

ABSTRACT
G-Protein Coupled Receptor (GPCR), Class C, Group 5, Member A (GPRC5A) has been implicated in several malignancies. The underlying mechanisms, however, remain poorly understood. Using a panel of human cell lines, we demonstrate that CRISPR/Cas9-mediated knockout and RNAi-mediated depletion of GPRC5A impairs cell adhesion to integrin substrates: collagens I and IV, fibronectin, as well as to extracellular matrix proteins derived from the Engelbreth-Holm-Swarm (EHS) mouse sarcoma (Matrigel). Consistent with the phenotype, knock-out of GPRC5A correlated with a reduced integrin β1 (ITGB1) protein expression, impaired phosphorylation of the focal adhesion kinase (FAK), and lower activity of small GTPases RhoA and Rac1. Furthermore, we provide the first evidence for a direct interaction between GPRC5A and a receptor tyrosine kinase EphA2, an upstream regulator of FAK, although its contribution to the observed adhesion phenotype is unclear. Our findings reveal an unprecedented role for GPRC5A in regulation of the ITGB1-mediated cell adhesion and it's downstream signaling, thus indicating a potential novel role for GPRC5A in human epithelial cancers.
KEYWORDS: ECM, EphA2, GPRC5A, integrin β1, matrix adhesion
Introduction
The epithelial tissue integrity and homeostasis depends on the proper cell-cell and cell-matrix adhesion.1-3 A defective cell-matrix interaction allows epithelial cells, normally resting on a layer of extracellular matrix known as a basal membrane, to detach, affecting the tissue architecture and allowing for cell migration and invasive behavior.2,3 Thus, an impaired regulation of the cell-matrix adhesion may facilitate cancer progression, as shown for various human cancers (reviewed in refs. 4, 5).
Numerous and functionally distinct molecules regulate adhesion to extracellular matrix (ECM) in epithelial cells. Integrins, the major group of ECM receptors at the epithelial cell surface, provide a primary connection between the cellular environment and cytoskeleton (reviewed in refs. 4, 6 and elsewhere). Upon binding to the extracellular matrix proteins, integrins' α and β subunits change their conformation and recruit adaptor and signaling proteins to activate Focal Adhesion Kinase (FAK), an intracellular master regulator of cell adhesion.7,8 FAK, activated via autophosphorylation at the Y397 residue, facilitates the assembly of focal adhesions (cell-matrix adhesion sites) and binds Src tyrosine kinase.8-10 The FAK/Src complex formation leads to a full activation of FAK through its phosphorylation at Y576, Y577, Y861, and Y925,9,11 and regulates the focal adhesions' turnover and activity of the Rho family small GTPases (reviewed in refs. 9, 12). In turn, Rho GTPases RhoA, Rac1, cdc42, and other members of the Rho family regulate the assembly of actin structures (e.g. stress fibers , lamellipodia and filopodia) and, thus, coordinate formation of cellular protrusions and cell spreading at different stages of the cell-matrix adhesion (reviewed in refs. 13, 14). Thus, integrin activation elicits a full range of molecular events underlying cell adhesion to the extracellular matrix.
Other membrane receptor molecules such as receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs) modulate the adhesion signaling via FAK/Src/Rho GTPases. For example, integrin ligation-induced trans-activation of the epithelial growth factor receptor (EGFR) facilitates activation of the Rac1 GTPase, and promotes lamellipodia formation and cell spreading.15 Another RTK, EPH receptor A2 (EphA2), associates with Src and FAK kinases and modulates FAK activity, Rho-mediated actin/myosin contractility, and cell spreading in a ligand-dependent manner in cancer cells.16,17 Multiple GPCRs may also impact adhesion by affecting signaling via integrin (e.g., CaSR18 and others19), FAK (β-AR,20 muscarinic M3 receptor21), Rho GTPase (GPR97,22 and others19), or Src (A2A adenosine receptor,23 PITPNM3,24 and others25). Therefore, interactions between the FAK/Src/Rho axis, RTKs, and GPCRs may significantly modulate epithelial cell adhesion to ECM.
G-Protein Coupled Receptor, Class C, Group 5, Member A (GPRC5A), an orphan GPCR, is highly expressed in many epithelial cancers including breast, colon, pancreatic, gastric, prostate, and testicular cancers.26 In breast and gastric cancers, GPRC5A protein expression correlates with the tumor grade and invasiveness.27,28 For patients with hepatocellular carcinoma and gastric adenocarcinoma, a high expression of GPRC5A protein is associated with a poor overall survival.28,29 Some experimental studies have demonstrated that inhibition of GPRC5A has a growth–suppressive effect on cancer cells, suggesting that GPRC5A plays a role in cell survival.30,31 However, the role of GPRC5A in regulating the invasiveness of cancer cells, including cell migration and cell-matrix adhesion, has not been investigated.
