

ABSTRACT
Vascular smooth muscle cell (SMC) switching between differentiated and dedifferentiated phenotypes is reversible and accompanied by morphological and functional alterations that require reconfiguration of cell-cell and cell-matrix adhesion networks. Studies attempting to explore changes in overall composition of the adhesion nexus during SMC phenotype transition are lacking. We have previously demonstrated that T-cadherin knockdown enforces SMC differentiation, whereas T-cadherin upregulation promotes SMC dedifferentiation. This study used human aortic SMCs ectopically modified with respect to T-cadherin expression to characterize phenotype-associated cell-matrix adhesion molecule expression, focal adhesions configuration and migration modes. Compared with dedifferentiated/migratory SMCs (expressing T-cadherin), the differentiated/contractile SMCs (T-cadherin-deficient) exhibited increased adhesion to several extracellular matrix substrata, decreased expression of several integrins, matrix metalloproteinases and collagens, and also distinct focal adhesion, adherens junction and intracellular tension network configurations. Differentiated and dedifferentiated phenotypes displayed distinct migrational velocity and directional persistence. The restricted migration efficiency of the differentiated phenotype was fully overcome by reducing actin polymerization with ROCK inhibitor Y-27632 whereas myosin II inhibitor blebbistatin was less effective. Migration efficiency of the dedifferentiated phenotype was diminished by promoting actin polymerization with lysophosphatidic acid. These findings held true in both 2D-monolayer and 3D-spheroid migration models. Thus, our data suggest that despite global differences in the cell adhesion nexus of the differentiated and dedifferentiated phenotypes, structural actin cytoskeleton characteristics per se play a crucial role in permissive regulation of cell-matrix adhesive interactions and cell migration behavior during T-cadherin-induced SMC phenotype transition.
KEYWORDS: 2-D monolayer, 3-D spheroid, actin cytoskeleton, adhesion, adhesion molecules, migration, phenotype modulation, T-cadherin, vascular smooth muscle cell
Introduction
Vascular smooth muscle cells (SMCs) retain a high degree of plasticity. In response to environmental guidance, SMCs can reversibly switch between 2 functionally and morphologically different phenotypes – contractile (quiescent) and synthetic (dedifferentiated). The functions of contractile and synthetic SMC phenotypes differ fundamentally, and cellular machinery and morphology during phenotype switch undergo remarkable changes.1 The contractile phenotype, which regulates hemodynamics and provides structural stability for the vessel, maintains a robust, relatively static cytoskeleton2 as well as mechanical stress-resilient adhesive interactions.3 In contrast, the synthetic phenotype, which is committed to repair and regeneration, requires a cellular architecture composed of dynamic cytoskeletal and adhesion molecule networks. The SMC dedifferentiation program can be initiated by a variety of extracellular stimuli such as growth factors, inflammatory cytokines or extracellular matrix (ECM) ligands.4,5 Signaling cascades activated by these stimuli result in loss of contractile competence and gain of new functions that include migration, ECM remodeling, and proliferation.5,6 Reconfiguration of cellular architecture, adhesive and cytoskeletal networks is necessary for this functional switch and is reflected by changes in cellular morphology in 2D, which is considered as a primary and classical hallmark of SMC phenotype switch.1,7
Most of our knowledge on cell adhesion properties during SMC phenotype transition is derived from reductionist studies which investigated the functional properties of any given adhesion molecule in certain biological contexts. Cellular adhesive functions, however, are enabled by the large and dynamic network of interconnected adhesion clusters. Studies attempting to explore changes in overall composition of the adhesion nexus during functional SMC phenotype switching are lacking. Moreover, while regulatory roles of adhesion receptor and actin cytoskeleton reconfiguration have been studied to some extent in non-muscle cells,8-11 they have been rarely addressed in the context of SMC phenotype transition.
Previous studies have shown that cellular anchorage and migration characteristics highly depend on the guidance properties of the ECM and cellular ability to respond to that guidance.12-14 The participation of the adhesome composition, cell-to-substratum and cell-to-cell interactions and actin cytoskeleton organization in the regulation of anchorage and migration characteristics of differentiated/contractile and dedifferentiated/migratory SMCs is not fully understood. We have previously shown that an atypical lipid-anchored adhesion and guidance receptor T-cadherin regulates classical hallmarks of SMC phenotype transition, including sensitivity to insulin, contractile competence, ECM remodeling,15 migration and proliferation.16 T-cadherin knockdown prevents, whereas T-cadherin upregulation promotes, SMC dedifferentiation through PI3K/Akt/mTOR and PI3K/Akt/GSK3β signaling axes.16 In the present study, we have exploited human aortic vascular SMCs with ectopically modified T-cadherin expression as a model to investigate phenotype switch-associated changes in SMC adhesion networks and migration characteristics. We aimed to identify cell-matrix adhesion molecules most affected by SMC phenotype transition, to characterize phenotype-specific migration modes, and to determine the contribution of the actin cytoskeleton to the differential migration modes.
Materials and methods
Cell culture and transduction
Primary human aortic SMCs (cat. # PCS-100–012) were obtained from ATCC (LGS Standards GmbH) and cultured in low glucose (5.5 mM) DMEM (cat. # D6046) (Sigma-Aldrich Chemie) supplemented with 5% FCS (cat. # 10500–064) and 5% of smooth muscle cell growth supplement (cat. # S-007–25) (both from Gibco, Thermo Fisher Scientific Life Technologies Europe). T-cadherin was stably overexpressed in SMCs (Tcad+-SMC) using pLVX-puro vector carrying full length human T-cadherin cDNA (empty pLVX-puro vector SMC transductants (E-SMC) served as control).16 Stable T-cadherin knockdown SMC transductants (shTcad-SMC) were generated using pLKO.1-puro vector carrying human T-cadherin shRNA (SMCs transduced with non-target shRNA (shC-SMC) served as control).16 SMCs were transduced at passage 5, puromycin-selected (3 µg/ml), and used at up to passage 8–10. Expression of T-cadherin was routinely monitored by immunoblotting (representative blot is shown in Supplemental Fig. S1A).
Static cell adhesion assay
The Chemicon fluorometric ECM cell adhesion array kit (cat. # ECM545) (Merck Millipore) was used to estimate adhesion to various ECM components. Subconfluent cultures were lifted using buffered EDTA solution (2.5 mM in PBS), plated into 96-well microtiter plate (5 × 105 cells/well, in triplicates) of rehydrated ECM strips and incubated for 1 h in the tissue culture incubator. Strips were then processed for fluorometric quantification (485/530) of cell adhesion. Fluorometric data for all transductants and their adhesion onto the different substrata are expressed relative to that obtained for the E-SMC on collagen I, which in any given experiment was arbitrarily set as 1.
Adhesion and extracellular matrix molecule profiling by qPCR arrays
Total RNA of SMCs transduced with lentiviral particles carrying T-cadherin shRNA or control shRNA and normally cultured (without passaging) for 4 d after the application of viral particles was extracted using High Pure RNA Isolation Kit (cat. # 11828665001) (Roche Diagnostics). RT2 First Strand Kit (cat. # 330401) was used for reverse transcription, quantitative real time PCR was performed using RT2 profiler PCR arrays (Human Extracellular Matrix & Adhesion molecules (cat. # PAHS-013ZE) and Human Focal Adhesions (cat. # PAHS-145 ZE)), RT2 SYBR Green ROX qPCR Mastermix (cat. # 330520) (all from Qiagen), and ABI Vii A7 Real-Time PCR system (Applied Biosystems, Thermo Fisher Scientific Life Technologies Europe). Obtained Ct values were uploaded on to the Qiagen data analysis web portal, the Ct cut-off 35 was set, normalization was done using manual selection from the panel of reference genes. Fold change/regulation was calculated by ΔΔCt method. The p-values were calculated by a Student's t-test of the replicate 2(- ΔCt) values for each gene, fold-changes larger than 2 with p-values less than 0.05 were considered statistically significant.
Immunoblotting
Cells were plated into 6-well dishes (2 × 105 cells/well) and cultured for 48 h. Whole cell lysates were prepared and analyzed by immunoblotting as described before.16 Primary antibodies against following proteins/epitopes were used: T-cadherin (cat.# AF3264) (R&D Systems Europe Ltd.), Akt (cat. # 9272), integrin β1 (cat. # 34971), (Cell Signaling Technologies, BioConcept Ltd.), phospho (p)-FAKTyr397 (cat # 44–624G), p-FAKTyr577 (cat.# 44–614ZG), (Invitrogen), FAK (cat. # 610087), N-cadherin (cat. #610920), (BD Biosciences), integrin β3 (cat. # ab7167) (Abcam). Secondary HRP-conjugated anti-species specific IgGs were from Southern Biotechnology (BioConcept Ltd.). Akt was used as the loading control. Representative blots are shown.
Immunofluorescence microscopy
Cells were plated into 24-well plates (2 × 104 cells/well) containing round 13-mm glass coverslips (VWR International) precoated with 0.5% gelatin (cat. # 40780500) (Merck Millipore) or fibronectin (5 µg/ml) (cat. # F2006) (Sigma-Aldrich) and cultured for 48 h (on gelatin) for paxillin; and N-cadherin, or 2 h (on fibronectin) for integrin α5β1 and ανβ3 staining. Cells on coverslips were rinsed with PBS, fixed in 4% paraformaldehyde, permeabilized and blocked with 0.3% Triton X-100/10%BSA. Cells were incubated with primary antibodies against either paxillin (cat. # P13520) (Cell Signaling Technologies), N-cadherin (cat.# 610920) (BD Biosciences), integrin α5β1 (cat. # A108) or αvβ3 (cat. # A109) (Thelios/Gibco) followed by Alexa Fluor 546 or Alexa Fluor 488 species-specific secondary IgG (Invitrogen). Counterstaining of nuclei and fibrous actin was achieved with Hoechst 33342 (cat. # B2261) (Sigma-Aldrich Chemie) and TRITC- (cat. # P1951) or Alexa Fluor 647-conjugated (cat. # ABNOU0298) phalloidin (Sigma-Aldrich or VWR International, respectively). Preparations were mounted on slides using Vectashield mounting medium (cat. # H-1000) (Vector Laboratories, Adipogen AG). Images were acquired under an Olympus BX63 fluorescent microscope, equipped with a DP80 camera and CellSens software (all from Olympus Switzerland).
