

Abstract
Breast cancer is the most prevalent malignancy in women and the second most common cause of cancer-related death worldwide. Despite major innovations in early detection and advanced therapeutics, up to 30% of women with node-negative breast cancer and 70% of women with node-positive breast cancer will develop recurrence. The recognition that breast tumors are infiltrated by a complex array of immune cells that influence their development, progression, and metastasis, as well as their responsiveness to systemic therapies has sparked major interest in the development of immunotherapies. In fact, not only the native host immune system can be altered to promote potent antitumor response, but also its components can be manipulated to generate effective therapeutic strategies. We present here a review of the major approaches to immunotherapy in breast cancers, both successes and failures, as well as new therapies on the horizon.
Keywords: adoptive transfer, breast cancer, cancer vaccine, cytokines, immunomodulation, immunotherapy, monoclonal antibody, oncolytic virotherapy
Introduction
Over the past half century, advancements in our understanding of breast cancer biology have transformed the current landscape of disease management, leading to improvements in early detection strategies, development of breast-conserving surgery techniques, utilization of cytotoxic chemotherapy regimens for the treatment of both local and metastatic disease, engineering of targeted therapies against the hormone pathway and human epidermal growth factor receptor 2 (HER2/neu), and employment of hormonally directed therapies as a preventive measure [1]. This evolution in breast cancer management has led to a one-third reduction in mortality since the year 1990 [2,3], yet breast cancer remains the most prevalent malignancy in women and second most common cause of cancer-related death worldwide [4,5]. Disease-related mortality stems primarily from primary (de novo) or secondary resistance to available systemic therapies. A number of novel approaches are being pursued to prevent or circumvent mechanisms of treatment resistance and hopefully improve long-term survival. The recognition of the role of the immune microenvironment in tumor biology has opened the door to an exciting era in oncology in which immune components are manipulated to harness the inherent ability of the host’s immune system to combat malignancy. In this review, immunotherapy approaches for the treatment of breast cancers will be explored with a focus on both successes and failures as well as new therapies on the horizon.
Immune microenvironment in breast cancer
Breast tumors are complex systems comprising two primary components: the cancer cells typically derived from malignant transformation of mammary ductal or lobular cells, and the surrounding stromal compartment composed of a variety of normal host cells (e.g., fibroblasts, immune cells, and cells of the vasculature) and extracellular matrix molecules that are conscripted to provide a biochemical and structural milieu supportive of tumor development, progression, and metastasis [6–10]. One major class of stromal host cells, the immune infiltrate, has garnered considerable attention for its exploitability in the treatment of many malignancies, including breast cancer [11].
Cancer-related inflammation has long been recognized by early pathologists, and in 1863 was even postulated to play a causative role in oncogenesis [12]. Although malignancy-associated inflammation is no longer considered a driver of tumorigenesis, chronic inflammation increases cancer risk and plays a major role in disease pathophysiology [11,13–15]. In breast malignancies, this immune infiltrate has been well studied. Immunohistochemical analyses in the early-to-mid-twentieth century showed a correlation between high levels of mononuclear immune cell infiltration and better prognosis, particularly in the medullary carcinoma histopathologic subtype of breast cancer [16,17]. Immunophenotyping studies in the 1980s further characterized breast tumor immune infiltrates, identifying cells derived from both myeloid (innate) and lymphoid (adaptive) lineages, including T lymphocytes (primarily CD8+ and CD4+ cells), macrophages, natural killer (NK) cells, and B lymphocytes [18,19].
Tumor-infiltrating lymphocytes (TILs) are one of the most prominent groups of immune cells in breast cancers [20] and consist of predominantly CD8+ cytotoxic T lymphocytes, CD4+ T helper lymphocytes, NK cells, and FOXP3+ T regulatory cells (Tregs) [21–23]. High levels of CD8+ T-cell infiltration into breast tumors have been correlated with improved outcomes [24–27], due in part to cancer cell-directed cytotoxicity. Similarly, high levels of NK-cell infiltration are associated with improved prognosis in breast cancer [28] by mediating CD8+ cytotoxic T-lymphocyte activity through the production of proinflammatory cytokines such as interferon (IFN)-γ [29–31].
In contrast, high levels of Tregs, which play pivotal roles in immune tolerance by suppressing T-cell activation in the normal physiologic state and killing or inactivating effector T cells in highly inflammatory states [32,33], have been shown to promote tumor progression and are associated with poor prognosis [34,35]. Furthermore, depletion of Tregs in preclinical models by blocking the interleukin-2 pathway has been shown to inhibit mammary tumor growth, likely by reprogramming the microenvironment from an immunosuppressive one to one that is amenable to other immunotherapies (e.g., checkpoint inhibitors) [36,37]. This is supported by patient data showing that reduction in FOXP3+ regulatory T cells following neoadjuvant chemotherapy is associated with pathologic complete responses [24].
The role of CD4+ T helper lymphocytes in breast cancer is less clear, as they have been noted to have both tumor-promoting and tumor-inhibiting properties, depending on their cytokine expression profile and microenvironmental context. Proinflammatory (Th1) CD4+ T lymphocytes, which typically express IFN-γ, have been shown to promote cytotoxic T-lymphocyte activity [38]. In contrast, anti-inflammatory (Th2) CD4+ T lymphocytes, which classically express interleukin-4 (IL-4), have been shown to promote metastasis in preclinical models [39,40]. Similarly, the Th17 subset of CD4+ helper T cells commonly infiltrates ER− and triple-negative malignancies and is associated with poor prognosis [41] owing to its promoting effects on cancer cell proliferation, metastasis, and chemoresistance [42,43].
Myeloid lineage cells may comprise up to 80% of the immune cell infiltrate in breast tumors [44], and consist primarily of macrophages, as well as smaller numbers of neutrophils, immature granulocytes, and myeloid-derived suppressor cells. In a classification scheme akin to that of T helper lymphocyte polarization, these macrophages and neutrophils are similarly classified as type 1 or 2 based on analogous cytokine profiles [21]. Breast tumor-associated macrophages (TAMs) are typically M2-polarized, expressing and responding to cytokines such as IL-4, and have been shown to promote angiogenesis [45], cancer cell intravasation into blood vessels [46], diminish responses to cytotoxic chemotherapies [39,47–49], and facilitate metastasis. High levels of TAM infiltration are associated with poorer outcomes [50,51]. In addition to TAMs, other myeloid lineage immune cells have also been identified in the breast tumor immune infiltrate. Mast cells promote angiogenesis in breast tumors and metastatic lesions [52], but their relationship with prognosis varies based on breast cancer subtype and tumor stage [53]. Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells that promote an immunosuppressive tumor microenvironment through multiple mechanisms, including expression of Th2-type cytokines, production of reactive oxygen species, and the metabolism of arginine [54].
The role of the B-lymphocyte infiltrate in breast malignancies is less understood. Studies in medullary carcinoma of the breast, in which high levels of B cell infiltration into tumors are associated with better outcomes, suggest that B lymphocytes undergo clonal proliferation and affinity maturation within primary tumors against both host- and tumor-directed antigens, which ultimately lead to activation of apoptosis [55,56]. These data are further supported by studies in intraductal carcinomas, in which similar results were observed [57,58].
Immunotherapy in breast cancer
The recognition of an inflammatory infiltrate in solid tumors and our growing understanding of the varied functions of tumor inflammation have raised the question of whether the immune system can be effectively exploited to promote tumor destruction. This led to the birth of the field of cancer immunotherapy, which is broadly defined as any therapeutic approach that utilizes a specific component of the immune system or enhances or augments the intrinsic host immune response to treat malignancy [59]. Two fundamental principles have been recognized as guiding the development of cancer immunotherapy. They are as follows: (1) both the innate and adaptive immune systems play an important role in cancer cell immunosurveillance and destruction by utilizing the same mechanisms in play during infection with foreign pathogens and (2) oncogenesis and progression occurs through the selection and outgrowth of cancer cells with low immunogenicity and the generation of an immunosuppressive microenvironment [60]. Therapeutic strategies have therefore focused on activating both innate and antibody-mediated immune responses to induce cancer cell death. These technologies include cytokines and growth factors, immunomodulatory agents, alteration of the tumor microenvironment, passive immunization with monoclonal antibodies, bispecific and multispecific antibodies, checkpoint inhibitors, vaccines, oncolytic viruses, and adoptive T-cell transfer [61,62] (Table 1).
Table 1.
Strategies for activation of the immune system against breast cancer.
| Class | Mechanism | Examples |
|---|---|---|
| Cytokines | Bind to cytokine receptors to initiate cell signaling pathways and stimulate immune cell trafficking and effector function | Interleukin-2 Interleukin-12 |
| Growth factors | Increase number of circulating granulocytes | G-CSF GM-CSF |
| Toll-like receptor agonists | Bind TLRs to activate antigen-presenting cells (dendritic cells) to upregulate expression of cytokines and co-stimulatory molecules to attract and stimulate effector immune cells (cytotoxic T lymphocytes) | Polyadenylic-polyuridylic acid (Poly A:U) Polyinosinic-polycytidylic acid (Poly I:C) |
| Immune checkpoint inhibitors | Antibody to CTLA-4, PD-1, or PD-L1 molecules releases T cells from inhibitory signals, thereby unleashing cytotoxic T-cell activity | Ipilimumab (CTLA-4 antibody) Nivolumab (PD-1 antibody) Pembrolizumab (PD-1 antibody) Atezolizumab (PD-L1 antibody) Avelumab (PD-L1 antibody) Durvalumab (PD-L1 antibody) |
| Bispecific, multispecific antibodies | Simultaneously interact with a cancer-specific epitope and stimulatory molecule(s) on effector cell(s) | HER2/CD3 bispecific antibodies |
| Adoptive cell transfer | Infusion of T cells stimulated or engineered to have antitumor effector functions | Chimeric antigen receptor (CAR) T cells expressing HER2/neu |
| Oncolytic viruses | Viruses with specific tropism for cancer cells that induce cancer cell death and activate tumor-directed immune responses | JX-594 (pexastimogene devacirepvec) vaccinia poxvirus expressing GM-CSF) |
| Vaccines | Active immunization against tumor-specific antigens | Nelipepimut-S vaccine against HER2/neu |
Therapeutic strategies to harness the innate and adaptive immune system against breast cancer cells include nonspecific immune system stimulation with cytokines, growth factors, and Toll-like receptor agonists, release of T cells from inhibitory PD-L1 signals, use of antibodies to transmembrane tyrosine kinase receptor HER2 to tag HER2+ breast cancer cells for immune-mediated destruction, stimulation of T cells within the HER2+ breast tumor microenvironment, active vaccination or in vitro reprogramming of T cells against HER2/neu, and injection of oncolytic viruses. See text for details.
G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HER2, human epidermal growth factor receptor 2; TLR, Toll-like receptor.
Cytokines and growth factors
Cytokines and growth factors are secreted or membrane-bound proteins produced by both innate and adaptive immune cells in response to a stimulus (e.g., a pathogen or cancer cell). They exert pleiotropic effects on components of the immune system by binding to specific cytokine receptors on many different effector cells, initiating signaling pathways to modulate cell trafficking, survival, proliferation, maturation, and function, thereby promoting or inhibiting tumor-directed responses while maintaining immunologic homeostasis and self-tolerance. These molecules can also exert effects on cancer cells, contributing to their proliferation, invasiveness, intravasation, metastasis, and chemoresistance [63–66]. Activating or inhibiting these signaling pathways has been a major focus in immunotherapy research.
Cytokine therapy is a therapeutic strategy that was first recognized in the late 1800s when inoculation of highly virulent streptococcal cultures was shown to induce remission in patients with inoperable, metastatic sarcoma [67]. Later successes using systemic IL-2 for the treatment of metastatic renal cell carcinoma and metastatic melanoma [68,69] paved the application of cytokine therapy to other malignancies. However, in breast cancer, systemic cytokine treatment has been less successful for the treatment of breast cancer. IFNα was the first cytokine noted to have a potentially beneficial effect in the treatment of breast cancer. In 1980, Gutterman et al. administered partially purified IFNα derived from human buffy coat preparations to 17 patients with recurrent, metastatic breast cancer and noted 7 patients had tumor regression with 6 patients achieving partial remission as defined by >50% objective decrease in tumor size [70]. A subsequent Phase II study in patients with recurrent metastatic breast cancer who had not received cytotoxic salvage chemotherapy was conducted to determine the efficacy of similarly derived, partially purified IFNα preparations as monotherapy, and it was confirmed that systemic cytokine administration was indeed capable of inducing a partial objective response in 5 of 23 patients with breast cancer and a measurable response in 6 of 23 patients [71]. However, subsequent Phase II trials utilizing purified, recombinant IFNα did not yield significant tumor responses in the treatment of metastatic breast cancers [72,73]. Studies with systemic administration of other recombinant interferons were similarly unsuccessful in breast cancer [74–76], likely owing to the lack of other cytokines and chemokines present in the original preparations. The addition of IL-2 to IFN therapy has also been ineffective [77].
Limiting factors in the successful application of cytokines include tachyphylaxis with subsequent administrations, ineffective stimulation of T-cell-mediated tumor-directed responses, and significant dose-limiting side effects with systemic therapy, including overwhelming fatigue and severe cytokine release syndromes. Strategies for improving immune activation and decreasing the systemic effects of cytokine therapy are underway in preclinical models and early-phase clinical trials. These approaches include intra-tumoral injection of cytokines [78], combination of cytokine therapy with systemic therapy [79,80], gene therapy with adenovirus vectors and oncolytic viruses expressing cytokines and chemokines under the direction of tissue-specific promotors [81,82], tumor-targeted super-antigen therapy utilizing components of bacterial toxins [83], and cytokine-antibody fusion molecules (reviewed [84]).
Systemic administration of growth factors has similarly found limited use for inducing remission of breast cancer. However, in the management of chemotherapy-induced toxicities, growth factors, particularly granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF), are routinely used for the prevention of neutropenia [85,86]. Another growing niche for growth factors in breast cancer therapy is as adjuvants to other immunotherapies, such as cancer-directed vaccines.