Here, we demonstrate that GPRC5A is required to sustain cell adhesion to distinct ECM components (fibronectin, collagen I, collagen IV, and Matrigel compounds). Impaired adhesion of GPRC5A knock-out cells to ECM components correlated with reduced protein levels of integrin β1, decreased activation of FAK and its reduced binding to the Src kinase. In accordance with impaired activation of the FAK/Src complex, GPRC5A knock-out cells revealed a reduced activity of the downstream Rho family small GTPases RhoA and Rac1, but not cdc42. A further phosphoproteomics analysis and biochemical experiments provided evidence for a physical interaction between GPRC5A and EphA2, an established FAK-regulating RTK. Although a possible mechanism underlying the adhesion defects in GPRC5A knock-out cells is not clear, our data for the first time implicate GPRC5A as a positive modulator of epithelial cell adhesion, and suggest a novel role of GPRC5A in several human cancers.
Results
Knock-out of GPRC5A leads to reduced cell adhesion and spreading, but not cell migration
In order to understand whether GPRC5A has a role in cell adhesion, we, first, knocked-out GPRC5A using the clustered regularly interspaced short palindromic repeats associated protein 9(CRISPR/Cas9)-mediated approach in 6 human cell lines of different origins: MDA-MB-231 (triple-negative breast cancer), MCF10A (a non-tumorigenic mammary epithelial cell line), MCF7 (breast adenocarcinoma), HeLa (cervical carcinoma), NCI-H292 (lung mucoepidermoid carcinoma), and Calu-1 (lung epidermoid carcinoma). We established 2 independent variants for each cell line using 2 different guide RNAs (sgRNAs) targeting GPRC5A (sgRNA sequences are shown in Supplementary Table ST1). A deep sequencing of the target site confirmed that both sgRNAs produced frameshift mutations in about 80% of the targeted cells almost certainly resulting in a non-functional truncated protein (Supplementary Figure S1A-C). The vast majority of the remaining 20% amplicons revealed in-frame deletions at the target site, which are also likely to produce a non-functional protein due to a critical position within the membrane domain of GPRC5A. Although unmodified wild type (WT) sequences could be detected in less than 1% of the targeted amplicons, no or negligibly little full-length GPRC5A protein could be detected by Western blotting (Supplementary Fig. S2) suggesting that deletion of GPRC5A was essentially complete.
Then, we asked whether the knock-out of GPRC5A could affect the cellular adhesion to ECM. We first compared the adhesion to one matrix component, Collagen type I (Col I, 0.1 mg/ml), for knock-out cell lines and their respective controls. We found that both variants of GPRC5A knock-out MDA-MB-231 cell lines adhered to Col I 1.5 - 2 times less efficiently that control cells (Fig. 1A). The greatest difference between WT and knock-out cells was observed 30 minutes after seeding the cells on Col I matrix and persisted up to 90 minutes (Fig. 1A). Similarly to MDA-MB-231, all other tested cell lines except Calu-1 showed a reduced adhesion to Collagen type I upon efficient knock-out of GPRC5A within 30 min after plating (Fig. 1B). Independent experiments utilizing a transient siRNA-mediated gene knock-down in MDA-MB-231 cells indicated that the adhesion phenotype is associated only with GPRC5A, but not its paralogs GPRC5B, GPRC5C, and GPRC5D (Supplementary Figure S3A-B). Furthermore, a constitutive shRNA-mediated depletion of GPRC5A in MDA-MB-231 cells lead to about 1.7-fold reduction in adhesion to Col I, while overexpression of a GPRC5A-coding cDNA construct rescued the adhesion defect (Supplementary Figure S3C-D). In aggregate, the data indicate that the effect of GPRC5A on cell adhesion is specific and does not depend on the method of gene targeting.
Figure 1.
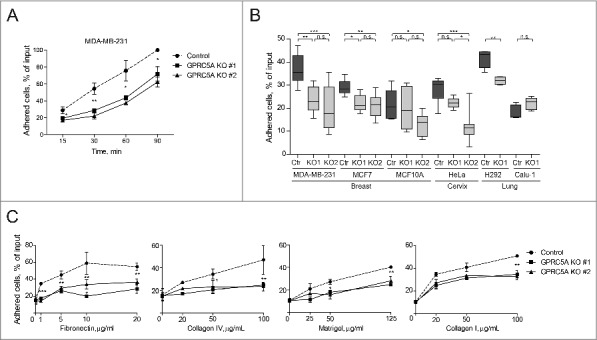
GPRC5A modulates cell adhesion to a range of ECM components in several cell lines. (A) Quantification of a cell adhesion assay demonstrating a time-dependent decrease in the ability of GPRC5A knockout (GPRC5A-KO) MDA-MB-231 cells to attach to Collagen I-coated (0.1 mg/mL) surface. (B) Most of the 6 cell lines of different origin show a reduced ability to attach to Collagen I-coated (0.1 mg/mL) surface within 30 min after plating upon GPRC5A knock-out. Black line in the box shows the mean. Whiskers show minimum and maximum values. (C) Cell adhesion assay showing the effect of GPRC5A knockout on the ability of MDA-MB-231 cells to attach to distinct ECM components in a dose-dependent manner. Error bars in (A) and (C) represent SEM from 3 independent experiments performed in 5 technical replicas each (N = 3, n = 5). Statistical significance was evaluated using ANOVA with Tukey post-hoc test: *, p < 0 .05; **, p < 0 .01; ***, p < 0 .001.