Image quantification of focal adhesions
Color images obtained by immunofluorescent microscopy were processed using ImageJ (NIH) software. Quantification of paxillin-stained focal adhesions was done by setting fixed threshold for single channel 8bit black and white images; FA count and size, or nuclei count was estimated using analyze particles function. FA count per cell was calculated dividing FA count by nuclear count from each field. Ten random fields for each group from each of 3 independent experiments were assessed. Quantification of α5β1 FAs was done by subjecting fixed size (1400μm2) regions of interest to Trainable Weka segmentation (ImageJ) and analyzing resulting probability maps by analyze particles function after prefixed thresholding (ImageJ). At least 15 random areas were analyzed for each group. Estimates of average FA size, number and circularity per area (1400 μm2) are presented.
Cell migration analysis in 2D and time-lapse videomicroscopy
Non-treated tissue culture dishes pre-coated with fibronectin (5 µg/ml) and then blocked with BSA (0.1%) were used. To analyze random migration of individual cells in 2D culture SMC transductants were plated at a density of 2.5 × 103 cells per well in 24-well plates. After allowing an adherence period of 2 h cell migration was recorded by time-lapse videomicroscopy. In the experiments, where cytoskeleton modulating compounds were used, cells were allowed to adhere to fibronectin for 90 min, followed by 30 min preincubation in the presence of either 1 μM blebbistatin (cat. # B0560) (Sigma-Aldrich), 5 μM Y-27632 (cat. # 688001) (Calbiochem/Merck Millipore) or 5 nM oleoyl-L-α-lysophosphatidic acid (LPA) (cat. # BML-LP100) (Enzo Life Sciences) before initiation of videomicroscopy. Phase contrast images at 4X magnification were taken every 30 min for 12 h using an Olympus IX-81 inverted time-lapse microscope equipped with a digital camera and CellSens software (both from Olympus) within a humidified incubation chamber with 5% CO2 at 37°C. For any given experimental condition, analysis of the migration characteristics of 72–100 randomly chosen cells of each transductant type was performed manually, using ImageJ (NIH) plugin MTrackJ. DiPer macros package was used to plot cell migration trajectories, mean square displacement (MSD), directional ratio and migration velocity.17 Quantitative cell migration differences between the transductants phenotype were estimated by comparing 3 migration parameters: MSD, directionality ratio and migration velocity. Descriptions and calculation methods for each of these parameters have been fully detailed.17 In brief, MSD was calculated as an overall readout for migration efficiency: MSD is a measure of the surface area explored by the cell over increasing time intervals and depends on migration directionality and velocity. Directionality ratio, or so called “straightness of the migration," is a measure of directional persistence, which usually is calculated by dividing the length of the straight line connecting the migration start- and end-points by the migration trajectory. Here directionality ratio was calculated as an index changing over increasing time. Migration velocity was calculated as the average speed over the experimental period.
Cell migration analysis in 3D
Migration assay in 3D was performed as described previously18 with minor modifications. Spheroids composed of 2.5 × 103 SMCs per spheroid were prepared using the “hanging drop” method end embedded within Matrigel (cat. #354234) (3 mg/ml; BD Biosciences/Corning) into 96-well dishes. Each condition was performed in triplicate and each gel contained 20–30 spheroids. Gels were overlaid with 200 μl normal SMC growth medium and incubated for up to 3 d. Images of spheroids were captured every day using an Olympus IX50 inverted fluorescence microscope. Expansion (outward migration) capacity was measured morphometrically using ImageJ software and calculated as the difference between the circumference of the outer invasion border and the circumference of the original spheroid. Every spheroid was analyzed, and the results were averaged.
Statistical analysis
All experiments were performed on at least 3 independent occasions and unless otherwise stated results are given as mean ± SD. GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis. Data presented in bar graphs were analyzed by one-way analysis of variance (ANOVA) followed by Tukey post-hoc testing for multiple comparisons. Data from random migration experiments yielding calculated MSD and directional ratio values were analyzed by nonlinear regression with least squares second order polynomial curve fitting (quadratic) with extra sum-of-squares F test as comparison method. P<0.05 was considered as threshold of significance.
Results
Ectopic T-cadherin-induced SMC phenotype switching alters SMC adhesion properties
We have previously shown that T-cadherin regulates SMC morphology and functional phenotype: T-cadherin-overexpression (Tcad+) in SMCs induced a dedifferentiated, very poorly spread phenotype (vs. their control empty vector (E) transductants), whereas T-cadherin-depleted SMCs (shTcad) exhibited a well-differentiated contractile, fully spread phenotype (vs. their control scrambled shRNA (shC) transductants).16 To investigate whether the morphological/spreading distinction between these phenotypes reflects a differential ability to adhere to the matrix, subconfluent cell monolayers were stained for focal adhesion (FA) adaptor protein paxillin. The differentiated shTcad SMCs spread most efficiently (Fig. 1A), formed the highest number (Fig. 1B) and size (Fig. 1C) of FAs, and exhibited a high abundance of the elongated, mature FA type (Fig. 1A, inset), indicating enhanced matrix anchorage function. In contrast, the dedifferentiated Tcad+-SMCs exhibited reduced spreading: they appeared thinner (Fig. 1A), formed less FAs per cell (Fig. 1B) and mature adhesions were rare, if present (Fig. 1A, inset), suggesting weaker matrix interactions.
Figure 1.
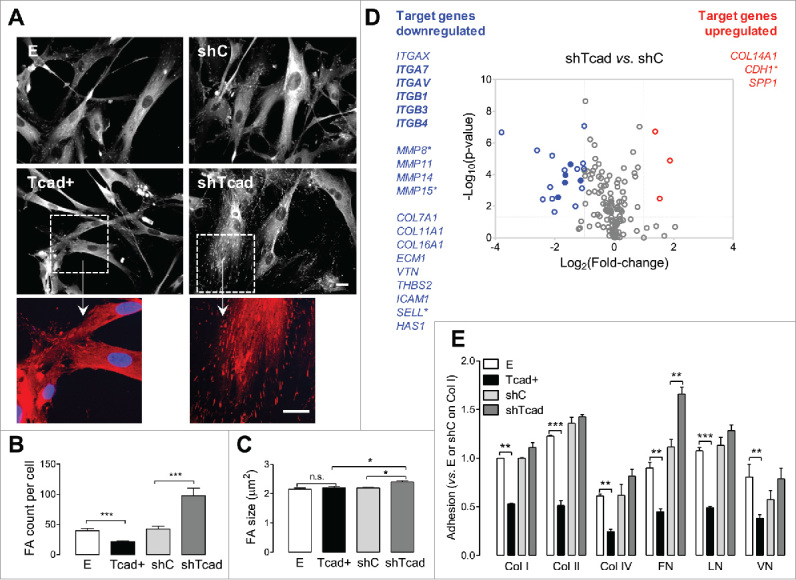
Modulation of T-cadherin expression changes SMC adhesion properties and adhesion molecule expression profile. SMC stably transduced with respect to T-cadherin upregulation (Tcad+) or deficiency (shTcad) and their controls (E or shC) were plated onto gelatin-precoated glass coverslips (A-C), standard tissue culture dishes (D), or Chemicon fluorometric ECM cell adhesion array strips (E). (A) Representative photomicrographs of SMC following fixation and immunofluorescence staining for paxillin 48 h after plating. High magnification fragments (boxed frames) are presented to highlight the difference in FAs between Tcad+- and shTcad-SMCs. Scale bars = 25 µm. Paxillin-stained images were processed for the quantification of (B) FA count/per cell and (C) FA size. (D) Comparative analysis of adhesion-relevant gene expression profiles in shTcad SMC vs. control shC SMC using Human Extracellular Matrix & Adhesion Molecules and Human Focal Adhesions gene arrays. The 2 profiling panels contained a total of 147 target genes, 17 of which were common to both panels. Total RNA was extracted 4 d after viral transduction and culture. Two vertical lines on the volcano plot mark 2-fold change boundaries and the horizontal line marks the statistical significance boundary (p < 0.05). Target genes with fold change ≥ 2 and statistical significance are marked with blue circles/text (downregulated genes) and red circles/text (upregulated genes). Target genes shared between the 2 arrays and commonly regulated are indicated with filled circles/bolded text. Target genes with relatively high average threshold cycle (Ct> 30) are marked with an asterisk. (E) Adhesion onto the indicated ECM components was quantified after 1 h incubation. Data are given as mean ± SD. Significant differences between E and Tcad+ SMC or shC and shTcad SMC are indicated with asterisks: **P <0.01; ***P <0.001.
Clustering of integrins is the initial step of cell adhesion and triggers auto-phosphorylation of focal adhesion kinase (FAK) at Tyr 397,19 which can be used as a molecular readout for probing the degree of cell spreading. In agreement with the findings obtained through paxillin immunofluorescence staining (Fig. 1A) levels of FAKTyr397 phosphorylation were highest in the well-spread shTcad SMCs and lowest in Tcad+ SMCs (Supplemental Fig. S1B). In contrast to the Tyr397 phosphorylation status, Src kinase-dependent phosphorylation of FAK on Tyr57719 was not significantly altered by either upregulation or knock-down of T-cadherin (Supplemental Fig. S1B).
