

ABSTRACT
Alterations in DNA methylation and gene expression in blood leukocytes are potential biomarkers of harm and mediators of the deleterious effects of tobacco exposure. However, methodological issues, including the use of self-reported smoking status and mixed cell types have made previously identified alterations in DNA methylation and gene expression difficult to interpret. In this study, we examined associations of tobacco exposure with DNA methylation and gene expression, utilizing a biomarker of tobacco exposure (urine cotinine) and CD14+ purified monocyte samples from 934 participants of the community-based Multi-Ethnic Study of Atherosclerosis (MESA). Urine cotinine levels were measured using an immunoassay. DNA methylation and gene expression were measured with microarrays. Multivariate linear regression was used to test for associations adjusting for age, sex, race/ethnicity, education, and study site. Urine cotinine levels were associated with methylation of 176 CpGs [false discovery rate (FDR)<0.01]. Four CpGs not previously identified by studies of non-purified blood samples nominally replicated (P value<0.05) with plasma cotinine-associated methylation in 128 independent monocyte samples. Urine cotinine levels associated with expression of 12 genes (FDR<0.01), including increased expression of P2RY6 (Beta ± standard error = 0.078 ± 0.008, P = 1.99 × 10−22), a gene previously identified to be involved in the release of pro-inflammatory cytokines. No cotinine-associated (FDR<0.01) methylation profiles significantly (FDR<0.01) correlated with cotinine-associated (FDR<0.01) gene expression profiles. In conclusion, our findings i) identify potential monocyte-specific smoking-associated methylation patterns and ii) suggest that alterations in methylation may not be a main mechanism regulating gene expression in monocytes in response to cigarette smoking.
KEYWORDS: CD14+ monocyte, Cigarette, cotinine, DNA methylation, gene expression, smoking, tobacco
Introduction
Tobacco smoking is a major risk factor for cardiovascular diseases, respiratory diseases, cancer, and death [1,2]. The development of smoking-related morbidity and mortality has been hypothesized to be mediated by increased activation of blood leukocytes and changes in oxidative stress, inflammation, and endothelial function [3,4]. However, the underlying molecular mechanisms are not fully understood. Identifying molecular features that are modified by tobacco smoking may have critical implications for our understanding of the development of smoking-associated diseases and may lead to the development of early biomarkers of harm for the deleterious effects of tobacco product exposure.
A number of previous studies have systematically investigated smoking-associated DNA methylation [5–12] and gene expression [13–15] profiles in blood samples. Alterations consistently observed in blood samples from smokers compared to non-smokers include DNA methylation profiles—within the aryl hydrocarbon receptor repressor (AHRR, cg05575921), on chromosome 6p21.33 (cg06126421), and within coagulation factor II (thrombin) receptor-like 3 (F2RL3, cg03636183) [7,11,12,16]—and expression profiles of genes such as leucine rich repeat neuronal 3 (LRRN3) and SAM and SH3 domain containing 1 (SASH1) [13–15,17,18]. Smoking-associated DNA methylation profiles in blood have been used as biomarkers to improve risk prediction of smoking-related morbidity and mortality, including lung cancer incidence and mortality [19–21], cardiovascular mortality, and all-cause mortality [22]. However, the biological causes and consequences of smoking-related alterations in DNA methylation and gene expression in blood samples are unclear. Blood is a heterogeneous mixture of immune cell types, and DNA methylation and gene expression profiles vary across cell types [23]. Tobacco smoke exposure can affect the proportions of individual immune cell types within blood [24]. Therefore, it remains unclear if smoking-associated molecular alterations identified in blood samples are artifacts of the different proportions of immune cell types in the blood of smokers compared to non-smokers, or if there are molecular alterations that result from smoking exposure only in certain subsets of immune cell types.
Studying the effects of smoking in specific subsets of immune cell types may provide insight into the early molecular features that contribute to smoking-related immune dysfunction and disease. Two recent small scale studies (≤34 participants) [25,26] presented evidence for distinct immune cell-type differences in smoking-related methylation of candidate genes previously identified to harbor smoking-associated methylation in blood [25,26]. For instance, methylation of AHRR (cg05575921), 6p21.33 (cg06126421), and F2RL3 (cg03636183) showed greater methylation differences between smokers and non-smokers in granulocytes and monocytes than in B cells or T cells [26]. Further insight into the causes and biological implications of smoking-related epigenetic modifications may be gained from systematic studies in purified cell types known to play a role in the pathophysiology of smoking-related diseases [25].
Herein, we aim to better understand the relationship between current tobacco smoking exposure and genome-wide measures of DNA methylation and gene expression in purified CD14+ monocytes samples. Monocytes are immune cells known to be influenced by tobacco smoke exposure [24] and hypothesized to play a role in the development of smoking-related cardiovascular disease [27]. To build upon previous findings, we utilized concentrations of a nicotine metabolite, cotinine, in urine, as a quantitative measure of current tobacco smoking exposure, and DNA methylation and gene expression measured concurrently in purified CD14+ monocyte samples from participants of a population-based cohort study in several cities across the United States, the Multi-Ethnic Study of Atherosclerosis (MESA). To increase the biological interpretation of smoking-associated DNA methylation in monocytes, we also report associations between DNA methylation and concurrently measured gene expression profiles in vivo.
Results
The study population included 934 MESA participants, with urinary cotinine concentrations and genome-wide profiling of DNA methylation and gene expression in CD14+ monocyte samples collected during the fifth exam (from 2010–2012). Population characteristics are listed in Table 1. Urinary cotinine concentrations above a threshold previously used to classify active smokers in MESA (>100 ng/ml [28]) were detected in 8.7% of the population, as shown in Figure 1, by self-reported smoking status, including 1.2% of participants reporting smoking fewer than 100 cigarettes in their lifetime and not smoking any cigarettes within the past 30 days (never smokers), 4.0% of participants reporting smoking 100 cigarettes in their lifetime but not smoking within the past 30 days (former smokers), and 92% of participants that reported smoking cigarettes within the past 30 days (current smokers). Urine cotinine levels indicative of active or secondhand smoke exposure (>the detectable threshold of 8 ng/ml) were detected in 5.5% of never smokers, 12.2% of former smokers, and 97% of current smokers.
Table 1.