We further tested the ability of GPRC5A knock-out MDA-MB-231 cells to adhere to other defined ECM components constituting the normal basal lamina, such as fibronectin and Collagen type IV,32 or a laminin-rich tumor-derived ECM compound Matrigel. Interestingly, GPRC5A knock-out affected cell adhesion to all those ECM components in a dose-dependent manner (Fig. 1C). We observed the greatest difference between control and GPRC5A knock-out cells for Fibronectin, while adhesion defects for Collagen type IV and Matrigel were less pronounced (Fig. 1C). Nevertheless, even for Collagen IV and Matrigel the effect of GPRC5A knock-out on cell adhesion reached statistical significance at the highest concentration of the matrix proteins (Fig. 1C). Together, these observations indicate that GPRC5A modulates epithelial cell adhesion to a broad range of ECM components.
After the initial attachment to ECM, epithelial cells spread out by extending actin-driven lamellipodia and filopodia-like protrusions.5,9,13 Therefore, we tested whether the cell spreading required GPRC5A. For this purpose, we plated GPRC5A knock-out and control MDA-MB-231 cells on a Collagen I-coated surface and measured the cell spreading as a ratio between the growing total cell area and the mostly constant nucleus area at distinct time points. Consistent with changes in cell adhesion, the differences in cell spreading between control and GPRC5A knock-out cells became apparent already 15 minutes upon cell seeding (Fig. 2A, B). Thirty minutes after plating on Collagen I GPRC5A knock-out cells on average spread about 1.5 times less efficiently than control cells (Fig. 2A, B). The number of flattened cells (for which the total/nucleus area ratio was greater than 3) was about 30% less for GPRC5A knock-out cells compared with control (Fig. 2C), suggesting that GPRC5A is involved in cell spreading.
Figure 2.
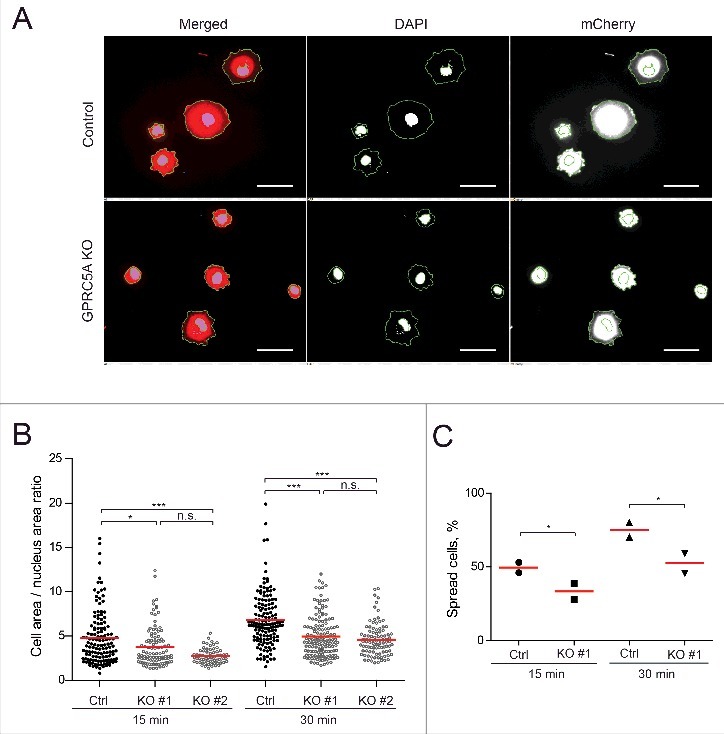
GPRC5A affects cell spreading. (A) GPRC5A-KO MDA-MB-231 cells demonstrate slower spreading on Collagen I-coated (0.1 mg/mL) surface compared with isogenic control. Representative images show nuclear (DAPI) and the whole cell area (mCherry) 30 min after plating. Scale bars correspond to 100 µm. (B) At least 60 cells were quantified for each sample and plotted as the nucleus / whole cell area ratios 15 or 30 min after plating for 2 independent experiments with 2 technical replicas in each (N = 2, n = 2). Red lines represent mean values. (C) The number of flattened cells (for which the total/nucleus area ratio was greater than 3) was smaller for GPRC5A knock-out cells (KO) compared with control (Ctrl). Statistical significance was evaluated using ANOVA with Tukey post-hoc test: *, p < 0 .05; **, p < 0 .01; ***, p < 0 .001.
