes and calcium transients are both disinhibited by PV-eNpHR3.0 silencing of FSIs. (N=5 PV-eNpHR3.0 mice, n=34 MSNs for electrophysiology, sign rank test P<10−4 Pre-Light, P<10−6 Post-Light; N=4 PV-eNpHR3.0 mice, n=235 neurons for calcium transients, sign rank test P<10−33 Pre-Light, P<10−36 Post-light). Comparison of spiking and calcium transients reveals greater disinhibition of calcium, consistent with a preferential role for FSIs in controlling MSN calcium signaling (rank sum test, P<0.01). (G) Modulation index (Light On)/(Light On + Light Off) for all neurons shows disinhibition of calcium transients during eNpHR3.0 suppression of FSIs, but not in control animals. (N=4 control mice, n=400 neurons, sign rank test against 0.5, P>0.1; N=4 test mice, n=235 neurons, sign rank test against 0.5, P<10−32). (H) Calcium transient peak amplitudes are increased during FSI silencing (N=4 control mice, n=120 neurons, P>0.2 N=4 PV-eNpHR3.0 mice, n=212 neurons, P<10−9). Average transient from exemplar neuron inset with Light Off (black) and Light On (Amber). All error bars are s.e.m. and center values are group means.
The FSI modulation of MSN calcium transients could arise from FSI control over MSN spiking (Fig 1k–n), from direct control over MSN membrane potential and voltage-gated calcium channels (Carter and Sabatini, 2004), or from a combination of both. To examine this, we prepared acute slices from adult mouse brains and obtained whole-cell current-clamp recordings from MSNs with the calcium-sensitive dye Oregon-Green BAPTA-6F (OGB-6F) included in the recording pipette (Fig S4a). Depolarizing current injections elicited action potentials in the MSN and concomitant elevations of the somatic calcium signal (Fig S4b), which scaled linearly with the number of action potentials in a burst (Fig S4c). We hypothesized that a reduction in inhibitory tone during FSI silencing could depolarize MSNs, and that this change in resting potential could alter the sources of calcium during subsequent action potentials (Carter and Sabatini, 2004). However, changing membrane potential with a sub-threshold current injection through the recording pipette (resting Vm = −86.5 +/− 1.9 mV; depolarized Vm = −64.9 +/− 1.0 mV) did not alter the relationship between spiking and calcium influx (Fig S4d–f). We next tested the impact of directly activating FSIs using the red-shifted opsin ChrimsonR (Klapoetke et al., 2014). Optogenetic activation of FSIs reduced the number of MSN action potentials by ~50% (Fig S4g–k) but did not affect the relationship between MSN spiking and calcium influx (Fig S4l). Overall, these data suggest that the change in firing pattern and increase in burstiness of MSNs during silencing or loss of striatal FSIs may account for the preferential increase in calcium transients. To test this, we inhibited FSIs for more sustained periods using 30-sec light pulses in a subset of in vivo physiology recordings. We simulated calcium signals from individual MSNs based on single-unit recordings during optogenetic silencing of FSIs using the decay constant of GCaMP6f recorded in acute slices (0.812 sec) and an approximation of the supralinear spike-to-calcium transformation measured for GCaMP6f (Akerboom et al., 2012; Chen et al., 2013) (Fig S4m). This revealed an increase in simulated calcium transients that exceeded the disinhibition in MSN spiking, consistent with our recording data (Fig S3d–k).
To examine the behavioral consequences of these changes in striatal network activity, we first observed mice moving around an open-field arena. Previous studies in both animals (Burguiere et al., 2013; Gernert et al., 2000; Gittis et al., 2011; Xu et al., 2016) and humans (Kalanithi et al., 2005; Kataoka et al., 2010) suggest that dysfunction of striatal FSIs results in abnormal involuntary movements. Surprisingly, in spite of the pronounced physiological changes in MSN spiking activity and calcium signaling (Figs 1,2), we observed no clear acute behavioral changes during either unilateral (Fig 3a,b) or bilateral (Fig 3c) optogenetic silencing of FSIs with eNpHR3.0. To ensure that this negative result did not arise from technical shortcomings, such as inadequate silencing of FSIs, we adopted a less ambiguous genetically-targeted lesioning approach by injecting an adeno-associated virus to drive Cre-dependent expression of caspase-3 (AAV-DIO-Casp3) in dorsal striatum PV-2A-Cre mice (Fig 3d–g, S1g,h) Unexpectedly, and in contrast to the prevailing hypotheses in the field (Gernert et al., 2000; Gittis et al., 2011; Kalanithi et al., 2005; Kataoka et al., 2010) (but see (Rapanelli et al., 2017; Xu et al., 2016)), selectively ablating striatal FSIs did not cause any apparent abnormal involuntary movements or dyskinesias, and open-field locomotion was unimpaired (Fig 3h). When tested more stringently with a series of standard motor performance assays including the balance beam (Fig 3i), rotarod (Fig 3j), and pole test (Fig 3k), FSI-lesioned mice displayed no motor deficits, indicating that a near-complete loss of striatal FSIs is not sufficient to produce movement disorders or impair online motor performance.
Figure 3. Neither acute nor chronic disruption of striatal FSIs affects motor performance.

(A) Configuration for optogenetic suppression of FSIs in vivo. (B,C) Optogenetic suppression of FSIs does not acutely affect rotations (N=9 mice per group; sign rank test P>0.1) or locomotion (N=9 mice per group, unpaired t-test P>0.5) in an open field. (D) Configuration for ablation of FSIs. (E,F) Immunostaining for parvalbumin (white) in coronal hemi-sections to quantify ablation of striatal FSIs by injection of AAV-DIO-Casp3 virus into dorsal striatum of PV-cre mice. White box in left panel is expanded in right panel. (G) Quantification of parvalbumin-expressing FSIs in dorsal striatum from control and FSI-lesioned mice (N=12 mice per group; rank sum test P<10−4). (H) Normal open field locomotion (N=10 mice per group; unpaired two-tailed t-test P>0.2), (I) normal balance beam performance (N=8 mice per group; unpaired two-tailed t-test P>0.7), and (J) unimpaired performance throughout learning on the rotarod in FSI-lesioned mice. Lighter lines are individual animals, darker lines are group averages. (N=12 mice per group, 2-way ANOVA PTrial<10−10, PGroup>0.5, PInteract>0.5). (K) Normal pole test time to descend in FSI-lesioned mice (N=8 mice per group; unpaired two-tailed t-test P>0.4). All error bars are s.e.m. and center values are group means.
The most direct evidence linking striatal FSI dysfunction to acute motor impairments was a recent study in which acute intrastriatal injection of the drug IEM-1460 (a blocker of calcium-permeable AMPA receptors) was found to preferentially suppress FSIs and induce acute dyskinesias in mice (Gittis et al., 2011). We therefore repeated this experiment in FSI-lesioned animals, using an intermediate dosage of IEM-1460, which efficiently drives axial twisting dyskinesias that can be quantified by counting of body rotations. Intrastriatal injection of IEM-1460 into striatal FSI-lesioned mice drove axial twisting and rotations that were indistinguishable from those induced in control animals (Fig S5a), demonstrating that IEM-1460 likely acts on additional striatal or extra-striatal circuit elements and that silencing of striatal FSIs by this drug is not sufficient to drive this behavioral effect.
The FSI modulation of MSN burst firing (Fig 1k–n) and calcium signaling (Fig 2) suggested that these interneurons may be primarily important for regulating plasticity and learning. To determine whether the disinhibitory effect on MSN calcium transients persisted in chronically FSI-lesioned mice, we used the head-mounted microendoscope-based miniature microscope to measure MSN calcium transients in untrained, freely moving mice. Consistent with acute optogenetic silencing of FSIs (Fig 2), FSI-lesioned mice exhibited a higher rate of MSN calcium transients than littermate controls (Fig 4a,b). Calcium signaling is a potent driver of synaptic plasticity in MSNs (Charpier and Deniau, 1997; Shen et al., 2008), and calcium-dependent signaling pathways and synaptic plasticity are integral for learning and memory throughout the brain. In particular, high levels of calcium are associated with the induction of LTP (Charpier and Deniau, 1997; Malenka et al., 1988; Shen et al., 2008). We therefore hypothesized that the elevated calcium transient rates in MSNs during acute (Fig 2) or chronic (Fig 4a,b) manipulation of striatal FSIs could dysregulate LTP induction in MSNs and interfere with plasticity mechanisms underlying learning. To test for changes in synaptic strength, we prepared acute slices 2–3 weeks after injection of the AAV-DIO-Casp3 virus and measured AMPA/NMDA ratios in MSNs from FSI-lesioned mice. The AMPA/NMDA ratio was significantly elevated in FSI-lesioned mice relative to littermate controls (Fig 4c–e), consistent with an increase in long-term potentiation (LTP) in MSNs. These experiments were performed in D1-tdTomato reporter mice, to fluorescently label direct pathway MSNs. Although the increase in AMPA/NMDA ratios appeared to be driven primarily by an increase in synaptic strength onto indirect pathway MSNs, this trend did not reach statistical significance (Fig S5b). These results provide direct evidence that ablation of striatal FSIs drives apparently non-specific synaptic plasticity throughout the striatal network. We therefore set out to test whether this intervention affects striatum-dependent learning.
Figure 4. FSI ablation causes increased calcium transient rate and elevated AMPA/NMDA ratios in neighboring MSNs.
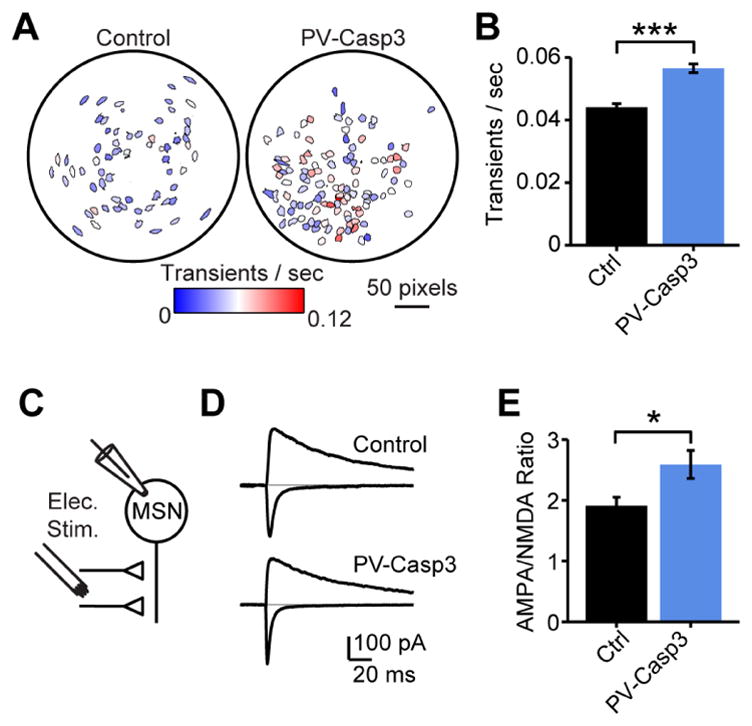
(A) Exemplar cell fields for in vivo calcium imaging from dorsal striatum of control and FSI-lesioned mice, colored by calcium transient rate in an open field. (B) Elevated rate of calcium transients in FSI-lesioned mice (N=4 control mice, n=164 neurons; N=3 FSI-lesioned mice, n=162 neurons; rank sum test P<10−11). (C) Acute slice recording configuration for AMPA/NMDA ratios. (D) Exemplar AMPA/NMDA currents at −75 mV and +40 mV from control and FSI-lesioned mice. (E) Group data for AMPA/NMDA ratio measurements from MSNs in control (N=5 mice, n=21 neurons) and FSI-lesioned (N=4 mice, n=20 neurons) mice showing altered plasticity after FSI lesions (rank sum test P<0.02).
We trained mice to perform a 3-nosepoke sequence task for liquid sucrose rewards (Fig 5a). Recent reports indicate that the striatum is an essential site of plasticity in a similar sequence task (Rothwell et al., 2015; Tecuapetla et al., 2016) and that optogenetic manipulation of striatal MSNs acutely alters this behavior (Tecuapetla et al., 2016). Consistent with our hypothesis that FSIs facilitate striatum-dependent learning of action selection strategies, FSI-lesioned mice made an increased number of choice errors early, but not late in training (Fig 5b,c, S5e–g, S6), whereas motor performance (Fig 5d) and motivation (Fig S5c,d) were unaffected. Bilateral optogenetic silencing of FSIs in expert mice did not impair execution (Fig 5e,f), accuracy (Fig 5f), or motivation (Fig 5h). Bilateral silencing early in training was attempted, but naïve tethered mice performed trials too slowly and irregularly to support meaningful assessment of acute FSI suppression on behavioral performance early in learning. The increased number of errors early, but not late in training (Fig 5b,c), and the lack of impairment during acute optogenetic silencing of FSIs in trained mice (Fig 5f), points to a specific impairment in learning, rather than performance of this striatum-dependent task.
Figure 5. Loss of striatal FSIs impairs sequence learning in naive animals but does not affect performance in expert mice.