Characteristics of MESA study participants.
| CD14+ monocytes (n = 934) | |
|---|---|
| Proportion or Mean (SD) | |
| Demographic | |
| Mean age (years) | 69 (9) |
| Gender (% female) | 53 |
| Race/ethnicity | |
| White (%) | 45 |
| Hispanic (%) | 34 |
| Black (%) | 21 |
| Self-reported smoking status | |
| Never smoker (%) | 45 |
| Former smoker (%) | 49 |
| Current smoker (%) | 6.7 |
| Urine cotinine levels | |
| Active smoking (% >100ng/ml) | 8.7 |
| Detectable (% >8ng/ml) | 14.8 |
SD, standard deviation
Figure 1.
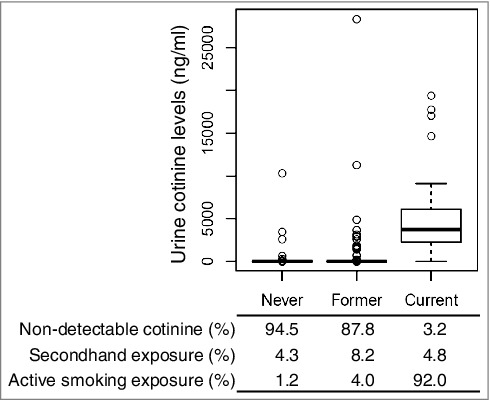
Cotinine concentrations by self-reported smoking status. A comparison of urine cotinine concentrations with self-reported smoking status reveals potential misclassification; cotinine detection limit: 8 ng/ml; active smoking levels: >100 ng/ml (urine). Boxplots show cotinine concentrations (ng/ml, y-axis) with self-reported smoking status (420 never smokers, 458 former smokers, 63 current smokers). The bold horizontal line represents the median, the top and bottom borders of the box represent the 75th and 25th percentiles, respectively, while the circles represent outliers.
Cotinine-Associated DNA Methylation in CD14+ Monocytes
Urine cotinine concentrations associated [false discovery rate (FDR)<0.01] with methylation of 176 CpG sites in 930 CD14+ monocyte samples, out of 484,817 CpGs measured using the Illumina HumanMethylation450 BeadChip (Figure 2 and Table S1). The strongest correlation detected was between urine cotinine and methylation of AHRR cg05575921 (partial correlation, prho = −0.55, Beta ± standard error (SE) = −0.23 ± 0.01, P = 1.04 × 10−73, FDR = 4.82 × 10−68). Log cotinine concentrations uniquely explained 29% of the variance of cg05575921 methylation, holding all other variables constant. Other top loci detected harboring cotinine-associated CpG methylation were intergenic regions on chromosome 2q37.1 (cg21566642 upstream of alkaline phosphatase placental like 2, ALPPL2) and on chromosome 6p21.33 (cg06126421 within the MHC class III region). Top cotinine-associated methylation sites located near protein coding genes included cg03636183 on chromosome 19 within F2R like thrombin/trypsin receptor 3 (F2RL3), cg09935388 on chromosome 1 within growth factor independent 1 transcription repressor (GFI1), cg21322436 on chromosome 7 within the promoter region of contactin associated protein-like 2 (CNTNAP2), and cg05284742 on chromosome 14 within inositol-tetrakisphosphate 1-kinase (ITPK1). The effect sizes resulting from the association between log urine cotinine levels and DNA methylation are depicted in Figure 3. Results between methylation and cotinine with an additional adjustment for urine creatinine levels were highly correlated with results without adjustment for urine creatinine levels (correlation = 0.988, Figure S1).
Figure 2.
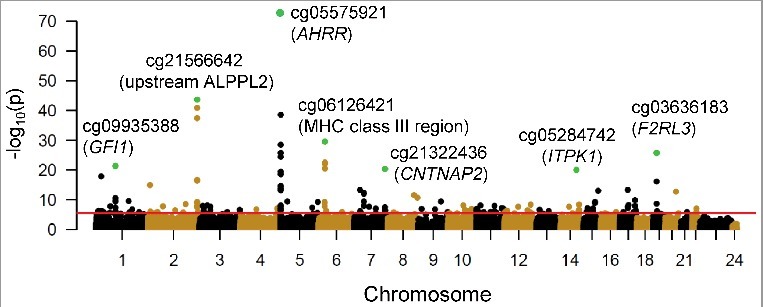
Cotinine concentration associations with DNA methylation in 930 CD14+ monocyte samples. A Manhattan plot shows the significance [-log10(P value), y-axis] resulting from the association between log urine cotinine levels (ng/ml) and DNA methylation for each of the 484,817 CpG sites investigated (represented as circles). CpGs are ordered by chromosome and chromosomal position (x-axis). The red line represents a false discovery rate of 0.01, corresponding to a P value ∼ 3.8 × 10−6. Green circles represent the most significant association detected at the top seven genomic loci harboring cotinine-associated methylation profiles; nearby genes include: growth factor independent 1 transcription repressor (GFI1), aryl-hydrocarbon receptor repressor (AHRR), contactin associated protein-like 2 (CNTNAP2), inositol-tetrakisphosphate 1-kinase (ITPK1), and F2R-like thrombin/trypsin receptor 3 (F2RL3).
Figure 3.
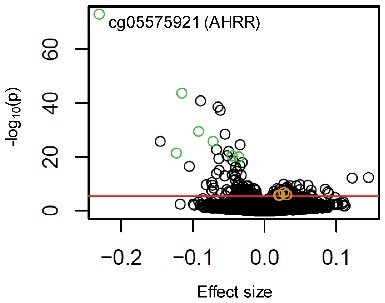
Effect size of association between urine cotinine levels and DNA methylation in monocytes. A volcano plot depicts the significance [-log10(P value), y-axis] vs. the effect size (beta, x-axis) from the associations between DNA methylation (M-value) and log urine cotinine levels (ng/ml) for each of the 484,817 CpG sites investigated (significant results not previously identified shown as golden circles; top seven most significant genomic loci shown as green circles; red line indicates genome-wide significance, FDR < 0.01).