Cell adhesion to ECM is tightly linked with epithelial cells' ability to migrate and invade the matrix, which, in turn, is an important feature of the malignant transformation13,14 Therefore, we questioned whether, along with cell adhesion, depletion of GPRC5A also affected cell migration. We tested the performance of serum-starved WT and GPRC5A-KO MDA-MB-231 cells in an imaging-based gradient-directed migration assay using collagen-coated ClearView Plates. Somewhat surprisingly, we found that GPRC5A knock-out MDA-MB-231 cells did not show any difference in migration toward serum compared with control cells (Supplementary Figure S4A). However, GPRC5A knock-out HeLa cells did show a moderate increase in cell migration in the same assay (Supplementary Figure S4B). This apparent inconsistency suggests that GPRC5A may affect the gradient-directed cell migration but the underlying mechanism is not tightly linked to the role of GPRC5A in cell adhesion.
GPRC5A knock-out cells demonstrate deregulated expression of integrin β1
Integrin receptors represent one of the principal molecule classes mediating adhesion to ECM.5,6,13 Within the integrin family, β1 integrin is involved in adhesion to a wide range of matrix molecules, including collagens, laminins, fibronectin, and vitronectin, while different α-integrins are more restrictive in their ECM specificity.33-35 Therefore, as GPRC5A knock-out cells exhibited a reduced adhesion to a wide range of ECM proteins, we chose first to address the expression of integrin β1. We measured the amount of the total integrin β1 protein in MDA-MB-231 cells grown for 48 h on the Collagen I matrix. Western blot analysis revealed about 50% reduction of integrin β1 protein (Fig. 3A, B) in GPRC5A knock-out cells relative to control. We obtained a similar result when GPRC5A was constitutively inhibited by a small hairpin RNA (shGPRC5A; Supplementary Figure S5A-B). Moreover, overexpression of GPRC5A-coding cDNA in shGPRC5A-carrying MDA-MB-231 cells restored the expression of integrin β1 protein (Supplementary Figure S5A-B), further supporting specificity of the phenotype. Next, we complemented the expression analysis of integrin β1 by quantifying the amount of its mRNA transcript, and, surprisingly, found that the level of GPRC5A transcript was elevated in both GPRC5A knock-out variants (Fig. 3C). Although we did not investigate the mechanism any further, it is likely that the elevated expression of the integrin β1 transcript is a compensatory effect in response to the decrease in integrin β1 protein in GPRC5A knock-out cells. In contrast to integrin β1, we did not detect any effect of GPRC5A knock-out on the integrin α2 protein, a partner for integrin β1 forming an α2β1 receptor to collagen34,35 (data not shown), which is consistent with a wider specificity of the GPRC5A-mediated adhesion defect.
Figure 3.
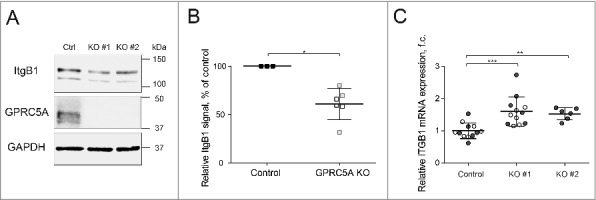
Knock-out of GPRC5A results in reduced β1 integrin protein expression. (A) Western blot demonstrating a reduction in the total integrin β1 (ItgB1) protein in GPRC5A knock-out MDA-MB-231 cells grown on Collagen I matrix (0.1 mg/mL) for 48 h. KO#1 and KO#2 refer to 2 independent small guide RNAs used for a CRISPR/Cas9-mediated GPRC5A knockout. (B) Quantification of integrin β1 protein from 3 independent experiments like the one shown in (A). Black lines show mean values. Error bars represent SEM. Statistical significance was evaluated using the Mann-Whitney non-parametric test. (C) RT-qPCR results demonstrate that, in contrast to the protein, the amount of the ItgB1 transcript is increased in GPRC5A knock-out cells. Statistical significance was evaluated using ANOVA with Tukey post-hoc test. *, p < 0 .05; **, p < 0 .01; ***, p < 0 .001.