(A) Task design for 3-port sequence task. (B,C) Ablating striatal FSIs with AAV-DIO-Casp3 reduces accuracy early but not late in learning (N=11 mice per group; two-way ANOVA PGroup<10−5; unpaired two-tailed t-test P<0.05 early; P>0.05 late). (D) FSI ablation does not affect motor performance (N=11 mice per group; two-way ANOVA PGroup>0.05). (E) Mice were pre-trained untethered for at least 5 days, followed by 1–2 days of habituation to optical fibers before experiencing two laser stimulation sessions. (F) Optogenetically suppressing FSIs on interleaved trials in expert mice does not affect error rate (paired two-tailed t-test P>0.05), (G) efficiency (sign rank test P>0.05) or (H) motivation in sequence task (paired two-tailed t-test P>0.05; N=9 mice per group all conditions). Optogenetic silencing for full trial duration (see methods). All error bars are s.e.m. and center values are group means.
Learning of complex motor tasks relies on the progressive refinement of neuronal ensembles aligned to specific task events in striatum (Barnes et al., 2005) and motor cortex (Peters et al., 2014). If disrupting FSI activity alters the specificity with which calcium transients signal specific task events, it would provide a potential explanation for the slower learning rate in FSI-lesioned animals. Using the head-mounted microendoscope-based miniature microscope, we found that many MSNs had calcium transients aligned to specific behavioral events during the 3-port sequence task (e.g. Fig 6a,b). Different MSNs responded to separate events throughout the task (Fig 6d), and calcium transients were primarily aligned to contralateral movements (Fig 6e). It has recently been reported that spatially clustered MSNs signal similar behavioral events (Barbera et al., 2016; Klaus et al., 2017). However, we found no evidence for this type of clustering (Fig 6f,g). While most MSNs maintained their event-aligned preference during FSI silencing (Fig 6h,I, S7i), we observed an increase in overall calcium transient rates together with a decrease in the selectivity with which calcium transients in individual MSNs encoded particular task events (Fig 6c,h–k). The proportional increase in overall calcium transients (Fig 6c) exceeded the proportional increase in peak rate of event-aligned calcium transients in significantly event-aligned neurons (Fig 6j), indicating that the net effect of FSI silencing was a drop in the task event-specificity of MSN calcium signaling. A direct comparison of event-aligned vs. overall calcium transient modulation for each significantly event-aligned neuron revealed that, for most neurons, the degree of modulation for event-aligned transients was less than that of overall, non-aligned transients (Fig 6k). An ROC analysis confirmed this observation by revealing reduced discriminability of event-aligned calcium transient rate from baseline after FSI silencing (Fig S7). Together, these results demonstrate that FSIs regulate MSN calcium transients in the context of striatum-dependent behaviors, and that their influence sharpens the relationship between those transients and specific task events. Consequently, impairments in FSI signaling reduce the efficiency with which calcium-dependent plasticity in MSNs can drive experience-dependent action selection learning in the striatal network.
Figure 6. FSIs shape MSN ensemble calcium signaling during sequence task.

(A,B) Calcium imaging from exemplar MSN during sequence task reveals task event-aligned calcium transients. (C) Optogenetically silencing FSIs increases overall calcium transient rate in significantly event-aligned neurons (N=4 mice per group; n=100 control and n=119 PV-eNpHR3.0 neurons; sign rank test P>0.05 for control and P<10−17 for PV-eNpHR3.0). (D) Fraction of MSNs responding to each task event, grouped by direction of initial movement relative to the recording site. (N=4 mice per group; n=36–116 neurons per mouse). (E) Same data as Panel D, grouped by movement shows most MSNs align to contralateral movement (sign rank test P<0.02). (F) Exemplar cell field colored by task-event preference. (G) Average spatial distance between cell pairs, grouped by whether neither neuron aligned to any event (No Resp.), only one aligned (One Resp.), both aligned to different events (Two Diff.), or both aligned to the same event (Two Same). (N=8 mice; n=210–4005 cell pairs per mouse, 2-way ANOVA Pmouse<0.02, Presp>0.8). (H,I) Recordings from two exemplar neurons reveal modulation of event-aligned transients during FSI silencing. (J) Optogenetically silencing FSIs increases event-aligned calcium transient rate in significantly event-aligned neurons (N=4 mice per group; n=100 control and n=119 PV-eNpHR3.0 neurons; sign rank test P>0.8 for control and P<10−14 for PV-eNpHR3.0). (K) Silencing FSIs drives greater increase in overall calcium transient rate than event-aligned transient rate (N=4 mice per group; n=119 PV-eNpHR3.0 neurons; sign rank test to compare the ratio (Event-Aligned Modulation)/(Overall Modulation) for each neuron against the null value of 1, P<0.01). All error bars are s.e.m. and center values are group means.
The learning deficits in FSI-lesioned mice (Fig 5b,c) are consistent with two non-exclusive possibilities: a partial loss of striatum-dependent learning that is overcome with prolonged training, or a more severe deficit in striatum-dependent learning that is circumvented by employing an alternate striatum-independent strategy. For example, striatal lesions are known to suppress egocentric choices (selecting actions relative to body position), while hippocampal lesions suppress allocentric (spatially-based) choices (Packard, 1999; Packard and McGaugh, 1996). We therefore designed a novel sequence task in which the start position of the sequence was cued by illuminating one of five ports. Initiation was usually (90% of trials) on one side of the box (“training”; Fig 7a–d), and port entry triggered illumination of the two neighboring ports. Mice learned by exploration that only proceeding to the center port triggered reward delivery (Fig 7a–d). Mice were occasionally (10% of trials) instructed to initiate from a different port (“probe”; Fig 7c). An allocentric strategy would guide mice to the same location in space (center port) on probe trials (Fig 7c, top), while an egocentric strategy would instruct the equivalent body movement (e.g. “turn right”), taking mice to the outside port (Fig 7c, bottom). Control mice predominantly adopted an egocentric strategy but FSI-lesioned mice were significantly less likely to do so (Fig 7c), consistent with a deficit in egocentric learning in FSI-lesioned mice.
Figure 7. Loss of FSIs impairs striatum-dependent egocentric learning.

(A,B) FSI-lesions do not impair accuracy on training trials in egocentric-allocentric operant task (N=8 mice per group; two-way ANOVA PGroup >0.9). (C,D) Probe trials initiated at different nosepokes reveal change in task strategy (N=8 mice per group; unpaired two-tailed t-test P<0.01). (E,F) FSI-lesioned mice perform equivalently at locating hidden platform in water T-maze (N=8 mice per group; two-way ANOVA PGroup>0.3). Arrows indicate sessions containing probe trials. (G,H) Probe trials initiated on opposite end of the maze reveal decreased utilization of egocentric strategy (N=8 mice per group; rank sum test P<0.002). All error bars are s.e.m. and center values are group means.
We next tested the generality of this egocentric learning deficit using a classical cross-maze test (Fig 7e–h). Mice started in the same location on each training trial and readily learned to identify the platform location (Fig 7e). After initial training, probe trials were introduced on a subset of sessions. In probe trials, mice started in the opposite arm and the initial starting arm was blocked off (Fig 7g). Importantly, transient pharmacological silencing (Packard and McGaugh, 1996) or activation (Packard, 1999) of either striatum or hippocampus reversibly alters task strategy in a manner consistent with the lesion data, demonstrating that on-line striatal activity is a critical determinant of task strategy. Control mice almost exclusively adopted an egocentric strategy, whereas FSI-lesioned mice were significantly less likely to do so (Fig 7h), pointing towards a general deficit in striatum-dependent egocentric learning in FSI-lesioned mice. Notably, the egocentric learning deficit was present in both the cross-maze assay (which is inherently stressful) and the novel operant task (which is largely stress-free), suggesting that this deficit is not related to stress or anxiety (Xu et al., 2016).
To test whether FSI-lesioned mice are simply unable to express an egocentric strategy, or whether they are delayed in learning, we extended the task by two days in a subset of mice. Because all other aspects of striatal circuitry, including MSN plasticity mechanisms, are unaltered in these mice, and the task-event related activity is intact but diluted by excess, aberrant calcium events (Fig 6), we hypothesized that the FSI-lesioned mice would eventually be able to adopt an egocentric strategy. Indeed, over three days of training, the FSI-lesioned mice gradually adopted more egocentric strategies in this task, even though this strategy was not being explicitly reinforced (data not shown, ANOVA Pday < 0.01). Our results are therefore consistent with FSIs enhancing the efficiency of striatum-dependent action selection learning.
DISCUSSION
We establish a mechanistic foundation for how FSIs control striatal network activity and motor behavior. Taken together, our results describe a unified model for the function of striatal FSIs from physiology to behavior, in which feed-forward inhibition regulates MSN bursting, calcium and plasticity to facilitate learning. Furthermore, unlike cortex and hippocampus where FSIs deliver feed-back as well as feed-forward inhibition, striatal FSIs only deliver feed-forward inhibition, providing unique insight into its importance that would not be accessible in other brain structures.
Circuit basis of feed-forward inhibition
Beyond control over spike timing (Pouille and Scanziani, 2001), previous studies have reported that FSI-mediated feed-forward inhibition linearly scales network responses and expands their dynamic range (Atallah et al., 2012; Lee et al., 2012; Pouille et al., 2009; Wilson et al., 2012), while neuromodulation of feed-forward inhibition regulates the signal-to-noise of synaptic (Kuo and Trussell, 2011) or spike transmission (Owen et al., 2013). Each of these mechanisms may be important for shaping network representations of behavioral events and controlling ensemble calcium signaling during learning (Agetsuma, 2017; Bissiere et al., 2003).
In canonical Hebbian plasticity, pairing of afferent input and postsynaptic spiking is expected to potentiate synaptic strength, most often through calcium-dependent synaptic plasticity (Malenka et al., 1988). We propose that loss of feed-forward inhibition following suppression or loss of striatal FSIs removes an essential brake on postsynaptic responses, which increases postsynaptic bursting and drives excess calcium influx during excitatory afferent input. This in turn drives aberrant and non-specific induction of synaptic plasticity throughout the network (Fig 4c–e) that can mask or interfere with corticostriatal LTP, which is implicated in similar sequence learning assays (Rothwell et al., 2015). At a finer level, local calcium signaling within individual spines or dendrites is known to induce focal synaptic plasticity, whose specificity is believed to be computationally important (Bloodgood and Sabatini, 2005; Harvey and Svoboda, 2007). The neuron-wide calcium transients recorded here likely arise from back-propagating bursts of action potentials that invade all spines and dendrites (Carter and Sabatini, 2004). Excess engagement of this neuron-wide calcium signaling may disrupt dendrite- and spine-specific plasticity traces and impair information storage at the level of individual neurons, as well as in the context of ensembles and networks of neurons.
Qualitatively similar physiological results have been reported previously in sensory cortical regions, where silencing FSIs broadened the tuning and reduced the specificity of pyramidal neuron responses to visual stimuli (Atallah et al., 2012; Lee et al., 2012; Wilson et al., 2012). However, in each of those cases the tuning was explicitly linked to on-line circuit and behavioral performance. In contrast, we find no immediate effect on behavioral performance and instead propose that the primary consequence of interfering with striatal FSIs is to reduce the specificity of calcium signaling and calcium-dependent plasticity in MSNs, which in turn impairs specific forms of striatum-dependent learning.
Disrupting FSIs affects learning but not performance
Our finding that lesioning striatal FSIs impairs acquisition, but not execution, of striatum-dependent action selection strategies is perhaps the most surprising result. This dichotomy raises an apparent paradox: if precisely organized MSN activity is required to drive efficient learning, then how is it possible that disrupting expression of that activity pattern by acutely and transiently suppressing FSIs (Fig 5) does not acutely impair choice accuracy in well-trained mice? Our results are consistent with two, mutually compatible possibilities. First, loss of FSIs may slow learning through accumulation of aberrant synaptic plasticity in the striatal network over many hundreds of trials without disrupting overall network activity enough to impair behavioral performance on any one trial. Second, reduced specificity of task-event encoding may have an out-sized effect on behavior early in learning, when the mouse is less certain which action to select, whereas later in training redundant encoding mechanisms may be sufficient to absorb a noisier or less specific signal task representation with minimal loss of behavioral performance. Consistent with both of these possibilities, our calcium imaging results show that overall task event-related activity is retained in the absence of FSIs, even though the specificity of calcium signaling is reduced in many individual MSNs (Fig 6, S7).
Studies in both striatum (Barnes et al., 2005) and motor cortex (Huber et al., 2012; Peters et al., 2014) highlight how network responses that are broadly tuned in naïve animals become more focused and reproducible with extensive training and suggest that this refinement may depend on local inhibitory microcircuitry (Chen et al., 2015). Furthermore, in other systems including ocular dominance plasticity in cortex (Fagiolini and Hensch, 2000; Kuhlman et al., 2013; Yazaki-Sugiyama et al., 2009) and experience-dependent plasticity in hippocampus (Donato et al., 2013), parvalbumin-expressing FSIs regulate network plasticity and learning. Our results support the hypothesis that FSIs facilitate learning by restricting bursting, calcium influx, and synaptic plasticity to appropriate temporal windows and task-relevant ensembles of neurons throughout the network.