Of the 176 CpGs with urine cotinine-associated (FDR<0.01) methylation detected in CD14+ monocyte samples, only a small portion (17 CpGs) were not previously reported to have methylation associated with smoking status or cotinine in whole blood samples[11,12,16], including 12 CpGs located near genes previously not identified to harbor smoking-associated methylation (FUT4, MAP2K6, FAM153B, CYTH1, RAD9B, PDE1C, HNRNPF, TSPAN4, ANKRD45, ZNF326, CORO1C, GLT1D1). For the 17 CpGs not previously detected in whole blood, urine cotinine concentrations weakly correlated with methylation levels (prho ranging: -0.16 – 0.17). No outliers were identified driving the significant associations detected between cotinine and methylation for these 17 CpGs (residual plots are provided in Figure S2). Notably, the majority (82%) of the potentially monocyte specific-associations were direct associations, while the majority (62%) of associations previously reported in whole blood samples were inverse associations.
Replication of cotinine-associated methylation sites not previously reported in whole blood samples was observed at a Bonferroni adjusted threshold (P<0.003) for one of the 17 CpG sites, cg13300301, located in the first exon of fucosyltransferase 4 (FUT4), utilizing serum cotinine concentrations and DNA methylation measured in 128 independently collected CD14+ monocyte samples. Nominal replication (P<0.05) was observed for four of the 17 CpGs not previously identified with smoking-associated methylation in whole blood samples. The effect sizes between log cotinine concentrations and methylation for these four CpGs are illustrated in Figure 3 (and listed in Table S2).
Cotinine-associated gene expression in CD14+ monocytes
Urine cotinine concentrations significantly associated (FDR<0.01) with mRNA expression of 12 genes in 934 CD14+ monocyte samples, out of the 10,898 expressed genes measured using the Illumina HumanHT-12 v4 Expression BeadChip (Figure 4 and Table S3). The most significantly associated gene was pyrimidinergic receptor P2Y6 (P2RY6), with log cotinine concentrations directly associating with mRNA levels (prho = 0.33, Beta ± SE = 0.078 ± 0.008, P = 1.99E-22, FDR = 2.42E-18). Log cotinine concentrations uniquely explained 9.5% of the variance of P2RY6, holding all other variables constant. Other top genes with cotinine-associated expression included MMP25 (prho = 0.28, Beta ± SE = 0.074 ± 0.009, P = 1.35E-15), SASH1 (prho = 0.24, Beta ± SE = 0.07 ± 0.01, P = 2.15E-12), and SEMA6B (prho = 0.22, Beta ± SE = 0.031 ± 0.005, P = 3.58E-10). Results between gene expression and cotinine with an additional adjustment for urine creatinine levels were highly correlated with results without adjustment for urine creatinine levels (correlation = 0.987, Figure S1).
Figure 4.
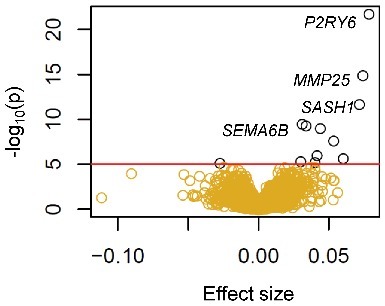
Cotinine concentration associations with gene expression in 934 monocyte samples. A volcano plot depicts the significance [-log10(P value), y-axis] vs. the effect size (x-axis) from the associations between mRNA expression (log2 transformed) and log urine cotinine levels (ng/ml) for each of the 10,898 gene transcripts investigated (shown as golden circles; red line and black circles indicates genome-wide significance, FDR < 0.01).
Eight of the genes with urine cotinine-associated (FDR<0.01) expression in CD14+ monocyte samples were previously reported to have differential gene expression in whole blood samples from 1,421 current smokers compared to 4,860 never smokers [15]. Four genes with urine cotinine-associated (FDR<0.01) expression in monocytes were not previously detected to have cotinine-associated expression in whole blood samples [15], including MMP25, SEMA6B, C2ORF55, and ZNF597. Previous studies of smoking-associated transcription in lymphocytes (B cells, T cells, and Natural Killer cells) [13] and monocytes [29] have identified 10 of the 12 cotinine-associated genes (MMP25, SEMA6B, P2RY6, SASH1, FUCA1, PID1, CLEC10A, ACOX2, FPR3, RGL1). Replication analysis in CD14+ monocyte samples independently collected from 10 current smokers and 10 non-smokers did not replicate (P<0.05) associations between plasma cotinine and expression of the two previously unidentified genes (C2ORF55 and ZNF597, Table S3).
In vivo functional validation of differentially methylated CpGs in relation to the urine cotinine
DNA methylation is a potential regulator of gene expression [30]. To prioritize the list of cotinine-associated methylation sites for potentially functional alterations, we performed a look-up of expression-associated methylation sites (eMS) reported by our previous study [31], which utilized the same monocyte samples. Twenty of the cotinine-associated methylation sites had methylation significantly associated (FDR<0.01) with gene expression of at least one nearby gene (within one megabase, Table S4). However, none of the genes identified harboring cotinine-associated eMS had expression associated with urine cotinine levels at a stringent genome-wide significance threshold (FDR<0.01). Nominal associations (P<0.05) between gene expression and cotinine were identified for five of the genes harboring cotinine-associated eMS (PTK2, NCF4, PDLIM7, PIWIL4, NT5C, see Table 2).
Table 2.
Associations between urine cotinine levels, DNA methylation, and cis-gene expression in CD14+ monocytes.
| Urine cotinine ∼ Methylation |
Methylation ∼ cis Gene expression |
Urine Cotinine ∼ Gene expression |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CpG ID (location) | Chr | Cor | P value | FDR | Gene | Cor | P value | FDR | Cor | P value | FDR |
| cg12075928 (PTK2 Body) | 8 | −0.22 | 1.64E-11 | 2.24E-07 | PTK2 | 0.13 | 6.74E-06 | 1.59E-03 | −0.12 | 5.74E-04 | 0.08 |
| cg20066188 (CYTH4 Body) | 22 | −0.18 | 7.54E-08 | 4.19E-04 | NCF4 | −0.12 | 2.00E-05 | 4.04E-03 | 0.10 | 3.50E-03 | 0.18 |
| cg12513616 (LOC100128340 body) | 5 | −0.20 | 4.55E-10 | 4.79E-06 | PDLIM7 | −0.12 | 1.21E-05 | 2.63E-03 | 0.10 | 4.30E-03 | 0.20 |
| cg13300301 (FUT4 1stExon) | 11 | 0.16 | 1.45E-06 | 4.54E-03 | PIWIL4 | −0.16 | 4.88E-09 | 2.32E-06 | −0.10 | 4.73E-03 | 0.20 |
| cg02186444 (ARMC7 Body) | 17 | 0.21 | 1.32E-10 | 1.70E-06 | NT5C | −0.21 | 4.71E-14 | 4.07E-11 | −0.09 | 0.01 | 0.29 |
CpGs whose degree of methylation significantly associated with urine cotinine levels in CD14+ monocyte samples (n = 930, FDR<0.01), and cis-gene expression (n = 1,264, genes ±1 MB; FDR<0.01), with urine cotinine associations also associated with gene expression in CD14+ monocyte samples (n = 934, P<0.05). Results are sorted by significance of urine cotinine and gene expression. Partial correlations (cor) were adjusted for age, sex, race, study site, and residual contamination with non-targeted cells.