T-cadherin-induced SMC phenotype switching modulates integrin and ECM molecule gene expression profiles
To investigate SMC phenotype-associated changes in adhesome at the transcriptional level, we performed gene expression profiling using the Focal Adhesions and Extracellular Matrix & Adhesion Molecule RT-PCR arrays. We compared shTcad-SMCs with the corresponding control shC-SMCs, because this pair showed the most robust differences in morphology, spreading and integrin clustering. The 2 profiling panels contained a total of 147 target genes, 17 of which were common to both panels. The average mRNA expression from 3 independent biologic experiments was significantly changed (p<0.05 and fold regulation above 2) for 22 genes from 147 tested targets (Fig. 1D) and Supplemental Table S1). As expected from pre-selected and validated target panel arrays, we observed relatively low fold change variability between individual measurements as can be seen from data scattering toward higher reliability areas on the volcano plot (Fig. 1D). Of the 17 target genes shared on the Focal Adhesions array and the Extracellular Matrix & Adhesion Molecule array 5 (ITGA7, ITGAV, ITGAX, ITGB1 and ITGB3) were significantly and commonly regulated (Supplemental Table S1). The close recapitulation of fold-change values demonstrates good overall reproducibility of the screening. Two other shared genes (ITGA6 and ITGB2) that were low-expression targets (Ct>30) were regulated in an opposing manner between the arrays, and therefore excluded from the final list of regulated genes and volcano plot. For this reason, other significantly regulated, but low-expression genes (CDH1, MMP8, MMP15 and SELL; Ct>30), were considered as “low confidence” targets (Fig. 1D). Of the 22 adhesion-relevant molecules significantly altered by T-cadherin knock-down, 3 were upregulated and 19 were downregulated (Fig. 1E). A listing of these molecules according to the specific functional groupings of the RT-PCR arrays as well as the number of available test targets for each functional group is given in Supplemental Table S2.
Among the significantly regulated genes, integrins emerged as the dominating functional group (6 from 21 integrins tested). Matrix metalloproteinases (3 from 18 ECM proteases tested) and collagens (4 from 12 collagens tested) were 2 other functional groups affected by T-cadherin knock-down. There were no changes in expression of the gene targets functionally linked to signaling through focal adhesion kinase, integrin linked kinase, G proteins, AKT/PI3K pathways, caveolins, cytoskeletal regulators and actin binding proteins (Supplemental Table S1).
To address whether the observed adhesion difference between contractile shTcad and dedifferentiated Tcad+ SMC phenotypes was ligand-specific and driven by regulation of specific matrix adhesion receptors, we examined adhesion to various ECM components using ECM cell adhesion array. Adhesion to all of the tested matrixes, namely collagen I (col I), collagen II (col II), collagen IV (col IV), fibronectin (Fn), laminin (Ln) and vitronectin (Vn), was reduced by at least 50% in the Tcad+SMCs (p at least < 0.01 vs. E-SMCs for all matrixes). There was a general trend for improved adhesion of shTcad -SMCs (vs. shC) onto each matrix substrata, although a significant difference was achieved only for adhesion onto fibronectin (Fig. 1E).
Expression of β1 and β3 integrin proteins and organization of α5β1 and ανβ3 integrin focal adhesion networks are distinct between the SMC phenotypes
Integrins β1 and β3 were among the target genes identified by qPCR profiling as being downregulated in the contractile shTcad-SMCs (Supplemental Table S1). Immunoblotting of whole cell lysates demonstrated a significant downregulation of integrin β1 and integrin β3 protein abundancies in shTcad-SMCs and also a significant, albeit more modest, upregulation in dedifferentiated Tcad+-SMCs (Fig. 2A, B).
Figure 2.
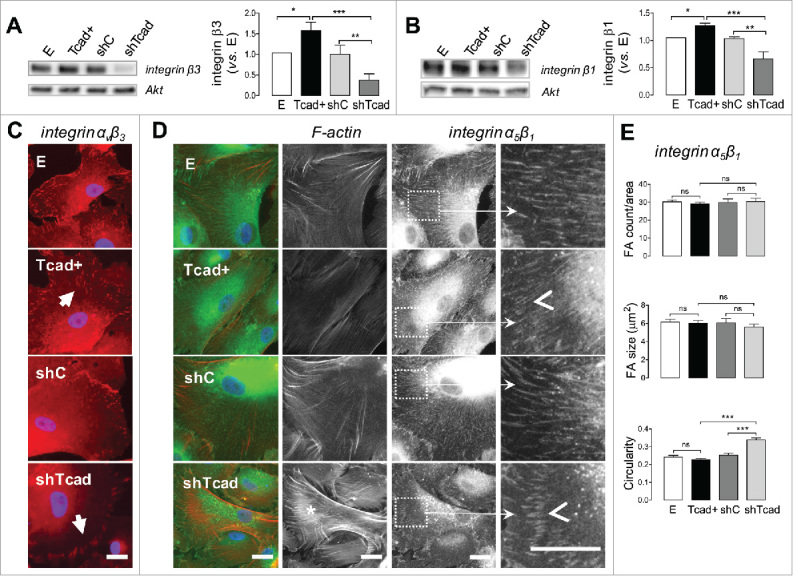
T-cadherin-based SMC phenotype switch alters morphology and distribution of integrin-ανβ3 and integrin-α5β1 specific cell-matrix adhesions. SMC transductants were plated onto fibronectin pre-coated tissue culture dishes or glass coverslips for immunoblotting or immunocytochemistry protocols, respectively. (A, B) Representative blots of integrin β3, integrin β1 (with Akt as internal loading control) are presented and quantification of abundancies in the different transductants is expressed relative to levels in control E SMC. Obtained data are presented as mean ± SD. One-way ANOVA followed by Tukey post-hoc testing was used for statistical analysis. Asterisks indicate significant differences. (*P <0.05; **P <0.01; ***P <0.001). (C, D) Representative images of SMCs after 2 h of spreading on fibronectin. Scale bars = 25 µm. (C) SMCs stained for integrin-ανβ3 (red), and nuclei (blue). Note the prominent FAs on the periphery of shTcad-SMCs and the smaller scattered FAs in the lamellar regions of Tcad+-SMCs (indicated by closed arrowheads). (D) SMCs stained for fibrous actin (F-actin; red), integrin α5β1 (green), and nuclei (blue). F-actin and integrin α5β1 channels are separately presented in grayscale to the right of the merged color images. Pronounced actin bundles characteristic of shTcad-SMCs are indicated by the asterisk *. To highlight differences in integrin α5β1-specific FA morphology high magnification fragments (white squares) are presented: open arrowheads point to the distinct morphometries of shTcad-SMCs (short, thick ordered FA arrays) and Tcad+-SMCs (longer, thin FA arrays). (E) Morphometric quantification of integrin α5β1-specific FAs in each transductant with respect to number/area, size and circularity. Obtained data are presented as mean ± SD. One-way ANOVA followed by Tukey post-hoc testing was used for statistical analysis. **P ***P <0.001;. ns, not significantly different.
Integrins β1 and β3 can dimerize with α integrins to yield several fibronectin-binding heterodimers. The review by Humphries at al., lists 9 functionally validated high affinity fibronectin binding heterodimers (ανβ3, ανβ6, ανβ1, α4β1, α5β1, α8β1, αIIbβ3, α4β7, αDβ2),20 and expression of at least 6 different fibronectin binding integrin heterodimers (ανβ3, ανβ1, α3β1, α4β1, α5β1, α8β1,) have been reported in vascular smooth muscle cells.3,21-23 Since adhesion differences between Tcad+-SMCs and shTcad-SMCs were most prominent for fibronectin (Fig. 1F) and integrin α5β1 and ανβ3/β1 are dominant receptors for SMC adhesion to fibronectin3,23 we chose to more closely examine α5β1 and ανβ3 heterodimer-mediated adhesion of SMCs on fibronectin. Moreover, these heterodimers are the most studied fibronectin binding receptors in the context of atherosclerosis/restenosis3 and other pathologies (e.g. aneurysm,24 hypertension25) characterized by vascular remodeling; their relevance for SMC phenotype transition has been repeatedly reported both in vitro26-28 and in vivo.21,29-31
Integrin ανβ3 staining of SMCs after 2 h spreading on fibronectin revealed a cellular periphery-defined presence of FAs, with most of the ανβ3-specific FAs being localized at the lamellipodium and the front lamella (Fig. 2C and Supplemental Fig. S2). Between SMC phenotypes, the most prominent differences in ανβ3-specific FAs were evident with respect morphometry and localization, rather than overall staining intensity, albeit weaker in shTcad-SMCs. Large clustered FAs, localized to the tips of lamellipodium, dominated in contractile shTcad-SMCs. In contrast, small, scattered and more abundant FAs were present in the differentiated SMCs, and FA localization was not restricted to the lamellipodial tips, but extended to the front-or even mid- (particularly in Tcad+-SMCs) lamellar regions (Fig. 2C).
Integrin α5β1 staining of spreading SMC resulted in characteristic staining pattern for this heterodimer: dense networks of fibrillar FAs were distributed through the whole cell spreading area between perinuclear regions and cell edges (Fig. 2D and Supplemental Fig. S2). As for ανβ3-specific FAs, we observed marked alterations in integrin α5β1-specific FA morphometry and localization between the dedifferentiated and contractile phenotypes. The differentiated phenotypes exhibited nearly uniform networks of long and thin FAs with the most differentiated T-cad+-SMCs presenting the thinnest and longest adhesions. In contrast, integrin α5β1-specific FAs of the contractile shTcad-SMCs appeared shorter and thicker or drop-shaped, and were occasionally concentrated in ordered arcs localized at the lamella (Fig. 2D). In dedifferentiated SMCs short thick FAs were rarely present and if so, were localized exclusively at the cell edges or the tips of lamellipodium. A difference in integrin α5β1-specific FAs shape was the major distinction between of Tcad+- and shTcad-SMCs, rather than FA count or overall size (Fig. 2E).