We also tested whether GPRC5A protein associated with integrin β1 in focal adhesion sites. We co-stained GPRC5A and markers of the focal adhesion sites Vinculin and Paxillin in MDA-MB-231 cells fixed 30 min or 6 h after plating on Matrigel. We found GPRC5A to reside in what appeared as intracellular vesicles and at the plasma membrane, where it was particularly abundant in F-actin-containing membrane protrusions (Supplementary Fig. S6). However, GPRC5A did not co-localize with Vinculin and Paxillin in these cells (Supplementary Figure S6 and data not shown) suggesting that GPRC5A is not directly involved in the focal adhesions' architecture. Altogether, our data suggest that GPRC5A knock-out negatively affects integrin β1 protein expression, consistent with a decrease in adhesion in GPRC5A knock-out cells.
Knock-out of GPRC5A compromises activation of FAK/Src signaling during cell adhesion
Integrin-mediated binding to ECM leads to activation of the focal adhesion kinase (FAK), which, in turn, regulates the assembly of focal adhesions and remodeling of the local cytoskeleton facilitating cell spreading.5,9,14 As knock-out of GPRC5A was associated with a decrease in integrin β1, we tested whether it also affected FAK signaling. First, we collected protein lysates of MDA-MB-231 cells at different time-points after plating on Collagen I (between 0 – 90 min) and analyzed them by Western blotting. While the total FAK protein level remained unchanged in control and GPRC5A knock-out cells throughout the experiment, the analysis revealed differences in the level of phosphorylated FAK. The amount of FAK phosphorylated at Tyr397 (pY397) peaked 30 minutes after cell seeding, at which point it was about 50% lower in GPRC5A knock-out cells compared with control (Fig. 4A, B). FAK phosphorylation at the Y397 residue reflects its initial autophosphorylation upon integrin ligation8,11 and its reduction is fully consistent with reduced levels of integrin β1 in GPRC5A knock-out cells.
Figure 4.
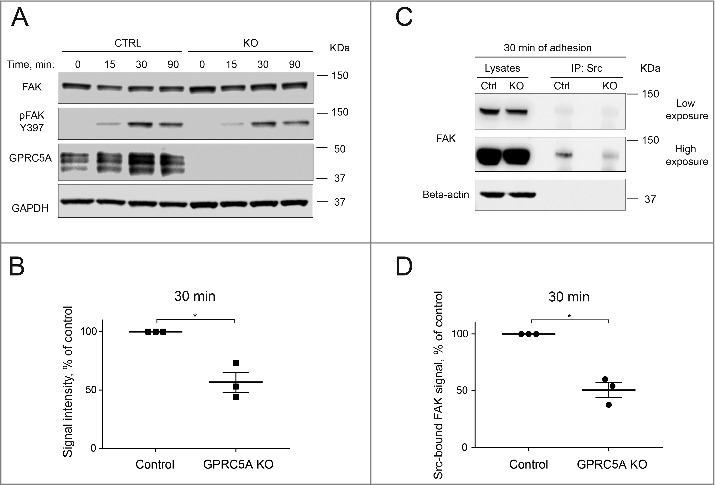
Knock-out of GPRC5A results in impaired activation of FAK in adhering cells. (A) A representative Western blot demonstrating FAK phosphorylation dynamics in MDA-MB-231 cells at different time points after plating on Collagen I (0.1 mg/mL). (B) Quantification of the Western blot shown in (A) for 30 min after plating. Phosphorylated FAK signal was normalized to the loading control (GAPDH) and total FAK signal. Data represent the mean ± SEM, N = 3. Statistical significance was evaluated using one-way ANOVA test. *, p < 0 .05. (C) Representative Western blot demonstrating the amount of FAK co-immunoprecipitated with Src in control and GPRC5A knock-out MDA-MB-231 cells 30 minutes after plating on Collagen I (0.1 mg/mL). (D) Quantification of the Western blot shown in (C). Src-co-immunoprecipitated FAK signal was normalized to loading control and total FAK signal of the input samples. Data represent means ± SEM, N = 3. Statistical significance was evaluated using one-way ANOVA test. *, p < 0 .05.
Y397-autophosphorylated FAK interacts with Src family kinases to form a signaling complex facilitating a full activation of FAK itself, and transmitting the signal further to downstream mediators of adhesion and spreading.9 In order to assess whether the reduced FAK phosphorylation affected the efficiency of FAK-Src interaction in GPRC5A knock-out cells, we performed a Src pull-down assay in control and GPRC5A knock-out MDA-MB-231 cells 30 min upon plating on Collagen I. We found that the amount of FAK co-immunoprecipitated with Src was about 2 times lower in GPRC5A knock-out cells compared with control (Fig. 4C, D), suggesting that GPRC5A is required for both proper activation of FAK and formation of its signaling complex with Src during the initial adhesion to ECM.