Discussion
Results from this study characterize tobacco exposure-related alterations in DNA methylation and gene expression in monocytes. The majority of the tobacco exposure-associated DNA methylation and gene expression profiles identified in monocytes were previously identified to be associated with self-reported smoking status in whole blood samples. A small number of previously unidentified methylation profiles were weakly associated with estimates of recent tobacco exposure, four of which nominally replicated with methylation associated with plasma cotinine in independently collected monocyte samples. Some of the genes identified with tobacco exposure-associated expression are potential mediators of inflammatory alterations in monocytes in response to cigarette exposure, such as P2RY6. Using concurrent profiling of DNA methylation and gene expression, relationships between tobacco exposure-associated DNA methylation and nearby gene expression levels were examined. None of the cotinine-associated genes had expression levels strongly correlated with nearby cotinine-associated methylation profiles, making the implications of smoking-associated methylation in monocytes unclear.
The majority of previous studies characterizing smoking-associated DNA methylation [5–12] patterns defined smoking exposure using self-reported smoking status. To limit the potential for misclassification bias resulting from self-reported exposure, three recent studies have utilized cotinine as a robust biomarker of recent tobacco exposure. The first study of smoking-associated DNA methylation to utilize cotinine concentrations to validate smoking status investigated DNA methylation in whole blood samples from 107 young African American men, and identified two CpGs with methylation significantly different between current and never smokers (cg05575921 and cg21161138) [9], both of which had cotinine-associated methylation in this study of monocytes (FDR<0.01). A more recent genome-wide investigation of serum cotinine-associated DNA methylation included 1,000 German men and women, which identified 40 CpGs with cotinine-associated methylation profiles at a genome-wide significance threshold [16], 75% of which were identified in this study of monocytes (FDR<0.01). A third study of smoking-associated blood methylation in a Korean population (n = 100) did not identify any cotinine-associated methylation sites at a genome-wide significant threshold [32]. In our genome-wide study, we used urine cotinine levels as a dose-dependent biomarker of recent tobacco exposure in a large, multi-ethnic population (n = 934). A comparison of self-reported smoking status and urine cotinine levels in our study revealed misclassification in self-reported smoking status.
In addition to utilizing urine cotinine levels as a biomarker of recent tobacco exposure, in this study DNA methylation and gene expression were measured in CD14+ monocyte samples to reduce heterogeneity that may have been present in previous studies that examined mixed cell samples. Of the 176 cotinine-associated methylation sites identified in monocytes, 17 were not previously identified by studies of smoking-associated methylation in whole blood samples [11,12,16]. Nominal replication was observed for four newly identified cotinine-associated methylation sites in independently collected monocyte samples (cg13300301, cg12323063, cg08252384, cg12586707). These methylation profiles represent potential monocyte specific alterations in methylation that may result from tobacco product exposure. Intriguingly, the potentially monocyte specific cotinine-associated methylation sites identified in this study all had increasing methylation with cotinine levels, in contrast to the majority of previously identified associations with methylation levels inversely associated with smoke exposure. It is possible that smoking leads to demethylation of genes more often than methylation of genes, and that methylation of genes may occur in a more cell-specific fashion compared to demethylation of genes. Future studies investigating smoking-associated changes in methylation across many purified cell types will be necessary to better characterize the potential cell-specific relationship between increasing and decreasing levels of methylation in response to smoking exposure.
Top cotinine-associated methylation sites identified in monocyte samples, such as those located in AHRR and F2RL3, were previously identified by studies of whole blood. Recent longitudinal studies have reported AHRR and F2RL3 methylation profiles measured in whole blood can improve risk prediction for all-cause mortality and CVD mortality beyond traditional risk factors [22], supporting these methylation profiles as potential early biomarkers of smoking-related harm. However, it remains unclear if these methylation alterations associated with smoking exposure play a functional role in the development of smoking-related diseases, or if they are providing a better measure of smoking exposure than the use of self-reported smoking history or biomarkers of recent exposure such as cotinine.
Significant alterations in gene expression levels in whole blood from current smokers compared to non-smokers have also been identified, and have been used to accurately discriminate smokers from non-smokers [9,14,17,18,29]. Smoking-associated alterations in gene expression have potential as early biomarkers of harm, which may mediate some of the biological responses to tobacco exposure. However, as gene expression signatures vary by cell type, it is unclear if the previously identified smoking-associated alterations in gene expression in whole blood reflected changing proportions of specific immune cell populations in smokers compared to non-smokers. To better understand the relationship between smoking and gene expression, we investigated gene expression associations with recent smoking exposure in CD14+ monocyte samples. Notably, a top gene previously identified with smoking-associated expression in whole blood samples, which was also included in algorithms to discriminate current smokers from non-smokers (LRRN3) [13–15,17,18], was not detectable in monocytes. Top genes identified with expression in monocytes associated with recent tobacco exposure included previously identified genes, such as P2RY6, MMP25, and SASH1. These genes may represent potential candidates for early biomarkers of harm, which may mediate some of the deleterious effects of tobacco smoke exposure, based on previous findings of these genes [33–36]. For instance, P2RY6 was reported to be related to contractile changes in vasculature after sub-chronic smoke exposure in mice [33]. P2RY6 has also been implicated in mediating release of pro-inflammatory cytokines and chemokines in monocytes [34] and macrophages [35] and promoting vascular inflammation [37]. MMP25 deficiency has been linked to an defective NF-κB activation and impaired innate immune response in mice [38]. Future monitoring of these alterations in gene expression, which are linked to inflammatory processes, may offer avenues for regulatory scientists and tobacco companies to measure early toxicity responses to tobacco product use. Longitudinal studies will be necessary to validate the identified genes, such as P2RY6, MMP25, and SASH1, as early biomarkers of harm.