FSIs inhibit MSNs in vivo
A recent study using optogenetic silencing of FSIs reported a surprising and counter-intuitive suppression of neighboring MSN firing – the opposite of the expected disinhibition (Lee et al., 2017). This study suggested that the suppression of MSN firing arises through disinhibition of other interneurons (Lee et al., 2017). However, because the penetrance of opsin expression was <40% in the PV-Cre mouse line used for that study, unlabeled FSIs might also have contributed to this effect. In contrast, the PV-2A-cre mouse line that we use here has high penetrance and selectivity (Fig S1g,h), permitting efficient silencing across the FSI population and revealing predominant disinhibition of neighboring MSNs at modest light power (~3 mW per hemisphere). These results give us confidence that the disinhibition of MSNs that we observe upon FSI silencing is a true demonstration of FSI-MSN inhibition in vivo. Importantly, in spite of the apparent qualitative similarity between our behavioral results and those results reported by Lee et al. (Lee et al., 2017), their results arise from silencing of striatum, rather than cell-type-specific effects of FSIs on striatal processing.
In contrast, the lack of apparent monosynaptic inhibition in cross-correlograms between simultaneously recorded FSI-MSN pairs in vivo has been described by multiple labs and is unlikely due to variability in experimental preparations. This raises the question of how optogenetic silencing of FSIs can cause robust disinhibition of MSNs (Figs 1, 2, S3a–c) if individual FSIs do not apparently suppress MSNs in vivo? The most straightforward explanation is that in MSNs, which have a notoriously low membrane resistance (Nisenbaum et al., 1994), more than one FSI may be required to detectably suppress MSN firing.
FSIs in striatum-dependent movement disorders
The most widespread and popular hypothesis regarding the role of striatal FSIs holds that loss (Gernert et al., 2000; Kalanithi et al., 2005; Kataoka et al., 2010) or suppression (Bronfeld and Bar-Gad, 2013; Gittis et al., 2011) of FSIs results in aberrant involuntary movements, tics, or dyskinesias. This attractive hypothesis is based on a simple physiological premise that FSIs are positioned to gate striatal responses to afferent input and therefore might be expected to suppress unwanted activity and involuntary movements. The physiological and behavioral evidence in support of this hypothesis, however, is largely circumstantial, and at least one recent study in which FSIs were lesioned using a genetic targeting strategy failed to uncover clear evidence for acute motor deficits (Xu et al., 2016). One recent study reports that co-ablation of FSIs and cholinergic interneurons is sufficient to drive spontaneous, stereotyped movements, while ablation of FSIs alone is not (Rapanelli et al., 2017). It therefore appears likely that dyskinesias and abnormal involuntary movements could arise from a combined deficit on fast-spiking and cholinergic interneuron signaling, while the physiological and behavioral deficits arising from loss of FSIs alone are much more specific. Indeed, the behavioral impairments we observe in FSI-lesioned mice appear to be remarkably restricted to learning of striatum-dependent action selection strategies. Our physiological results, including increased MSN spike bursting and calcium signaling, as well as elevated AMPA/NMDA ratios in MSNs and decreased specificity of task-event-related calcium signaling are all consistent with a primary role for FSIs in shaping ensemble responses and plasticity to improve the efficiency of striatum-dependent learning. Taken together, our results raise the possibility that basal ganglia diseases linked to interneuron dysfunction may primarily arise from aberrant engagement of network plasticity.
Data Availability
All data and software are available upon request.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| chicken anti-YFP | Aves Labs Inc. | GFP-1020 |
| rabbit anti-parvalbumin | Swant Inc. | PV 27 |
| donkey anti-chicken 488 | Jackson Immunoresearch | 703-545-155 |
| donkey anti-rabbit 647 | Invitrogen | A-31573 |
| Bacterial and Virus Strains | ||
| AAV5-EF1α-Flex-taCasp3-TEVp | UNC Vector Core | N/A |
| AAV5-EF1α-DIO-eYFP | UPenn Vector Core | N/A |
| AAV8-hSyn-eGFP | UNC Vector Core | N/A |
| AAV5-EF1α-DIO-eNpHR3.0-YFP | UNC Vector Core | N/A |
| AAV5-EF1α-DIO-eNpHR3.0-mCherry | UNC Vector Core | N/A |
| AAV5-Syn-GCaMP6f | UPenn Vector Core | N/A |
| AAV5-EF1α-DIO-mCherry | UNC Vector Core | N/A |
| AAV5-Syn-Flex-ChrimsonR-tdTomato | UNC Vector Core | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: PA-2A-cre | The Jackson Laboratory | Jax stock #012358 |
| Mouse: Drd1a-tdTomato | The Jackson Laboratory | Jax stock #016204 |
| Mouse: Lhx6-GFP | GENSAT | MGI #3839374 |
| Software and Algorithms | ||
| MATLAB | The MathWorks | https://www.mathworks.com/products/matlab |
| Fiji (ImageJ) | NIH | http://fiji.sc |
| Constrained Non-negative Matrix Factorization (CNMF) | (Zhou et al., 2016) | N/A |
| Mosaic version 1.2.0 | Inscopix Inc. | https://www.inscopix.com |
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Anatol Kreitzer (anatol.kreitzer@gladstone.ucsf.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
104 adult transgenic or wild-type mice on a C57BL/6 background aged 50 to 365 days were used in the experiments. N=22 PV-2A-cre mice (15 male, 7 female; Jackson Stock #012358) were used for FSI ablation behavioral studies. N=6 PV-2A-cre mice crossed to Drd1a-tdTomato (2 male, 4 female; Jackson Stock #012358 & #016204) were used for eNpHR3.0-driven optogenetic suppression of FSIs in acute slice recordings of feed-forward inhibition. N=7 PV-2A-cre (7 female; Jackson Stock #012358) were used for in vivo electrophysiology recording. N=15 PV-2A-cre mice (N=7 male, N=8 female; Jackson Stock #012358) were used for in vivo calcium imaging. N=9 PV-2A-cre mice (N=4 male, N=5 female; Jackson Stock #012358) were used for acute slice measurements of AMPA/NMDA ratios. N=6 mice (N=5 male; N=1 female; Jackson Stock #012358) were used for acute slice calcium imaging. N=10 PV-2A-cre (N=7 male; N=3 female; Jackson Stock #012358) were used for intracranial IEM-1460 infusion into FSI-lesioned mice. N=18 PV-2A-cre mice (8 male, 10 female; Jackson Stock #012358) were used for behavioral assays involving optogenetic control of FSIs with eNpHR3.0. N=11 Drd1a-tdTomato mice crossed to Lhx6-GFP (4 male, 7 female; Jackson Stock #016204 and MGI #3839374) were used for acute slice recordings of feed-forward inhibition and paired interneuron-MSN recordings. Sample size for primary behavioral experiments (e.g. cross-maze) was determined by estimate of effect size and variance in an initial cohort (N≥4 mice per group), and independent replication of that effect in at least one additional, independent cohort containing an equal or greater number of mice. Sample size for physiological recording and in vivo imaging were determined based on previously published studies, and statistical significance was calculated using post-hoc tests.
No randomization of animals was implemented. Experimenters were blinded to animal genotype in the cross maze and motor performance assays, but not in operant behavior. Operant behavior experimental control, recording and analysis were automated to remove possibility for experimenter bias. Age and sex of mice was balanced across cohorts, and littermate controls were used in each experiment.
METHOD DETAILS
Stereotactic surgery
All procedures were in accordance with protocols approved by the UCSF Institutional Animal Care and Use Committee. Mice were maintained on a 12/12 light/dark cycle and fed ad libitum. Experiments were carried out during the dark cycle. All surgeries were carried out in aseptic conditions while mice were anaesthetized with isoflurane (5% for induction, 0.5–1.5% afterward) in a manual stereotactic frame (Kopf). Buprenorphine HCl (0.1 mg kg−1, intraperotineal injection) and Ketoprofen (5 mg kg−1, subcutaneous injection) were used for postoperative analgesia. Mice were allowed to recover for 14 – 42 days before experiments.
Virus injection
For cell-type-specific ablation of parvalbumin-expressing fast-spiking interneurons in the dorsal striatum, we injected 500 nL of adeno-associated virus serotype 5 (AAV5) carrying modified procaspase-3 (taCasp3) together with a tobacco etch virus protease (TEVp) in a double-floxed inverted open reading frame under the control of the EF1α promoter (AAV5-EF1α-Flex-taCasp3-TEVp). Expression of taCasp3 triggers cell-autonomous apoptosis with minimal toxicity to adjacent non-Cre-expressing cells (Yang et al., 2013). All viruses were obtained from UNC Vector Core or University of Pennsylvania Vector Core. A control virus carrying only the fluorophore eYFP (AAV5-EF1α-DIO-eYFP) was injected into littermate control animals. The virus was injected bilaterally into dorsal striatum of adult animals in a stereotactic surgery as described above, at coordinates +1.0 anteroposterior (AP), +/−2.3 mediolateral (ML), and −2.5 dorsoventral (DV), measured from bregma on the skull surface. Virus infection spanned most of dorsal striatum: −0.5 to +1.25 AP, +/−1 to 2.8 ML and −1.5 to −3.5 DV. Injections were performed using a 5 μl Hamilton syringe through a hydraulic pump (Harvard Instruments). The needle was held in place for 1 min before the start of injection, injection speed was 100 nL min−1, and the injection needle was raised 5 minutes after completion of virus delivery.
For AMPA-NMDA ratio measurements, a fluorescent reporter virus (AAV8-hSyn-eGFP) was mixed 1:2 with either AAV5-EF1a-Flex-taCasp3-TEVp or control saline and 0.75 μL were injected bilaterally into PV-2A-cre mice at +1.0 AP, +/−2.3 ML, −2.3 DV. For optogenetic silencing of FSIs in acute slices, open field behavior and in vivo physiology, we injected 1000 nL of AAV carrying halorhodopsin (eNpHR3.0) in a double-floxed inverted open reading frame (DIO) under the control of the EF1α promoter and tagged with the eYFP fluorophore (AAV5-EF1α-DIO-eNpHR3.0-YFP) at the coordinates +1.0 AP, +/−2.2 ML, −2.5 DV from bregma. For acute slice calcium imaging experiments paired with optogenetic activation of FSIs, PV-2A-cre mice were injected with AAV5-Syn-Flex-ChrimsonR-tdTomato, 0.75 μL – 1 μL, bilaterally into dorsolateral striatum at +1.0 AP, +/− 2.3 ML, −2.5 DV ~4 weeks before preparation of acute slices for recording.
For optogenetic control of FSIs during calcium imaging in vivo, two viruses were combined and injected in the same surgery. For these experiments, we injected 500 nL of AAV carrying halorhodopsin (eNpHR3.0) in a double-floxed inverted open reading frame (DIO) under the control of the EF1α promoter and tagged with the mCherry fluorophore (AAV5-EF1α-DIO-eNpHR3.0-mCherry), together with 250 nL of AAV carrying the calcium indicator GCaMP6f under the control of the synapsin promotor (AAV5-Syn-GCaMP6f). In control animals, the halorhodopsin virus was replaced by an equivalent volume of control virus carrying the mCherry fluophore alone (AAV5-EF1α-DIO-mCherry).
Intracranial drug infusion
For intracranial drug infusion experiments, cannulae (Plastics One, Inc) were chronically implanted into dorsolateral striatum (+1.0 AP, −2.3 ML, −2.5 DV from bregma) during the same surgery as virus injection as described above. Animals were allowed to recover for 4 weeks prior to behavioral experiments. On counter-balanced days, animals were placed into the open field arena for a brief, 5 min habituation session, prior to intracranial drug infusion in the home-cage. Infusions volume was 0.5μL of IEM-1460 at a concentration of 1.5mM in 0.9% saline, delivered continuously over 2 min at a rate of 2.5 μL/min. Control infusions on counter-balanced days consisted of 0.9% saline alone. Infusion cannulae were left in place for approximately 1 min following the infusion, and then removed as the animals were placed back into the open field arena for 30 min of continuous observation. Automated behavioral tracking (Noldus Inc.) was employed to track rotations. One mouse failed to develop dyskinesias or contralateral rotations following IEM-1460 injection. Visual inspection revealed the imnjection cannula was damaged and this mouse was excluded from further analysis.
Optogenetic ferrule implants
For optogenetic silencing of FSIs in vivo during open field behavior experiments, virus was injected as described above, and 400 μm fiber optic ferrules with 0.48 NA were implanted in the same surgery at coordinates +1.0 AP, +/−2.2 ML, −2.3 DV from bregma.
Chronic array implants
To record single unit electrophysiological activity from medium spiny neurons and fast-spiking interneurons in vivo in awake, freely moving mice, we implanted multi-electrode arrays into striatum. Under anaesthesia in a stereotactic surgery, the scalp was opened and a hole was drilled in the skull (+0.5 to +1.5 mm AP, −2.5 to −1.5 mm ML from bregma) and dura was removed from the craniectomy. Virus was injected as described above. Two skull screws were implanted in the opposing hemisphere. Dental adhesive (C&B Metabond, Parkell) was used to fix the skull screws in place and coat the surface of the skull. An array of 32 microwires (4 × 8 array, 35 μm tungsten wires, 150 μm spacing between wires, 150 – 200 μm spacing between rows; Innovative Physiology) was combined with a 200 μm diameter optical fiber and lowered into the striatum (2.5 mm below the surface of the brain) and cemented in place with dental acrylic (Ortho-Jet, Lang Dental). After the cement dried, the scalp was sutured shut. Animals were allowed to recover for at least seven days before striatal recordings were made.