Disruption of both cytokine and growth factor signaling pathways has also been a major area of immunotherapy research. Neutralizing antibodies against cytokines and certain chemokines have been shown to be effective in preclinical models and are currently in early-phase clinical trials with results forthcoming [87].
Immunomodulatory agents
Immunomodulatory agents are chemicals capable of altering immune responses, angiogenesis, and cancer cell proliferation. The earliest immunomodulatory agents focused on generating potent innate immune responses to stimulate lasting cytotoxic CD8+ T-cell effector functions through the activation of Toll-like receptors (TLRs). TLR agonists are nonspecific molecules capable of binding these pattern-recognition receptors, thereby activating transcriptional pathways regulated by nuclear factor (NF)-κB and activating complement pathways, opsonization, and phagocytosis, inducing apoptosis and stimulating cytotoxic T-cell responses [88].
Preclinical animal studies in the 1960s and 1970s showed that administration of polyadenylic-polyuridylic acid (poly A:U), a double-stranded RNA polynucleotide that stimulates TLR3, after mastectomy in spontaneous tumor and orthotopic transplant models of breast cancer significantly delayed relapse [89,90]. Furthermore, administration of poly A:U to newborn mice that developed spontaneous mammary tumors resulted in reduced incidence of malignancy [91]. This agent entered clinical trials in France during the 1980s [92–94] as an adjuvant therapy in surgically resectable breast cancer and showed significantly improved overall survival in node-positive patients. Head-to-head studies comparing adjuvant cytotoxic therapy with cyclophosphamide, methotrexate, and fluorouracil (CMF) to adjuvant poly A:U demonstrated a significantly improved disease-free survival and reduced incidence of metastasis in patients with limited disease (i.e., 1–3 positive nodes) with positive TLR3-expression [94]. Similar results were obtained when a combination of adjuvant locoregional radiation and poly A:U therapy was compared to adjuvant chemotherapy with CMF [95,96]. Despite these promising results, adjuvant poly A:U has not been approved for the treatment of breast cancer, likely owing to its applicability to a very small population of women with limited disease. Another TLR3 agonist, polyinosinic-polycytidylic acid (poly I:C), has been shown to induce tumor necrosis [97], reduce metastases [98], and induce long-lasting CD8+ T-cell responses in combination with CpG oligodeoxynucleotides (a TLR9 agonist) [99] in preclinical models. The use of poly I:C has been limited owing to significant toxic effects, specifically severe cytokine release syndromes.
Cytokine-release syndromes with the systemic administration of TLR agonists have largely limited their use as monotherapy in clinical scenarios. However, research into modifying delivery and structural components to reduce their side-effect profiles and their use as immunotherapeutic adjuvants is ongoing. Several TLR agonists, including monophosphoryl lipid A and OK-432 (TLR4 agonists) [100], poly ICLC (TLR3 agonist that is a compounded mixture of poly I:C and poly-lysine), and CpG oligodeoxynucleotide (TLR9 agonist), are being studied as vaccine enhancers. They are also being studied as adjuncts to systemic or radiation therapy [99,101], as well as local therapeutics, for example, topical imiquimod (TLR7 agonist) as therapy for skin metastases in breast cancer [102].
Other immunomodulatory agents inhibit immunosuppressive molecules. For example, indoleamine 2,3-dioxygenase (IDO) is an enzyme that metabolizes tryptophan to produce metabolites that stimulate the expansion of Treg populations, thereby suppressing cytotoxic T-cell activity [103]. In a transgenic model of breast cancer, indoximod (an inhibitor of IDO) in combination with cytotoxic systemic therapy induced tumor regression [104]. These studies have resulted in the initiation of clinical trials utilizing indoximod with cytotoxic therapy for the treatment of breast cancer [105,106].
Altering the tumor microenvironment
The complexity of the tumor microenvironment and the many roles its constituents play in breast cancer development, progression, metastasis, and response to chemotherapeutic agents has altered the tumor microenvironment a particularly exciting area of research. Efforts to target the immune-stroma include those aimed at preventing recruitment, altering polarization, inhibiting activation, and depleting certain tumor-infiltrating immune cells that may contribute to tumor pathogenesis. TAMs are of particular interest, in part because they can be polarized by Th2 cytokines to adopt an M2 phenotype with tumor-promoting, immunosuppressive functions [107]. Moreover, they comprise a significant proportion of tumor-infiltrating leukocytes in breast cancers, correlating with poor prognosis [108,109]. Colony-stimulating factor-1 (CSF-1) is a regulatory growth factor crucial for macrophage survival, proliferation, and effector functions [110], and neutralizing this molecule or its cognate receptor CSF-1R has been a particularly attractive approach to targeting TAMs. Indeed, blocking antibodies against CSF-1R leads to the depletion of TAMs and FOXP3+ Treg populations, an increase in the ratio of CD8+:CD4+ T cells, and delays in tumor growth in TAM-rich tumor models [111]. Neutralizing antibodies against CSF-1 in combination with paclitaxel effectively reduces metastatic burden in a mouse model of luminal B breast cancer in part by augmenting CD8+ T-cell activity [109]. There are currently three monoclonal antibodies targeting CSF-1R in early-phase trials: emactuzumab administered alone or in combination with paclitaxel (NCT01494688) or in combination with the PD-L1 inhibitor atezolizumab (NCT02323191) for unspecified advanced solid tumors, LY3022855 as monotherapy for unspecified treatment-refractory solid tumors (NCT01346358) or specifically for treatment-refractory breast or prostate cancer (NCT02265536), and AMG 820 as monotherapy in unspecified advanced solid malignancies (NCT01444404).
Preventing myeloid cell recruitment is another strategy for interfering with TAM function. Studies in murine models of breast cancer have shown that the interaction between the chemokine CCL2 (C-C motif chemokine ligand 2) and its receptor CCR2 (C-C chemokine receptor 2) promotes recruitment of TAMs into tumors and metastatic sites, promoting tumor angiogenesis, cancer cell extravasation, and metastasis, and inhibiting CCL2/CCR2-mediated recruitment delays relapse, improves survival, and decreases metastatic burden [49,112,113]. Despite promise as a therapeutic strategy in preclinical models, CCL2 antagonism has had limited success. The anti-CCL2 monoclonal antibody carlumab, while generally safe in a Phase I trial for advanced solid malignancies [114], ultimately did not demonstrate in vivo ability to block the CCL2/CCR2 pathway or exert antitumor effects in metastatic castrate-resistant prostate cancer, resulting in suspension of further planned studies with carlumab [115]. Similar results were obtained when an antibody against CCR2 was used in patients with advanced malignancy and bony metastases [116].
Immune checkpoint Inhibition
The detection of immune checkpoints that prevent autoimmunity when normal immune responses are triggered, the identification of specific cell types and molecules involved in this process, and the understanding of roles that each of these cells and molecules play in promoting an immunosuppressive tumor microenvironment have led to the development of blocking monoclonal antibodies aimed at improving the host immune response to cancer. By antagonizing these molecules, potent anticancer T-cell responses may be reactivated. This class of therapies, called immune checkpoint inhibitors, has revolutionized the treatment of various malignancies, including malignant melanoma, renal cell carcinoma, non-Hodgkin’s lymphoma and smoking-associated non-small-cell lung cancer [117].
Immune checkpoint inhibitors function by interfering with two separate T-cell inhibitory pathways: cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed death 1 (PD-1). Traditionally, activation of T-cell responses requires two functional synapses between antigen-presenting cells (APCs) and T cells: (1) binding of an antigen-containing major histocompatibility complex (MHC) molecule with a T-cell receptor (TCR) and (2) binding of the APC cell surface molecule B7 to the T-cell co-stimulatory molecule CD28. These two interactions result in upregulation of IL-2 expression by the activated T cell, leading to proliferation and stimulation of effector function. CTLA-4 and PD-1 are cell surface molecules expressed by T cells that are structurally homologous to CD28, but which act as co-inhibitory molecules when bound to their respective ligands. CTLA-4 acts a co-inhibitory signal to prevent IL-2 expression by T cells when complexed with B7 on an APC. PD-1, which is expressed on a wide variety of cells that include leukocytes and parenchymal cells, binds to its ligands PD-L1 and PD-L2 on APCs. Expression of CTLA-4 depends on potency of the immunologic trigger, while expression of PD-1 depends on the duration of the immunologic response and is associated with T-cell exhaustion. Increased expression of CTLA-4 and PD-L1 in tumor tissues allows for the evasion of tumor-directed T-cell responses [118,119] (Figure 1).
Figure 1.

Immune checkpoint inhibitors.
A. Normal T-cell activation requires two functional synapses: binding of an antigen-containing MHC molecule with a T-cell receptor and binding of the co-stimulatory molecule CD28 found on T cells with B7, found on antigen-presenting cells. B. CTLA-4 is a co-inhibitory molecule present on normal T cells. Binding of CTLA-4 with B7 inhibits activation of T cells. Blocking antibodies against CTLA-4 prevents its binding with B7, thereby allowing for CD28 interaction with B7 and T-cell activation. C. PD-1 is a co-inhibitory molecule present on normal T cells. Its ligand, PD-L1, is upregulated in cancer cells. Blocking antibodies against either PD-1 or PD-L1 allow for T-cell activation.
There are currently six immune checkpoint inhibitors with FDA-approved indications for clinical use: the CTLA-4 blocking antibody ipilimumab, the PD-1 blocking antibodies nivolumab and pembrolizumab, and the PD-L1 neutralizing antibodies atezolizumab, avelumab, and durvalumab. While there are very rare indications for the use of checkpoint inhibitors in breast cancer treatment, there is evidence to suggest that these therapies may be effective, particularly in breast cancer subtypes with large immune infiltrates, including HER2+ and TNBC. These data include increased mutational rates resulting in a larger burden of tumor-associated neoantigens [120], recruitment of tumor-infiltrating lymphocytes [121], and PD-L1 expression [122,123]. Indeed, several early-phase trials are underway with checkpoint inhibitors in monotherapy (KEYNOTE-012 [124] and KEYNOTE-086 [125] evaluating pembrolizumab for TNBC, KEYNOTE-028 evaluating pembrolizumab for PD-L1+ ER+ HER2− breast cancer [126], and the JAVELIN trial evaluating avelumab for locally advanced or metastatic breast cancers) or in combination with cytotoxic therapy (nanoparticle albumin-bound paclitaxel with atezolizumab [127–129] and the I-SPY-2 trial evaluating paclitaxel with or without pembrolizumab for 12 weeks followed by doxorubicin and cyclophosphamide for 8–12 weeks [130]). Preliminary results from early-phase trials with pembrolizumab [125] or atezolizumab [131] monotherapy for TNBC indicate that patients who initially respond to therapy have durable treatment responses. It does, however, appear that these responses are dependent on high levels of tumor-infiltrating lymphocytes [125,131], and this response wanes with increasing exposure to systemic cytotoxic chemotherapy [132] (Table 2).
Table 2.
Clinical trials utilizing immune checkpoint inhibitors for the treatment of breast cancer.
| Trial | Inhibitor | Therapeutic strategy | No. pts. | ORR | Median time to response (Range) | Median DoR (months) | Toxicities |
|---|---|---|---|---|---|---|---|
| KEYNOTE-012 Phase I [124] |
Pembrolizumab | Single agent in advanced PD-L1+ TNBC | 32 | 18.5% | 17.9 weeks (7.3 to 32.4 weeks) | Common: arthralgia, fatigue, myalgia, Nausea 15.6% ≥grade 3 toxicity |
|
| KEYNOTE-028 Phase I [126] |
Pembrolizumab | Single agent in ER+, HER2−, locally advanced or metastatic disease, ECOG 0–1, and failure or inability to receive standard therapy | 25 | 12% (95% CI, 2.5–31.2) | 8 weeks (8.7 to >44 weeks) | Common: nausea, fatigue, arthralgia, anorexia, mucositis, pruritus, rash, blurred vision 16% ≥grade 3 toxicity |
|
| KEYNOTE-086 Phase II [125] |
Pembrolizumab | Single agent in advanced PD-L1+ triple negative breast cancer | 170 | 5% regardless of PD-L1 expression | – | – | 12% ≥grade 3 toxicity |
| KEYNOTE-173 Phase Ib [209] |
Pembrolizumab | Neoadjuvant with chemotherapy for locally advanced TNBC (Cohort A: nab-paclitaxel followed by doxorubicin and cyclophosphamide Cohort B: nab-paclitaxel and carboplatin followed by doxorubicin and cyclophosphamide) |
Cohort A: 10 Cohort B: 10 |
Cohort A: 80%, prior to surgery Cohort B: 100%, prior to surgery |
– | – | Myelosuppression: Cohort A: 3/10 Cohort B: 4/10 Cohort A: 80% ≥grade 3 toxicity Cohort B: 100% ≥grade 3 toxicity |
| I-SPY 2 Phase 2 [130] |
Pembrolizumab | Invasive breast cancer with neoadjuvant 12 weeks of paclitaxel with or without pembrolizumab followed by 4 cycles of doxorubicin and cyclophosphamide | 69 | HR+/HER−: 7/25 (28.0%) compared to 13/88 (14.8%) controls TNBC: 15/21 (71.4%) compared to 16/83 (19.3%) controls |
– | – | – |
| JAVELIN Phase Ib [129] |
Avelumab | Metastatic breast cancer refractory to therapy or with progression after standard-of-care therapy | 168 | 3.0% (overall) 5.2% (TNBC) |
– | – | 13.7% ≥grade 3 toxicity |
|
NCT01375842 Phase Ia [131] |
Atezolizumab | TNBC | 63 | 10% (95% CI, 5–17%) | – | – | – |
| IMpassion130 Phase Ib [127] |
Atezolizumab | Combination with nab-paclitaxel in metastatic TNBC treated with ≤3 prior lines of therapy | 32 | 42% (95% CI, 22–63%) | 21 (3 to 26+) | Common: neutropenia |
Other immune checkpoints have also been identified, including two inhibitory signals that are co-expressed by activated T cells and upregulated with PD-1 on chronically stimulated tumor-infiltrating lymphocytes – lymphocyte activation gene 3 (LAG3) [133] and T-cell immunoglobulin and mucin domain 3 (TIM-3) [134] – and a co-stimulatory receptor in the TNF family of receptors called OX40 that is capable of promoting CD8+ T-cell proliferation and activity, inhibiting generation of Tregs, and expanding memory CD4+ T-cell populations [135]. Early-phase clinical trials with these agents are underway and have been reviewed elsewhere [136].