Previous studies of smoking-associated methylation have lacked concurrent profiling of gene expression; therefore, the functional consequences of smoking-associated methylation remain unclear. Using a multi-omic approach, results from this study revealed 20 cotinine-associated methylation sites whose methylation levels reflected expression patterns of nearby genes. The majority (∼80%) of significant associations between methylation and nearby gene expression levels were inverse associations, supporting the previous observations that methylation is typically inversely associated with nearby gene expression levels. Five of the genes harboring cotinine-associated eMS had expression nominally associated with urine cotinine levels. Four of them had expression profiles associating with cotinine levels in the direction of the effect that was expected based on the directions of correlation between cotinine and methylation, and methylation and gene expression, including downregulation of PTK2, PIWIL4, and NT5C and upregulation of NCF4. One of the potentially monocyte-specific CpGs (cg13300301) had methylation directly associated with cotinine and inversely associated with expression of piwi like RNA-mediated gene silencing (PIWIL4), a gene with lower expression nominally associating with cotinine levels. PIWIL4 encodes an Argonaute family protein that is involved with binding small noncoding RNAs called PIWI-interacting RNAs (piRNAs) and regulating gene expression [39]. PIWIL4 has been suggested to be involved in cancer formation and or progression, and PIWIL4 knockdown can reduce cell migration and proliferation, and increase apoptosis in a breast cancer cell line (MDA-MB-231) [40]. Given the weak correlations observed between cotinine, methylation, and gene expression and the cross-sectional nature of the measurements, it is not clear if cg13300301 methylation is a potential mediator of tobacco exposure-related effects on PIWIL4 expression in monocytes.
The majority of tobacco exposure-associated DNA methylation profiles are not associated with gene expression in monocytes. Therefore, the relationships between smoking-associated alterations DNA methylation and nearby gene expression levels remain unclear. Notably, in this study AHRR was not detectable using microarrays; however, we previously detected AHRR expression in monocytes using RNA-sequencing and RT-PCR, technologies more sensitive to low abundance genes [26,41]. We found AHRR methylation was significantly associated with AHRR expression (P = 1.4 × 10−17) and carotid plaque scores (P = 3.1 × 10−10), suggesting that some DNA methylation alterations may indeed be related to functional changes in gene expression and may serve as potential early biomarkers of cardiovascular disease [41].
Interpretation of the relationships between smoking, DNA methylation, and gene expression characterized by this study are limited by a number of factors. The cross-sectional analysis of smoking exposure, DNA methylation, and gene expression limits our ability to rule out other influences on DNA methylation and gene expression that might confound the results. Additionally, cotinine concentrations are subject to considerable individual variability in the rates of nicotine metabolism and elimination [42] and do not reflect long-term or cumulative smoking exposure. In the discovery analysis, cotinine levels were measured in urine using immunoassay, which is a less sensitive and specific way to measure cotinine compared to chromatography-based analytical methods [43]. Another potential limitation is that, unlike the discovery analysis, the replication analysis was based on cotinine measured in plasma; however, given the high correlation between cotinine concentrations measured in urine and plasma, cotinine measured in either of these fluids can be used as a proxy of recent nicotine intake [44]. Also, this study only examined DNA methylation and gene expression successfully measured by microarray. Many CpGs were not included on the microarray, and other technologies such as RNA-sequencing may be better suited to measure expression of low-abundance genes, such as AHRR, that may be missed using microarrays. Additionally, because the effect sizes detected for potentially monocyte-specific smoking-related alterations in methylation tended to be weak, it is possible that we were underpowered in our replication analysis to replicate other significant associations detected between cotinine and methylation in the discovery cohort. Lastly, although our study utilized highly purified monocyte samples, it is possible that the CD14+ selection was not sufficient to purify out subtype populations that may change in concentration due to tobacco smoking exposure.
In conclusion, the majority of the smoking-associated methylation and gene expression profiles we identified in CD14+ monocyte samples were previously identified in studies of whole blood, including the largest effect sizes identified. A small number of CpG sites in monocytes were identified with methylation weakly associated with a biomarker of smoking, which represent potential monocyte-specific smoking-associated methylation profiles. Some top gene expression profiles associated with smoking in monocytes have biologically plausible roles in the deleterious effects of smoking and represent candidate genes for future studies of biomarkers of harm. Alterations in gene expression associated with smoking did not appear to be related to smoking-associated methylation. The functional consequence of smoking-associated methylation remains unclear. Future longitudinal studies will be necessary to better understand the potential for smoking-associated methylation and gene expression to serve as early biomarkers of harm.
Methods
Study design and data collection
MESA was designed to investigate the prevalence, correlates, and progression of subclinical cardiovascular disease (CVD) in a population cohort of 6,814 participants. MESA enrolled participants aged 45–84 free of CVD at baseline from July 2000 to July 2002 at six field centers (Baltimore, MD; Chicago, Illinois; Los Angeles, CA; New York, NY; St. Paul, MN, and Winston-Salem, NC). Since its inception, five clinic visits collected extensive clinical, socio-demographic, lifestyle, behavior, laboratory, nutrition, and medication data [45]. During the fifth exam, urine cotinine concentrations and genome-wide methylomic and transcriptomic profiles in CD14+ purified monocytes were measured in a subset of 934 participants from four MESA field centers [Baltimore, MD; New York, NY; St. Paul, MN; and Winston-Salem, NC (April 2010-February 2012)] [31]. The study protocol was approved by the Institutional Review Board at each site. All participants signed informed consent.
Smoking exposure assessment
Urinary cotinine concentrations were measured from spot samples via immunoassay [46] (Immulite 2000 Nicotine Metabolite Assay; Diagnostic Products Corp., Los Angeles, CA) at the National Institute for Occupational Safety and Health (NIOSH) core laboratory in a random subsample of MESA participants enrolled in the MESA Lung Ancillary Study. Intra-assay coefficient of variation was 2.02%, and the minimum detectable concentration was 10 ng/mL. Undetectable values of cotinine were assigned as the detection threshold (7.07 ng/ml). To address potential complications in measured cotinine levels arising from urinary dilution, urinary creatinine levels were included in statistical analyses. Urinary creatinine was measured by the rate Jaffe reaction [Vitros 950IRC instrument (Johnson & Johnson Clinical Diagnostics Inc)] [47]. The range of urinary creatinine levels was 0.05–16.5 mg/dL, with a CV range of 2.5–2.9%.