In vivo electrophysiology recordings
Voltage signals from each site on a 32-channel microwire array were recorded in awake, freely moving mice in an open field arena. Signals were band-pass-filtered, such that activity between 300 and 6,000 Hz was analyzed as spiking activity. This data was amplified, processed and digitally captured using commercial hardware and software (Plexon or SpikeGadgets). Single units were discriminated with principal component analysis (Plexon Offline Sorter, or SpikeGadgets MatClust). Two criteria were used to ensure quality of recorded units: (1) recorded units smaller than 100 μV (~3 times the noise band) were excluded from further analysis and (2) recorded units in which more than 1% of interspike intervals were shorter than 2 ms were excluded from further analysis. FSIs and MSNs were distinguished based on waveform and firing rate. To determine waveform characteristics, the average waveform from each single unit was up-sampled from 30 kHz to 300 kHz using spline fitting (Matlab). Putative FSIs exhibited a peak-trough distance < 367 μsec and a firing rate > 2 Hz. Putative MSNs exhibited a peak-trough distance > 367 μsec and an average firing rate < 10 Hz. Units not meeting either of these criteria (short waveform with low firing rate) were excluded from further analysis.
Green light pulses 1 sec or 30 sec in duration and ~3 mW in brightness were delivered through an 200 μm optical fiber contained within the implanted microwire recording array with a duty cycle of 25% for 60 min (900 pulses for 1 sec; 30 pulses for 30 sec). For each unit, the laser responses were calculated using 1 sec light pulses. The Light On response was the average firing rate during the 1 sec period in which the laser was on, whereas the Light Off response was the average firing rate over the 1 sec before and 1 sec after the Light On period. The Modulation Index was calculated as (Light On)/(Light On + Light Off), such that a value of 0.5 = no modulation, 1 = activation of spiking during Light On, and 0 = silencing of spiking during Light On. The Fano Factor was calculated using variance in spike rates across 900 sequential laser pulses. The number of spikes for each neuron on each trial was calculated for three separate bins: Pre (1 sec prior to light onset), “Light” (full 1 sec duration of light pulse) and “Post” (1 sec follow light offset). The Fano Factor for each neuron was separately calculated for each of these time bins as σ2/μ where σ=standard deviation in number of spikes per trial and μ=average number of spikes per trial.
Bursts were detected from the same data set with 1 sec light pulses by post-hoc inspection of spike trains from individual MSNs. Only MSNs with a firing rate >0.25 Hz were considered (N=34 out of 64 PV-eNpHR3.0 MSNs; N=8 out of 25 control MSNs). Burst frequency and duration criteria were defined according to our own measurements (Fig S5m) and previous published reports (Kerr and Plenz, 2002), using values predicted to drive somatic and dendritic calcium transients: at least 8 spikes at an average firing rate of at least 20 Hz, and in which no single inter-spike-interval exceeds 100 msec. Only neurons with at least 10 detected bursts were considered for this analysis. The time-point of each burst was defined as the average of the times of the spikes contained in that burst.
Simulated calcium signals were calculated using recordings from the 30 sec light pulse sessions. The simulated calcium signal for each MSN was calculated at a sampling rate of 15 Hz, identical to the sampling rate of in vivo calcium imaging sessions. The calcium response from each spike in the MSN spike train was simulated sequentially by adding a decaying exponential onto the overall calcium signal (τ=0.812 sec; fit from decay constant of acute slice calcium imaging experiments with GCaMP6f). The amplitude of the calcium transient was determined as a function of the simulated calcium value at the onset of that spike using the saturation supralinear function plotted in Fig S6a–c, as an approximation of the measured supralinear responses in acute slices (Fig S5m). Calcium transients occurring when the initial simulated calcium exceeded the maximum value of 15 were assigned a minimum amplitude of 0.01. The resulting calcium train was then smoothed with a 0.5 sec square filter, and peaks were detected as transients exceeding average + 4 standard deviations of the simulated calcium signal for that MSN, with a minimum inter-peak interval of 0.5 sec.
Head-mounted microscope implants
To record calcium signals in vivo in freely-moving mice, animals were implanted with a gradient refractive index lens into dorsal striatum. Under anaesthesia in a stereotactic surgery, the scalp was opened and a hole was drilled in the skull (+0.5 to +1.5 mm AP, −2.5 to −1.5 mm ML from bregma) and dura was removed from the craniectomy. Virus injections were made as described above. A GRIN lens (Inscopix, Inc; 1.0 mm diameter, ~4.0mm length) was slowly lowered over ~30 min to final coordinates +1.0 mm AP, −2.0 mm ML, −2.2 mm DV and secured in place with dental adhesive (C&B Metabond, Parkell), which was used to coat the surface of the skull, and a headcap was formed with dental acrylic (Ortho-Jet, Lang Dental). Animals were allowed to recover for 3 – 6 weeks, before being placed back under anaesthesia to allow a microscope baseplate to be added to the top surface of the implant. Animals were allowed 2 – 5 days of recovery after the second (non-invasive) procedure, before recording. Recordings were made 8 – 16 weeks following initial virus injection.
Cross maze behavior
Mice were singly housed and habituated to the testing room for 2 days prior to the start of the experiment. Training and testing were performed under normal (white) light conditions to encourage use of distal cues (placed at the ends of the arms). All trials were recorded/tracked using Noldus Ethovision. Trials consisted of two types: Response Learning (Days 1–5) and Probe Trials (sessions 6, 9 and 10). For response learning, mice were placed in the start arm of the water T-maze and required to locate the hidden escape platform at the end of either the right or left arm of the maze. The platform remained in the same location for each mouse, and the location was counter-balanced on the left or right side across mice such that an equal number of experimental mice were trained to each location. Mice were given 6 trials a day in 2 separate sessions (3 hr intersession interval) of 3 trials each (5 minute intertrial interval) for 5 consecutive days. After finding the platform, mice were allowed to remain on the platform for an additional 10 sec. Mice failing to find the platform within 60 sec were guided to the platform location and allowed to remain for 10 seconds. Errors were defined as the mouse entering the incorrect arm or re-entering the start arm before locating the hidden platform.
Probe trials were performed on a maximum of one trial within each session, with the remaining two trials in that session acting as regular response learning trials. For probe trials, the platform was removed from the maze, and the barrier moved to the opposite arm of the maze such that it blocked off the start arm on regular trials. The mouse started opposite the start arm on regular trials, and was allowed to swim until it fully entered one of the two choice arms. Immediately upon entry into a choice arm, the mouse was scooped out of the water by the experimenter and its response was recorded without allowing the mouse to reach the end of the arm or turn around to investigate the opposite arm.
Open field behavior tracking
Locomotion was tracked in a brightly lit open-field arena using an overhead camera and post-hoc tracking software (Noldus, Inc). Videos of behavior were recorded from overhead and side views, and mice were monitored continuously for any abnormal involuntary movements.
Optogenetic control of open-field behavior
Mice implanted bilaterally with fiber optic ferrules in dorsal striatum. Mice were tethered to flexible optical fibers connected to a two-channel fiber optic commutator (Doric Lenses). Locomotion and behavior were tracked and recorded as described above. Optogenetic light stimulation was delivered with 30 sec pulses of ~3 mW green light at a 25% duty cycle for 30 min either bilaterally or unilaterally on subsequent days and compared across three time windows: “Pre” (30 sec prior to light onset), “Light” (5 – 30 sec after light onset), “Post” (5 – 30 sec after light offset). Green light (532 nm) was generated by TTL-gating of solid-state lasers (Shanghai Laser and Optics Century) connected to 400 μm diameter fiber optic cables.
Accelerating Rotarod
Rotarod testing was performed under normal light, with animals brought into testing room and given at least 30 minutes to acclimate before testing. On the first day, one to five mice were simultaneously placed on the rotarod apparatus with the rod rotating at the constant speed of 16 rpm (rotations per minute) for three trials. The trial ended when the mouse fell down or when 5 minutes was completed. The inter-trial interval for each mouse was 15 – 20 minutes. On the second and third day of testing, 1–5 mice were simultaneously placed on the rotarod apparatus with the rod rotating at an accelerated speed from 4 – 40 rpm. The trial ended when the mouse fell down or when the 5 minutes is completed. There were 3 trials in the morning and 3 trials in the afternoon, making a total of 6 trials a day. The inter-trial interval for each mouse was between 15 – 20 minutes, with a 2 hr break between the morning session and the afternoon session. To assay acute motor performance, the average latency to fall was averaged over the final day (6 trials per mouse). No differences between FSI-lesioned and control mice were present throughout training.
Pole Test
Pole test descending was performed under normal light, with animals were brought into testing room and given at least 30 minutes to acclimate before testing. Mice were placed facing upward atop a vertical wooden pole 50 cm long (1 cm diameter.) The base of the pole was placed in the home cage. For initial training, there were 2 trials per day with each trial lasting a maximum of 60 seconds. The latency for the mouse to turn around and climb down was recorded. Testing was performed on the third day. The best performance over the five trials on the testing day was used for analysis. If a mouse slipped, fell, or slid down the pole, the time for that trial was discarded. The best performance score is reflective of the highest level of performance where mistakes were not made.
Balance Beam Test
Balance beam testing was performed under normal light, with animals brought into testing room and given at least 30 minutes to acclimate before testing. Testing took place over 3 days with 3 trials per day. The first day included two additional training trials. On the first training trial of the first day, mice were placed on the middle of the largest diameter beam facing a dark box and guided to the enclosure. On the second training trial on the first day, mice were placed on the end of the beam facing the dark box and guided to the enclosure. On the third to fifth trials (testing trials) on the first day, mice were placed on the end of the beam facing the dark box and traversed the beam unguided. On the second day, mice were placed at the end of the medium diameter beam, facing the dark box. On the third day, mice were placed at the end of the smallest diameter beam, facing the dark box. Latency to traverse the beam into the dark box, number of foot slips, and the number of falls were recorded. Trials were limited to a maximum latency of 60 seconds.
Acute slice recordings
Mice were euthanized with a lethal dose of ketamine and xylazine followed by transcardial perfusion with 8 mL of ice cold artificial corticospinal fluid (ACSF) containing (in mM): NaCl (79), KCl (2.3), NaHCO3 (23), Sucrose (68), NaH2PO4 (1.1), MgCl2 (6), D-Glucose (12), CaCl2 (0.5). Para-horizontal (250 μm thick) containing dorsal striatum were then prepared at an angle 30° off of horizontal with a vibratome (Leica) in the same solution before incubation in 33°C ACSF containing (in mM): NaCl (125), NaHCO3 (26), NaH2PO4 (1.25), KCl (2.5), MgCl2 (1), CaCl2 (2), and D-Glucose (12.5), continuously bubbled with 95/5% O2/CO2. After 60 minutes of recovery, slices were either kept at room temperature or transferred to a recording chamber superfused with recording ACSF (4–6 mL min−1) at 33°C.
Whole-cell voltage clamp recordings for measurement of AMPA/NMDA ratios were obtained using an internal solution containing (in mM) CsMeSO3 (110), CsCl (15), NaCl (8) BAPTA-Cs4 (10), HEPES (10), Mg-ATP (2), Na-GTP (0.3), QX-314 (5), and picrotoxin (50 μM) was included in the bath. Recordings were strictly restricted to within the virus infection zone delineated by the fluorescent reporter virus that was co-injected together with the DIO-Casp3 virus. Whole cell recordings were obtained from MSNs and allowed to stabilize at −75 mV for at least 5 minutes prior to measurements. During the stabilization period, EPSCs were evoked with a glass monopolar stimulating electrode placed adjacent recorded MSN. Once stable EPSCs −200 pA to −500pA in amplitude were achieved at −75 mV, the holding potential was alternated between −75 mV and +40 mV to record AMPA/NMDA ratios. All holding potentials are reported after correction for ~15 mV junction potential measured empirically using EPSC reversal potential. Whole-cell voltage clamp recordings for isolation of monosynaptic EPSCs and di-synaptic feed-forward IPSCs were obtained using an internal solution containing (in mM): CsMeSO3 (135), NaCl (8), EGTA (0.5), HEPES (10), Mg-ATP (2), Na-GTP (0.3), QX-314 (5). Current-clamp recordings were obtained with an internal solution containing (in mM): K-Gluconate (135), NaCl (10), MgCl2 (2), EGTA (0.5), HEPES (10), Mg-ATP (2), Na-GTP (0.3). For interneuron-MSN paired recording measurements of synaptic strengths, the MSN recording was obtained using a voltage-clamp solution described above, while the pipette for the interneuron was tip-filled with a current-clamp solution and back-filled with a voltage clamp solution. This allowed an initial characterization of the firing properties of the interneuron, followed by voltage-clamp of the interneuron after allowing ~10 minutes for the back-filled voltage clamp internal solution to dialyze into the cell. Optogenetic light stimulation was delivered to acute slices using green light at approximately 3 mW mm−2 generated by an LED (Prizmatix) coupled to the microscope epi-fluorescence excitation light beam path. For feed-forward inhibition experiments, only neurons exhibiting >1ms lag of IPSC relative to EPSC onset, and a large-amplitude IPSC with rapid rise-time were considered. A subset of recordings with ~zero IPSC lag relative to EPSC and slower IPSC rise-time were discarded, on the presumption that this IPSC arose from leakage of the stimulating electrode current into striatum to drive monosynaptic IPSCs from intra-striatal fibers and therefore did not represent true feed-forward inhibition.