One of the most distinguishing architectural characteristics of differentiated SMCs is their robust actin cytoskeleton, capable of generating high traction forces essential for contractile function. FA growth during cell spreading highly depends on the applied traction force.32 Indeed, ordered arcs of thick α5β1-specific FAs in the contractile shTcad-SMC phenotype were localized at the terminal tips of large actin bundles (Fig. 2D). Bundles of dense parallel actin fibers were infrequently observed in E- and shC-SMCs and mostly absent in the highly dedifferentiated Tcad+-SMC phenotype (Fig. 2D, and Supplemental Fig. S3).
Configuration of adherens junctions is altered following ectopic T-cadherin-induced SMC phenotype switching
Coordination between cell-matrix and cell-cell adhesion networks during functional cell phenotype transitions have been suggested and supported by the experimental evidences.33 Given the prominent cell-matrix adhesion and cytoskeletal distinctions between the phenotypes, we expected to document adjustments in intercellular tension distribution through a demonstration of changes in adherens junction (AJ) configuration. Classical cadherins and their connection to the actin cytoskeleton play a crucial role in formation of stable AJs. Immunofluorescence staining of confluent monolayers for N-cadherin (Fig. 3A), the major classical cadherin in SMCs,34 showed junctional staining typical for cultured SMCs, whereby the cell-cell interface was rich in cadherin fingers,35,36 also called “strikingly elongated AJs."35 AJ morphology was markedly different between the dedifferentiated Tcad+-SMC and contractile shTcad-SMC phenotypes (Fig. 3A). AJ morphology in Tcad+-SMCs was visibly characterized by the formation of fewer and shorter cadherin fingers and smaller cadherin finger clusters, and junctional staining typical for cryptic lamellipodia was evident at the cell-cell borders. In contrast, AJ morphology in shTcad-SMCs was characterized by the formation of well pronounced cadherin finger-rich cellular junctions, with cadherin fingers being longer, highly compact and reaching deeply into the cell body. Cadherin finger clusters in shTcad were larger and often constituted the total perimeter of the cell, which was rarely evident in the control SMCs and almost never present in Tcad+-SMCs. The phenotypes did not differ with respect to N-cadherin protein abundance (Fig. 3B).
Figure 3.
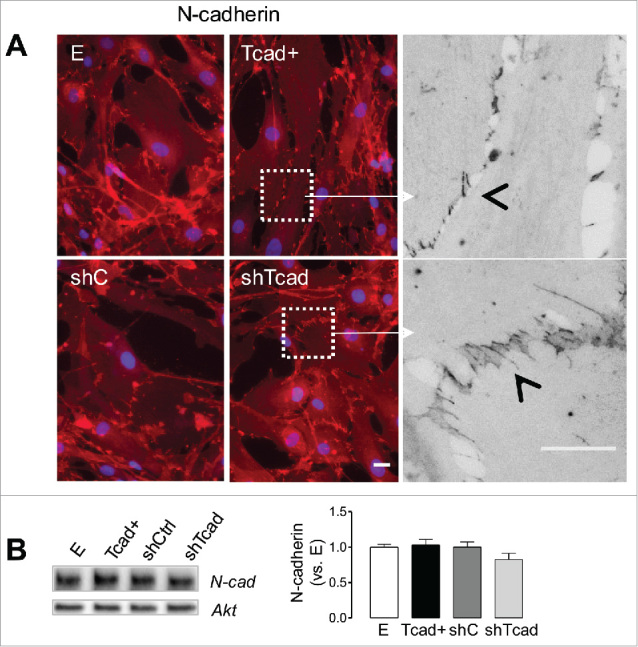
T-cadherin-associated SMC phenotype switch alters morphological properties of N-cadherin positive AJs. SMC transductants were plated onto fibronectin pre-coated glass coverslips or tissue culture dishes for immunofluorescence staining or immunoblotting protocols, respectively (A) Representative images of SMCs stained for N-cadherin in confluent cell monolayers. Scale bars = 25 µm. Higher magnification fragments (white squares) from Tcad+- and shC-SMCs (in grayscale) illustrate the very different patterns of N-cadherin distribution at cell-cell junctions. Cell-cell interface and cadherin fingers are indicated by the open arrow heads. (B) Representative immunoblots of N-cadherin (with Akt as internal loading control) are presented and quantification of abundancies in the different transductants is expressed relative to levels in control E SMC. Obtained data are presented as mean ± SD.
Random migration capacity of SMC is regulated by the structural actin cytoskeleton characteristics and actomyosin activity
A classical characteristic of SMC dedifferentiation is a loss of contractile machinery and decay of actin stress fiber networks. Supplemental Fig. S3 recapitulates our previous findings16 that stress fibers in dedifferentiated Tcad+-SMCs are weak and peripherally distributed, whereas they are very robust and well organized along the major cell axis in contractile shTcad-SMCs. Since cell tension generated by stress fibers is essential for cell movement and anchorage to the ECM37-39 we investigated the impact of phenotype-associated changes in cytoskeleton structure on SMC migration.
We first evaluated random migration in 2D. This setup enables estimation of cell motility parameters that are exclusively dependent on cell response to ECM and eliminates cell-cell interaction-dependent component of the process. Random migration of individual SMCs on fibronectin in 2D over a 12 h period was monitored using time-lapse videomicroscopy (Fig. 4A). Dedifferentiated Tcad+-SMCs migrated most efficiently (highest mean square displacement (MSD)) by maintaining best directional persistence (highest directionality ratio) and migrating most rapidly (Fig. 4B-D). Contractile shTcad-SMCs exhibited lowest migration efficiently and velocity and weakest directional persistence.
Figure 4.
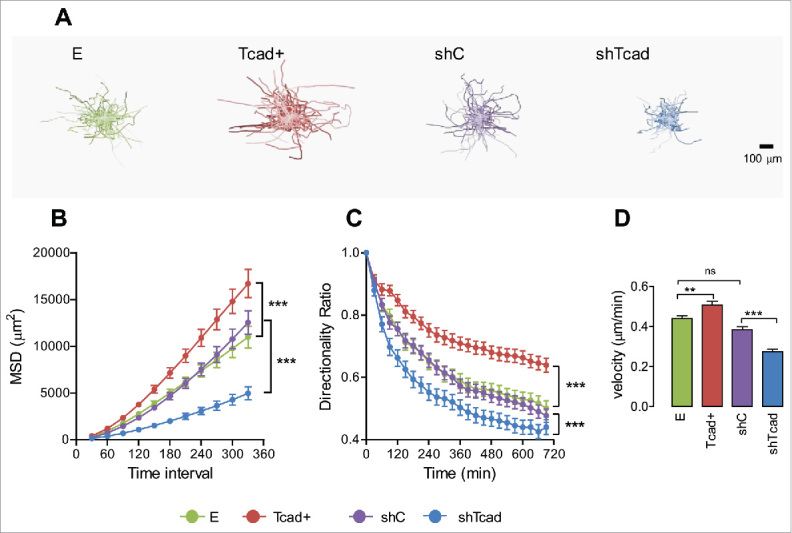
Dedifferentiated and contractile SMC phenotypes exhibit different modes of random cell migration in 2D monolayer culture. Migration of SMC transductants sparsely seeded onto fibronectin pre-coated culture was recorded by time lapse videomicroscopy. (A) Upper panels show typical plots of trajectories from migration origin (n = 100 cells/transductant). (B) Mean square displacement (MSD) was calculated as a measure of migration efficiency. (C) The directionality ratio (or straightness ratio) was calculated as a measure of directional persistence: each data point represents the directionality ratio at the given specific time point. Statistical analysis of data for directionality ratio and MSD was performed using nonlinear regression analysis. Significant differences between E and Tcad+ and between shC and shTcad are indicated (****P < 0.0001). E and shC were not different. (D) Migration velocity. Each data point (mean ± SEM) represents measurements on 100 cells in 3 independent experiments. One-way ANOVA followed by Tukey post-hoc testing was used for statistical analysis. Asterisks indicate significant differences (**P < 0.01; ***P < 0.001). ns, not significantly different.
To test whether migration competence of SMCs might be determined by the structural characteristics of the actin cytoskeleton we recorded random SMC migration in the presence of actin cytoskeleton-modulating pharmacological compounds.
The first series of experiments tested established tension force-reducing and stress fiber-disrupting compounds, namely blebbistatin, a specific inhibitor of myosin II adenosine triphosphatase (ATPase) activity,,40,41 and Y-27632, a selective inhibitor of Rho-associated, coiled-coil containing protein kinase (ROCK), and was performed on stress-fiber enriched shTcad-SMCs (and corresponding control shC-SMCs). Efficacy of these compounds on stress-fiber disruption in SMCs is presented in Supplemental Fig. S4. Blebbistatin increased the migration efficiency (MSD) of shTcad-SMCs only (Fig. 5A), through a greater enhancement of directional persistence in these cells as compared with shC-SMCs (e.g., change in directionality ratio (ΔDR) 0.16 vs. ΔDR of 0.04, respectively at 390 min; Fig. 5B). Blebbistatin did not alter migration velocities (Fig. 5C). Y-27632 strongly increased the migration efficiency (MSD) of shTcad-SMCs (Fig. 5A), likely through a greater enhancement of both their directional persistence (e.g., ΔDR of 0.14 vs. ΔDR of 0.06, for shTcad-SMCs and shC-SMCs, respectively at 390 min; Fig. 5B) and overall migration velocity (Fig. 5C).
Figure 5.