GPRC5A knock-out cells demonstrate reduced activity of Rho GTPases during adhesion
Among the downstream targets of FAK/Src signaling, Rho GTPases play a critical role in regulation of cytoskeleton rearrangements required for cell-substrate adhesion.14,36 We hypothesized that, if FAK activation was impaired, the function of Rho GTPases might also be perturbed in adhering GPRC5A-KO cells. To test this, we used a small G-protein pull-down activation assay to measure the amount of active GTP-bound Rac1, RhoA and cdc42 GTPases in control and GPRC5A knock-out MDA-MB-231 cells 30 minutes after plating on Collagen I-coated surface. We found that the normalized GTP-bound RhoA and Rac1 signal intensities decreased about 5- and 2-fold in GPRC5A knock-out cells relative to control, respectively (Fig. 5A, B), indicating a reduction in the activity of RhoA and Rac1. In contrast, the difference in cdc42 GTPase activity between control and GPRC5A knock-out MDA-MB-231 cells was insignificant (Fig. 5A, B). Therefore, at least during adhesion to Collagen I, GPRC5A seems to affect primarily the activity of RhoA GTPase and, to a lesser extent, of Rac1, but not Cdc42, which is consistent with reduced FAK activation and impaired adhesion to matrix.
Figure 5.
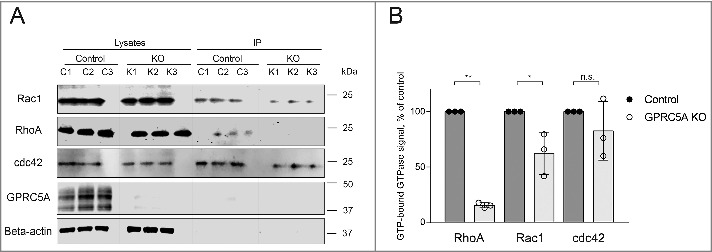
Knock-out of GPRC5A modulates the activity of RhoA and Rac1 GTPases. (A) Western blot demonstrating the amount of RhoA, Rac1, and cdc42 GTPases immunoprecipitated in GTP-bound state from the cells collected 30 minutes after plating on Collagen I (0.1 mg/mL). Three control (C1-3) and 3 knock-out (K1-3) samples were collected independently (N = 3). (B) Quantification of the experiment shown in (A). The signal from precipitated GTP-bound GTPases was normalized to the total amount of a corresponding GTPase protein and the loading control, and presented as % of control. Bars represent mean ± SEM, N = 3. Statistical significance was evaluated using one-way ANOVA test. *, p < 0 .05.
GPRC5A binds EphA2, a regulator of FAK activity
Our phenotypic observations so far suggested that epithelial cells require GPRC5A for adhesion to ECM. Therefore, we aimed to find possible molecular partners, which might link GPRC5A to integrin/FAK/Src/Rho adhesion signaling axes. We performed a Mass Spectrometry-based search for adhesion signaling molecules whose phosphorylation was altered in different matrix adhesion conditions in GPRC5A knock-out cells. We collected protein lysates from control and GPRC5A knock-out MDA-MB-231 cells 30 minutes or 24 h upon plating on Collagen I, and performed immunoprecipitation using anti-phospho-tyrosine or non-immune IgG (negative control) antibodies. The analysis identified 5 proteins possibly affected by GPRC5A knock-out (Supplementary Figure S8A-B), including FAK kinase. One of these proteins was receptor tyrosine kinase EphA2, previously shown to regulate FAK activation and matrix interactions in epithelial cancer cells.16,17,37,38 Therefore, we tested whether GPRC5A and EphA2 might physically interact. We performed a protein pull-down using anti-GPRC5A antibody in the lysates of Collagen I-attached MDA-MB-231 cells. Indeed, we detected a specific EphA2 band in GPRC5A immunoprecipitates from MDA-MB-231 cell line (Fig. 6 and Supplementary Figure S7 show 2 independent experiments), suggesting a novel interacting partner for GPRC5A. Notably, several human cancers reveal a correlation between expression of GPRC5A and EPHA2 (Supplementary Figure S9A-C). This is the first evidence to suggest a direct interaction between GPRC5A and EphA2, which might provide a mechanistic link between GPRC5A and the integrin/FAK/Src/Rho adhesion signaling axes and help to understand the role of GPRC5A in epithelial cancers. However, this will be a subject of a separate study.
Figure 6.
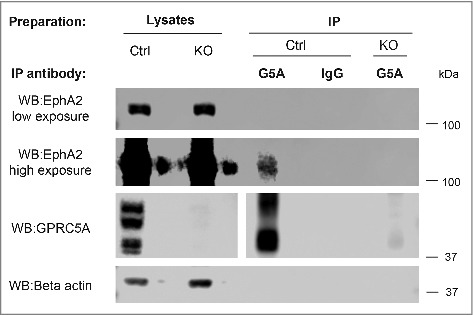
GPRC5A interacts with EphA2 tyrosine kinase. MDA-MB-231 cells were cultured on Collagen I – coated surface for 48 h. Proteins from MDA-MB-231 control (Ctrl) or GPRC5A knockout (KO) cells were immunoprecipitated. (IP) using either rabbit anti-GPRC5A polyclonal antibodies (G5A), or rabbit non-immune IgG (IgG) as a control as described in Materials and Methods. Both protein lysates and eluted protein complexes were separated on the same SDS-PAGE gel and analyzed by Western blotting (WB).