Participants who reported smoking fewer than 100 cigarettes in their lifetimes were classified as never smokers. Among participants who reported smoking greater than 100 cigarettes in their lifetime, those reporting not smoking during the last 30 days were classified as former smokers.
Purification of CD14+ monocytes
Blood was collected in sodium heparin-containing Vacutainer CPT™ cell separation tubes (Becton Dickinson, Rutherford, NJ) to separate peripheral blood mononuclear cells from other elements within two hours from blood draw. Subsequently, monocytes were isolated with anti-CD14 monoclonal antibody coated magnetic beads, using an autoMACS automated magnetic separation unit (Miltenyi Biotec, Bergisch Gladbach, Germany). Initially, flow cytometry analysis of 18 specimens was performed, including samples from all four MESA field centers, which were found to be consistently >90% pure.
DNA/RNA extraction
DNA and RNA were isolated from samples simultaneously using the AllPrep DNA/RNA Mini Kit (Qiagen, Inc., Hilden, Germany). DNA and RNA QC metrics included optical density (OD) measurements, using a NanoDrop spectrophotometer and evaluation of the integrity of 18 S and 28 S ribosomal RNA using the Agilent 2100 Bioanalyzer with RNA 6000 Nano chips (Agilent Technology, Inc., Santa Clara, CA) following manufacturer's instructions. RNA with RNA Integrity (RIN) scores >9.0 was used for global expression microarrays. The median of RIN for our 1,264 samples was 9.9.
Genome-wide methylation and gene expression quantification
Illumina HumanMethylation450 BeadChips and HiScan reader were used to perform the genome-wide methylation analysis, as previously described [31]. This methylation data has been deposited in the NCBI Gene Expression Omnibus and is accessible through GEO Series accession number GSE56046. The Illumina HumanHT-12 v4 Expression BeadChip and Illumina Bead Array Reader were used to perform the genome-wide gene expression analysis, following the Illumina expression protocol. GSE56047, as previously described [31].
Quality control and pre-processing of microarray data
Data pre-processing and quality control (QC) analyses were performed in R (http://www.r-project.org/) using Bioconductor (http://www.bioconductor.org/) packages, as previously described [31]. We included 2% blind duplicates. Correlations among technical replicates exceeded 0.997. Multidimensional scaling plots showed the five common control samples were highly clustered together and identified three outlier samples, which were excluded subsequently.
The Illumina HumanMethylation450 BeadChip included probes for 485,577 CpG sites. Probes excluded from analysis included probes with “detected” methylation levels in <90% of MESA samples using a detection P value cut-off of 0.05 (695 probes excluded) and control probes which assay highly-polymorphic single nucleotide polymorphisms (SNPs) rather than DNA methylation (65 probes excluded) [48], resulting in a total of 484,817 CpG sites included in the analyses.
The Illumina HumanHT-12 v4 Expression BeadChip included >47,000 probes for >30,000 genes (with unique Entrez gene IDs). Statistical analyses excluded probes with non-detectable expression in ≥90% of MESA samples (using a detection P value cut-off of 0.0001); probes overlapping repetitive elements or regions, probes with low variance across the samples (<10th percentile), or probes targeting putative and/or not well-characterized genes, i.e. gene names starting with KIAA, FLJ, HS, MGC, or LOC, 14,619 mRNA transcripts from 10,989 unique genes were included in the analysis.
To estimate residual sample contamination, we generated separate enrichment scores for neutrophils, B cells, T cells, and natural killer cells, as described previously [31]. We adjusted for residual sample contamination with non-monocyte cell types in all the analyses.
Study population
This study population included 934 MESA participants with urinary cotinine concentrations and gene expression/DNA methylation data in CD14+ monocytes collected during the fifth MESA Exam, from 2010 – 2012 (53% female; 45% Caucasian, 34% Hispanic, 21% African-American; Asian-American participants of MESA were not included in this ancillary study due to the limited sample size within MESA).
Statistical analysis
Using R statistical software, linear regression was used to examine the association between the independent variable (log transformed urine cotinine concentration) and the dependent variable (DNA methylation or gene expression). For DNA methylation, individual models were run for each of the 484,817 CpGs (‘M value’—log ratio of the methylated to the unmethylated intensity in monocyte samples [49], adjusted for chip and sample position on chip). For gene expression, individual models were run for each of the 14,619 mRNA transcripts from 10,989 genes (log2 transformed). Model covariates included age, sex, ethnicity/race, education (3 levels: <high school diploma, high school degree/diploma, >high school degree), study site, and estimated residual sample contamination with non-monocyte cell types (neutrophils, B cells, T cells, and natural killer cells). P values were adjusted for multiple testing using the q-value false discovery rate (FDR) [50] method. Sensitivity analysis was performed examining associations between methylation and cotinine with an additional adjustment for urinary creatinine levels.
We estimate 80% power to detect 3.2% variance in log2 mRNA expression levels (mean = 5.7, SD = 0.4), and 4.0% variance in DNA methylation M-values (mean = 2, SD = 0.7), explained by log urine cotinine concentrations (SD = 1.7, n = 934), based on a power calculation using a simple linear regression (alpha = 0.05) with a Bonferroni correction to adjust for 10,898 genes and 484,817 CpG sites, respectively (using Quanto v1.2).
Replication analysis
CpGs with methylation in CD14+ monocytes significantly associated with urine cotinine (FDR<0.01) in the discovery population (MESA) that were not previously reported to be associated with self-reported smoking status [11,12] or plasma cotinine [16] in whole blood samples were included in replication analyses. Replication utilized serum cotinine concentrations measured in an independent multi-ethnic population (n = 128) recruited by the National Institute of Environmental Health Sciences (NIEHS) Clinical Research Unit, with DNA methylation measured in CD14+ monocyte samples using the Illumina HumanMethylation450 microarray. Linear regression was performed with DNA methylation M-values as the dependent variables and log plasma cotinine concentrations as the independent variables. Covariates included age, sex, race, and estimates of sample concentrations of non-monocyte cell types (CD4+ T cells, CD8+ T cells, B cells, natural killer cells, neutrophils, and eosinophils).