Bispecific and multispecific antibodies
In addition to releasing the brakes on T-cell responses, antibodies can be engineered to directly activate potent tumor-directed responses at the site of the target. One such tool is the bispecific antibody (BsAb), which simultaneously interacts with cancer cell-specific epitopes and a stimulatory molecule on an effector cell population. This molecule creates an immunologic synapse capable of activating a potent innate or adaptive immune response [137]. In breast cancer, several iterations of BsAbs have been generated. HER2 is particularly attractive as the target for the tumor-oriented specificity and is frequently used [138,139]; however, other targets have also been employed, including epithelial cell adhesion molecule (EpCAM) [140,141] and epithelial growth factor receptor (EGFR) [142]. The initial effector cell target was the macrophage CD64 (Fcγ receptor I) to activate macrophage-mediated killing. Early-phase clinical trials using these antibodies were largely unsuccessful for several reasons, including the need for systemic cytokine or growth factor administration to boost macrophage responsiveness in some instances and the associated side-effect profile of these enhancing therapies, induction of autoantibodies against the administered BsAbs, and a lack of objective clinical response [143–146].
Although macrophage activation has had a limited effect on tumor-directed immunity in humans, T-cell activation has been a more promising area of research. Newer generations of BsAbs have effector cell specificities targeting CD3 on T cells [147] and elicit potent CD8+ cytotoxic T-cell activity both in vitro and in vivo in preclinical models of breast cancer [139,148–151]. Furthermore, pre-stimulating and arming T cells with BsAbs [152], as well as combining them with checkpoint inhibitors, are promising areas of further research.
Another approach to immune activation has been the use of multispecific antibodies, which are capable of binding to three or more epitopes and have also been utilized to activate tumor-directed immune responses. For example, the trifunctional monoclonal antibody ertumaxomab, which recognizes HER1, CD3, and Fcγ receptor types I and III, has been shown to induce lysis of human breast cancer cells in vitro [153] and is capable of eliciting a strong immune response in humans as evidenced by early-phase clinical trials [154].
Vaccines and oncolytic viruses
Cancer vaccines are a form of active immunization against tumor-specific antigens designed to stimulate immune responses directed against cancer cells, rather than prevent disease as in the canonical sense of the term ‘vaccine’. Many different approaches have been utilized to generate cancer vaccines, including whole tumor cells, whole cell lysates, peptides, glycosylated antigens, nucleic acids, and viruses [155,156]. When these vaccines are delivered, they elicit innate immune responses resulting in phagocytosis and processing of antigens by APCs. APCs insert these antigens into major MHC class I molecules to trigger T-cell-mediated cytotoxicity or MHC class II molecules to trigger T helper cell responses and humoral immunity. Adjunct molecules can also be used to induce more potent and durable responses [157,158].
In breast cancer, the most commonly employed epitope is HER2. While many different platforms have been used to generate anti-HER2 vaccines, the most promising is NeuVaxTM, a synthetic peptide analogue of HER2 called nelipepimut-S, which is capable of binding to CD8+ cytotoxic T cells and is administered with the immune adjuvant GM-CSF [159]. In Phase I and II trials, nelipepimut-S in combination with GM-CSF following surgical intervention with adequate lymph node dissection and completion of neoadjuvant or adjuvant radiation or chemotherapy improved disease-free survival [160], although results from a Phase III trial were less optimistic, with the study being suspended for futility following an interim safety and futility analysis. Several other trials with nelipepimut-S are in progress, including combination therapy with trastuzumab for ER−/PR−, lymph node-negative HER2+ or lymph node-positive HER2+ disease following standard-of-care therapy (NCT01570036), in high-risk HER2+ breast cancer populations (NCT02297698), and in ductal carcinoma in situ (NCT02636582). Other driver mutation-targeted vaccines currently in early-phase trials include AVX901, a virus-like replicon particle based on an attenuated Venezuelan equine encephalitis virus engineered to express HER2 [161] and INO-1400, a synthetic DNA vaccine targeting the hTERT oncogenic protein with or without a plasmid expressing IL-12 (NCT02960594). Vaccines against carbohydrates, which are expressed in a wide variety of solid tumor types, including breast cancer, are also being studied. One such example is MAG-Tn3, a multiple antigenic glycopeptide vaccine conjugated with tetanus toxoid that targets the universal carbohydrate tumor antigen Tn (α-D-N-acetylgalactosamine linked with serine or threonine), which is highly expressed in breast tumors [162]. It is currently in Phase I evaluation for patients with localized breast cancer at high risk for relapse (NCT02364492). Another example is OPT-822/OPT-821, a synthetic glycoprotein containing the universally expressed tumor carbohydrate Globo H bound to the carrier protein keyhole limpet hemocyanin (KLH). In an international, randomized, double-blind, placebo-controlled Phase II/III trial, patients who mounted an IgG response detectable at titers of ≥1:160 to the vaccine showed significant progression-free survival (HR, 0.71 [95% CI, 0.52–0.97] P=0.029) and interim overall survival (HR, 0.57 [95% CI, 0.33–0.97] P=0.04 for OS) [163].
Oncolytic viruses are genetically engineered viruses with cancer cell-specific tropism resulting in the infection and destruction of cancer cells while sparing host tissues. These viruses induce cancer cell death through multiple mechanisms, including virus-mediated cancer cell cytotoxicity, infection of surrounding endothelial cells resulting in destruction of tumor vasculature, and activation of tumor-directed immune responses [164,165]. Oncolytic virus-based vaccines that are approved for other malignancies are now in early-phase trial for the treatment of breast cancer. These include talimogene laherparepvec (T-vec), approved for use in locally recurrent, non-resectable melanoma, and JX-594 (pexastimogene devacirepvec), which has an FDA orphan drug designation for hepatocellular carcinoma. T-vec is a herpes simplex virus-1 (HSV-1) with deletions of ICP34.5 and ICP47, virulence factors that play critical roles in viral replication and inhibition of host immune responses, and engineered to produce GM-CSF [166]. It is currently in clinical trial as monotherapy for the treatment of locally recurrent breast cancer (NCT02658812), as neoadjuvant therapy in TNBC (NCT02779855), and in combination with the PD-L1 inhibitor atezolizumab for metastatic TNBC (NCT03256344). JX-594 is a replication-competent vaccinia poxvirus with deleterious mutations in its viral thymidine kinase gene, resulting in tropism for cancers with overexpression of thymidine kinase, and which expresses human GM-CSF to stimulate antitumor immune responses [167]. It is currently in trial as a combination therapy with metronomic cyclophosphamide in advanced breast cancer in France (NCT02630368).
Adoptive transfer of immune cells
Adoptive transfer is the collection and ex vivo manipulation of immune cells to stimulate antitumor activity, with reinfusion of these cells back into patients [168]. Evidence that adoptive transfer had potential as an effective anticancer therapy was first shown in syngeneic murine tumor transplant models in 1955 [169], with subsequent successes in rodent sarcoma and lymphoma models in the 1970s [170–172]. However, it was a series of studies in the early 1980s that provided the groundwork for the development of adoptive transfer technologies. Peripheral blood lymphocytes (PBL) collected from healthy donors and grown in the presence of PBLs from patients with malignancy were capable of lysing both previously cultured and freshly harvested human cancer cells [173]. Similar results were obtained using PBLs collected from patients with malignancy [174] and tumor-infiltrating leukocytes [175] expanded ex vivo with IL-2. Adoptive transfer of PBLs collected from cancer patients that were expanded ex vivo (lymphokine-activated killer cells) and coadministered with IL-2 resulted in regression of metastatic disease and improved survival in preclinical models of metastatic melanoma and colon adenocarcinoma [176–178], providing the impetus for further research and initiation of clinical trials [179–181].
After early clinical successes were observed in melanoma and renal cell carcinoma with the combination of lymphokine-activated killer (LAK) cells and IL-2, efficacy of this regimen in other malignancies was evaluated, including breast. However, this therapeutic strategy ultimately yielded minimal responses in advanced breast tumors, as well as most other epithelial malignancies evaluated [182,183]. In vitro three-dimensional cell culture models of breast cancer data suggested that the lack of response to adoptive transfer therapies was due to decreased ability of immune cells to infiltrate and adhere to tumors and was also related to the fact that environmental signals inhibited the proliferation of infiltrating leukocytes. These data provided a basis for understanding the lack of clinical efficacy [184,185].
While adoptive transfer of PBLs and TILs expanded ex vivo has largely remained unsuccessful in breast cancer, studies focusing particularly on TILs have shifted toward identifying and expanding effector cells in TIL populations that recognize specific, immunogenic, non-synonymous mutations that can be used for adoptive transfer. A particularly effective strategy for identifying these cells relies on the use of whole exome sequencing to detect clonal mutations in whole tumor tissue samples, inducing expression of oligopeptides bearing these mutations by immune cells (typically B cells or dendritic cells) ex vivo, coculturing these antigen-presenting cells with TILs, and sorting activated lymphocytes by flow cytometry for confirmation of cytotoxic activity and clonal expansion [186,187]. A recent case report showed that combining this strategy with pre-transfer lympho-depleting chemotherapy and a single dose of the immune checkpoint inhibitor pembrolizumab was capable of generating durable antitumor immunologic responses and measurable regression in a patient with metastatic, chemotherapy-refractory ER+, HER2− breast cancer [188].
A more common approach to adoptive therapy technologies that is rapidly becoming a major area of research is the genetic engineering of T cells capable of producing highly specific and potent antitumor responses. Techniques for rapidly generating T-cell specificity that allows for immediate initiation of downstream cell signaling cascades and effector functions, so-called armed T cells, are the transduction of retroviruses or lentiviruses encoding bispecific and multispecific antibodies [139,149,150] and chimeric antigen receptors (CARs). Chimeric antigen receptors are genetically engineered hybrid T-cell receptors typically consisting of a single-chain variable fragment (scFv) of the antibody-based B-cell receptor to confer antigen specificity plus the T-cell receptor CD3ζ transmembrane and intracellular signaling domains linked to one or more intracellular co-stimulatory domains (Figure 2) [189]. While many breast cancer-specific antigens have been identified, HER2 is again the most frequently targeted antigen for CAR design. T cells armed with HER2-specific CARs demonstrate potent in vitro T-cell-mediated cancer cell cytotoxicity and are capable of inducing regression of tumors in rodent mammary tumor models [190–192]. As these T-cell responses typically wane with time, other approaches have been utilized to prolong T-cell activity. For example, transposon technology has been used to express a HER2 CAR in Epstein–Barr virus (EBV)-specific cytotoxic T lymphocytes, which proliferate in response to chronic host infection with EBV, thereby affording continued in vivo T-cell expansion while redirecting effector activity toward HER2+ cancer cells [193]. There are currently two early-phase clinical trials assessing safety and efficacy of adoptive transfer of autologous HER2 CAR-expressing T cells in breast cancer patients (NCT01935843, NCT02547961).
Figure 2.
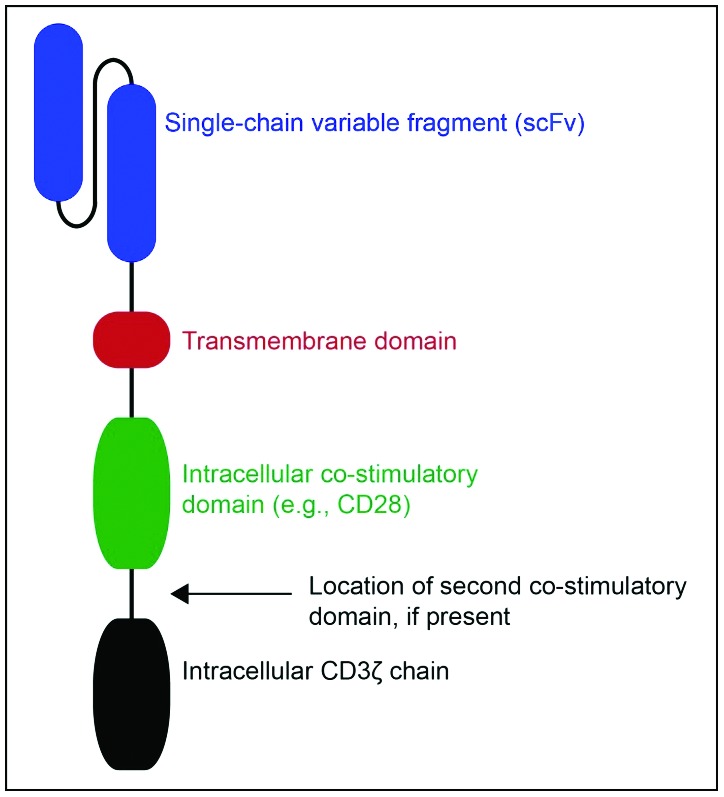
General structure of chimeric antigen receptor.
The second generation chimeric antigen receptor (pictured) comprises three primary components: a single chain variable fragment (scFV) that recognizes a specific tumor antigen (e.g., HER2), an intracellular co-stimulatory domain (commonly CD28), and the intracellular CD3ζ chain. Third generation CARs may have a second co-stimulatory domain between the second generation co-stimulatory domain and the CD3ζ chain (typically 4-1BBB or OX40).