Discussion
Using several independent approaches, such as CRISPR/Cas9-mediated genetic knock-out and a transient or constitutive siRNA- or shRNA-mediated gene knock-down, respectively, we demonstrate for the first time that GPRC5A modulates epithelial cell adhesion to a range of defined components of ECM. The effect on cell adhesion was confined only to GPRC5A, but not other members of this GPCR family, and reproduced in 3 different breast epithelial and one cervical carcinoma cell lines. Interestingly, 2 lung cancer cell lines that we tested revealed either a minimal or no significant difference between GPRC5A isogenic knock-out and control lines suggesting tissue-specific effects for GPRC5A. Indeed, GPRC5A seems to play a unique role in the lung, as its high expression in the normal lung epithelia39,40 is required to suppress lung carcinogenesis by several mechanisms.41-47 In contrast, GPRC5A is rather weakly expressed in the normal breast and several other epithelial tissues, but becomes overexpressed in respective cancers(30,48 and reviewed in ref. 26). It remains to be elucidated whether GPRC5A has tissue-specific roles in cell-matrix adhesion, and whether it is relevant to progression of any cancer type.
At the molecular level, GPRC5A deficiency was associated with a weaker expression of β1 integrin, reduced activating phosphorylation of FAK at Y397 residue, compromised binding between FAK and Src kinases, and diminished GTPase activity of Rac1 and RhoA, but not cdc42. All these molecules are well known mediators of cell-ECM adhesion and, together, may well explain the observed adhesion defect of GPRC5A-deficient cells. Among them, β1 integrin arguably serves as the key target of the GPRC5A-mediated modulation of adhesion . However, we failed to detect a direct interaction between GPRC5A and integrin β1 in pull-down experiments (data not shown) suggesting that GPRC5A could affect integrin β1 rather indirectly. We also found no evidence that the decrease in integrin β1 protein might be associated with an enhanced degradation by proteasomes, lysosomes, or cytoplasmic proteases (data not shown). On the other hand, the fact that, in contrast to the protein, the ITGB1 transcript was moderately increased in GPRC5A-deficient cells makes it possible that GPRC5A regulates integrin β1 at the level of mRNA translation. Indeed, a very recent study by Wang and colleagues elegantly demonstrates an unexpected direct link between GPRC5A and the translation initiation complex.45 The study shows that GPRC5A directly binds the eIF4F complex and thereby suppresses the translation of membrane-bound proteins.45
Our finding that EphA2 receptor tyrosine kinase can directly interact with GPRC5A suggests yet another possible mechanism linking GPRC5A with the integrin/FAK/Src/Rho pathway. EphA2 is known to trigger a temporary loss of FAK phosphorylation.16,37 Depending on the cellular and genetic context, EphA2 may also promote or suppress RhoA activity during cell-cell and cell-matrix interactions.49-52 In line with our finding, another study using a mass spectroscopy analysis found both GPRC5A and EphA2 enriched in the substrate-attached membrane material,53 suggesting that GPRC5A and EphA2 may localize in the same biologically functional domains on a plasma membrane. Nevertheless, the potential mode of GPRC5A-EphA2 interaction and its functional consequences remains unclear. However, it was shown that interaction between GPRC5A and RTKs can modulate their signaling.47,54 Moreover, the observed correlation between GPRC5A and EphA2 expression in several epithelial tumors (Supplementary Figure S9A-C) suggests their joint role in cancer. Indeed, a critical role for EphA2 in epithelial tumor metastases and invasiveness is well-studied.38,49,55 Our data suggest that GPRC5A may be involved in tumor progression due to its role in cell-matrix adhesion and, possibly, in association with EphA2 receptor.
Materials and methods
Cell culture
All cell lines were obtained from the ATCC. HeLa, MCF7, and MDA-MB-231 cell lines were propagated in DMEM medium (Lonza) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 1x PenStrep antibiotic supplement (all from Life Technologies) at 37°C in the 5% CO2 atmosphere. MCF10A cells were cultured in DMEM/F12 medium (Life Technologies) supplemented with the 5% horse serum, 20 ng/ml EGF, 0.5 µg/ml hydrocortisone, 0.1 µg/ml cholera toxin, 10 µg/ml insulin (all from Sigma Aldrich) and 1×Penicillin-Streptomycin (Life Technologies) . NCI-H292 and Calu-1 were cultured in RPMI medium, supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 1x PenStrep antibiotic supplement.