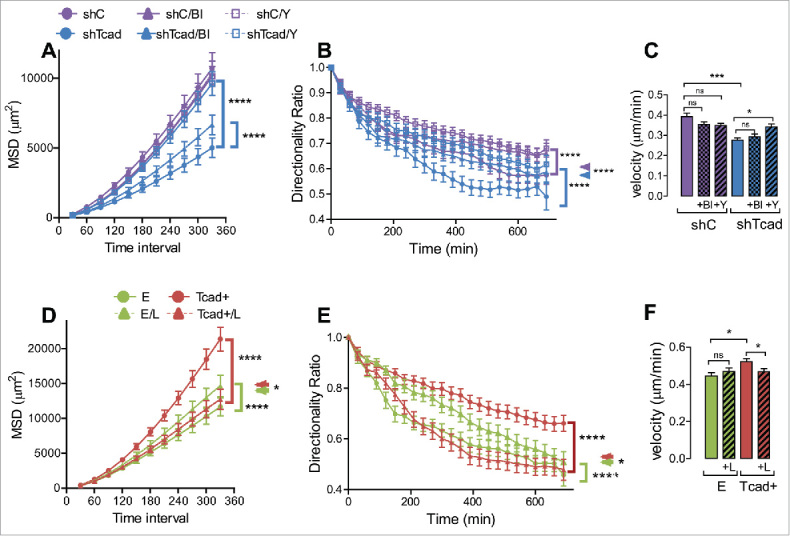
Pharmacological modulation of mechanical actin cytoskeleton properties affects the parameters of random migration. Migration experiments were performed without (control) or with inclusion of polymerized actin content-reducing compound Y-27632 (Y: 5 µM) or actomyosin activity inhibiting blebbistatin (Bl: 1 µM). In the case of shTcad-SMCs (purple) and shC-SMCs (blue) transductants (A-C), or actin polymerization-promoting compound LPA (L: 5 nM) in the case of Tcad+-SMCs (red) and E-SMCs (green) (D-F). Statistical analysis of MSD (A, D) and directionality ratio (B, E) was performed using nonlinear regression analysis. (A) Asterisked brackets indicate significant effects (P < 0.0001) of Bl or Y on the MSD of shTcad-SMCs compared with their untreated counterpart. (B) Asterisked brackets indicate significant effects (P < 0.0001) of either Bl or Y on shC-SMCs (purple bracket) and shTcad-SMCs (blue bracket) as compared with their own corresponding (untreated) control. Asterisked double arrowhead denotes significant differences (P < 0.0001) between shC-SMCs and shTcad-SMCs under each condition (untreated, + Bl or +Y). (D, E) Asterisked brackets indicate significant effects (P < 0.0001) of LPA on E-SMCs (green bracket) and Tcad+-SMCs (red bracket) as compared with their own corresponding (untreated) controls. Asterisked double arrowhead denotes significant a difference (*P < 0.05) between E-SMCs and Tcad+-SMCs in the presence of LPA. (C, F) One-way ANOVA followed by Tukey post-hoc testing was used for statistical analysis of velocity (*P < 0.05, ***P < 0.001, ns, not significant).
The second series of experiments tested lysophosphatidic acid (LPA), an indirect RhoA GTPase activator and inducer of stress fiber formation,42 and was performed on stress fiber-deficient Tcad+-SMCs (and corresponding control E-SMCs). Efficacy of stress fiber-induction by LPA in SMCs is illustrated in Supplemental Figure S5. LPA reduced the migration efficiency (MSD) of Tcad+-SMCs (Fig. 5D) through negative effects on directional persistence (Fig. 5E) and velocity (Fig. 5F). LPA slightly increased migration efficiency (MSD) of shC-SMCs, due to improved directional persistence (Fig. 5D).
Changes in the actin cytoskeleton structure and actomyosin activity affect SMC migration in 3D matrix
To examine migration of the SMC phenotypes in a 3-dimensional (3D) context which would more closely resemble in vivo medial SMC tissue organization we used a spheroid culture model that we have previously used for endothelial cells18 and tumor cells.43,44 Migration out from the multicellular spheroids into fibronectin-rich Matrigel was observable within 24 h (Fig. 6A, B). Both single cell and collective modes of migration, were apparent in all transductants. However, Tcad+-SMCs migrated out from the spheroids predominantly in a collective manner, whereas shTcad-SMCs formed fewer protrusions and appeared outside the spheroid body as single cells or as small cell clusters (Fig. 6A, B). With continued culture, and in a reflection of the early outgrowth patterns, Tcad+- and shTcad-SMC spheroids exhibited distinct migration efficiencies. Tcad+-SMC spheroids exhibited the greatest outward expansion capacity (Fig. 6C); they formed the densest network of protrusions and outgrowing multicellular processes were the longest and appeared more branched (Fig. 6B). shTcad-SMC spheroids exhibited the smallest outward migration capacity (Fig. 6C); they formed the sparsest network of outgrowth and protrusions were shorter, appeared poorly branched, and often collapsed in round cellular clusters (Fig. 6B), suggesting an inability to migrate collectively.
Figure 6.
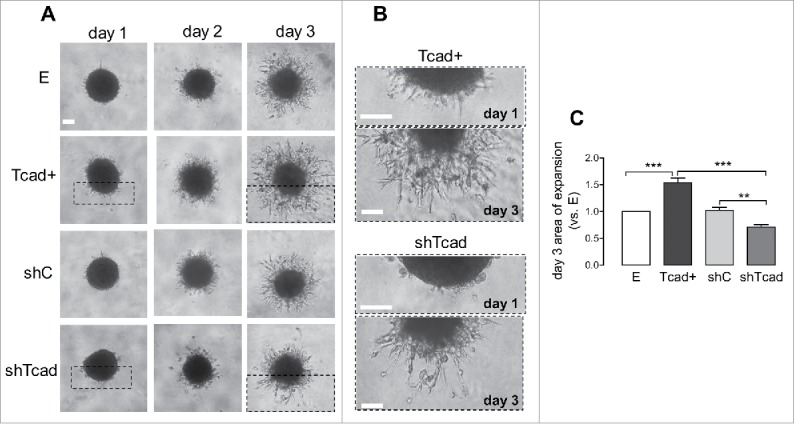
Dedifferentiated and contractile SMC phenotypes exhibit alterations in migration mode in 3D SMC-spheroid culture. SMC-spheroids were embedded in Matrigel and outward migration monitored over 3 d by image capture. (A) Representative images of spheroids on each day. (B) High magnification fragments (dashed boxes) of full-frame images of Tcad+ and shTcad SMC-spheroids on days 1 and 3 are shown to illustrate the difference in their mode of outward migration. Scale bars = 100 µm. (C) Morphometric quantification of expansion (outward migration) capacity. Expansion capacities of the different transductants are expressed relative to that in control E SMC. Obtained data are given as mean ± SD. One-way ANOVA followed by Tukey post-hoc testing was used for statistical analysis. Asterisks indicate significant differences (**P < 0.01; ***P < 0.001).
Effects of actin cytoskeleton-modulating compounds were also tested in our 3D model. Y-27632 increased outward expansion of shTcad-SMC (by ≈4-fold) and shC-SMC (by ≈2-fold) spheroid bodies (Fig. 7A). Y-27632 endowed the shTcad-SMCs with an improved capacity to migrate collectively, based on visual assessment of longer protrusions and a denser network of outgrowth as compared with untreated condition. Blebbistatin increased outward expansion of the shTcad-SMC spheroids, also in association with an apparent better collective migration capacity (Fig. 7B). LPA inhibited expansion of Tcad+-SMC spheroids (Fig. 7C).
Figure 7.
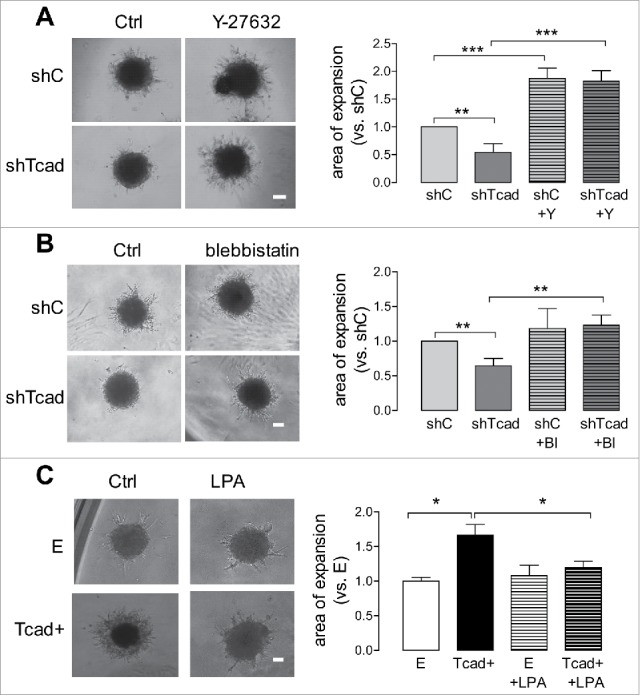
Pharmacological modulation of mechanical actin cytoskeleton properties affects SMC migration in 3D. SMC-spheroid migration experiments were performed without (control) or with inclusion of Y-27632 (Y: 5 µM) or blebbistatin (Bl: 1µM) in the case of shTcad-SMCs and shC-SMCs (A-C), or LPA (L: 5 nM) in the case of Tcad+-SMCs and E-SMCs. Representative images of spheroids after 3 d culture are shown. Scale bars = 100 µm. Outward migration was quantified after 3 d. Expansion capacities of the shTcad- or Tcad+-SMCs are expressed relative to that in their respective controls (shC- or E-SMCs). Obtained data are given as mean ± SD. One-way ANOVA followed by Tukey post-hoc testing was used for statistical analysis. Asterisks indicate significant differences (*P < 0.05; **P < 0.01; ***P < 0.001).