For acute slice calcium imaging experiments, the current clamp internal solution was supplemented with 50 μM Oregon Green BAPTA-6F (Molecular Probes). MSNs were allowed to stabilize for 5 min before starting recording. Injection durations were varied between 20 – 540 ms to evoke varying numbers of action potentials, and the injection amplitude for each recording was set to reliably drive a single action potential with the 20 ms injection. Depolarizing current injections of +200 pA to +850 pA were used to drive action potentials. For experiments investigating the effect of resting potential on MSN spike-to-calcium transformations, an initial current injection step (Iinit) of +100 pA to +450 pA was sustained for ~2 sec to drive a ~20 mV, sustained, sub-threshold depolarization of the MSN membrane before a second, briefer pulse of approximately equivalent amplitude (Ipulse) was delivered for the reported pulse duration (20 ms – 540 ms). On interleaved trials, a single current injection of amplitude Iinit + Ipulse was used to drive an equivalent number of action potentials from the MSN resting potential. For acute slice calcium imaging experiments including optogenetic activation of FSIs, all trials included the Iinit ~2 sec sub-threshold depolarizing current step prior to current injection to drive action potentials. On interleaved trials, red light pulses (605 nm, 0.1 – 0.25 mW mm−2) were delivered on interleaved trials. In order to simulate coincident excitatory drive onto presynaptic FSIs and postsynaptic MSNs, the time course of activation of FSIs was matched to the time course of the brief 20 ms – 540 ms current injection into the post-synaptic MSN. Oregon Green BAPTA-6F was excited with blue light (454 – 491 nm, 0.25 mW mm−2), and green emission light (510 nm – 555 nm) was collected with a cooled CCD camera (Photometrics) using an Olympus microscope and 40X, 0.8NA objective. Blue and red excitation light were generated by independently controlled LED systems (Thorlabs). On a sub-set of recordings, mice were injected with a virus encoding a the genetically encoded calcium indicator GCaMP6f (AAV5-Syn-GCaMP6f) 8 – 10 weeks prior to preparation of acute slices for recording. In these recordings, Oregon Green BAPTA-6F was omitted from the recording pipette.
Three port sequence task
Mice were given food and water ad libitum before water deprivation in preparation for training. Water-restricted mice were placed in a box containing three ports and trained to perform a three-poke sequence to trigger delivery of water sweetened with 10% sucrose at one of the pokes. Prior to the start of the three-poke task, naïve mice received two days of shaping in which they learned to perform the first two pokes in the task (data not shown). During these shaping days, entry into the center port triggered a solenoid click and delivery of a small amount of sucrose-water to the second port. Subsequent entry to the second port triggered a second click and delivery of the remainder of the reward for that trial.
After shaping, mice were transitioned to the three-poke sequence task. Entry into the center port at the start of a trial was signaled by an auditory “pip”. Entry into the second port was signaled by a brief solenoid click, and entry into the third port triggered reward delivery in that port. Errors were not punished or signaled. After initiation of a trial, mice were allowed 30 sec to complete the trial regardless of the number of errors committed. Port entry was detected using custom-built ports with an infrared beam across the mouth of the port. Reward delivery was controlled by solenoids (NResearch) gating gravity-fed fluid delivery within the ports. To maintain motivation and ensure naïve mice received adequate water on each day of the task, however, the reward amount was approximately doubled and mice were restricted to perform no more than 200 trials on the first day of training. Over days 2–7, the reward amount was held constant and all mice were restricted to no more than 500 trials per day. Two mice (one FSI-lesioned mouse and one YFP control mouse) were excluded based on failure to complete adequate number of trials within the allotted behavioral sessions.
Behavior was controlled and recorded using MBED microcontrollers (Adafruit) interfaced to computers running StateScript behavior control software (SpikeGadgets). For optogenetic experiments, this system was used to control closed-loop green light delivery (532 nm) by TTL-gating of solid state lasers (Shanghai Laser and Optics Century) connected to 400 μm diameter fiber optic cables on a randomly assigned subset (~30%) of trials. For optogenetic stimulation, the light was triggered with a randomly assigned (0 – 400 ms) delay following exit from the reward port on the preceding trial, and remained on for up to 8 sec or until completion (reward exit) on that trial. To calculate “Restricted First Poke Errors”, which count only Choice Errors while excluding Execution Errors, only the fraction of First Poke Errors occurring before the average time for each mouse to complete a trial in that session were considered.
Sequence task to distinguish egocentric and allocentric learning
To distinguish egocentric from allocentric learning, mice were trained on a complementary three-poke sequence task. In this task, mice were placed in a modified box containing 6 ports, including 5 ports arrayed along the front wall, and a single reward port in the rear of the box. The availability of a new trial was signaled by illumination of a single port along the front wall following exit from the reward port on the previous trial. Mice learned over 5 days of initial shaping to approach the illuminated port (either Port 2 or Port 4 illuminated with 50% probability) in order to trigger reward delivery at the port in the rear of the box.
After 5 days of shaping, the task was changed so that, although all trials still initiated with illumination of either Port 2 or Port 4, mice were instructed to initiate 90% of trials (“Training Trials”) on one of these ports and the remaining 10% of randomly interleaved trials (“Probe Trials”) at the other. Instead of directly triggering reward delivery, successful entry into the initiation port now triggered illumination of the two neighboring ports on the front panel, and the mouse was forced to choose between these two ports. On training trials, choosing the center port triggered reward delivery, whereas choosing the outside port did not. Irrespective of reward delivery, mice were required to enter the poke at the rear of the box to trigger initiation of the next trial. Mice were therefore trained to perform a three-poke sequence that resembled the more simple three-poke task described above.
To determine the task strategy being employed to complete this sequence, we examined the 10% of probe trials. To avoid introducing a bias, entry into either the center or the outside poke after initiation on probe trials triggered a reward delivery in the rear of the box. Choosing the center port on probe trials corresponded to the same location in space as training trials, and was scored as an allocentric choice, whereas choosing the outside port on probe trials corresponded to the same body movement as training trials, and was scored as an egocentric choice. Only probe trials in which the mouse correctly triggered the instructed initiation port were considered for the analysis. The side of the box (Port 2 or Port 4) used for training trials was counter-balanced across mice.
Histology
Animals were euthanized with a lethal dose of ketamine and xylazine (400 mg ketamine plus 20 mg xylazine per kilogram of body weight, i.p.) and transcardially perfused with PBS, followed by 4% paraformaldehyde (PFA). Following perfusion, brains were transferred into 4% PFA for 16 – 24 h and then moved to a 30% sucrose solution in PBS for 2 – 3 d (all at 4°C). Brains were then frozen and cut into 30 μm coronal sections with a sliding microtome (Leica Microsystems, model SM2000R) equipped with a freezing stage (Physitemp) and mounted on slides. Slides were blocked for 1 hour in 10% Normal Donkey Serum (NDS) in 0.5% PBST then incubated overnight in primary antibody (1:500 chicken anti-YFP, Aves Labs Inc, GFP-1020; and 1:500 rabbit anti-parvalbumin, Swant Inc., PV 27), 3% NDS in 0.5% PBST. The following day, they were washed 3 times for 10 minutes each in 0.5% PBST and incubated for 1 hour in secondary antibody (1:1000 donkey anti-chicken 488, Jackson Immunoresearch 703-545-155; and 1:1000 donkey anti-rabbit 647, Invitrogen, A-31573), 3% NDS in 0.5% PBST and 1:2000 DAPI. After this, slides were washed for 10 minutes in 0.5% PBST and 2 more 10 min periods with 1:1 PBS. Slides were then washed with 0.05% lithium carbonate and alcohol, rinsed with diH2O, and coverslipped with Cytoseal 60. Slides were scanned on a 6D high throughput microscope (Nikon, USA), globally gamma-adjusted to reduce background, and pseudocolored using freely available Fiji software. Automated registration of images (Nikon) was performed, and cell counts were manually acquired using open source code Fiji run on the ImageJ platform (NIH, Bethesda, MD).
QUANTIFICATION AND STATISTICAL ANALYSIS
In vivo imaging and analysis
Calcium imaging was performed using a head-mounted, microendoscope-based microscope to image through a chronically implanted GRIN lens placed above dorsal striatum (Inscopix, Inc). Blue light (435–460 nm) for excitation of the GCaMP6f calcium indicator was delivered at ~0.5 mW mm−2 for continuous recording throughout the session, while amber (590–650 nm) light for intermittent activation of eNpHR3.0 was delivered at ~3.5 mW mm−2. GCaMP6f emission signal (490–540 nm) was acquired continuously at 15 Hz and spatially down-sampled prior to motion correction. The motion-corrected video was then processed using Constrained Nonnegative Matrix Factorization (CNMF) to isolate signals from individual neurons (Zhou et al., 2016). As was evident from visual inspection of the raw video, and similar to signals described in striatum (Barbera et al., 2016) and other brain regions (Jennings et al., 2015; Ziv et al., 2013), the individual neuron calcium signals resulting from this analysis were punctuated by brief, discrete, positive transients. These transients were detected post-hoc using a threshold of 5 times the standard deviation of the full signal.
For open-field experiments featuring combined in vivo calcium imaging and optogenetic silencing of FSIs (Fig 2), amber light stimulation was delivered in 5 sec pulses with 16.67% duty cycle for 22.5 min (45 pulses) and an initial period of 2.5 minutes at the start of each recording was discarded to allow for stabilization of excitation LED temperatures. Light responsiveness was calculated by calculating a Modulation Index for each neuron, corresponding to the number of calcium transients occurring with the (Light On)/(Light On + Light Off). The Light On period corresponded to the full 5 sec duration of each optogenetic amber light pulse, while the Light Off period corresponded to the 5 sec period immediately following the extinguishing of the amber light.
As with any 1-photon imaging method, we expected to find some degree of spatial cross-talk between neighboring neurons. To quantify this cross-talk, we measured the fraction of time individual pairs of simultaneously recorded neurons spent above the spike detection threshold, relative to the fraction of time expected by chance. We then plotted this correlation against the distance between those neurons in the image plane and discovered an elevated degree of cross-talk only on neurons within 10 pixels of one another in the image (Fig S9g). This corresponded to ~1.7% of neurons in our recordings (Fig S9h).
Analysis of task event-aligned in vivo imaging results
Calcium transients recorded with the head-mounted microscope were aligned to four separate task events on each trial: (1) entry into Poke 1 to initiate the trial, (2) entry into Port 2, (3) entry into Port 3 (reward collection), and (4) exit from the reward port. Peak transient rates were calculated using a 400 ms window sliding in 20 ms increments across a range spanning +/− 2 sec relative to the task event. The event-aligned transient rate was determined to be the largest of these peak transient rates, and was calculated separately such that the peak Light On and Light Off conditions were permitted to correspond to different task events. To minimize the impact of the elevated calcium transient rate immediately after animals were plugged into the microscope and placed into the behavior box, all calcium transients prior to the first task-driven laser pulse were discarded from analysis of the overall calcium transient rate (Fig 6c).
We used random shuffling of transient times to determine statistical significance of calcium transient task event-alignment independent of light-induced changes in overall transient rate. Specifically, for each neuron a collection of ~1000 simulated sessions was generated, with each session containing an equivalent number of trials to the real data set. A histogram of shuffled transient rates was calculated from each simulated (shuffled) session, resulting in a collection of 10,000 simulated histogram bins (1000 sessions, 10 bins per session) that empirically describe the transient rate properties in that neuron. Histograms were calculated separately for Light On and Light Off conditions. Neurons with a peak task response exceeding at least 9,990 of these bins were determined to respond significantly to the task at the P<0.001 level (not corrected for multiple comparisons). To discount false positives derived from very low baseline transient rates or high levels of “burstiness”, only neurons that contained transients on at least three separate trials were considered as significant responses. If the transient rate in a neuron significantly exceeded chance when aligned to more than one task event, the task responsiveness was assigned to the event with the highest amplitude event-aligned transient rate.
The task-event preference vector for each neuron was calculated using the event-aligned transient rate for each neuron within +/− 1 sec of each task event (R1, R2, R3, R4), then normalizing by the length of this vector to generate a unit vector in a 4-dimensional space. This task-event preference was calculated independently for Light On and Light Off trials for each neuron. Only neurons containing at least 30 calcium transients aligned to at least one task event in either the Light On or Light Off condition were considered. To avoid artificially large apparent task-event preferences driven by small numbers of calcium transients, in neurons containing fewer than 4 calcium transients in either Light On or Light Off conditions, the unit vector for that condition was set to [0.5 0.5 0.5 0.5], corresponding to no preference.