Adoptive transfer of other immune cell populations, particularly NK cells and dendritic cells (DCs), has also been an active area of research. NK cells act through multiple mechanisms, including receptor-mediated cytotoxicity, ADCC, activation of death receptor-mediated apoptotic pathways, and expression of cytokines that promote adaptive immune responses [194]. NK cells collected from circulating peripheral blood mononuclear cells (PBMCs) in breast cancer patients exert potent cancer cell-directed cytotoxicity in vitro and significantly reduce cancer cell engraftment and growth in murine xenograft models of breast cancer metastasis [195]. NK cells can also be modified to express tumor antigen-directed CARs, leading to potent receptor-mediated cytotoxicity. For example, a modified NK-92 cell line derivative that expresses a CAR with an scFv targeting HER2 and the CD3ζ and CD28 co-stimulatory signaling domains is capable of potent and selective killing of cancer cells expressing HER2 in vitro and, upon adoptive transfer, is capable of homing to orthotopic HER2+ murine mammary tumors and reducing metastasis formation in murine models of pulmonary metastasis [196]. Another modification of the NK-92 cell line designed to simultaneously express the cytokine IL-15 and a CAR with an scFv targeting EpCAM and the CD3ζ and CD28 co-stimulatory signaling domains was also shown to selectively kill breast cancer cells in vitro [197].
Dendritic cells are the primary innate immune cell population responsible for activating adaptive T-cell-mediated cytotoxic and humoral responses. They are typically derived from the ex vivo differentiation of PBMCs, bone marrow-derived monocytes, or CD34+ hematopoietic stem cells with GM-CSF and IL-4 with or without TNF-α. In breast cancers, several mechanisms have been employed to load DCs with antigens, including infusion with whole tumor lysates [198], incubation with oligopeptides homologous to portions of known tumor antigens [199,200], transduction of viral vectors engineered to express tumor antigens [201,202], and fusion of activated DCs with cancer cells [203]. Despite their ability to activate T cells in vitro and promising evidence in murine breast cancer models, DC-based immunotherapies in monotherapy have been largely unsuccessful in early-phase trials [199,200,202].
Strategies employed to enhance the effects of adoptive transfer with DCs (so-called DC vaccines) are combining DC infusion with cytokine-induced killer cells (CIKs) and depleting Treg populations. CIKs are a heterogeneous population of MHC nonrestricted lymphocytes, expanded in vitro, that typically express both CD3 and CD56, but also include small subsets of CD3−, CD56+ NK cells and CD3+, CD56− T cells. All three of these subsets are activated by DCs, and those CIKs with dual expression of CD3 and CD56 are capable of both NK and antigen-specific T-cell activity when activated [204]. Indeed, in a Phase I/II trial, combining autologous transfer of ex vivo-selected and expanded DC/CIK with high dose chemotherapy improved both progression-free and overall survival in patients with metastatic breast cancer [205]. Promising results have also been seen with daclizumab, a monoclonal antibody directed against CD25, that downregulates expression of both CD25 and FOXP3 in Tregs, resulting in reprogramming of these immune cells and allowing for priming and boosting of CD8+ T-cell activity when combined with DC-based adoptive transfer in patients with metastatic breast cancer [37].
Challenges and future perspectives
Breast tumors are infiltrated by a diverse array of immune cells that shape and influence the progression of this heterogeneous group of malignancies. Increasing understanding of the basic biological mechanisms of this complex immune microenvironment and the recognition that components of the immune system and their effector functions can be augmented and potentiated to effectively target and destroy breast tumors has driven the rapid development and evolution of breast cancer immunotherapies.
Increasing sophistication of immune system-based technologies has resulted in enhanced specificity, leading to better side-effect profiles and improved outcomes. However, several challenges remain. Tumors have consistently found methods to adapt and develop resistance against cytotoxic chemotherapeutics, and emerging evidence indicates that this is true for immunotherapies as well [206]. Given the complexities of the immune system, combination therapies will need to be employed to circumvent immune-mediated resistance. These strategies include simultaneously combining immunotherapy with cytotoxic agents, augmenting both the innate and adaptive immune systems, tandem immunotherapies, and combining immune checkpoint inhibition with DC-based vaccines [207], among others. Combinatorial methods will necessarily need to be approached with caution, as global activation of the immune system has the potential for serious adverse effects, including severe cytokine release syndromes. Therefore, reliable mechanisms for efficiently and specifically activating or disinhibiting multiple tumor-directed responses in vivo will need to be further cultivated. Finally, while many of these approaches are technically feasible, immunotherapies are often cost prohibitive and will require development of streamlined, high-throughput technologies, particularly for those therapies that rely on adoptive transfer.
Despite these challenges, rapid advances in breast cancer immunotherapies are showing significant promise for the treatment of many subtypes, including TNBCs. In TNBCs, TIL infiltration correlates positively with response to immune checkpoint inhibitors, and these responses are long lasting [131,208]. As this response appears to be more effective when immunotherapy is administered as part of first-line therapy [126] and with the identification of biomarkers for response to immunotherapy, strategies harnessing the immune system may alter the landscape of current approaches for TNBCs.
Footnotes
Disclosure and potential conflicts of interest: The authors have declared that there are no conflicts of interest. The International Committee of Medical Journal Editors (ICMJE) Potential Conflicts of Interests form for the authors are available for download at: http://www.drugsincontext.com/wp-content/uploads/2018/01/dic.212520-COI.pdf
Funding declaration: No financial support was received.
Correct attribution: Copyright © 2018 Nakasone ES, Hurvitz SA, McCann KE. https://doi.org/10.7573/dic.212520. Published by Drugs in Context under Creative Commons License Deed CC BY NC ND 4.0.
Article URL: http://www.drugsincontext.com/harnessing-the-immune-system-in-the-battle-against-breast-cancer
Provenance: invited; externally peer reviewed.
Peer review comments to author: 9 January 2018
Drugs in Context is published by BioExcel Publishing Ltd. Registered office: Plaza Building, Lee High Road, London, England, SE13 5PT.
BioExcel Publishing Limited is registered in England Number 10038393. VAT GB 252772009.
For all manuscript and submissions enquiries, contact the Editorial office dic.editorial@bioexcelpublishing.com
For all permissions, rights and reprints, contact David Hughes david.hughes@bioexcelpublishing.com
References
- 1.Sledge GW, Mamounas EP, Hortobagyi GN, Burstein HJ, Goodwin PJ, Wolff AC. Past, present, and future challenges in breast cancer treatment. J Clin Oncol. 2016;32:1979–86. doi: 10.1200/JCO.2014.55.4139. http://dx.doi.org/10.1200/JCO.2014.55.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62. doi: 10.3322/caac.21203. http://dx.doi.org/10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 3.DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin. 2017;67(6):439–48. doi: 10.3322/caac.21412. http://dx.doi.org/10.3322/caac.21412. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. http://dx.doi.org/10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 5.Cardoso F, Harbeck N, Fallowfield L, Kyriakides S, Senkus E ESMO Guidelines Working Group. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23:vii11–vii19. doi: 10.1093/annonc/mds232. http://dx.doi.org/10.1093/annonc/mds232. [DOI] [PubMed] [Google Scholar]
- 6.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. http://dx.doi.org/10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Place AE, Jin Huh S, Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 2011;13:277. doi: 10.1186/bcr2912. http://dx.doi.org/10.1186/bcr2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spaw M, Anant S, Thomas SM. Stromal contributions to the carcinogenic process. Mol Carcinog. 2017;56:1199–213. doi: 10.1002/mc.22583. http://dx.doi.org/10.1002/mc.22583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–9. doi: 10.1038/35077241. http://dx.doi.org/10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 10.Yuan Y, Jiang Y-C, Sun C-K, Chen Q-M. Role of the tumor microenvironment in tumor progression and the clinical applications (review) Oncol Rep. 2016;35:2499–515. doi: 10.3892/or.2016.4660. http://dx.doi.org/10.3892/or.2016.4660. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. http://dx.doi.org/10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Virchow RLK. Translated from the 2d Edition of the original by Frank Chance With notes and numerous emendations, principally from MS Notes of the author. Philadelphia, USA: JB Lippincott; 1863. Cellular Pathology as Based Upon Physiological and Pathological Histology. http://dx.doi.org/10.5962/bhl.title.32770. [Google Scholar]
- 13.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–59. doi: 10.1056/NEJM198612253152606. http://dx.doi.org/10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 14.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. http://dx.doi.org/10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 15.Dvorak HF. Tumors: wounds that do not heal – redux. Cancer Immunol Res. 2015;3:1–11. doi: 10.1158/2326-6066.CIR-14-0209. http://dx.doi.org/10.1158/2326-6066.CIR-14-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore OS, Foote FW. The relatively favorable prognosis of medullary carcinoma of the breast. Cancer. 1949;2:635–42. doi: 10.1002/1097-0142(194907)2:4<635::aid-cncr2820020411>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 17.Berg JW. Inflammation and prognosis in breast cancer; a search for host resistance. Cancer. 1959;12:714–20. doi: 10.1002/1097-0142(195907/08)12:4<714::aid-cncr2820120414>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka H, Shimoda T, Uchida K, Suzuki T, Ischikawa E. Immunohistochemical study on the distribution and significance of mononuclear cells in human breast carcinoma. Acta Pathol Jpn. 1986;36:1455–68. doi: 10.1111/j.1440-1827.1986.tb02817.x. [DOI] [PubMed] [Google Scholar]
- 19.Bhan AK, DesMarais CL. Immunohistologic characterization of major histocompatibility antigens and inflammatory cellular infiltrate in human breast cancer. J Natl Cancer Inst. 1983;71:507–16. [PubMed] [Google Scholar]
- 20.Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, Zi X, Kwak M, Bergholtz H, Su Y, Ding L, Russnes HG, Richardson AL, Babski K, Min Hui Kim E, McDonnell CH, 3rd, Wagner J, Rowberry R, Freeman GJ, Dillon D, Sorlie T, Coussens LM, Garber JE, Fan R, Bobolis K, Allred DC, Jeong J, Park SY, Michor F, Polyak K. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discov. 2017;7:1098–115. doi: 10.1158/2159-8290.CD-17-0222. http://dx.doi.org/10.1158/2159-8290.CD-17-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9:539. doi: 10.1186/bcr1746. http://dx.doi.org/10.1186/bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogiya R, Niikura N, Kumaki N, Bianchini G, Kitano S, Iwamoto T, Hayashi N, Yokoyama K, Oshitanai R, Terao M, Morioka T, Tsuda B, Okamura T, Saito Y, Suzuki Y, Tokuda Y. Comparison of tumor-infiltrating lymphocytes between primary and metastatic tumors in breast cancer patients. Cancer Sci. 2016;107:1730–5. doi: 10.1111/cas.13101. http://dx.doi.org/10.1111/cas.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leong PP, Mohammad R, Ibrahim N, Ithnin H, Abdullah M, Davis WC, Seow HF. Phenotyping of lymphocytes expressing regulatory and effector markers in infiltrating ductal carcinoma of the breast. Immunol Lett. 2006;102:229–36. doi: 10.1016/j.imlet.2005.09.006. http://dx.doi.org/10.1016/j.imlet.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, Chauffert B, Fumoleau P, Ghiringhelli F. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin Cancer Res. 2008;14:2413–20. doi: 10.1158/1078-0432.CCR-07-4491. http://dx.doi.org/10.1158/1078-0432.CCR-07-4491. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Chen X, Zhou E, Chen G, Qian K, Wu X, Miao X, Tang Z. Intratumoral CD8+ cytotoxic lymphocyte is a favorable prognostic marker in node-negative breast cancer. Plos One. 2014;9:e95475. doi: 10.1371/journal.pone.0095475. http://dx.doi.org/10.1371/journal.pone.0095475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y, Lan S, Zheng Q. Prognostic significance of infiltrating immune cell subtypes in invasive ductal carcinoma of the breast. Tumori. 2017;0 doi: 10.5301/tj.5000624. https://doi.org/10.5301/tj.5000624. [DOI] [PubMed] [Google Scholar]