Collectively, the data obtained in 2D and 3D migration models support that myosin generated traction force plays only a limited role in cell anchorage, while intrinsic cellular contractility and tensile stress generated by actin cytoskeleton plays a dominant role in determining cell migration capacity during phenotype switching.
Discussion
The past decades have seen significant progress in understanding molecular and functional hallmarks of SMC phenotypic modulation, the complex mechanisms involving the regulatory circuits of transcription factors controlling contractile gene expression and crosstalk of intracellular signaling pathways.45-48 However, there have been no studies that have interrogated global differences in adhesive properties between differentiated and dedifferentiated SMC phenotypes. Here, we have characterized changes in the cell adhesion nexus that occur during SMC phenotype switching induced by T-cadherin upregulation (which promotes dedifferentiation)16 and T-cadherin depletion (which enforce differentiation).16,49 We demonstrate that the phenotype transition process involves several components of the adhesion nexus: adhesion molecule expression, cell-matrix adhesion, organization of intercellular contacts and control of intracellular tension forces generated by the actin cytoskeleton. T-cadherin-induced phenotype switching in SMCs was accompanied by altered cell adhesion to several ECM substrates, changes in expression profiles of adhesion-related genes, and reorganization of α5β1- and integrin ανβ3-mediated FAs and N-cadherin-based AJs. Further, the differentiated and dedifferentiated phenotypes exhibited profound differences with respect to their directional persistence and velocity of migration in 2D and their ability to migrate collectively in 3D. These differences were attenuated by actin polymerization- and intracellular tension-modifying compounds, indicating a key role for the actin cytoskeleton in transitions between static or migratory phenotypes.
Classical SMC phenotype transition is characterized by significant changes in cell morphology and spreading. 50,11,51 In agreement with this we observed striking differences in cell-matrix interactions between differentiated/contractile and dedifferentiated/migratory phenotypes induced by ectopic overexpression or silencing of T-cadherin (Tcad+ and shTcad, respectively). Dedifferentiated SMCs showed defective spreading and a compromised ability to form mature FAs, the type of FAs which dominated in the contractile phenotype. This qualitative observation was further supported by quantitative FA size comparison and FAKTyr397 phosphorylation: the contractile phenotype exhibited the largest FAs and highest level of FAKTyr397 phosphorylation, which reflects functional FA differences in contractile versus dedifferentiated SMC phenotypes (i.e. the shift from anchorage-optimized, to migration-permitting adhesions).
Cell adhesion mode and strength determine cell migration properties and are dependent on 2 major factors, namely adhesion receptor-ligand density and intrinsic contractility of the cell.52,53 Our qPCR adhesion molecule profiling in the contractile/differentiated phenotype induced by T-cadherin silencing revealed changes in expression of several integrins, matrix metalloproteases and collagens. Interestingly, although this phenotype showed better spreading and adhesion to ECM, it was characterized by an overall lower classical adhesion molecule expression as compared with dedifferentiated T-cadherin overexpressing cells. Moreover, change in cell-matrix adhesion competence was not specific to a particular type of ECM, but observed on all probed ECM types. It is often assumed that changes in adhesion molecule profile are primarily required to modify SMC preferences for certain ECM components.3 However, when plated on 6 different matrices (collagen- I, II, IV, fibronectin, laminin, and vitronectin), neither contractile nor dedifferentiated phenotypes showed major preference for any specific matrix component. This could be interpreted to indicate that an overall downregulation in adhesion capacity during SMC dedifferentiation is needed to enable a functional switch from an anchorage-optimized to a migration-optimized mode of adhesion.13,54 However, it remains unclear as to whether any specific matrix substrate can crucially regulate the functional switch during phenotype transition.
Our findings also suggest that a change in adhesion molecule profile might not be a primary factor regulating SMC adhesion and migration properties in the early stages of SMC phenotype switching. Our study provides several lines of evidence to indicate that intracellular tension generated by the alterations in actin cytoskeleton architecture may play an equally important role in regulating the functional mode of SMC adhesion. FA function, distribution and stability largely reflect the intracellular tension distribution landscape during cell spreading and are dependent on cytoskeletal tension and traction force.10,11,32 We observed prominent phenotype-specific differences in integrin α5β1- and ανβ3-based FA distribution and morphometry during cell spreading on fibronectin. In the contractile shTcad phenotype integrin ανβ3-based FA, which usually locate to the cellular areas of the highest tension,10 were prominently concentrated at the free cell edges as large adhesion complexes. In contrast, ανβ3-specific FA distribution in the dedifferentiated Tcad+ phenotype was typical for polarized, migrating cells. In the dedifferentiated phenotype α5β1-integrin specific FAs acquired a typical elongated morphometry known to engage in matrix remodeling,55 whereas in the contractile phenotype an additional large, drop-shaped clustered FA population localized at the tips of the dense bundles of the actin fibers and was restricted to lamella, indicative of the areas where forces exerted on the ECM is high.32 These high tension areas most likely represent cell anchorage sites, which the dedifferentiated phenotype is lacking due to the functional re-specification. Together, these observations suggest that changes in adhesome during SMC phenotypic modulation also includes spatial FA redistribution, which might be driven by cellular responses to changing cellular tension. Our initial intention of immunofluorescence staining for ανβ3- and α5β1-heterodimers was to compare the abundance of the 2 receptors between the SMC phenotypes. However, and even without parameters of abundance, highly prominent differences in subcellular ανβ3- and α5β1-specific FA distribution and morphometry between the phenotypes revealed the importance of cellular architecture reconfiguration and redistribution of intracellular tension during SMC phenotype transition. To our best knowledge, this line of investigation has never been performed and reported in the context of SMC phenotype transition. A limitation of our study is the selection of only 2 fibronectin receptors; it would be interesting to examine whether other fibronectin-binding integrins expressed on SMCs (e.g., ανβ1, α3β1, α4β1, α8β1)3,21,22 also display phenotype-specific alterations in distribution and morphometry.
A second line of evidence for cytoskeletal tension guidance in regulation of functional SMC adhesion derives from analysis of the distribution of N-cadherin at AJs. In 2D SMCs form serrated and irregular AJs enriched with cadherin fingers.35,56 Cadherin finger formation is driven by the intracellular tension generated by actomyosin activity.57 In agreement with the data on integrin α5β1- and ανβ3-specific FA morphometry and distribution, AJ characteristics of the contractile SMCs (abundant and rich in long cadherin fingers forming long clustered structures) reflect high intracellular traction force whereas AJs of the dedifferentiated phenotype (smoother with sparsely distributed, shorter cadherin fingers) reflect lower intracellular traction force. These morphological differences in AJs suggest functional divergence between the phenotypes with respect to collective cell behavior. Cadherin finger abundance in AJs has been associated with SMC collectivization required for synchronized SMC contraction, while linearization of AJs was observed in migratory SMC phenotype.35 We have not compared the strength of AJs between the phenotypes. However, a tension force-resistant AJ type is probably needed to preserve tissue integrity during collective SMC contraction. On the other hand, a mechanical weakening and increased dynamics of AJs might be expected in dedifferentiated SMC phenotype. Mechanically weaker AJs should favor effective migration to the site of injury by allowing easier dismantling of static connections between the cells, while still providing coordination for collective cell migration. Since the chief function of classical cadherins is to maintain stable cell-cell connections (and tissue integrity), high abundance of cadherin fingers perpendicular to the cell-cell interface in shTcad cells geometrically prolongs the effective perimeter of cadherin interaction zones, and therefore should result in mechanically stronger AJs. Although we did not test whether analogical differences in AJ morphology between the phenotypes are also present in 3D, or how AJ morphology affects collective cell migration in 3D, it is tempting to speculate that stronger, cadherin finger-rich AJs prevent effective collective cell migration into surrounding ECM, while linearization of AJs in dedifferentiated phenotype should promote such behavior.
The third line of evidence for a crucial role of actin cytoskeleton architecture and intracellular tensile force in regulating the functional mode of SMC adhesion during phenotype transition derives from experiments with pharmacological manipulation of actin stress fiber formation. Actin is a primary cytoskeletal component required for generation of intracellular tension, and it can produce tensile force by polymerization, or by association with myosin.58 Our single cell (2D) and collective cell (3D) migration analysis showed that despite the global differences in adhesion properties between contractile and dedifferentiated phenotypes, migratory function of dedifferentiated SMCs could be abolished by LPA which enhances actin polymerization and augmenting actin cytoskeleton “stiffness” and intracellular tensile force, whereas the contractile phenotype could be released from its adhesive anchors by ROCK inhibitor Y-27632 which reduces fibrous actin density via inhibition of actin polymerization.
RhoA and ROCK activity modulation can also regulate the differentiation status of SMC by transcriptional regulation: RhoA and ROCK activate pro-differentiation acting myocardin-related transcription factor entry to the nucleus, and initiate contractile protein expression.59-61 Accordingly, ROCK blockade and reduction of fibrous actin content in the cell promoted SMC dedifferentiation.61 However, we associate activation of migratory function in the contractile phenotype by ROCK inhibitor and its inhibition in the dedifferentiated phenotype by LPA treatment with changes in mechanical actin cytoskeleton properties rather than with transcriptional regulation. Changes in parameters of SMC random migration (MSD and directionality ratio) after a short 30 min pre-treatment period were evident at within at least 120 min of initiating videomicroscopy. The possible impact of transcriptional ROCK-dependent phenotype regulation in the 3D-spheroid expansion model is, on the other hand, more relevant, and cannot be ruled out completely, since the observation period was considerably longer (1–3 days).