The overall modulation index for each neuron was calculated as the average rate of calcium transients occurring with optogenetic stimulation (Light On)/(Light On + Light Off), including the entire trial duration for each trial. The event-aligned modulation index was calculated equivalently, but for only the 400 ms event-aligned window surrounding the peak event-aligned calcium transient rate for that neuron. To compare the modulation of overall calcium transients against event-aligned transients, we plotted the event-aligned modulation index against the overall modulation index and found that most neurons fell below the unity line, corresponding to decreased modulation of event-aligned transients relative to overall calcium transients. This was confirmed by performing a sign rank test of the ratio of (event-aligned modulation index)/(overall modulation index) for all neurons against the null value of 1. A value of 1 for this ratio would indicate that event-aligned calcium transients are not differentially modulated, and that optogenetic suppression of FSIs does not affect the selectivity with which calcium transients signal specific task events. Our finding that this ratio is significantly less than 1 is consistent with a decrease in selectivity with which calcium transients align to task event, and may help to explain why the increase in calcium transients following FSI silencing impairs, rather than facilitates task-dependent learning.
To perform Receiver Operator Characteristic (ROC) analysis of calcium transient task specificity, the preferred event-aligned transient window was first calculated for each neuron from peri-event histograms as described above for the Light Off and Light On trials. We then calculated the fraction of trials on which each neuron had an transient in its event-aligned window (True Positives) and the fraction of trials on which each neuron had an transient in a randomly selected “Shuffled” window (False Positives). ROC curves were calculated by varying the threshold (fraction of trials containing an transient) and comparing the fraction of neurons above that threshold for Light Off and Light On conditions separately. Two-fold cross-validation was performed by removing a randomly selected 50% of trials and re-calculating ROC curves for light off and light on conditions 1000 times. The “true” value for the AUC in the light on condition, calculated using all of the trials (vertical, dashed amber line in Fig S9b,e), was less than of these shuffled light off AUC values (solid black line), resulting in an empirically calculated value of P<0.016. In contrast, the same analysis revealed that optogenetic stimulation amber LED did not affect the AUC in mCherry control animals (P>0.4).
Supplementary Material
(A,B) Recording configuration using para-horizontal acute slices to preserve cortico-striatal connectivity for measurement of feed-forward inhibition. (C) Onset (20% of peak) of feed-forward IPSCs follows onset of EPSCs with a time-delay of 1–3 ms. x-axis is latency from stimulus onset. (N=10 mice, n=26 MSNs). (D) AMPA-R antagonist NBQX (10 μM) blocks excitatory synaptic transmission, suppressing both monosynaptic EPSC and di-synaptic feed-forward IPSC in all MSNs (N=2 mice, n=10 MSNs). (E,F) Equivalent suppression of EPSC and feed-forward IPSC in direct- and indirect-pathway MSNs (N=2 mice, n=5 MSNs per group). (G) Quantification of eNpHR3.0 expression within virus infection zone measuring penetrance (# neurons co-expressing both eNpHR3.0 and endogenous Parvalbumin protein divided by total # of neurons expressing Parvalbumin) and selectivity (# neurons co-expressing eNpHR3.0 and Parvalbumin divided by total number of eNpHR3.0-expressing neurons). (H) Lesion extent summarized and overlayed from 12 PV-cre mice injected with AAV-DIO-Casp3. Brains were sectioned and stained for endogenous parvalbumin. Lesion borders were manually traced and overlayed (red) at 5 locations spanning the rostral-caudal extent of striatum. (I) Recording configuration for interneuron-MSN pairs. (J) Tip-filling interneuron recording pipette with current-clamp internal solution allows initial identification of interneuron subtype by current injections. Lhx6-GFP mice label two distinct interneuron subtypes (FSIs and Persistent Low-Threshold Spiking interneurons). (K) Back-filling pipettes with voltage-clamp internal solution allows subsequent measurement of excitatory inputs onto interneuron and neighboring MSN in response to shared excitatory input. (L) FSIs receive stronger excitatory input than neighboring MSNs while PLTS interneurons receive weaker input. (N=4, mice n=10 FSI-MSN pairs, n=8 FSI-PLTS pairs; paired two-tailed t-test, P<10−4 for FSIs, P<10−3 for PLTS). All error bars are s.e.m. and center values are group means.
(A) Exemplar MSN recording of feed-forward inhibition showing suppression by IEM-1460 (50 μM). (B) Timecourse of IEM-1460 (50 μM) suppression of feed-forward IPSC and recovery by washout. (C,D) Group data for IEM-1460 suppression of feed-forward IPSC and recovery by washout. (N=6 mice, n=8 MSNs; Before-IEM and IEM-Wash for IPSC, sign rank test P<0.01; Before-IEM and Before-wash for EPSC, sign rank test P>0.2). (E) Selective suppression of feed-forward IPSCs but not EPSCs onto D1-tmt-positive direct pathway dMSNs by IEM-1460 (50 μM). (F) Selective suppression of feed-forward IPSCs but not EPSCs onto D1-tmt-negative indirect pathway iMSNs by IEM-1460 (50 μM). (G) Equivalent lack of suppression of EPSCs onto direct- and indirect-pathway MSNs by IEM-1460 (N=6 mice, n=5 dMSNs, n=3 iMSNs; rank sum test P>0.05). (H) Equivalent suppression of IPSCs onto direct- and indirect-pathway MSNs by IEM-1460. (N=6 mice, n=5 dMSNs, n=3 iMSNs; rank sum test P>0.05). (I) PV-eNpHR3.0 optogenetic suppression of feed-forward IPSCs onto direct-pathway MSNs. (J) PV-eNpHR3.0 optogenetic suppression of feed-forward IPSCs onto indirect pathway MSNs. (K) Equivalent lack of effect of PV-eNpHR3.0 on monosynaptic EPSCs onto direct- and indirect-pathway MSNs. (N=4 mice, n=6 dMSNs; rank sum test P>0.6). (L) Equivalent suppression of feed-forward IPSCs by PV-eNpHR3.0 in direct- and indirect-pathway MSNs. (N=5 mice, n=6 iMSNs; rank sum test P>0.5). All error bars are s.e.m. and center values are group means.
(A) Modulation index for FSIs (top) and MSNs (bottom) from in vivo physiological recordings from control mice moving freely in an open field shows no modulation (sign rank test against 0.5; P>0.1 MSNs, P>0.4 FSIs). (B) Modulation index for FSIs (top) and MSNs (bottom) in PV-eNpHR3.0 mice shows silencing of FSIs (sign rank test against 0.5; P<10−8 for MSNs, P<0.005 for FSIs). (C) Comparison of modulation index for spiking and calcium transients reveals greater disinhibition of calcium transients than individual spike rates during optogenetic suppression of FSIs (N=5 PV-eNpHR3.0 mice and n=34 MSNs for electrophysiology; N=2 mCherry control mice and n=10 MSNs for control electrophysiology; N=4 PV-eNpHR3.0 mice and n=234 neurons for imaging; N=4 mCherry control mice and n=400 neurons for control imaging. Rank sum test spikes control vs spikes PV-eNpHR3.0 P<0.003. Rank sum test transients control vs. transients PV-eNpHR3.0 P<10−42. Rank sum test PV-eNpHR3.0 spikes vs. PV-eNpHR3.0 transients P<10−3). Spiking data is the same as that shown in Fig 1N. (D) Calcium responses to individual spikes were simulated with a decaying exponential (τ =0.812 sec fit from GCaMP6f data in Fig S5) and an amplitude set with a saturating non-linear transformation to approximate the non-linear responses in Fig S5m without diverging at high burst rates. Calcium transient amplitudes were set at a minimum value of 0.1 when initial simulated calcium was >15. (E) Simulated calcium responses to 50 Hz bursts containing 1–40 spikes. (F) Amplitudes of simulated bursts in panel E reflect saturating nonlinear transformation. (G) Exemplar simulated calcium trace calculated from a portion of the spike train of a single putative MSN recorded in vivo (top) shows bursting aligned to optogenetic silencing of neighboring FSIs (amber blocks). Shuffled spike train with equivalent average firing rate produces many fewer calcium bursts (bottom), highlighting importance of MSN firing pattern in generating calcium bursts. Note difference in scale between true and shuffled simulated calcium signals. (H) Raster plot of recorded spikes from exemplar MSN plotted in panel G. (I) Raster plot of simulated calcium transients from the same recording. (J) Modulation index of recorded spikes and simulated calcium transients from all MSNs recorded with this protocol (N=5 PV-eNpHR3.0 mice, n=60 MSNs, sign rank test against null value of 0.5, P<10−6 for spikes, P<10−4 for simulated calcium transients; N=2 control mice and n=15 control MSNs, sign rank against 0.5, P>0.05 for spikes and P>0.4 for calcium transients). (K) Directly comparing modulation index of recorded spikes and simulated calcium transients reveals significantly greater modulation of simulated calcium transients (N=5 mice, n=60 MSNs, sign rank test P<0.01). All error bars are s.e.m. and center values are group means.
(A) Recording configuration for acute slice calcium imaging with Oregon-Green BAPTA-6F (50 μM) in recording pipette. (B) Exemplar MSN current clamp recording traces (top) with current injection of varying duration. Calcium signal (ΔF/F) plotted below. (C) Normalized group data reveals Oregon-Green BAPTA-6F linearly reports action potential-induced calcium influx (N=5 mice, n=27 MSNs). Responses normalized to average ΔF/F from sweeps in containing >10 spikes. (D) Tonic current injection altered the resting potential by +21.6 mV (N=3 mice, n=11 MSNs, two-tailed t-test P<10−6). (E,F) Spike-to-calcium relationships remains linear and unaltered despite changing resting potential. (N=3 mice, n=11 neurons, sign rank test P>0.2). (G) Recording configuration for acute slice calcium imaging with optogenetic manipulation of FSIs. (H) Exemplar MSN calcium signal recorded by subtracting baseline image from image acquired at the peak calcium signal. (I) Exemplar current clamp recordings (top) and calcium signals (bottom) evoked by 180 ms current injection under control conditions (left) and during optogenetic activation of FSIs (right). Optogenetic stimulation of FSIs triggered simultaneously with current injection to MSN to mimic coincident excitatory input. (J) Spike-to-calcium relationship remains linear despite optogenetic activation of FSIs. (K) ChrimsonR activation of FSIs reduces number of action potentials evoked by current injection into MSNs (N=4 mice, n=16 neurons; 2-way ANOVA with interaction Popto <10−17, PstimDur<10−50, Pinteract<10−13). (L) ChrimsonR activation of FSIs does not affect the spike-to-calcium transformation in MSNs (N=4 mice, n=16 neurons; two-tailed t-test P>0.6). (M) GCaMP6f calcium responses scale supralinearly with number of spikes in a burst (N=1 mouse, n=6 MSNs). Normalization across recordings as in Panel (C). All error bars are s.e.m. and center values are group means.
(A) Unilateral intracranial injection of IEM-1460 (0.5μL, 1.5mM – intermediate dosage) into dorsolateral striatum induced dyskinetic axial twisting, quantified by rotational bias (contra turns)/(ipsi + contra turns) in both control and FSI-lesioned mice (N=5 mice per group, 2-way ANOVA PGroup>0.2; unpaired two-tailed t-test between groups P>0.1 each condition). (B) AMPA/NMDA ratios recorded in D1-tdTomato reporter mice to identify subtype of the recorded MSN reveal most of the increase in AMPA/NMDA ratios in PV-Casp3 lesioned mice is driven by indirect pathway iMSNs, but this trend is not statistically significant by 2-way ANOVA. iMSNs relative to tmt+ dMSNs. (N=9 mice per group; n=10 control dMSNs, n=11 control iMSNs; n=9 PV-Casp3 dMSNs, n=11 PV-Casp3 iMSNs, two-way ANOVA PPathway>0.2, PCasp3<0.03, Pinteract>0.05). (C) Fraction of trials completed (sequence completed before 30 sec maximum duration following trial initiation), and (D) inter-trial interval are unaffected in FSI-lesioned mice, indicating unimpaired motivation. (E) Schematic describing two types of error following trial initiation. (F) Increased fraction of Poke 2 Errors in FSI-lesioned mice. (G) Increased fraction of Poke 3 Errors in FSI-lesioned mice. (N=11 mice per group. two-way ANOVA PGroup values reported on charts). All error bars are s.e.m. and center values are group means.
(A) Schematic of “Choice Error” in which the mouse proceeds immediately to the incorrect second port after initiation. (B) Schematic of “Execution Error” in which the mouse approaches the correct second port but fails to break the infrared beam before proceeding to the third port. (C) Latency histograms reveal increased errors at short latencies in FSI-lesioned mice (arrows on Sessions #1–3), consistent with a predominance of Choice Errors.