Genes with expression significantly associated with urine cotinine (FDR<0.01) not previously reported to be associated with smoking status were included in replication analysis. Replication was performed using gene expression measured using Affymetrix Whole transcriptome array in 10 smokers and 10 non-smokers recruited by the NIEHS Clinical Research Unit under protocol 10-E-0063.
In vivo functional annotation analysis
DNA methylation sites significantly associated with urine cotinine concentrations were investigated for association with nearby gene expression profiles by performing a look-up in the results from our previous analysis [31] of the same samples. Briefly, to identify DNA methylation associated with nearby gene expression, we fit separate linear regression models with the M-value for each CpG site (adjusted for methylation chip and position effects) as a predictor of transcript expression for any gene within 1 Mb of the CpG in question. Covariates were age, sex, and race/ethnicity, and study site.
Supplementary Material
Funding Statement
This work was supported by the National Heart, Lung, And Blood Institute (NHLBI) of the National Institutes of Health (NIH) under Grant Number P50HL120163 and in part by the Intramural Research Program of the National Institute of Environmental Health Sciences. NIEHS (projects: ZO1-ES100475 and Z01 ES046008). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Center for Tobacco Products. The MESA Epigenomics & Transcriptomics Study was funded by NHLBI grant R01HL101250, R01 DK103531-01, and R01 HL135009-01 to Wake Forest University Health Sciences. Cotinine measurements in the MESA study were funded by the MESA Lung Study (R01-HL077612 and R01-HL093081). The research described in this publication was funded in part by the US Environmental Protection Agency through RD831697 to the University of Washington (MESA Air); it has not been subjected to the Agency's required peer and policy review and therefore does not necessarily reflect the views of the Agency and no official endorsement should be inferred. MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. Support for MESA is provided by contracts N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-001079, UL1-TR-000040, and DK063491.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Acknowledgments
The authors would like to thank the other investigators, fellows, staff, and participants of the MESA study, the NIEHS Clinical Research Unit, and the American Heart Association Tobacco Regulation and Addition Center (A-TRAC) for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
References
- [1].U.S. Department of Health and Human Services The health consequences of smoking: 50 years of progress. A report of the surgeon general. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2014. [Google Scholar]
- [2].US Burden of Disease Collaborators The state of US health, 1990–2010: burden of diseases, injuries, and risk factors. JAMA. 2013 August 14;310(6):591–606. doi: 10.1001/jama.2013.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Blann AD, Kirkpatrick U, Devine C, et al. The influence of acute smoking on leucocytes, platelets and the endothelium. Atherosclerosis. 1998 October 5;141(1):133–139. doi: 10.1016/S0021-9150(98)00163-4. [DOI] [PubMed] [Google Scholar]
- [4].Csiszar A, Podlutsky A, Wolin MS, et al. Oxidative stress and accelerated vascular aging: implications for cigarette smoking. Front Biosci (Landmark Ed). 2009;14:3128–3144. doi: 10.2741/3440. PMID:19273262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012 November;7(11):1331–1338. doi: 10.4161/epi.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dogan MV, Shields B, Cutrona C, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151. doi: 10.1186/1471-2164-15-151. PMID:24559495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zeilinger S, Kuhnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812. doi: 10.1371/journal.pone.0063812. PMID:23691101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Monick MM, Beach SR, Plume J, et al. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet. 2012 March;159B(2):141–151. doi: 10.1002/ajmg.b.32021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Philibert RA, Beach SR, Lei MK, et al. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5(1):19. doi: 10.1186/1868-7083-5-19. PMID:24120260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wan ES, Qiu W, Baccarelli A, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012 July 1;21(13):3073–82. doi: 10.1093/hmg/dds135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gao X, Jia M, Zhang Y, et al. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015 October 16;7:113. doi: 10.1186/s13148-015-0148-3 eCollection 2015. doi: 10.1186/s13148-015-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016 Oct;9(5):436–447. doi: 10.1161/CIRCGENETICS.116.001506. PMID:27651444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Charlesworth JC, Curran JE, Johnson MP, et al. Transcriptomic epidemiology of smoking: the effect of smoking on gene expression in lymphocytes. BMC Medical Genomics. 2010;3(1):1–11. doi: 10.1186/1755-8794-3-29. PMID:20092628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Beineke P, Fitch K, Tao H, et al. A whole blood gene expression-based signature for smoking status. BMC Medical Genomics. 2012;5(1):1–9. doi: 10.1186/1755-8794-5-58. PMID:22221319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Huan T, Joehanes R, Schurmann C, et al. A Whole-Blood Transcriptome meta-analysis identifies gene expression signatures of cigarette smoking. Hum Mol Genet. 2016 Nov 1;25(21):4611–4623. doi: 10.1093/hmg/ddw288. PMID:28158590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang Y, Florath I, Saum KU, et al. Self-reported smoking, serum cotinine, and blood DNA methylation. Environ Res. 2016 January 28;146:395–403. doi: 10.1016/j.envres.2016.01.026. [DOI] [PubMed] [Google Scholar]
- [17].Martin F, Talikka M, Hoeng J, et al. Identification of gene expression signature for cigarette smoke exposure response – from man to mouse. Hum Exp Toxicol. 2015 December 1;34(12):1200–11. doi: 10.1177/0960327115600364. [DOI] [PubMed] [Google Scholar]
- [18].Poussin C, Belcastro V, Martin F, et al. Crowd-sourced verification of computational methods and data in systems toxicology: a case study with a heat-not-burn candidate modified risk tobacco product. Chem Res Toxicol. 2017 Apr 17;30(4):934–945. doi: 10.1021/acs.chemrestox.6b00345. PMID:28085253 [DOI] [PubMed] [Google Scholar]
- [19].Zhang Y, Schöttker B, Ordóñez-Mena J, et al. F2RL3 methylation, lung cancer incidence and mortality. Int J Cancer. 2015 October 1;137(7):1739–1748. doi: 10.1002/ijc.29537. [DOI] [PubMed] [Google Scholar]