- 27.Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, Green AR. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol. 2011;29:1949–55. doi: 10.1200/JCO.2010.30.5037. http://dx.doi.org/10.1200/JCO.2010.30.5037. [DOI] [PubMed] [Google Scholar]
- 28.Verma C, Kaewkangsadan V, Eremin JM, Cowley GP, Ilyas M, El-Sheemy MA, Eremin O. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): differential restoration of blood profiles by NAC and surgery. J Transl Med. 2015;13:180. doi: 10.1186/s12967-015-0535-8. http://dx.doi.org/10.1186/s12967-015-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandal A, Viswanathan C. Natural killer cells: in health and disease. Hematol Oncol Stem Cell Ther. 2015;8:47–55. doi: 10.1016/j.hemonc.2014.11.006. http://dx.doi.org/10.1016/j.hemonc.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Tu MM, Mahmoud AB, Wight A, Mottashed A, Bélanger S, Rahim MM, Abou-Samra E, Makrigiannis AP. Ly49 family receptors are required for cancer immunosurveillance mediated by natural killer cells. Cancer Res. 2014;74:3684–94. doi: 10.1158/0008-5472.CAN-13-3021. http://dx.doi.org/10.1158/0008-5472.CAN-13-3021. [DOI] [PubMed] [Google Scholar]
- 31.Tu MM, Rahim MMA, Sayed C, Mahmoud AB, Makrigiannis AP. Immunosurveillance and immunoediting of breast cancer via class I MHC receptors. Cancer Immunol Res. 2017;5(11):1016–28. doi: 10.1158/2326-6066.CIR-17-0056. http://dx.doi.org/10.1158/2326-6066.CIR-17-0056. [DOI] [PubMed] [Google Scholar]
- 32.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. http://dx.doi.org/10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–30. doi: 10.1016/j.smim.2011.10.002. http://dx.doi.org/10.1016/j.smim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Xu L, Xu W, Qiu S, Xiong S. Enrichment of CCR6+Foxp3+ regulatory T cells in the tumor mass correlates with impaired CD8+ T cell function and poor prognosis of breast cancer. Clin Immunol. 2010;135:466–75. doi: 10.1016/j.clim.2010.01.014. http://dx.doi.org/10.1016/j.clim.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe MAE, Oda JMM, Amarante MK, Cesar Voltarelli J. Regulatory T cells and breast cancer: implications for immunopathogenesis. Cancer Metastasis Rev. 2010;29:569–79. doi: 10.1007/s10555-010-9247-y. http://dx.doi.org/10.1007/s10555-010-9247-y. [DOI] [PubMed] [Google Scholar]
- 36.Knutson KL, Dang Y, Lu H, Lukas J, Almand B, Gad E, Azeke E, Disis ML. IL-2 immunotoxin therapy modulates tumor-associated regulatory T cells and leads to lasting immune-mediated rejection of breast cancers in neu-transgenic mice. J Immunol. 2006;177:84–91. doi: 10.4049/jimmunol.177.1.84. https://dx.doi.org/10.4049/jimmunol.177.1.84. [DOI] [PubMed] [Google Scholar]
- 37.Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ, Jr, Colligon TA, Trosko JA, Leinbach LI, Pletcher CH, Tweed CK, DeMichele A, Fox KR, Domchek SM, Riley JL, Vonderheide RH. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4:134ra62. doi: 10.1126/scitranslmed.3003330. http://dx.doi.org/10.1126/scitranslmed.3003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oldford SA, Robb JD, Codner D, Gadag V, Watson PH, Drover S. Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients. Int Immunol. 2006;18:1591–602. doi: 10.1093/intimm/dxl092. http://dx.doi.org/10.1093/intimm/dxl092. [DOI] [PubMed] [Google Scholar]
- 39.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. http://dx.doi.org/10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Q, Qin J, Zhong L, Gong L, Zhang B, Zhang Y, Gao WQ. CCL5-Mediated Th2 immune polarization promotes metastasis in luminal breast cancer. Cancer Res. 2015;75:4312–21. doi: 10.1158/0008-5472.CAN-14-3590. http://dx.doi.org/10.1158/0008-5472.CAN-14-3590. [DOI] [PubMed] [Google Scholar]
- 41.Yang L, Qi Y, Hu J, Tang L, Zhao S, Shan B. Expression of Th17 cells in breast cancer tissue and its association with clinical parameters. Cell Biochem Biophys. 2012;62:153–9. doi: 10.1007/s12013-011-9276-3. http://dx.doi.org/10.1007/s12013-011-9276-3. [DOI] [PubMed] [Google Scholar]
- 42.Zhu X, Mulcahy LA, Mohammed RA, Lee AH, Franks HA, Kilpatrick L, Yilmazer A, Paish EC, Ellis IO, Patel PM, Jackson AM. IL-17 expression by breast-cancer-associated macrophages: IL-17 promotes invasiveness of breast cancer cell lines. Breast Cancer Res. 2008;10:R95. doi: 10.1186/bcr2195. http://dx.doi.org/10.1186/bcr2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cochaud S, Giustiniani J, Thomas C, Laprevotte E, Garbar C, Savoye AM, Curé H, Mascaux C, Alberici G, Bonnefoy N, Eliaou JF, Bensussan A, Bastid J. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Sci Rep. 2013;3:3456. doi: 10.1038/srep03456. http://dx.doi.org/10.1038/srep03456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Sullivan C, Lewis CE. Tumour-associated leucocytes: friends or foes in breast carcinoma. J Pathol. 1994;172:229–35. doi: 10.1002/path.1711720302. http://dx.doi.org/10.1002/path.1711720302. [DOI] [PubMed] [Google Scholar]
- 45.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278. http://dx.doi.org/10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 46.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823. http://dx.doi.org/10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 47.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA. 2010;107:8363–8. doi: 10.1073/pnas.0911378107. http://dx.doi.org/10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–79. doi: 10.1101/gad.180331.111. http://dx.doi.org/10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, Park J, Sinha P, Bissell MJ, Frengen E, Werb Z, Egeblad M. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. http://dx.doi.org/10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medrek C, Pontén F, Jirström K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 2012;12:306. doi: 10.1186/1471-2407-12-306. http://dx.doi.org/10.1186/1471-2407-12-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klingen TA, Chen Y, Aas H, Wik E, Akslen LA. Tumor associated macrophages are strongly related to vascular invasion, non-luminal subtypes and interval breast cancer. Hum Pathol. 2017;69:72–80. doi: 10.1016/j.humpath.2017.09.001. http://dx.doi.org/10.1016/j.humpath.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Kankkunen JP, Harvima IT, Naukkarinen A. Quantitative analysis of tryptase and chymase containing mast cells in benign and malignant breast lesions. Int J Cancer. 1997;72:385–8. doi: 10.1002/(sici)1097-0215(19970729)72:3<385::aid-ijc1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 53.Cimpean AM, Tamma R, Ruggieri S, Nico B, Toma A, Ribatti D. Mast cells in breast cancer angiogenesis. Crit Rev Oncol Hematol. 2017;115:23–26. doi: 10.1016/j.critrevonc.2017.04.009. http://dx.doi.org/10.1016/j.critrevonc.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 54.Markowitz J, Wesolowski R, Papenfuss T, Brooks TR, Carson WE., 3rd Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res Treat. 2013;140:13–21. doi: 10.1007/s10549-013-2618-7. http://dx.doi.org/10.1007/s10549-013-2618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansen MH, Nielsen H, Ditzel HJ. The tumor-infiltrating B cell response in medullary breast cancer is oligoclonal and directed against the autoantigen actin exposed on the surface of apoptotic cancer cells. Proc Natl Acad Sci USA. 2001;98:12659–64. doi: 10.1073/pnas.171460798. http://dx.doi.org/10.1073/pnas.171460798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coronella JA, Spier C, Welch M, Trevor KT, Stopeck AT, Villar H, Hersh EM. Antigen-driven oligoclonal expansion of tumor-infiltrating B cells in infiltrating ductal carcinoma of the breast. J Immunol. 2002;169:1829–36. doi: 10.4049/jimmunol.169.4.1829. https://dx.doi.org/10.4049/jimmunol.169.4.1829. [DOI] [PubMed] [Google Scholar]
- 57.Nzula S, Going JJ, Stott DI. Antigen-driven clonal proliferation, somatic hypermutation, and selection of B lymphocytes infiltrating human ductal breast carcinomas. Cancer Res. 2003;63:3275–80. [PubMed] [Google Scholar]
- 58.Simsa P, Teillaud J-L, Stott DI, Toth J, Kotlan B. Tumor-infiltrating B cell immunoglobulin variable region gene usage in invasive ductal breast carcinoma. Pathol Oncol Res. 2005;11:92–97. doi: 10.1007/BF02893374. [DOI] [PubMed] [Google Scholar]
- 59.Fox BA, Schendel DJ, Butterfield LH, Aamdal S, Allison JP, Ascierto PA, Atkins MB, Bartunkova J, Bergmann L, Berinstein N, Bonorino CC, Borden E, Bramson JL, Britten CM, Cao X, Carson WE, Chang AE, Characiejus D, Choudhury AR, Coukos G, de Gruijl T, Dillman RO, Dolstra H, Dranoff G, Durrant LG, Finke JH, Galon J, Gollob JA, Gouttefangeas C, Grizzi F, Guida M, Håkansson L, Hege K, Herberman RB, Hodi FS, Hoos A, Huber C, Hwu P, Imai K, Jaffee EM, Janetzki S, June CH, Kalinski P, Kaufman HL, Kawakami K, Kawakami Y, Keilholtz U, Khleif SN, Kiessling R, Kotlan B, Kroemer G, Lapointe R, Levitsky HI, Lotze MT, Maccalli C, Maio M, Marschner JP, Mastrangelo MJ, Masucci G, Melero I, Melief C, Murphy WJ, Nelson B, Nicolini A, Nishimura MI, Odunsi K, Ohashi PS, O’Donnell-Tormey J, Old LJ, Ottensmeier C, Papamichail M, Parmiani G, Pawelec G, Proietti E, Qin S, Rees R, Ribas A, Ridolfi R, Ritter G, Rivoltini L, Romero PJ, Salem ML, Scheper RJ, Seliger B, Sharma P, Shiku H, Singh-Jasuja H, Song W, Straten PT, Tahara H, Tian Z, van Der Burg SH, von Hoegen P, Wang E, Welters MJ, Winter H, Withington T, Wolchok JD, Xiao W, Zitvogel L, Zwierzina H, Marincola FM, Gajewski TF, Wigginton JM, Disis ML. Defining the critical hurdles in cancer immunotherapy. J Transl Med. 2011;9:214. doi: 10.1186/1479-5876-9-214. http://dx.doi.org/10.1186/1479-5876-9-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–27. doi: 10.1038/nri1936. http://dx.doi.org/10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 61.Wright SE. Immunotherapy of breast cancer. Expert Opin Biol Ther. 2012;12:479–90. doi: 10.1517/14712598.2012.665445. http://dx.doi.org/10.1517/14712598.2012.665445. [DOI] [PubMed] [Google Scholar]
- 62.Morrissey KM, Yuraszeck TM, Li CC, Zhang Y, Kasichayanula S. Immunotherapy and novel combinations in oncology: current landscape, challenges, and opportunities. Clin Transl Sci. 2016;9:89–104. doi: 10.1111/cts.12391. http://dx.doi.org/10.1111/cts.12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li C-Y, Huang Q, Kung H-F. Cytokine and immuno-gene therapy for solid tumors. Cell Mol Immunol. 2005;2:81–91. [PubMed] [Google Scholar]
- 64.Kim-Schulze S, Taback B, Kaufman HL. Cytokine therapy for cancer. Surg Oncol Clin N Am. 2007;16:793–818. viii. doi: 10.1016/j.soc.2007.07.011. http://dx.doi.org/10.1016/j.soc.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 65.Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel) 2011;3:3856–93. doi: 10.3390/cancers3043856. http://dx.doi.org/10.3390/cancers3043856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao M, Brummer G, Acevedo D, Cheng N. Cytokine regulation of metastasis and tumorigenicity. Adv Cancer Res. 2016;132:265–367. doi: 10.1016/bs.acr.2016.05.005. http://dx.doi.org/10.1016/bs.acr.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 67.Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991;262:3–11. [PubMed] [Google Scholar]
- 68.Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–13. http://dx.doi.org/10.1001/jama.1994.03510360033032. [PubMed] [Google Scholar]
- 69.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192:5451–8. doi: 10.4049/jimmunol.1490019. http://dx.doi.org/10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gutterman JU, Blumenschein GR, Alexanian R, Yap HY, Buzdar AU, Cabanillas F, Hortobagyi GN, Hersh EM, Rasmussen SL, Harmon M, Kramer M, Pestka S. Leukocyte interferon-induced tumor regression in human metastatic breast cancer, multiple myeloma, and malignant lymphoma. Ann Intern Med. 1980;93:399–406. doi: 10.7326/0003-4819-93-3-399. http://dx.doi.org/10.7326/0003-4819-93-3-399. [DOI] [PubMed] [Google Scholar]
- 71.Borden EC, Holland JF, Dao TL, Gutterman JU, Wiener L, Chang Y-C, Patel J. Leukocyte-derived interferon (alpha) in human breast carcinoma. The American Cancer Society phase II trial. Ann Intern Med. 1982;97:1–6. doi: 10.7326/0003-4819-97-1-1. http://dx.doi.org/10.7326/0003-4819-97-1-1. [DOI] [PubMed] [Google Scholar]
- 72.Sherwin SA, Mayer D, Ochs JJ, Abrams PG, Knost JA, Foon KA, Fein S, Oldham RK. Recombinant leukocyte A interferon in advanced breast cancer. Results of a phase II efficacy trial. Ann Intern Med. 1983;98:598–602. doi: 10.7326/0003-4819-98-5-598. [DOI] [PubMed] [Google Scholar]
- 73.Quesada JR, Hawkins M, Horning S, Alexanian R, Borden E, Merigan T, Adams F, Gutterman JU. Collaborative phase I-II study of recombinant DNA-produced leukocyte interferon (clone A) in metastatic breast cancer, malignant lymphoma, and multiple myeloma. Am J Med. 1984;77:427–32. doi: 10.1016/0002-9343(84)90097-4. https://dx.doi.org/10.1016/0002-9343(84)90097-4. [DOI] [PubMed] [Google Scholar]