We have exploited 2 in vitro models which evaluate different aspects of the migration process. The spheroid expansion assay models collective SMC migration within the tissue. In contrast to random migration in 2D, effective collective cell expansion through the ECM requires effective communication not only with the surrounding matrix, but also between the cells themselves.62 The close reproducibility between our 2D and 3D migration models suggests that intrinsic cellular contractility and tensile stress generated by actin cytoskeleton play a dominant role in determining cell migration capacity during phenotype switching.
In addition to characterizing the regulation of anchorage-migration switch by actin cytoskeleton during SMC phenotypic modulation, this study has also expanded knowledge of molecular mechanisms regulated by T-cadherin in SMCs. Apart from T-cadherin, 3 other members of the superfamily are currently identified in vascular SMCs including classical Type I N- and R-cadherins and FAT1. T-cadherin and N-cadherin both belong to a heterogeneous group of guidance molecules orchestrating spatial and temporal morphogenetic processes such as navigation of projecting neurons during neural development.63,64 N-cadherin has been the most extensively characterized cadherin member with regard to regulation of SMC migration, proliferation and survival.34 Our recent studies identified T-cadherin as a key regulator of SMC contractile signaling, matrix remodeling and differentiation status via Akt/mTOR and GSK3β transduction axes.15,16 Herein we demonstrate that T-cadherin-induced phenotype switching in SMCs is accompanied not only by reconfiguration of α5β1- and integrin ανβ3-based FAs and N-cadherin-based AJs, but also by alterations in the directional persistence of migration in 2D and in the ability to collectively migrate in 3D. Thus, we speculate that T-cadherin may serve as a guidance cue for appropriate cellular navigation and reorganization within SMC layers during regenerative or pathological vascular tissue remodeling.
In conclusion, our results suggest that mechanical properties of the actin cytoskeleton and its ability to generate tensile force per se can regulate SMC functional phenotype. This mechanical regulatory circuit might be a key factor which enables the behavioral switch from static cell anchorage to migration. Such regulation resembles the model of biphasic cell migration response to increasing adhesion strength,12,13 which in the case of SMC phenotype transition can occur due to the change in intrinsic cell contractility, while the ECM ligand density and its stiffness remain constant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
AF, TJR and PE designed the study and experiments. AF and EK conducted the experiments. AF, TJR, EK and MP analyzed data; BD generated lentiviral vectors and viral particles. While each author contributed to the writing of the manuscript, AF and TJR are largely responsible for its writing.
Supplementary Material
Funding Statement
This work was supported by the Swiss National Science Foundation (grant no. 310030_159589 to TJR), the Stiftung für Herz- und Kreislaufkrankheiten (grant to TJR), and a fellowship grant (to AF) “Förderung exzellenter Nachwuchsforschender," Forschungsfonds 2015, University of Basel.
References
- [1].Campbell G, Campbell J, Manderson J, Horrigan S, Rennick R. Arterial smooth muscle. A multifunctional mesenchymal cell. Arch Pathol Lab Med 1988; 112:977-86 [PubMed] [Google Scholar]
- [2].Yamin R, Morgan KG. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J Physiol 2012; 590:4145-54; PMID:22687615; https://doi.org/ 10.1113/jphysiol.2012.232306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res 2001; 52:372-86; PMID:11738054; https://doi.org/ 10.1016/S0008-6363(01)00399-6 [DOI] [PubMed] [Google Scholar]
- [4].Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp 2007; 283:174-91; discussion 91-3, 238-41; PMID:18300422 [DOI] [PubMed] [Google Scholar]
- [5].Beamish JA, He P, Kottke-Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev 2010; 16:467-91; PMID:20334504; https://doi.org/ 10.1089/ten.teb.2009.0630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arteriosclerosis Thromb Vasc Biol 2011; 31:1495-505; PMID:21677292; https://doi.org/ 10.1161/ATVBAHA.110.221135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol 2015; 209:13-22; PMID:25869663; https://doi.org/ 10.1083/jcb.201412052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gupta M, Sarangi BR, Deschamps J, Nematbakhsh Y, Callan-Jones A, Margadant F, Mège RM, Lim CT, Voituriez R, Ladoux B. Adaptive rheology and ordering of cell cytoskeleton govern matrix rigidity sensing. Nat Commun 2015; 6:7525; PMID:26109233; https://doi.org/ 10.1038/ncomms8525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Devedec SE, Balcioğlu HE, He S, Snaar-Jagalska BE, Vreugdenhil E, et al.. beta1 integrin inhibition elicits a prometastatic switch through the TGFbeta-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci Signal 2014; 7:ra15; PMID:24518294; https://doi.org/ 10.1126/scisignal.2004751 [DOI] [PubMed] [Google Scholar]
- [10].Schiller HB, Hermann MR, Polleux J, Vignaud T, Zanivan S, Friedel CC, Raducanu A, Gottschalk KE, Théry M, Mann M, et al.. beta1- and alphav-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat Cell Biol 2013; 15:625-36; PMID:23708002; https://doi.org/ 10.1038/ncb2747 [DOI] [PubMed] [Google Scholar]
- [11].Balcioglu HE, van Hoorn H, Donato DM, Schmidt T, Danen EH. The integrin expression profile modulates orientation and dynamics of force transmission at cell-matrix adhesions. J Cell Sci 2015; 128:1316-26; PMID:25663698; https://doi.org/ 10.1242/jcs.156950 [DOI] [PubMed] [Google Scholar]
- [12].Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol 2005; 204:198-209; PMID:15669099; https://doi.org/ 10.1002/jcp.20274 [DOI] [PubMed] [Google Scholar]
- [13].Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 2006; 125:1361-74; PMID:16814721; https://doi.org/ 10.1016/j.cell.2006.05.029 [DOI] [PubMed] [Google Scholar]
- [14].Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol 2009; 10:21-33; PMID:19197329; https://doi.org/ 10.1038/nrm2593 [DOI] [PubMed] [Google Scholar]
- [15].Frismantiene A, Pfaff D, Frachet A, Coen M, Joshi MB, Maslova K, Bochaton-Piallat ML, Erne P, Resink TJ, Philippova M. Regulation of contractile signaling and matrix remodeling by T-cadherin in vascular smooth muscle cells: constitutive and insulin-dependent effects. Cell Signal 2014; 26:1897-908; PMID:24815187; https://doi.org/ 10.1016/j.cellsig.2014.05.001 [DOI] [PubMed] [Google Scholar]
- [16].Frismantiene A, Dasen B, Pfaff D, Erne P, Resink TJ, Philippova M. T-cadherin promotes vascular smooth muscle cell dedifferentiation via a GSK3beta-inactivation dependent mechanism. Cell Signal 2016; 28:516-30; PMID:26907733; https://doi.org/ 10.1016/j.cellsig.2016.02.014 [DOI] [PubMed] [Google Scholar]
- [17].Gorelik R, Gautreau A. Quantitative and unbiased analysis of directional persistence in cell migration. Nat Protocols 2014; 9:1931-43; PMID:25033209; https://doi.org/ 10.1038/nprot.2014.131 [DOI] [PubMed] [Google Scholar]
- [18].Philippova M, Banfi A, Ivanov D, Gianni-Barrera R, Allenspach R, Erne P, Resink T. Atypical GPI-anchored T-cadherin stimulates angiogenesis in vitro and in vivo. Arterioscler Thromb Vasc Biol 2006; 26:2222-30; PMID:16873731; https://doi.org/ 10.1161/01.ATV.0000238356.20565.92 [DOI] [PubMed] [Google Scholar]
- [19].Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 2005; 6:56-68; PMID:15688067; https://doi.org/ 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- [20].Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci 2006; 119:3901-3; PMID:16988024; https://doi.org/ 10.1242/jcs.03098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zargham R, Thibault G. α8β1 Integrin expression in the rat carotid artery: involvement in smooth muscle cell migration and neointima formation. Cardiovasc Res 2005; 65:813-22; PMID:15721861; https://doi.org/ 10.1016/j.cardiores.2004.11.021 [DOI] [PubMed] [Google Scholar]
- [22].Zargham R. Tensegrin in context. Cell Adh Migr 2010; 4:485-90; https://doi.org/ 10.4161/cam.4.4.12403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002; 110:673-87; PMID:12297042; https://doi.org/ 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- [24].Cheuk BL, Cheng SW. Differential expression of integrin alpha5beta1 in human abdominal aortic aneurysm and healthy aortic tissues and its significance in pathogenesis. J Surg Res 2004; 118:176-82; PMID:15100006; https://doi.org/ 10.1016/S0022-4804(03)00351-2 [DOI] [PubMed] [Google Scholar]
- [25].Heerkens EH, Izzard AS, Heagerty AM. Integrins, vascular remodeling, and hypertension. Hypertension 2007; 49:1-4; PMID:17145983; https://doi.org/ 10.1161/01.HYP.0000252753.63224.3b [DOI] [PubMed] [Google Scholar]
- [26].Barillari G, Albonici L, Incerpi S, Bogetto L, Pistritto G, Volpi A, Ensoli B, Manzari V. Inflammatory cytokines stimulate vascular smooth muscle cells locomotion and growth by enhancing alpha5beta1 integrin expression and function. Atherosclerosis 2001; 154:377-85; PMID:11166770; https://doi.org/ 10.1016/S0021-9150(00)00506-2 [DOI] [PubMed] [Google Scholar]