- [20].Zhang Y, Elgizouli M, Schöttker B, et al. Smoking-associated DNA methylation markers predict lung cancer incidence. Clinical Epigenetics. 2016;8(1):127. doi: 10.1186/s13148-016-0292-4. PMID:27924164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang Y, Breitling LP, Balavarca Y, et al. Comparison and combination of blood DNA methylation at smoking-associated genes and at lung cancer-related genes in prediction of lung cancer mortality. Int J Cancer. 2016 December 1;139(11):2482–2492. doi: 10.1002/ijc.30374. [DOI] [PubMed] [Google Scholar]
- [22].Zhang Y, Schöttker B, Florath I, et al. Smoking-associated DNA methylation biomarkers and their predictive value for all-cause and cardiovascular mortality. Environ Health Perspect. 2016 January;124(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kundaje A, Meuleman W, Ernst J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015 February 19;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bergmann S, Siekmeier R, Mix C, et al. Even moderate cigarette smoking influences the pattern of circulating monocytes and the concentration of sICAM-1. Respir Physiol. 1998 December;114(3):269–275. doi: 10.1016/S0034-5687(98)00098-X. [DOI] [PubMed] [Google Scholar]
- [25].Bauer M, Fink B, Thurmann L, et al. Tobacco smoking differently influences cell types of the innate and adaptive immune system-indications from CpG site methylation. Clin Epigenetics. 2016 August 3;7:83. doi: 10.1186/s13148-016-0249-7 eCollection;%2015.:83-0249. doi: 10.1186/s13148-016-0249-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Su D, Wang X, Campbell MR, et al. Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PLoS One. 2016 December 9;11(12):e0166486. doi: 10.1371/journal.pone.0166486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011 July;31(7):1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lovasi GS, Diez Roux AV, Hoffman EA, et al. Association of environmental tobacco smoke exposure in childhood with early emphysema in adulthood among nonsmokers: the MESA-lung study. Am J Epidemiol. 2010 January 1;171(1):54–62. doi: 10.1093/aje/kwp358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zeller T, Wild P, Szymczak S, et al. Genetics and beyond–the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5(5):e10693. doi: 10.1371/journal.pone.0010693. PMID:20502693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. PMID:22641018 [DOI] [PubMed] [Google Scholar]
- [31].Liu Y, Ding J, Reynolds LM, et al. Methylomics of gene expression in human monocytes. Hum Mol Genet. 2013 December 15;22(24):5065–74. doi: 10.1093/hmg/ddt356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee MK, Hong Y, Kim SY, et al. DNA methylation and smoking in Korean adults: epigenome-wide association study. Clinical Epigenetics. 2016;8(1):103. doi: 10.1186/s13148-016-0266-6. PMID:27688819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Haanes KA, Kruse LS, Wulf-Johansson H, et al. Contractile changes in the vasculature after subchronic smoking: a comparison between wild type and surfactant Protein D knock-out mice. Nicotine Tob Res. 2016 May 1;18(5):642–646. doi: 10.1093/ntr/ntv243. [DOI] [PubMed] [Google Scholar]
- [34].Cox MA, Gomes B, Palmer K, et al. The pyrimidinergic P2Y6 receptor mediates a novel release of proinflammatory cytokines and chemokines in monocytic cells stimulated with UDP. Biochem Biophys Res Commun. 2005 May 6;330(2):467–473. doi: 10.1016/j.bbrc.2005.03.004. [DOI] [PubMed] [Google Scholar]
- [35].Garcia RA, Yan M, Search D, et al. P2Y6 receptor potentiates pro-inflammatory responses in macrophages and exhibits differential roles in atherosclerotic lesion development. PLoS One. 2014 October 31;9(10):e111385. doi: 10.1371/journal.pone.0111385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Weidmann H, Touat-Hamici Z, Durand H, et al. SASH1, a new potential link between smoking and atherosclerosis. Atherosclerosis. 2015 October;242(2):571–579. doi: 10.1016/j.atherosclerosis.2015.08.013. [DOI] [PubMed] [Google Scholar]
- [37].Riegel AK, Faigle M, Zug S, et al. Selective induction of endothelial P2Y6 nucleotide receptor promotes vascular inflammation. Blood. 2011 February 24;117(8):2548–2555. doi: 10.1182/blood-2010-10-313957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Soria-Valles C, Gutiérrez-Fernández A, Osorio FG, et al. MMP-25 metalloprotease regulates innate immune response through NF-KB signaling. J Immunol. 2016 June 17;197(1):296. doi: 10.4049/jimmunol.1600094. [DOI] [PubMed] [Google Scholar]
- [39].Girard A, Sachidanandam R, Hannon GJ, et al. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006 July 13;442(7099):199–202. [DOI] [PubMed] [Google Scholar]
- [40].Wang Z, Liu N, Shi S, et al. The role of PIWIL4, an argonaute family protein, in breast cancer. J Biol Chem. 2016 May 13;291(20):10646–10658. doi: 10.1074/jbc.M116.723239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Reynolds LM, Wan M, Ding J, et al. DNA Methylation of the Aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis. Circ Cardiovasc Genet. 2015 October;8(5):707–716. doi: 10.1161/CIRCGENETICS.115.001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Swan GE, Benowitz NL, Lessov CN, et al. Nicotine metabolism: the impact of CYP2A6 on estimates of additive genetic influence. Pharmacogenet Genomics. 2005 February;15(2):115–125. doi: 10.1097/01213011-200502000-00007. [DOI] [PubMed] [Google Scholar]
- [43].Avila-Tang E, Al-Delaimy WK, Ashley DL, et al. Assessing secondhand smoke using biological markers. Tob Control. 2013 May;22(3):164–171. doi: 10.1136/tobaccocontrol-2011-050298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Benowitz NL, Hukkanen J, Jacob P., III Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 2009;192:29–60. doi: 10.1007/978-3-540-69248-5_2. PMID:19184645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bild DE, Bluemke DA, Burke GL, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. 2002 November 1;156(9):871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- [46].Rodriguez J, Jiang R, Johnson WC, et al. The association of pipe and cigar use with cotinine levels, lung function, and airflow obstruction: a cross-sectional study. Ann Intern Med. 2010 February 16;152(4):201–210. doi: 10.7326/0003-4819-152-4-201002160-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zemaitis P, Liu K, Jacobs DR, et al. Cumulative systolic BP and changes in Urine Albumin-to-Creatinine Ratios in nondiabetic participants of the multi-ethnic study of atherosclerosis. Clin J Am Soc Nephrol. 2014 November 7;9(11):1922–1929. doi: 10.2215/CJN.02450314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pidsley R, Wong CCY, Volta M, et al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. PMID:23631413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010 November 30;11:587. doi: 10.1186/1471-2105-11-587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003 August 5;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.