- 74.Barreras L, Vogel CL, Koch G, Marcus SG. Phase II trial of recombinant beta (IFN-betaser) interferon in the treatment of metastatic breast cancer. Invest New Drugs. 1988;6:211–5. doi: 10.1007/BF00175400. [DOI] [PubMed] [Google Scholar]
- 75.Bruntsch U, Groos G, Tigges FJ, Hofschneider H, Gallmeier WM. Lack of response in nine patients with breast cancer treated with fibroblast interferon. Cancer Chemother Pharmacol. 1984;13:39–42. doi: 10.1007/BF00401445. [DOI] [PubMed] [Google Scholar]
- 76.Muss HB, Caponera M, Zekan PJ, Jackson DV, Jr, Stuart JJ, Richards F, Cooper MR, Levin EA, Reich SD, Capizzi RL. Recombinant gamma interferon in advanced breast cancer: a phase II trial. Invest New Drugs. 1986;4:377–81. doi: 10.1007/BF00173511. [DOI] [PubMed] [Google Scholar]
- 77.Kimmick G, Ratain MJ, Berry D, Woolf S, Norton L, Muss HB Cancer and Leukemia Group B. Subcutaneously administered recombinant human interleukin-2 and interferon alfa-2a for advanced breast cancer: a phase II study of the Cancer and Leukemia Group B (CALGB 9041) Invest New Drugs. 2004;22:83–89. doi: 10.1023/b:drug.0000006178.32718.22. [DOI] [PubMed] [Google Scholar]
- 78.Sabel MS, Skitzki J, Stoolman L, Egilmez NK, Mathiowitz E, Bailey N, Chang WJ, Chang AE. Intratumoral IL-12 and TNF-alpha-loaded microspheres lead to regression of breast cancer and systemic antitumor immunity. Ann Surg Oncol. 2004;11:147–56. doi: 10.1245/aso.2004.03.022. http://dx.doi.org/10.1245/ASO.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 79.Repka T, Chiorean EG, Gay J, Herwig KE, Kohl VK, Yee D, Miller JS. Trastuzumab and interleukin-2 in HER2-positive metastatic breast cancer: a pilot study. Clin Cancer Res. 2003;9:2440–6. http://clincancerres.aacrjournals.org/content/9/7/2440.long. [PubMed] [Google Scholar]
- 80.Bekaii-Saab TS, Roda JM, Guenterberg KD, Ramaswamy B, Young DC, Ferketich AK, Lamb TA, Grever MR, Shapiro CL, Carson WE., 3rd A phase I trial of paclitaxel and trastuzumab in combination with interleukin-12 in patients with HER2/neu-expressing malignancies. Mol Cancer Ther. 2009;8:2983–91. doi: 10.1158/1535-7163.MCT-09-0820. http://dx.doi.org/10.1158/1535-7163.MCT-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stewart AK, Lassam NJ, Quirt IC, Bailey DJ, Rotstein LE, Krajden M, Dessureault S, Gallinger S, Cappe D, Wan Y, Addison CL, Moen RC, Gauldie J, Graham FL. Adenovector-mediated gene delivery of interleukin-2 in metastatic breast cancer and melanoma: results of a phase 1 clinical trial. Gene Ther. 1999;6:350–63. doi: 10.1038/sj.gt.3300833. http://dx.doi.org/10.1038/sj.gt.3300833. [DOI] [PubMed] [Google Scholar]
- 82.Chaurasiya S, Hew P, Crosley P, Sharon D, Potts K, Agopsowicz K, Long M, Shi C, Hitt MM. Breast cancer gene therapy using an adenovirus encoding human IL-2 under control of mammaglobin promoter/enhancer sequences. Cancer Gene Ther. 2016;23:178–87. doi: 10.1038/cgt.2016.18. http://dx.doi.org/10.1038/cgt.2016.18. [DOI] [PubMed] [Google Scholar]
- 83.Yousefi F, Siadat SD, Saraji AA, Hesaraki S, Aslani MM, Mousavi SF, Imani Fooladi AA. Tagging staphylococcal enterotoxin B (SEB) with TGFaL3 for breast cancer therapy. Tumour Biol. 2016;37:5305–16. doi: 10.1007/s13277-015-4334-x. http://dx.doi.org/10.1007/s13277-015-4334-x. [DOI] [PubMed] [Google Scholar]
- 84.Valedkarimi Z, Nasiri H, Aghebati-Maleki L, Majidi J. Antibody-cytokine fusion proteins for improving efficacy and safety of cancer therapy. Biomed Pharmacother. 2017;95:731–42. doi: 10.1016/j.biopha.2017.07.160. http://dx.doi.org/10.1016/j.biopha.2017.07.160. [DOI] [PubMed] [Google Scholar]
- 85.Wingard JR, Elmongy M. Strategies for minimizing complications of neutropenia: prophylactic myeloid growth factors or antibiotics. Crit Rev Oncol Hematol. 2009;72:144–54. doi: 10.1016/j.critrevonc.2009.01.003. http://dx.doi.org/10.1016/j.critrevonc.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 86.Crawford J, Allen J, Armitage J, Blayney DW, Cataland SR, Heaney ML, Htoy S, Hudock S, Kloth DD, Kuter DJ, Lyman GH, McMahon B, Steensma DP, Vadhan-Raj S, Westervelt P, Westmoreland M National Comprehensive Cancer Network. Myeloid growth factors. J Natl Compr Canc Netw. 2011;9:914–32. doi: 10.6004/jnccn.2011.0075. http://dx.doi.org/10.6004/jnccn.2011.0075. [DOI] [PubMed] [Google Scholar]
- 87.Palucka AK, Coussens LM. The basis of oncoimmunology. Cell. 2016;164:1233–47. doi: 10.1016/j.cell.2016.01.049. http://dx.doi.org/10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adams S. Toll-like receptor agonists in cancer therapy. Immunotherapy. 2009;1:949–64. doi: 10.2217/imt.09.70. http://dx.doi.org/10.2217/imt.09.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Braun W, Plescia OJ, Raskova J, Webb D. Basic proteins and synthetic polynucleotides as modifiers of immunogenicity of syngeneic tumor cells. Isr J Med Sci. 1971;7:72–82. [PubMed] [Google Scholar]
- 90.Lacour F, Spira A, Lacour J, Prade M. Polyadenylic-polyuridylic acid, an adjunct to surgery in the treatment of spontaneous mammary tumors in C3H-He mice and transplantable melanoma in the hamster. Cancer Res. 1972;32:648–9. http://cancerres.aacrjournals.org/content/canres/32/3/648.full.pdf. [PubMed] [Google Scholar]
- 91.Lacour F, Delage G, Chianale C. Reduced incidence of spontaneous mammary tumors in C3H/He mice after treatment with polyadenylate-polyuridylate. Science. 1975;187:256–7. doi: 10.1126/science.163037. [DOI] [PubMed] [Google Scholar]
- 92.Lacour J, Lacour F, Spira A, Michelson M, Petit JY, Delage G, Sarrazin D, Contesso G, Viguier J. Adjuvant treatment with polyadenylic-polyuridylic acid (Polya.Polyu) in operable breast cancer. Lancet. 1980;2:161–4. doi: 10.1016/s0140-6736(80)90057-4. https://dx.doi.org/10.1016/S0140-6736(80)90057-4. [DOI] [PubMed] [Google Scholar]
- 93.Lacour J, Lacour F, Spira A, Michelson M, Petit JY, Delage G, Sarrazin D, Contesso G, Viguier J. Adjuvant treatment with polyadenylic-polyuridylic acid in operable breast cancer: updated results of a randomised trial. Br Med J (Clin Res Ed) 1984;288:589–92. doi: 10.1136/bmj.288.6417.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lacour J, Lacour F, Ducot B, Spira A, Michelson M, Petit JY, Sarrazin D, Contesso G. Polyadenylic-polyuridylic acid as adjuvant in the treatment of operable breast cancer: recent results. Eur J Surg Oncol. 1988;14:311–6. [PubMed] [Google Scholar]
- 95.Lacour J, Laplanche A, Delozier T, Berlie J, Mourali N, Julien JP, De Gislain C, Namer M, Petit JC, Denis V. Polyadenylic-polyuridylic acid plus locoregional and pelvic radiotherapy versus chemotherapy with CMF as adjuvants in operable breast cancer. A 6 1/2 year follow-up analysis of a randomized trial of the French Federation of Cancer Centers (F.F.C.C.) Breast Cancer Res Treat. 1991;19:15–21. doi: 10.1007/BF01975200. [DOI] [PubMed] [Google Scholar]
- 96.Laplanche A, Alzieu L, Delozier T, Berlie J, Veyret C, Fargeot P, Luboinski M, Lacour J. Polyadenylic-polyuridylic acid plus locoregional radiotherapy versus chemotherapy with CMF in operable breast cancer: a 14 year follow-up analysis of a randomized trial of the Fédération Nationale des Centres de Lutte contre le Cancer (FNCLCC) Breast Cancer Res Treat. 2000;64:189–91. doi: 10.1023/a:1006498121628. [DOI] [PubMed] [Google Scholar]
- 97.Potmesil M, Goldfeder A. Inhibitory effect of polyinosinic:polycytidylic acid on the growth of transplantable mouse mammary carcinoma. Proc Soc Exp Biol Med. 1972;139:1392–7. doi: 10.3181/00379727-139-36370. [DOI] [PubMed] [Google Scholar]
- 98.Lee AE, Rogers LA, Longcroft JM, Jeffery RE. Reduction of metastasis in a murine mammary tumour model by heparin and polyinosinic-polycytidylic acid. Clin Exp Metastasis. 1990;8:165–71. doi: 10.1007/BF00117789. http://dx.doi.org/10.1007/BF00117789. [DOI] [PubMed] [Google Scholar]
- 99.Charlebois R, Bertrand A, Allard D, Buisseret L, Turcotte M, Pommey S, Chrobak P, Stagg J. PolyI:C and CpG synergize with anti-ErbB2 mAb for treatment of breast tumors resistant to immune checkpoint inhibitors. Cancer Res. 2017;77:312–9. doi: 10.1158/0008-5472.CAN-16-1873. http://dx.doi.org/10.1158/0008-5472.CAN-16-1873. [DOI] [PubMed] [Google Scholar]
- 100.Curigliano G, Romieu G, Campone M, Dorval T, Duck L, Canon JL, Roemer-Becuwe C, Roselli M, Neciosup S, Burny W, Callegaro A, de Sousa Alves PM, Louahed J, Brichard V, Lehmann FF. A phase I/II trial of the safety and clinical activity of a HER2-protein based immunotherapeutic for treating women with HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2016;156:301–10. doi: 10.1007/s10549-016-3750-y. http://dx.doi.org/10.1007/s10549-016-3750-y. [DOI] [PubMed] [Google Scholar]
- 101.Li L, Wang W, Pan H, Ma G, Shi X, Xie H, Liu X, Ding Q, Zhou W, Wang S. Microwave ablation combined with OK-432 induces Th1-type response and specific antitumor immunity in a murine model of breast cancer. J Transl Med. 2017;15:23. doi: 10.1186/s12967-017-1124-9. http://dx.doi.org/10.1186/s12967-017-1124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, Hochman T, Shuman N, Axelrod D, Speyer J, Novik Y, Tiersten A, Goldberg JD, Formenti SC, Bhardwaj N, Unutmaz D, Demaria S. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. 2012;18:6748–57. doi: 10.1158/1078-0432.CCR-12-1149. http://dx.doi.org/10.1158/1078-0432.CCR-12-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74. doi: 10.1038/nm934. http://dx.doi.org/10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 104.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–9. doi: 10.1038/nm1196. http://dx.doi.org/10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 105.Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, Ismail-Khan R, Minton S, Vahanian NN, Link C, Sullivan DM, Antonia S. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5:8136–46. doi: 10.18632/oncotarget.2357. http://dx.doi.org/10.18632/oncotarget.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Soliman HH, Minton SE, Han HS, Ismail-Khan R, Neuger A, Khambati F, Noyes D, Lush R, Chiappori AA, Roberts JD, Link C, Vahanian NN, Mautino M, Streicher H, Sullivan DM, Antonia SJ. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 2016;7:22928–38. doi: 10.18632/oncotarget.8216. http://dx.doi.org/10.18632/oncotarget.8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. http://dx.doi.org/10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 108.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625–9. http://cancerres.aacrjournals.org/content/56/20/4625.long. [PubMed] [Google Scholar]
- 109.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirström K, West BL, Coussens LM. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. http://dx.doi.org/10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. http://dx.doi.org/10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 111.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, Rüttinger D. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–59. doi: 10.1016/j.ccr.2014.05.016. http://dx.doi.org/10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 112.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5. doi: 10.1038/nature10138. http://dx.doi.org/10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, Bentires-Alj M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130–3. doi: 10.1038/nature13862. http://dx.doi.org/10.1038/nature13862. [DOI] [PubMed] [Google Scholar]
- 114.Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, Wang G, Tromp BJ, Puchalski TA, Balkwill F, Berns B, Seetharam S, de Bono JS, Tolcher AW. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71:1041–50. doi: 10.1007/s00280-013-2099-8. http://dx.doi.org/10.1007/s00280-013-2099-8. [DOI] [PubMed] [Google Scholar]
- 115.Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, Elsayed Y, Dawkins F, de Bono JS. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31:760–8. doi: 10.1007/s10637-012-9869-8. http://dx.doi.org/10.1007/s10637-012-9869-8. [DOI] [PubMed] [Google Scholar]
- 116.Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L. Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol. 2015;6:12. doi: 10.3389/fimmu.2015.00012. http://dx.doi.org/10.3389/fimmu.2015.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14. doi: 10.1016/j.cell.2015.03.030. http://dx.doi.org/10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. http://dx.doi.org/10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98–106. doi: 10.1097/COC.0000000000000239. http://dx.doi.org/10.1097/COC.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743–50. doi: 10.1101/gr.165985.113. http://dx.doi.org/10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Denkert C, von Minckwitz G, Darb-Esfahani S, Ingold Heppner B, Klauschen F, Furlanetto J, Pfitzner B, Huober J, Schmitt W, Blohmer J-U, Kümmel S, Engels K, Lederer B, Schneeweiss A, Hartmann A, Jakisch C, Untch M, Hanusch C, Weber K, Loibl S. Abstract S1-09: evaluation of tumor-infiltrating lymphocytes (TILs) as predictive and prognostic biomarker in different subtypes of breast cancer treated with neoadjuvant therapy – a metaanalysis of 3771 patients. Cancer Res. 2017;77:S1–09. http://dx.doi.org/10.1158/1538-7445.SABCS16-S1-09. [Google Scholar]