- [27].Blindt R, Krott N, Hanrath P, vom Dahl J, van Eys G, Bosserhoff AK. Expression patterns of integrins on quiescent and invasive smooth muscle cells and impact on cell locomotion. J Mol Cell Cardiol 2002; 34:1633-44; PMID:12505061; https://doi.org/ 10.1006/jmcc.2002.2113 [DOI] [PubMed] [Google Scholar]
- [28].Ikari Y, Yee KO, Schwartz SM. Role of alpha5beta1 and alphavbeta3 integrins on smooth muscle cell spreading and migration in fibrin gels. Thromb Haemost 2000; 84:701-5; PMID:11057873 [PubMed] [Google Scholar]
- [29].Kappert K, Blaschke F, Meehan WP, Kawano H, Grill M, Fleck E, Hsueh WA, Law RE, Graf K. Integrins alphavbeta3 and alphavbeta5 mediate VSMC migration and are elevated during neointima formation in the rat aorta. Basic Res Cardiol 2001; 96:42-9; PMID:11215531; https://doi.org/ 10.1007/s003950170076 [DOI] [PubMed] [Google Scholar]
- [30].Pickering JG, Chow LH, Li S, Rogers KA, Rocnik EF, Zhong R, Chan BM. alpha5beta1 integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury. Am J Pathol 2000; 156:453-65; PMID:10666375; https://doi.org/ 10.1016/S0002-9440(10)64750-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Slepian MJ, Massia SP, Dehdashti B, Fritz A, Whitesell L. Beta3-integrins rather than beta1-integrins dominate integrin-matrix interactions involved in postinjury smooth muscle cell migration. Circulation 1998; 97:1818-27; PMID:9603537; https://doi.org/ 10.1161/01.CIR.97.18.1818 [DOI] [PubMed] [Google Scholar]
- [32].Stricker J, Aratyn-Schaus Y, Oakes PW, Gardel ML. Spatiotemporal constraints on the force-dependent growth of focal adhesions. Biophys J 2011; 100:2883-93; PMID:21689521; https://doi.org/ 10.1016/j.bpj.2011.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Weber GF, Bjerke MA, DeSimone DW. Integrins and cadherins join forces to form adhesive networks. J Cell Sci 2011; 124:1183-93; PMID:21444749; https://doi.org/ 10.1242/jcs.064618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sun Z, Parrish AR, Hill MA, Meininger GA. N-cadherin, a vascular smooth muscle cell-cell adhesion molecule: function and signaling for vasomotor control. Microcirculation 2014; 21:208-18; PMID:24521477; https://doi.org/ 10.1111/micc.12123 [DOI] [PubMed] [Google Scholar]
- [35].Balint B, Yin H, Chakrabarti S, Chu MW, Sims SM, Pickering JG. Collectivization of Vascular Smooth Muscle Cells via TGF-beta-Cadherin-11-Dependent Adhesive Switching. Arterioscler Thromb Vasc Biol 2015; 35:1254-64; PMID:25767275; https://doi.org/ 10.1161/ATVBAHA.115.305310 [DOI] [PubMed] [Google Scholar]
- [36].Jones M, Sabatini PJ, Lee FS, Bendeck MP, Langille BL. N-cadherin upregulation and function in response of smooth muscle cells to arterial injury. Arterioscler Thromb Vasc Biol 2002; 22:1972-7; PMID:12482821; https://doi.org/ 10.1161/01.ATV.0000036416.14084.5A [DOI] [PubMed] [Google Scholar]
- [37].Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 2009; 10:778-90; PMID:19851336; https://doi.org/ 10.1038/nrm2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 2003; 4:446-56; PMID:12778124; https://doi.org/ 10.1038/nrm1128 [DOI] [PubMed] [Google Scholar]
- [39].Katoh K, Kano Y, Noda Y. Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J R Soc Interface 2011; 8:305-11; PMID:20826475; https://doi.org/ 10.1098/rsif.2010.0419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 2003; 299:1743-7; PMID:12637748; https://doi.org/ 10.1126/science.1081412 [DOI] [PubMed] [Google Scholar]
- [41].Wang HH, Tanaka H, Qin X, Zhao T, Ye LH, Okagaki T, Katayama T, Nakamura A, Ishikawa R, Thatcher SE, et al.. Blebbistatin inhibits the chemotaxis of vascular smooth muscle cells by disrupting the myosin II-actin interaction. Am J Physiol Heart Circ Physiol 2008; 294:H2060-8; PMID:18296570; https://doi.org/ 10.1152/ajpheart.00970.2007 [DOI] [PubMed] [Google Scholar]
- [42].Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992; 70:389-99; PMID:1643657; https://doi.org/ 10.1016/0092-8674(92)90163-7 [DOI] [PubMed] [Google Scholar]
- [43].Pfaff D, Philippova M, Buechner SA, Maslova K, Mathys T, Erne P, Resink TJ. T-cadherin loss induces an invasive phenotype in human keratinocytes and squamous cell carcinoma (SCC) cells in vitro and is associated with malignant transformation of cutaneous SCC in vivo. Br J Dermatol 2010; 163:353-63; PMID:20394625; https://doi.org/ 10.1111/j.1365-2133.2010.09801.x [DOI] [PubMed] [Google Scholar]
- [44].Maslova K, Kyriakakis E, Pfaff D, Frachet A, Frismantiene A, Bubendorf L, Ruiz C, Vlajnic T, Erne P, Resink TJ, et al.. EGFR and IGF-1R in regulation of prostate cancer cell phenotype and polarity: opposing functions and modulation by T-cadherin. FASEB J 2015; 29:494-507; PMID:25381040; https://doi.org/ 10.1096/fj.14-249367 [DOI] [PubMed] [Google Scholar]
- [45].Campbell JH, Campbell GR. Smooth muscle phenotypic modulation–a personal experience. Arterioscler Thromb Vasc Biol 2012; 32:1784-9; PMID:22815344; https://doi.org/ 10.1161/ATVBAHA.111.243212 [DOI] [PubMed] [Google Scholar]
- [46].Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 2012; 95:156-64; PMID:22406749; https://doi.org/ 10.1093/cvr/cvs115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg 2007; 45 Suppl A:A25-32; PMID:17544021; https://doi.org/ 10.1016/j.jvs.2007.03.001 [DOI] [PubMed] [Google Scholar]
- [48].Shi N, Chen SY. Smooth muscle cell differentiation: model systems, regulatory mechanisms, and vascular diseases. J Cell Physiol 2015; 231(4):777-87 [DOI] [PubMed] [Google Scholar]
- [49].Fujishima Y, Maeda N, Matsuda K, Masuda S, Mori T, Fukuda S, Sekimoto R, Yamaoka M, Obata Y, Kita S, et al.. Adiponectin association with T-cadherin protects against neointima proliferation and atherosclerosis. FASEB J 2017; 31(4):1571-83; PMID:28062540 [DOI] [PubMed] [Google Scholar]
- [50].Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004; 84:767-801; PMID:15269336; https://doi.org/ 10.1152/physrev.00041.2003 [DOI] [PubMed] [Google Scholar]
- [51].Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res 2006; 69:614-24; PMID:16266693; https://doi.org/ 10.1016/j.cardiores.2005.08.002 [DOI] [PubMed] [Google Scholar]
- [52].Gardel ML, Sabass B, Ji L, Danuser G, Schwarz US, Waterman CM. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. J Cell Biol 2008; 183:999-1005; PMID:19075110; https://doi.org/ 10.1083/jcb.200810060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Schwartz MA, Horwitz AR. Integrating adhesion, protrusion, and contraction during cell migration. Cell 2006; 125:1223-5; PMID:16814706; https://doi.org/ 10.1016/j.cell.2006.06.015 [DOI] [PubMed] [Google Scholar]
- [54].DiMilla PA, Barbee K, Lauffenburger DA. Mathematical model for the effects of adhesion and mechanics on cell migration speed. Biophys J 1991; 60:15-37; PMID:1883934; https://doi.org/ 10.1016/S0006-3495(91)82027-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Huveneers S, Truong H, Fässler R, Sonnenberg A, Danen EHJ. Binding of soluble fibronectin to integrin α5β1 – link to focal adhesion redistribution and contractile shape. J Cell Sci 2008; 121:2452-62; PMID:18611961; https://doi.org/ 10.1242/jcs.033001 [DOI] [PubMed] [Google Scholar]
- [56].Gabella G. Morphology of smooth muscle. In: Kao CY, Carsten ME, eds. Cellular Aspects of Smooth Muscle Function. Cambridge: Cambridge University Press, 1997:1-47 [Google Scholar]
- [57].Brevier J, Montero D, Svitkina T, Riveline D. The asymmetric self-assembly mechanism of adherens junctions: a cellular push-pull unit. Phys Biol 2008; 5:016005; PMID:18379019; https://doi.org/ 10.1088/1478-3975/5/1/016005 [DOI] [PubMed] [Google Scholar]
- [58].Carey SP, Charest JM, Reinhart-King CA. Forces During Cell Adhesion and Spreading: Implications for Cellular Homeostasis. In: Gefen A, ed. Cellular and Biomolecular Mechanics and Mechanobiology. Berlin, Heidelberg: Springer Berlin Heidelberg, 2011:29-69 [Google Scholar]
- [59].Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004; 428:185-9; PMID:15014501; https://doi.org/ 10.1038/nature02382 [DOI] [PubMed] [Google Scholar]
- [60].Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 2010; 11:353-65; PMID:20414257; https://doi.org/ 10.1038/nrm2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem 2001; 276:341-7; PMID:11035001; https://doi.org/ 10.1074/jbc.M005505200 [DOI] [PubMed] [Google Scholar]
- [62].Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol 2010; 188:11-9; PMID:19951899; https://doi.org/ 10.1083/jcb.200909003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Philippova M, Joshi MB, Kyriakakis E, Pfaff D, Erne P, Resink TJ. A guide and guard: the many faces of T-cadherin. Cell Signal 2009; 21:1035-44; PMID:19399994; https://doi.org/ 10.1016/j.cellsig.2009.01.035 [DOI] [PubMed] [Google Scholar]
- [64].Cadherins Ranscht B.: molecular codes for axon guidance and synapse formation. Int J Dev Neurosci 2000; 18:643-51; PMID:10978842; https://doi.org/ 10.1016/S0736-5748(00)00030-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.