(A) Normalized histograms of neurons containing calcium transients on a given fraction of trials within 200 ms of peak event-aligned transient rate (true positive), compared to neurons containing calcium transients in randomly selected time bin (false positive) for control mice (N=4 mice, n=273 neurons). (B) Cross-validation for ROC analysis (2-fold; 1000 iterations) with AUC values from full distribution (vertical dashed lines) reveals no change in control mice (fraction Light Off iterations less than true Light On >0.4; fraction Light On iterations greater than true Light Off value >0.4). (C) ROC curves calculated from control mice reveal unchanged discrimination by MSN calcium transients with optogenetic LED illumination (N=4 mice; n=273 neurons). (D) Normalized histograms of neurons containing calcium transients on a given fraction of trials within 200 ms of peak event-aligned transient rate (true positive), compared to fraction of neurons containing calcium transients in randomly selected time bin (false positive) for PV-eNpHR3.0 mice (N=4 mice, n=237 neurons). (E,F) ROC curves calculated using histograms from PV-eNpHR3.0 mice reveal reduced discrimination of task events by MSN calcium transients with optogenetic silencing of FSIs (N=4 mice; n=237 neurons). Significance calculated by bootstrapping analysis (2 fold, 1000 iterations; P<0.02). Fraction Light Off iterations less than true Light On <0.02; Fraction Light On iterations greater than true Light Off < 0.02. (G) Estimation of cross-talk between neighboring calcium units based on spatial distance. Similarity score calculated for each pair as mean(A & B) divided by (mean(A) * mean(B)), where A is a vector representing time points unit A spends above detection threshold, and B is the equivalent vector for unit B. Score of 1 corresponds to uncorrelated units, while higher scores reflect increased correlation. (H) Histogram of spatial distances between cell pairs in all control recordings (N=8 mice, n=555 MSNs) reveals only ~1.7% of total population of cell pairs experienced cross talk above the event detection threshold. (I) Similarity in task-event preference unit vectors (see methods) in Light On and Light Off conditions is not significantly affected by optogenetic silencing of FSIs (N=4 mice per group; n=273 control and n=237 PV-eNpHR3.0 neurons; unpaired two-tailed t-test P>0.05). All error bars are s.e.m. and center values are group means.
Highlights.
Fast-spiking interneurons (FSIs) mediate feed-forward control of bursting
Feed-forward control of bursting restricts calcium entry and plasticity
Restricting plasticity in striatal principal cells facilitates sequence learning
Data suggest a unified model for FSI function in gain control and plasticity
Acknowledgments
We thank Delanie Shulte and Benjamin Margolin for assistance with genotyping, histology, and microscopy, and the A.C.K. laboratory for comments on the manuscript. The authors wish to acknowledge Inscopix, Inc. for a scientific collaboration and providing access to nVoke integrated imaging and optogenetics system. Thank you to the Michael Gill and the Gladstone Behavioral Core and Nikon Imaging Core at UCSF for assistance with specific experiments. This work was funded by NIH R01 NS078435, F32 NS083369 (to S.F.O.) and K99 MH110597 (to S.F.O.), and RR018928 (to the Gladstone Institutes)
Footnotes
AUTHOR CONTRIBUTIONS
S.F.O. designed and performed experiments, analyzed data and wrote the manuscript. J.D.B. and A.C.K. designed experiments and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors have no competing financial interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agetsuma MH, Jordan P, Tao Kentaro, Fujisawa Shigeyoshi, Yuste Rafael. Parvalbumin-Positive Interneurons Regulate Neuronal Ensembles in Visual Cortex. Cerebral Cortex. 2017:1–15. doi: 10.1093/cercor/bhx169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73:159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhurin KI, Mac V, Golshani P, Masmanidis SC. Temporal correlations among functionally specialized striatal neural ensembles in reward-conditioned mice. J Neurophysiol. 2016;115:1521–1532. doi: 10.1152/jn.01037.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera G, Liang B, Zhang L, Gerfen CR, Culurciello E, Chen R, Li Y, Lin DT. Spatially Compact Neural Clusters in the Dorsal Striatum Encode Locomotion Relevant Information. Neuron. 2016;92:202–213. doi: 10.1016/j.neuron.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes TD, Kubota Y, Hu D, Jin DZ, Graybiel AM. Activity of striatal neurons reflects dynamic encoding and recoding of procedural memories. Nature. 2005;437:1158–1161. doi: 10.1038/nature04053. [DOI] [PubMed] [Google Scholar]
- Berke JD. Uncoordinated firing rate changes of striatal fast-spiking interneurons during behavioral task performance. J Neurosci. 2008;28:10075–10080. doi: 10.1523/JNEUROSCI.2192-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissiere S, Humeau Y, Luthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- Blitz DM, Regehr WG. Timing and specificity of feed-forward inhibition within the LGN. Neuron. 2005;45:917–928. doi: 10.1016/j.neuron.2005.01.033. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Neuronal activity regulates diffusion across the neck of dendritic spines. Science. 2005;310:866–869. doi: 10.1126/science.1114816. [DOI] [PubMed] [Google Scholar]
- Bronfeld M, Bar-Gad I. Tic disorders: what happens in the basal ganglia? Neuroscientist. 2013;19:101–108. doi: 10.1177/1073858412444466. [DOI] [PubMed] [Google Scholar]
- Burguiere E, Monteiro P, Feng G, Graybiel AM. Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science. 2013;340:1243–1246. doi: 10.1126/science.1232380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Eidelberg E. Commissural projection to the dentate gyrus of the rat: evidence for feed-forward inhibition. Brain Res. 1981;230:346–350. doi: 10.1016/0006-8993(81)90413-3. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Charpier S, Deniau JM. In vivo activity-dependent plasticity at cortico-striatal connections: evidence for physiological long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:7036–7040. doi: 10.1073/pnas.94.13.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Kim AN, Peters AJ, Komiyama T. Subtype-specific plasticity of inhibitory circuits in motor cortex during motor learning. Nat Neurosci. 2015;18:1109–1115. doi: 10.1038/nn.4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504:272–276. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Hensch TK. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 2000;404:183–186. doi: 10.1038/35004582. [DOI] [PubMed] [Google Scholar]
- Gage GJ, Stoetzner CR, Wiltschko AB, Berke JD. Selective activation of striatal fast-spiking interneurons during choice execution. Neuron. 2010;67:466–479. doi: 10.1016/j.neuron.2010.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernert M, Hamann M, Bennay M, Loscher W, Richter A. Deficit of striatal parvalbumin-reactive GABAergic interneurons and decreased basal ganglia output in a genetic rodent model of idiopathic paroxysmal dystonia. J Neurosci. 2000;20:7052–7058. doi: 10.1523/JNEUROSCI.20-18-07052.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Leventhal DK, Fensterheim BA, Pettibone JR, Berke JD, Kreitzer AC. Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. J Neurosci. 2011;31:15727–15731. doi: 10.1523/JNEUROSCI.3875-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223–2234. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Scanziani M. Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat Neurosci. 2006;9:807–815. doi: 10.1038/nn1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber D, Gutnisky DA, Peron S, O’Connor DH, Wiegert JS, Tian L, Oertner TG, Looger LL, Svoboda K. Multiple dynamic representations in the motor cortex during sensorimotor learning. Nature. 2012;484:473–478. doi: 10.1038/nature11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Ung RL, Resendez SL, Stamatakis AM, Taylor JG, Huang J, Veleta K, Kantak PA, Aita M, Shilling-Scrivo K, et al. Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell. 2015;160:516–527. doi: 10.1016/j.cell.2014.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalanithi PS, Zheng W, Kataoka Y, DiFiglia M, Grantz H, Saper CB, Schwartz ML, Leckman JF, Vaccarino FM. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci U S A. 2005;102:13307–13312. doi: 10.1073/pnas.0502624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka Y, Kalanithi PS, Grantz H, Schwartz ML, Saper C, Leckman JF, Vaccarino FM. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol. 2010;518:277–291. doi: 10.1002/cne.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Plenz D. Dendritic calcium encodes striatal neuron output during upstates. J Neurosci. 2002;22:1499–1512. doi: 10.1523/JNEUROSCI.22-05-01499.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. Independent optical excitation of distinct neural populations. Nat Methods. 2014;11:338–346. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Martins GJ, Paixao VB, Zhou P, Paninski L, Costa RM. The Spatiotemporal Organization of the Striatum Encodes Action Space. Neuron. 2017;95:1171–1180. e1177. doi: 10.1016/j.neuron.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nature neuroscience. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, Trachtenberg JT. A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature. 2013;501:543–546. doi: 10.1038/nature12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo SP, Trussell LO. Spontaneous spiking and synaptic depression underlie noradrenergic control of feed-forward inhibition. Neuron. 2011;71:306–318. doi: 10.1016/j.neuron.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Holley SM, Shobe JL, Chong NC, Cepeda C, Levine MS, Masmanidis SC. Parvalbumin Interneurons Modulate Striatal Output and Enhance Performance during Associative Learning. Neuron. 2017;93:1451–1463. e1454. doi: 10.1016/j.neuron.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kwan AC, Zhang S, Phoumthipphavong V, Flannery JG, Masmanidis SC, Taniguchi H, Huang ZJ, Zhang F, Boyden ES, et al. Activation of specific interneurons improves V1 feature selectivity and visual perception. Nature. 2012;488:379–383. doi: 10.1038/nature11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Xu ZC, Wilson CJ. Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. Journal of neurophysiology. 1994;71:1174–1189. doi: 10.1152/jn.1994.71.3.1174. [DOI] [PubMed] [Google Scholar]
- Owen SF, Tuncdemir SN, Bader PL, Tirko NN, Fishell G, Tsien RW. Oxytocin enhances hippocampal spike transmission by modulating fast-spiking interneurons. Nature. 2013;500:458–462. doi: 10.1038/nature12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard MG. Glutamate infused posttraining into the hippocampus or caudate-putamen differentially strengthens place and response learning. Proc Natl Acad Sci U S A. 1999;96:12881–12886. doi: 10.1073/pnas.96.22.12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard MG, McGaugh JL. Inactivation of hippocampus or caudate nucleus with lidocaine differentially affects expression of place and response learning. Neurobiol Learn Mem. 1996;65:65–72. doi: 10.1006/nlme.1996.0007. [DOI] [PubMed] [Google Scholar]
- Peters AJ, Chen SX, Komiyama T. Emergence of reproducible spatiotemporal activity during motor learning. Nature. 2014;510:263–267. doi: 10.1038/nature13235. [DOI] [PubMed] [Google Scholar]
- Planert H, Szydlowski SN, Hjorth JJ, Grillner S, Silberberg G. Dynamics of synaptic transmission between fast-spiking interneurons and striatal projection neurons of the direct and indirect pathways. J Neurosci. 2010;30:3499–3507. doi: 10.1523/JNEUROSCI.5139-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Marin-Burgin A, Adesnik H, Atallah BV, Scanziani M. Input normalization by global feedforward inhibition expands cortical dynamic range. Nature neuroscience. 2009;12:1577–1585. doi: 10.1038/nn.2441. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science (New York, NY. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Rapanelli M, Frick LR, Xu M, Groman SM, Jindachomthong K, Tamamaki N, Tanahira C, Taylor JR, Pittenger C. Targeted Interneuron Depletion in the Dorsal Striatum Produces Autism-like Behavioral Abnormalities in Male but Not Female Mice. Biol Psychiatry. 2017;82:194–203. doi: 10.1016/j.biopsych.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Hayton SJ, Sun GL, Fuccillo MV, Lim BK, Malenka RC. Input- and Output-Specific Regulation of Serial Order Performance by Corticostriatal Circuits. Neuron. 2015;88:345–356. doi: 10.1016/j.neuron.2015.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC, Buzsaki G. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci. 2012;15:769–775. doi: 10.1038/nn.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science (New York, NY. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal RM, Gaspar RC, Keefe KA, Wilcox KS. A rat brain slice preparation for characterizing both thalamostriatal and corticostriatal afferents. J Neurosci Methods. 2007;159:224–235. doi: 10.1016/j.jneumeth.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Saulnier JL, Begue A, Feng DD, Huang KW, Sabatini BL. Principles of Synaptic Organization of GABAergic Interneurons in the Striatum. Neuron. 2016;92:84–92. doi: 10.1016/j.neuron.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Jin X, Lima SQ, Costa RM. Complementary Contributions of Striatal Projection Pathways to Action Initiation and Execution. Cell. 2016;166:703–715. doi: 10.1016/j.cell.2016.06.032. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Runyan CA, Wang FL, Sur M. Division and subtraction by distinct cortical inhibitory networks in vivo. Nature. 2012;488:343–348. doi: 10.1038/nature11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Li L, Pittenger C. Ablation of fast-spiking interneurons in the dorsal striatum, recapitulating abnormalities seen post-mortem in Tourette syndrome, produces anxiety and elevated grooming. Neuroscience. 2016;324:321–329. doi: 10.1016/j.neuroscience.2016.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CF, Chiang MC, Gray DC, Prabhakaran M, Alvarado M, Juntti SA, Unger EK, Wells JA, Shah NM. Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell. 2013;153:896–909. doi: 10.1016/j.cell.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki-Sugiyama Y, Kang S, Cateau H, Fukai T, Hensch TK. Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature. 2009;462:218–221. doi: 10.1038/nature08485. [DOI] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nature reviews. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- Zhou P, Resendez SL, Stuber GD, Kass RE, Paninski L. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. 2016 doi: 10.7554/eLife.28728. arXiv 1605.07266v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Burns LD, Cocker ED, Hamel EO, Ghosh KK, Kitch LJ, El Gamal A, Schnitzer MJ. Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci. 2013;16:264–266. doi: 10.1038/nn.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A,B) Recording configuration using para-horizontal acute slices to preserve cortico-striatal connectivity for measurement of feed-forward inhibition. (C) Onset (20% of peak) of feed-forward IPSCs follows onset of EPSCs with a time-delay of 1–3 ms. x-axis is latency from stimulus onset. (N=10 mice, n=26 MSNs). (D) AMPA-R antagonist NBQX (10 μM) blocks excitatory synaptic transmission, suppressing both monosynaptic EPSC and di-synaptic feed-forward IPSC in all MSNs (N=2 mice, n=10 MSNs). (E,F) Equivalent suppression of EPSC and feed-forward IPSC in direct- and indirect-pathway MSNs (N=2 mice, n=5 MSNs per group). (G) Quantification of eNpHR3.0 expression within virus infection zone measuring penetrance (# neurons co-expressing both eNpHR3.0 and endogenous Parvalbumin protein divided by total # of neurons expressing Parvalbumin) and selectivity (# neurons co-expressing eNpHR3.0 and Parvalbumin divided by total number of eNpHR3.0-expressing neurons). (H) Lesion extent summarized and overlayed from 12 PV-cre mice injected with AAV-DIO-Casp3. Brains were sectioned and stained for endogenous parvalbumin. Lesion borders were manually traced and overlayed (red) at 5 locations spanning the rostral-caudal extent of striatum. (I) Recording configuration for interneuron-MSN pairs. (J) Tip-filling interneuron recording pipette with current-clamp internal solution allows initial identification of interneuron subtype by current injections. Lhx6-GFP mice label two distinct interneuron subtypes (FSIs and Persistent Low-Threshold Spiking interneurons). (K) Back-filling pipettes with voltage-clamp internal solution allows subsequent measurement of excitatory inputs onto interneuron and neighboring MSN in response to shared excitatory input. (L) FSIs receive stronger excitatory input than neighboring MSNs while PLTS interneurons receive weaker input. (N=4, mice n=10 FSI-MSN pairs, n=8 FSI-PLTS pairs; paired two-tailed t-test, P<10−4 for FSIs, P<10−3 for PLTS). All error bars are s.e.m. and center values are group means.