- 122.Ali HR, Glont SE, Blows FM, Provenzano E, Dawson SJ, Liu B, Hiller L, Dunn J, Poole CJ, Bowden S, Earl HM, Pharoah PD, Caldas C. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann Oncol. 2015;26:1488–93. doi: 10.1093/annonc/mdv192. http://dx.doi.org/10.1093/annonc/mdv192. [DOI] [PubMed] [Google Scholar]
- 123.Sabatier R, Finetti P, Mamessier E, Raynaud S, Cervera N, Lambaudie E, Jacquemier J, Viens P, Birnbaum D, Bertucci F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget. 2015;6:5449–64. doi: 10.18632/oncotarget.3216. http://dx.doi.org/10.18632/oncotarget.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol. 2016;34:2460–7. doi: 10.1200/JCO.2015.64.8931. http://dx.doi.org/10.1200/JCO.2015.64.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, Cescon DW, Iwata H, Campone M, Nanda R, Hui R, Curigliano G, Toppmeyer, O’Shaughnessy J, Loi S, Paluch-Shimon S, Card D, Zhao J, Karantza V, Cortes J. Phase 2 study of pembrolizumab (pembro) monotherapy for previously treated metastatic triple-negative breast cancer (mTNBC): KEYNOTE-086 cohort A. JCO. 2017;35:1008. doi: 10.1093/annonc/mdy517. http://ascopubs.org/doi/abs/10.1200/JCO.2017.35.15_suppl.1088. [DOI] [PubMed] [Google Scholar]
- 126.Rugo HS, Delord J-P, Im S-A, Ott PA, Piha-Paul SA, Bedard PL, Sachdev J, Le Tourneau C, van Brummelen E, Varga A, Saraf S, Pietrangelo D, Karantza V, Tan A. Abstract S5-07: preliminary efficacy and safety of pembrolizumab (MK-3475) in patients with PD-L1–positive, estrogen receptor-positive (ER+)/HER2-negative advanced breast cancer enrolled in KEYNOTE-028. Cancer Res. 2016;76:S5–07. http://dx.doi.org/10.1158/1538-7445.SABCS15-S5-07. [Google Scholar]
- 127.Adams S, Diamond JR, Hamilton EP, Pohlmann PR, Tolaney SM, Molinero L. Phase Ib trial of atezolizumab in combination with nab-paclitaxel in patients with metastatic triple-negative breast cancer (mTNBC) JCO. 2016;34:1009. http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.1009. [Google Scholar]
- 128.Emens LA, Adams S, Loi S, Schneeweiss A, Rugo HS, Winer EP. IMpassion130: a Phase III randomized trial of atezolizumab with nab-paclitaxel for first-line treatment of patients with metastatic triple-negative breast cancer (mTNBC) JCO. 2016;34:TPS1104. http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.TPS1104#affiliationsContainer. [Google Scholar]
- 129.Dirix LY, Takacs I, Nikolinakos P, Jerusalem G, Arkenau H-T, Hamilton EPP, von Heydebreck A, Grote H-J, Chin K, Lippman ME. Abstract S1-04: Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase Ib JAVELIN solid tumor trial. Cancer Res. 2016;76:S1–04. doi: 10.1007/s10549-017-4537-5. http://dx.doi.org/10.1158/1538-7445.SABCS15-S1-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nanda R, Liu MC, Yau C, Asare S, Hylton N, Van’t Veer L. Pembrolizumab plus standard neoadjuvant therapy for high-risk breast cancer (BC): results from I-SPY 2. JCO. 2017;35:506. http://ascopubs.org/doi/abs/10.1200/JCO.2017.35.15_suppl.506#affiliationsContainer. [Google Scholar]
- 131.Schmid P, Cruz C, Braiteh FS, Eder JP, Tolaney S, Kuter I, Nanda R, Chung C, Cassier P, Delord J-P, Gordon M, Li Y, Liu B, O’Hear C, Faso M, Molinero L, Emens L. Abstract 2986: Atezolizumab in metastatic TNBC (mTNBC): long-term clinical outcomes and biomarker analyses. Cancer Res. 2017;77:2986. http://dx.doi.org/10.1158/1538-7445.AM2017-2986. [Google Scholar]
- 132.Safonov A, Jiang T, Bianchini G, Győrffy B, Karn T, Hatzis C, Pusztai L. Abstract S1-07: Immune sculpting of the triple negative breast cancer genome. Cancer Res. 2017;77:S1–07. doi: 10.1158/0008-5472.CAN-16-3478. [DOI] [PubMed] [Google Scholar]
- 133.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27. doi: 10.1158/0008-5472.CAN-11-1620. http://dx.doi.org/10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276:97–111. doi: 10.1111/imr.12520. http://dx.doi.org/10.1111/imr.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–39. doi: 10.1084/jem.20071341. http://dx.doi.org/10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moreno Ayala MA, Gottardo MF, Asad AS, Zuccato C, Nicola A, Seilicovich A, Candolfi M. Immunotherapy for the treatment of breast cancer. Expert Opin Biol Ther. 2017;17:797–812. doi: 10.1080/14712598.2017.1324566. http://dx.doi.org/10.1080/14712598.2017.1324566. [DOI] [PubMed] [Google Scholar]
- 137.Choi BD, Cai M, Bigner DD, Mehta AI, Kuan CT, Sampson JH. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin Biol Ther. 2011;11:843–53. doi: 10.1517/14712598.2011.572874. http://dx.doi.org/10.1517/14712598.2011.572874. [DOI] [PubMed] [Google Scholar]
- 138.Weiner LM, Clark JI, Ring DB, Alpaugh RK. Clinical development of 2B1, a bispecific murine monoclonal antibody targeting c-erbB-2 and Fc gamma RIII. J Hematother. 1995;4:453–6. doi: 10.1089/scd.1.1995.4.453. http://dx.doi.org/10.1089/scd.1.1995.4.453. [DOI] [PubMed] [Google Scholar]
- 139.Sen M, Wankowski DM, Garlie NK, Siebenlist RE, Van Epps D, LeFever AV, Lum LG. Use of anti-CD3 x anti-HER2/neu bispecific antibody for redirecting cytotoxicity of activated T cells toward HER2/neu+ tumors. J Hematother Stem Cell Res. 2001;10:247–60. doi: 10.1089/15258160151134944. http://dx.doi.org/10.1089/15258160151134944. [DOI] [PubMed] [Google Scholar]
- 140.Flieger D, Kufer P, Beier I, Sauerbruch T, Schmidt-Wolf IG. A bispecific single-chain antibody directed against EpCAM/CD3 in combination with the cytokines interferon alpha and interleukin-2 efficiently retargets T and CD3+CD56+ natural-killer-like T lymphocytes to EpCAM-expressing tumor cells. Cancer Immunol Immunother. 2000;49:441–8. doi: 10.1007/s002620000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Amann M, D’Argouges S, Lorenczewski G, Brischwein K, Kischel R, Lutterbuese R, Mangold S, Rau D, Volkland J, Pflanz S, Raum T, Münz M, Kufer P, Schlereth B, Baeuerle PA, Friedrich M. Antitumor activity of an EpCAM/CD3-bispecific BiTE antibody during long-term treatment of mice in the absence of T-cell anergy and sustained cytokine release. J Immunother. 2009;32:452–64. doi: 10.1097/CJI.0b013e3181a1c097. http://dx.doi.org/10.1097/CJI.0b013e3181a1c097. [DOI] [PubMed] [Google Scholar]
- 142.Wallace PK, Romet-Lemonne JL, Chokri M, Kasper LH, Fanger MW, Fadul CE. Production of macrophage-activated killer cells for targeting of glioblastoma cells with bispecific antibody to FcgammaRI and the epidermal growth factor receptor. Cancer Immunol Immunother. 2000;49:493–503. doi: 10.1007/s002620000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Borghaei H, Alpaugh RK, Bernardo P, Palazzo IE, Dutcher JP, Venkatraj U, Wood WC, Goldstein L, Weiner LM. Induction of adaptive Anti-HER2/neu immune responses in a Phase 1B/2 trial of 2B1 bispecific murine monoclonal antibody in metastatic breast cancer (E3194): a trial coordinated by the Eastern Cooperative Oncology Group. J Immunother. 2007;30:455–67. doi: 10.1097/CJI.0b013e31803bb421. http://dx.doi.org/10.1097/CJI.0b013e31803bb421. [DOI] [PubMed] [Google Scholar]
- 144.Valone FH, Kaufman PA, Guyre PM, Lewis LD, Memoli V, Ernstoff MS, Wells W, Barth R, Deo Y, Fisher J, Phipps K, Graziano R, Meyer L, Mrozek-Orlowski M, Wardwell K, Guyre V, Morley TL, Arvizu C, Wallace P, Fanger MW. Clinical trials of bispecific antibody MDX-210 in women with advanced breast or ovarian cancer that overexpresses HER-2/neu. J Hematother. 1995;4:471–5. doi: 10.1089/scd.1.1995.4.471. http://dx.doi.org/10.1089/scd.1.1995.4.471. [DOI] [PubMed] [Google Scholar]
- 145.Pullarkat V, Deo Y, Link J, Spears L, Marty V, Curnow R, Groshen S, Gee C, Weber JS. A phase I study of a HER2/neu bispecific antibody with granulocyte-colony-stimulating factor in patients with metastatic breast cancer that overexpresses HER2/neu. Cancer Immunol Immunother. 1999;48:9–21. doi: 10.1007/s002620050543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fury MG, Lipton A, Smith KM, Winston CB, Pfister DG. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol Immunother. 2008;57:155–63. doi: 10.1007/s00262-007-0357-5. http://dx.doi.org/10.1007/s00262-007-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? MAbs. 2009;1:539–47. doi: 10.4161/mabs.1.6.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shalaby MR, Carter P, Maneval D, Giltinan D, Kotts C. Bispecific HER2 x CD3 antibodies enhance T-cell cytotoxicity in vitro and localize to HER2-overexpressing xenografts in nude mice. Clin Immunol Immunopathol. 1995;74:185–92. doi: 10.1006/clin.1995.1027. [DOI] [PubMed] [Google Scholar]
- 149.Zhou Y, Gou L-T, Guo Z-H, Liu H-R, Wang J-M, Zhou S-X, Yang J-L, Li X-A. Fully human HER2/cluster of differentiation 3 bispecific antibody triggers potent and specific cytotoxicity of T lymphocytes against breast cancer. Mol Med Rep. 2015;12:147–54. doi: 10.3892/mmr.2015.3441. http://dx.doi.org/10.3892/mmr.2015.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lopez-Albaitero A, Xu H, Guo H, Wang L, Wu Z, Tran H, Chandarlapaty S, Scaltriti M, Janjigian Y, de Stanchina E, Cheung N-K. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. Oncoimmunology. 2017;6:e1267891. doi: 10.1080/2162402X.2016.1267891. http://dx.doi.org/10.1080/2162402X.2016.1267891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Urbanska K, Lynn RC, Stashwick C, Thakur A, Lum LG, Powell DJ., Jr Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J Transl Med. 2014;12:347. doi: 10.1186/s12967-014-0347-2. http://dx.doi.org/10.1186/s12967-014-0347-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Grabert RC, Cousens LP, Smith JA, Olson S, Gall J, Young WB, Davol PA, Lum LG. Human T cells armed with Her2/neu bispecific antibodies divide, are cytotoxic, and secrete cytokines with repeated stimulation. Clin Cancer Res. 2006;12:569–76. doi: 10.1158/1078-0432.CCR-05-2005. http://dx.doi.org/10.1158/1078-0432.CCR-05-2005. [DOI] [PubMed] [Google Scholar]
- 153.Jäger M, Schoberth A, Ruf P, Hess J, Lindhofer H. The trifunctional antibody ertumaxomab destroys tumor cells that express low levels of human epidermal growth factor receptor 2. Cancer Res. 2009;69:4270–6. doi: 10.1158/0008-5472.CAN-08-2861. http://dx.doi.org/10.1158/0008-5472.CAN-08-2861. [DOI] [PubMed] [Google Scholar]
- 154.Kiewe P, Hasmüller S, Kahlert S, Heinrigs M, Rack B, Marmé A, Korfel A, Jäger M, Lindhofer H, Sommer H, Thiel E, Untch M. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin Cancer Res. 2006;12:3085–91. doi: 10.1158/1078-0432.CCR-05-2436. http://dx.doi.org/10.1158/1078-0432.CCR-05-2436. [DOI] [PubMed] [Google Scholar]
- 155.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3(8):630–41. doi: 10.1038/nri1150. http://dx.doi.org/10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 156.Myc LA, Gamian A, Myc A. Cancer vaccines. Any future? Arch Immunol Ther Exp (Warsz) 2011;59:249–59. doi: 10.1007/s00005-011-0129-y. http://dx.doi.org/10.1007/s00005-011-0129-y. [DOI] [PubMed] [Google Scholar]
- 157.Clem AS. Fundamentals of vaccine immunology. J Glob Infect Dis. 2011;3:73–8. doi: 10.4103/0974-777X.77299. http://dx.doi.org/10.4103/0974-777X.77299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sayour EJ, Mitchell DA. Manipulation of innate and adaptive immunity through cancer vaccines. J Immunol Res. 2017;2017:3145742. doi: 10.1155/2017/3145742. http://dx.doi.org/10.1155/2017/3145742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mittendorf EA, Holmes JP, Ponniah S, Peoples GE. The E75 HER2/neu peptide vaccine. Cancer Immunol Immunother. 2008;57:1511–21. doi: 10.1007/s00262-008-0540-3. http://dx.doi.org/10.1007/s00262-008-0540-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mittendorf EA, Clifton GT, Holmes JP, Schneble E, van Echo D, Ponniah S, Peoples GE. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol. 2014;25:1735–42. doi: 10.1093/annonc/mdu211. http://dx.doi.org/10.1093/annonc/mdu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang X, Wang J-P, Maughan MF, Lachman LB. Alphavirus replicon particles containing the gene for HER2/neu inhibit breast cancer growth and tumorigenesis. Breast Cancer Res. 2005;7:R145–55. doi: 10.1186/bcr962. http://dx.doi.org/10.1186/bcr962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Laubreton D, Bay S, Sedlik C, Artaud C, Ganneau C, Dériaud E, Viel S, Puaux AL, Amigorena S, Gérard C, Lo-Man R, Leclerc C. The fully synthetic MAG-Tn3 therapeutic vaccine containing the tetanus toxoid-derived TT830-844 universal epitope provides anti-tumor immunity. Cancer Immunol Immunother. 2016;65:315–25. doi: 10.1007/s00262-016-1802-0. http://dx.doi.org/10.1007/s00262-016-1802-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Huang C-S, Yu AL, Tseng L-M, Chow LWC, Hou M-F, Hurvitz SA. Randomized phase II/III trial of active immunotherapy with OPT-822/OPT-821 in patients with metastatic breast cancer. JCO. 2016;34:1003. http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.1003. [Google Scholar]