(A) Exemplar MSN recording of feed-forward inhibition showing suppression by IEM-1460 (50 μM). (B) Timecourse of IEM-1460 (50 μM) suppression of feed-forward IPSC and recovery by washout. (C,D) Group data for IEM-1460 suppression of feed-forward IPSC and recovery by washout. (N=6 mice, n=8 MSNs; Before-IEM and IEM-Wash for IPSC, sign rank test P<0.01; Before-IEM and Before-wash for EPSC, sign rank test P>0.2). (E) Selective suppression of feed-forward IPSCs but not EPSCs onto D1-tmt-positive direct pathway dMSNs by IEM-1460 (50 μM). (F) Selective suppression of feed-forward IPSCs but not EPSCs onto D1-tmt-negative indirect pathway iMSNs by IEM-1460 (50 μM). (G) Equivalent lack of suppression of EPSCs onto direct- and indirect-pathway MSNs by IEM-1460 (N=6 mice, n=5 dMSNs, n=3 iMSNs; rank sum test P>0.05). (H) Equivalent suppression of IPSCs onto direct- and indirect-pathway MSNs by IEM-1460. (N=6 mice, n=5 dMSNs, n=3 iMSNs; rank sum test P>0.05). (I) PV-eNpHR3.0 optogenetic suppression of feed-forward IPSCs onto direct-pathway MSNs. (J) PV-eNpHR3.0 optogenetic suppression of feed-forward IPSCs onto indirect pathway MSNs. (K) Equivalent lack of effect of PV-eNpHR3.0 on monosynaptic EPSCs onto direct- and indirect-pathway MSNs. (N=4 mice, n=6 dMSNs; rank sum test P>0.6). (L) Equivalent suppression of feed-forward IPSCs by PV-eNpHR3.0 in direct- and indirect-pathway MSNs. (N=5 mice, n=6 iMSNs; rank sum test P>0.5). All error bars are s.e.m. and center values are group means.
(A) Modulation index for FSIs (top) and MSNs (bottom) from in vivo physiological recordings from control mice moving freely in an open field shows no modulation (sign rank test against 0.5; P>0.1 MSNs, P>0.4 FSIs). (B) Modulation index for FSIs (top) and MSNs (bottom) in PV-eNpHR3.0 mice shows silencing of FSIs (sign rank test against 0.5; P<10−8 for MSNs, P<0.005 for FSIs). (C) Comparison of modulation index for spiking and calcium transients reveals greater disinhibition of calcium transients than individual spike rates during optogenetic suppression of FSIs (N=5 PV-eNpHR3.0 mice and n=34 MSNs for electrophysiology; N=2 mCherry control mice and n=10 MSNs for control electrophysiology; N=4 PV-eNpHR3.0 mice and n=234 neurons for imaging; N=4 mCherry control mice and n=400 neurons for control imaging. Rank sum test spikes control vs spikes PV-eNpHR3.0 P<0.003. Rank sum test transients control vs. transients PV-eNpHR3.0 P<10−42. Rank sum test PV-eNpHR3.0 spikes vs. PV-eNpHR3.0 transients P<10−3). Spiking data is the same as that shown in Fig 1N. (D) Calcium responses to individual spikes were simulated with a decaying exponential (τ =0.812 sec fit from GCaMP6f data in Fig S5) and an amplitude set with a saturating non-linear transformation to approximate the non-linear responses in Fig S5m without diverging at high burst rates. Calcium transient amplitudes were set at a minimum value of 0.1 when initial simulated calcium was >15. (E) Simulated calcium responses to 50 Hz bursts containing 1–40 spikes. (F) Amplitudes of simulated bursts in panel E reflect saturating nonlinear transformation. (G) Exemplar simulated calcium trace calculated from a portion of the spike train of a single putative MSN recorded in vivo (top) shows bursting aligned to optogenetic silencing of neighboring FSIs (amber blocks). Shuffled spike train with equivalent average firing rate produces many fewer calcium bursts (bottom), highlighting importance of MSN firing pattern in generating calcium bursts. Note difference in scale between true and shuffled simulated calcium signals. (H) Raster plot of recorded spikes from exemplar MSN plotted in panel G. (I) Raster plot of simulated calcium transients from the same recording. (J) Modulation index of recorded spikes and simulated calcium transients from all MSNs recorded with this protocol (N=5 PV-eNpHR3.0 mice, n=60 MSNs, sign rank test against null value of 0.5, P<10−6 for spikes, P<10−4 for simulated calcium transients; N=2 control mice and n=15 control MSNs, sign rank against 0.5, P>0.05 for spikes and P>0.4 for calcium transients). (K) Directly comparing modulation index of recorded spikes and simulated calcium transients reveals significantly greater modulation of simulated calcium transients (N=5 mice, n=60 MSNs, sign rank test P<0.01). All error bars are s.e.m. and center values are group means.
(A) Recording configuration for acute slice calcium imaging with Oregon-Green BAPTA-6F (50 μM) in recording pipette. (B) Exemplar MSN current clamp recording traces (top) with current injection of varying duration. Calcium signal (ΔF/F) plotted below. (C) Normalized group data reveals Oregon-Green BAPTA-6F linearly reports action potential-induced calcium influx (N=5 mice, n=27 MSNs). Responses normalized to average ΔF/F from sweeps in containing >10 spikes. (D) Tonic current injection altered the resting potential by +21.6 mV (N=3 mice, n=11 MSNs, two-tailed t-test P<10−6). (E,F) Spike-to-calcium relationships remains linear and unaltered despite changing resting potential. (N=3 mice, n=11 neurons, sign rank test P>0.2). (G) Recording configuration for acute slice calcium imaging with optogenetic manipulation of FSIs. (H) Exemplar MSN calcium signal recorded by subtracting baseline image from image acquired at the peak calcium signal. (I) Exemplar current clamp recordings (top) and calcium signals (bottom) evoked by 180 ms current injection under control conditions (left) and during optogenetic activation of FSIs (right). Optogenetic stimulation of FSIs triggered simultaneously with current injection to MSN to mimic coincident excitatory input. (J) Spike-to-calcium relationship remains linear despite optogenetic activation of FSIs. (K) ChrimsonR activation of FSIs reduces number of action potentials evoked by current injection into MSNs (N=4 mice, n=16 neurons; 2-way ANOVA with interaction Popto <10−17, PstimDur<10−50, Pinteract<10−13). (L) ChrimsonR activation of FSIs does not affect the spike-to-calcium transformation in MSNs (N=4 mice, n=16 neurons; two-tailed t-test P>0.6). (M) GCaMP6f calcium responses scale supralinearly with number of spikes in a burst (N=1 mouse, n=6 MSNs). Normalization across recordings as in Panel (C). All error bars are s.e.m. and center values are group means.
(A) Unilateral intracranial injection of IEM-1460 (0.5μL, 1.5mM – intermediate dosage) into dorsolateral striatum induced dyskinetic axial twisting, quantified by rotational bias (contra turns)/(ipsi + contra turns) in both control and FSI-lesioned mice (N=5 mice per group, 2-way ANOVA PGroup>0.2; unpaired two-tailed t-test between groups P>0.1 each condition). (B) AMPA/NMDA ratios recorded in D1-tdTomato reporter mice to identify subtype of the recorded MSN reveal most of the increase in AMPA/NMDA ratios in PV-Casp3 lesioned mice is driven by indirect pathway iMSNs, but this trend is not statistically significant by 2-way ANOVA. iMSNs relative to tmt+ dMSNs. (N=9 mice per group; n=10 control dMSNs, n=11 control iMSNs; n=9 PV-Casp3 dMSNs, n=11 PV-Casp3 iMSNs, two-way ANOVA PPathway>0.2, PCasp3<0.03, Pinteract>0.05). (C) Fraction of trials completed (sequence completed before 30 sec maximum duration following trial initiation), and (D) inter-trial interval are unaffected in FSI-lesioned mice, indicating unimpaired motivation. (E) Schematic describing two types of error following trial initiation. (F) Increased fraction of Poke 2 Errors in FSI-lesioned mice. (G) Increased fraction of Poke 3 Errors in FSI-lesioned mice. (N=11 mice per group. two-way ANOVA PGroup values reported on charts). All error bars are s.e.m. and center values are group means.
(A) Schematic of “Choice Error” in which the mouse proceeds immediately to the incorrect second port after initiation. (B) Schematic of “Execution Error” in which the mouse approaches the correct second port but fails to break the infrared beam before proceeding to the third port. (C) Latency histograms reveal increased errors at short latencies in FSI-lesioned mice (arrows on Sessions #1–3), consistent with a predominance of Choice Errors.
(A) Normalized histograms of neurons containing calcium transients on a given fraction of trials within 200 ms of peak event-aligned transient rate (true positive), compared to neurons containing calcium transients in randomly selected time bin (false positive) for control mice (N=4 mice, n=273 neurons). (B) Cross-validation for ROC analysis (2-fold; 1000 iterations) with AUC values from full distribution (vertical dashed lines) reveals no change in control mice (fraction Light Off iterations less than true Light On >0.4; fraction Light On iterations greater than true Light Off value >0.4). (C) ROC curves calculated from control mice reveal unchanged discrimination by MSN calcium transients with optogenetic LED illumination (N=4 mice; n=273 neurons). (D) Normalized histograms of neurons containing calcium transients on a given fraction of trials within 200 ms of peak event-aligned transient rate (true positive), compared to fraction of neurons containing calcium transients in randomly selected time bin (false positive) for PV-eNpHR3.0 mice (N=4 mice, n=237 neurons). (E,F) ROC curves calculated using histograms from PV-eNpHR3.0 mice reveal reduced discrimination of task events by MSN calcium transients with optogenetic silencing of FSIs (N=4 mice; n=237 neurons). Significance calculated by bootstrapping analysis (2 fold, 1000 iterations; P<0.02). Fraction Light Off iterations less than true Light On <0.02; Fraction Light On iterations greater than true Light Off < 0.02. (G) Estimation of cross-talk between neighboring calcium units based on spatial distance. Similarity score calculated for each pair as mean(A & B) divided by (mean(A) * mean(B)), where A is a vector representing time points unit A spends above detection threshold, and B is the equivalent vector for unit B. Score of 1 corresponds to uncorrelated units, while higher scores reflect increased correlation. (H) Histogram of spatial distances between cell pairs in all control recordings (N=8 mice, n=555 MSNs) reveals only ~1.7% of total population of cell pairs experienced cross talk above the event detection threshold. (I) Similarity in task-event preference unit vectors (see methods) in Light On and Light Off conditions is not significantly affected by optogenetic silencing of FSIs (N=4 mice per group; n=273 control and n=237 PV-eNpHR3.0 neurons; unpaired two-tailed t-test P>0.05). All error bars are s.e.m. and center values are group means.
Data Availability Statement
All data and software are available upon request.