- 164.Russell SJ, Peng K-W, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–70. doi: 10.1038/nbt.2287. http://dx.doi.org/10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. 2014;4:74. doi: 10.3389/fonc.2014.00074. http://dx.doi.org/10.3389/fonc.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, McGrath Y, Thomas SK, Thornton M, Bullock P, Love CA, Coffin RS. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. doi: 10.1038/sj.gt.3301885. http://dx.doi.org/10.1038/sj.gt.3301885. [DOI] [PubMed] [Google Scholar]
- 167.Merrick AE, Ilett EJ, Melcher AA. JX-594, a targeted oncolytic poxvirus for the treatment of cancer. Curr Opin Investig Drugs. 2009;10:1372–82. [PubMed] [Google Scholar]
- 168.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. http://dx.doi.org/10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mitchison NA. Studies on the immunological response to foreign tumor transplants in the mouse. I. The role of lymph node cells in conferring immunity by adoptive transfer. J Exp Med. 1955;102:157–77. doi: 10.1084/jem.102.2.157. http://dx.doi.org/10.1084/jem.102.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Borberg H, Oettgen HF, Choudry K, Beattie EJ. Inhibition of established transplants of chemically induced sarcomas in syngeneic mice by lymphocytes from immunized donors. Int J Cancer. 1972;10:539–47. doi: 10.1002/ijc.2910100312. [DOI] [PubMed] [Google Scholar]
- 171.Bernstein ID. Passive transfer of systemic tumor immunity with cells generated in vitro by a secondary immune response to a syngeneic rat gross virus-induced lymphoma. J Immunol. 1977;118:122–8. http://www.jimmunol.org/content/118/1/122.long. [PubMed] [Google Scholar]
- 172.Cheever MA, Greenberg PD, Fefer A. Tumor neutralization, immunotherapy, and chemoimmunotherapy of a Friend leukemia with cells secondarily sensitized in vitro: II. Comparison of cells cultured with and without tumor to noncultured immune cells. J Immunol. 1978;121:2220–7. [PubMed] [Google Scholar]
- 173.Strausser JL, Mazumder A, Grimm EA, Lotze MT, Rosenberg SA. Lysis of human solid tumors by autologous cells sensitized in vitro to alloantigens. J Immunol. 1981;127:266–71. https://link.springer.com/content/pdf/10.1007/BF00199454.pdf. [PubMed] [Google Scholar]
- 174.Lotze MT, Grimm EA, Mazumder A, Strausser JL, Rosenberg SA. Lysis of fresh and cultured autologous tumor by human lymphocytes cultured in T-cell growth factor. Cancer Res. 1981;41:4420–25. http://cancerres.aacrjournals.org/content/41/11_Part_1/4420.long. [PubMed] [Google Scholar]
- 175.Yron I, Wood TA, Spiess PJ, Rosenberg SA. In vitro growth of murine T cells. V. The isolation and growth of lymphoid cells infiltrating syngeneic solid tumors. J Immunol. 1980;125:238–45. http://www.jimmunol.org/content/125/1/238.long. [PubMed] [Google Scholar]
- 176.Rosenstein M, Eberlein T, Kemeny MM, Sugarbaker PH, Rosenberg SA. In vitro growth of murine T cells. VI. Accelerated skin graft rejection caused by adoptively transferred cells expanded in T cell growth factor. J Immunol. 1981;127:566–71. http://www.jimmunol.org/content/127/2/566.long. [PubMed] [Google Scholar]
- 177.Mazumder A, Rosenberg SA. Successful immunotherapy of natural killer-resistant established pulmonary melanoma metastases by the intravenous adoptive transfer of syngeneic lymphocytes activated in vitro by interleukin 2. J Exp Med. 1984;159:495–507. doi: 10.1084/jem.159.2.495. http://jem.rupress.org/content/jem/159/2/495.full.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rosenberg SA. Immunotherapy of cancer by systemic administration of lymphoid cells plus interleukin-2. J Biol Response Mod. 1984;3:501–11. [PubMed] [Google Scholar]
- 179.Okuno K, Takagi H, Nakamura T, Nakamura Y, Iwasa Z, Yasutomi M. Treatment for unresectable hepatoma via selective hepatic arterial infusion of lymphokine-activated killer cells generated from autologous spleen cells. Cancer. 1986;58:1001–6. doi: 10.1002/1097-0142(19860901)58:5<1001::aid-cncr2820580502>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 180.Schoof DD, Gramolini BA, Davidson DL, Massaro AF, Wilson RE, Eberlein TJ. Adoptive immunotherapy of human cancer using low-dose recombinant interleukin 2 and lymphokine-activated killer cells. Cancer Res. 1988;48:5007–10. [PubMed] [Google Scholar]
- 181.Rosenberg SA, Lotze MT, Yang JC, Topalian SL, Chang AE, Schwartzentruber DJ, Aebersold P, Leitman S, Linehan WM, Seipp CA. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. J Natl Cancer Inst. 1993;85:622–32. doi: 10.1093/jnci/85.8.622. [DOI] [PubMed] [Google Scholar]
- 182.West WH. Continuous infusion recombinant interleukin-2 (rIL-2) in adoptive cellular therapy of renal carcinoma and other malignancies. Cancer Treat Rev. 1989;16( Suppl A):83–9. doi: 10.1016/0305-7372(89)90027-3. [DOI] [PubMed] [Google Scholar]
- 183.Sparano JA, Fisher RI, Weiss GR, Margolin K, Aronson FR, Hawkins MJ, Atkins MB, Dutcher JP, Gaynor ER, Boldt DH. Phase II trials of high-dose interleukin-2 and lymphokine-activated killer cells in advanced breast carcinoma and carcinoma of the lung, ovary, and pancreas and other tumors. J Immunother Emphasis Tumor Immunol. 1994;16:216–23. doi: 10.1097/00002371-199410000-00006. [DOI] [PubMed] [Google Scholar]
- 184.Gharib M, Mainguené C, Tamboise E, Tamboise A, Beaupain R. Lymphokine-activated killer cells induce differentiation in MCF-7 breast carcinoma nodules but not in mastosis nodules maintained in three-dimensional culture. Tumour Biol. 1994;15:90–100. doi: 10.1159/000217879. https://dx.doi.org/10.1159/000217879. [DOI] [PubMed] [Google Scholar]
- 185.Cardillo M, Yankelevich B, Mazumder A, Lupu R. Heregulin induces increase in sensitivity of an erbB-2-overexpressing breast cancer cell type to lysis by lymphokine-activated killer cells. Cancer Immunol Immunother. 1996;43:19–25. doi: 10.1007/s002620050298. https://link.springer.com/content/pdf/10.1007/s002620050298.pdf. [DOI] [PubMed] [Google Scholar]
- 186.Assadipour Y, Zacharakis N, Crystal JS, Prickett TD, Gartner JJ, Somerville RPT, Xu H, Black MA, Jia L, Chinnasamy H, Kriley I, Lu L, Wunderlich JR, Zheng Z, Lu YC, Robbins PF, Rosenberg SA, Goff SL, Feldman SA. Characterization of an immunogenic mutation in a patient with metastatic triple-negative breast cancer. Clin Cancer Res. 2017;23:4347–53. doi: 10.1158/1078-0432.CCR-16-1423. http://dx.doi.org/10.1158/1078-0432.CCR-16-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Parkhurst M, Gros A, Pasetto A, Prickett T, Crystal JS, Robbins P, Rosenberg SA. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin Cancer Res. 2017;23:2491–505. doi: 10.1158/1078-0432.CCR-16-2680. http://dx.doi.org/10.1158/1078-0432.CCR-16-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zacharakis N, Trebska-McGowan K, Somerville R, Lu Y-C, Pasetto A, Black M, Chinnasamy H, Xu H, Gartner JJ, Prickett TD, Robbins PF, Rosenberg SA, Goff SL, Feldman SA. Abstract 4982: Regression of metastatic breast cancer after adoptive cell transfer of tumor infiltrating lymphocytes and checkpoint blockade. Cancer Res. 2017;77:4982. http://dx.doi.org/10.1158/1538-7445.AM2017-4982. [Google Scholar]
- 189.Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics. 2016;3:16014. doi: 10.1038/mto.2016.14. http://dx.doi.org/10.1038/mto.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol. 1993;151:6577–82. http://www.jimmunol.org/content/151/11/6577.long. [PubMed] [Google Scholar]
- 191.Sun M, Shi H, Liu C, Liu J, Liu X, Sun Y. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res. 2014;16:R61. doi: 10.1186/bcr3674. http://dx.doi.org/10.1186/bcr3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183:5563–74. doi: 10.4049/jimmunol.0900447. http://dx.doi.org/10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L, Dotti G, Gottschalk SM, Wilson MH, Rooney CM. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther. 2011;19:2133–43. doi: 10.1038/mt.2011.131. http://dx.doi.org/10.1038/mt.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J. 2015;21:486–91. doi: 10.1097/PPO.0000000000000156. http://dx.doi.org/10.1097/PPO.0000000000000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shenouda MM, Gillgrass A, Nham T, Hogg R, Lee AJ, Chew MV, Shafaei M, Aarts C, Lee DA, Hassell J, Bane A, Dhesy-Thind S, Ashkar AA. Ex vivo expanded natural killer cells from breast cancer patients and healthy donors are highly cytotoxic against breast cancer cell lines and patient-derived tumours. Breast Cancer Res. 2017;19:76. doi: 10.1186/s13058-017-0867-9. http://dx.doi.org/10.1186/s13058-017-0867-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bönig H, Köhl U, Kloess S, Köhler S, Holtgreve-Grez H, Jauch A, Schmidt M, Schubert R, Kühlcke K, Seifried E, Klingemann HG, Rieger MA, Tonn T, Grez M, Wels WS. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23:330–8. doi: 10.1038/mt.2014.219. http://dx.doi.org/10.1038/mt.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61:1451–61. doi: 10.1007/s00262-012-1212-x. http://dx.doi.org/10.1007/s00262-012-1212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Coveney E, Wheatley GH, Lyerly HK. Active immunization using dendritic cells mixed with tumor cells inhibits the growth of primary breast cancer. Surgery. 1997;122:228–34. doi: 10.1016/s0039-6060(97)90013-1. https://www.researchgate.net/publication/10707410_Active_Immunization_Using_Dendritic_Cells_Mixed_With_Tumor_Cells_Inhibits_The_Growth_Of_Lymphomas. [DOI] [PubMed] [Google Scholar]
- 199.Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 2000;96:3102–8. http://www.bloodjournal.org/content/96/9/3102.long?sso-checked=true. [PubMed] [Google Scholar]
- 200.Morse MA, Deng Y, Coleman D, Hull S, Kitrell-Fisher E, Nair S, Schlom J, Ryback ME, Lyerly HK. A Phase I study of active immunotherapy with carcinoembryonic antigen peptide (CAP-1)-pulsed, autologous human cultured dendritic cells in patients with metastatic malignancies expressing carcinoembryonic antigen. Clin Cancer Res. 1999;5:1331–8. http://clincancerres.aacrjournals.org/content/5/6/1331.long. [PubMed] [Google Scholar]
- 201.Chen Y, Emtage P, Zhu Q, Foley R, Muller W, Hitt M, Gauldie J, Wan Y. Induction of ErbB-2/neu-specific protective and therapeutic antitumor immunity using genetically modified dendritic cells: enhanced efficacy by cotransduction of gene encoding IL-12. Gene Ther. 2001;8:316–23. doi: 10.1038/sj.gt.3301396. http://dx.doi.org/10.1038/sj.gt.3301396. [DOI] [PubMed] [Google Scholar]
- 202.Pecher G, Häring A, Kaiser L, Thiel E. Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunol Immunother. 2002;51:669–73. doi: 10.1007/s00262-002-0317-z. http://dx.doi.org/10.1007/s00262-002-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gong J, Avigan D, Chen D, Wu Z, Koido S, Kashiwaba M, Kufe D. Activation of antitumor cytotoxic T lymphocytes by fusions of human dendritic cells and breast carcinoma cells. Proc Natl Acad Sci USA. 2000;97:2715–8. doi: 10.1073/pnas.050587197. http://dx.doi.org/10.1073/pnas.050587197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhang Q, Liu X-Y, Zhang T, Xhang X-F, Xhao L, Long F, Liu Z-K, Wang E-H. The dual-functional capability of cytokine-induced killer cells and application in tumor immunology. Hum Immunol. 2015;76:385–91. doi: 10.1016/j.humimm.2014.09.021. http://dx.doi.org/10.1016/j.humimm.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 205.Ren J, Di L, Song G, Yu J, Jia J, Zhu Y, Yan Y, Jiang H, Liang X, Che L, Zhang J, Wan F, Wang X, Zhou X, Lyerly HK. Selections of appropriate regimen of high-dose chemotherapy combined with adoptive cellular therapy with dendritic and cytokine-induced killer cells improved progression-free and overall survival in patients with metastatic breast cancer: reargument of such contentious therapeutic preferences. Clin Transl Oncol. 2013;15:780–8. doi: 10.1007/s12094-013-1001-9. http://dx.doi.org/10.1007/s12094-013-1001-9. [DOI] [PubMed] [Google Scholar]
- 206.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23. doi: 10.1016/j.cell.2017.01.017. http://dx.doi.org/10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Czerniecki BJ, Koski GK, Koldovsky U, Xu S, Cohen PA, Mick R, Nisenbaum H, Pasha T, Xu M, Fox KR, Weinstein S, Orel SG, Vonderheide R, Coukos G, DeMichele A, Araujo L, Spitz FR, Rosen M, Levine BL, June C, Zhang PJ. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007;67:1842–52. doi: 10.1158/0008-5472.CAN-06-4038. http://dx.doi.org/10.1158/0008-5472.CAN-06-4038. [DOI] [PubMed] [Google Scholar]
- 208.Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A. Phase 2 study of pembrolizumab (pembro) monotherapy for previously treated metastatic triple-negative breast cancer (mTNBC): KEYNOTE-086 cohort A. JCO. 2017;35:1008. doi: 10.1093/annonc/mdy517. http://ascopubs.org/doi/abs/10.1200/JCO.2017.35.15_suppl.1008. [DOI] [PubMed] [Google Scholar]
- 209.Schmid P, Park YH, Muñoz-Couselo E, Kim S-B, Sohn J, Im S-A. Pembrolizumab (pembro) + chemotherapy (chemo) as neoadjuvant treatment for triple negative breast cancer (TNBC): preliminary results from KEYNOTE-173. JCO. 2017;35:556. http://ascopubs.org/doi/abs/10.1200/JCO.2017.35.15_suppl.556#affiliationsContainer. [Google Scholar]