CRISPR/Cas9-mediated GPRC5A knock-out
Oligonucleotides encoding guide RNAs were purchased from SigmaAldrich. Lentiviral vectors for sgRNA expression were generated by cloning oligonucleotides encoding sgRNA into LentiGuide plasmid (#52963, Addgene) as described.56 293T cells were transfected with LentiGuide or LentiCas9 lentiviral plasmids and packaging plasmids pCMV-VSV-G (#8454, Addgene) and pCMV-dR8.2 (#8455, Addgene)57 using Lipofectamine 2000 transfection reagent (Life Technologies). Supernatants were collected on the second day after transfection. For infection, cells were seeded at a density of 5*104 cells/cm2 in 24 well plates, culture media was changed 2 hours after seeding to the medium containing lentiviral particles (MOI=5) and 8 μg/ml polybrene. Next day, media was replaced for Blasticidine (LentiCas9; 5-12 μg/ml) or Puromycine (LentiGuide; 1-2 μg/ml) containing media and cells were selected for 7 and 4 d respectively.
Next generation sequencing
GPRC5A target region was amplified using Phusion DNA polymerase (Thermo Fisher Scientific) and G5A-NGS-F1 and G5A-NGS-R1 primers (Supplementary Table ST2). After the initial denaturation at 98°C for 5 min, 34 polymerase chain reaction (PCR) cycles were performed as follows 98°C for 30 s, 64°C for 30 s, and 72°C for 30 s, and completed with a final elongation at 72°C for 10 min. PCR products were purified using Gel and PCR purification kit (Macherey-Nagel). A second round of PCR amplification was performed to introduce Illumina sequencing adaptors using P5 index X and P7 index X primers (Supplementary Table ST2) and the following cycling conditions (denaturation at 98°C for 30s, 30 cycles at 98°C x 10 s, 59°C x 30 s, 72°C x 15 s, followed by elongation at 72°C for 10 min. Amplification products were purified from a 2% agarose gel as above and sequenced using Illumina MiSeq (Illumina).
RNA interference
siRNAs targeting human GPRC5A (siGPRC5A_2 against target sequence 5′-GAGGCTAAAGATCACCCTAAA-3′, cat# SI00058604; and siGPRC5A_5 targeting sequence 5′-CAACTCAAGTTTAGACCCTTA-3′, cat# SI02225734), GPRC5B (siGPRC5B_2 against target sequence 5′-CGCAAACTAAAGCAAAGCTAA-3′, cat# SI02642066; and siGPRC5B_5 targeting sequence 5′-CTCGCCCTGTTCCTACACTTA-3′, cat# SI00114639), GPRC5C (siGPRC5C_3 against target sequence 5′-GAGCATGTTCGTGGAGAACAA-3′, cat# SI00122542; and siGPRC5C_4 targeting sequence 5′-CCTGGTAGAGGTCATCATCAA-3′, cat# SI00122556), GPRC5D (siGPRC5D_5 against target sequence 5′-CAGAGGTATGATGTTTGTGAA-3′, cat# SI02643389; and siGPRC5D_6 targeting sequence 5′-ATCATCGAGCTCAATCAACAA-3′, cat# SI03047184), and non-targeting control siRNA (cat# Ctrl_AllStars_1, target sequence not disclosed) were purchased from Qiagen. MDA-MB-231 cells were transfected at 20 nM final siRNA concentration using Lipofectamin RNAiMAX reagent (Life Technologies) according to manufacturer's instructions and as described elsewhere.58 Alternatively, a stable knockdown of GPRC5A was achieved via lentivirally transduced shRNA (clone TRCN0000005628, recognizing sequence 5′-GCCCTTAATCTTGCTGTTATT-3′ in the 3′-UTR of GPRC5A), obtained from the Biomedicum Helsinki Functional Genomics Unit . GPRC5A was stably overexpressed using a cDNA (RefSeq ID NM_003979.3) cloned into pLenti-C-Myc-DDK lentiviral vector (cat#RC200118L1, OriGene Tehcnologies).
Quantitative Reverse Transcription-PCR
Total RNA was isolated using NucleoSpin RNA II kit (Macherey-Nagel). 160 ng of total RNA were used as a template for cDNA synthesis using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Real-Time qPCR was carried out using the SYBR Green detection method on a Bio-Rad C1000 cycler (BioRad). Primer sequences are listed in the Supplementary Table ST3. Data were analyzed with the CFX Manager software (Bio-Rad). All experiments were repeated at least twice in triplicates.
Cell adhesion assay
Adhesion assay was performed as described in.59 96-well plates (Thermo Fischer Scientific) were coated either with a thin layer of Matrigel diluted with a serum-free medium (BD Bioscience), collagen type I (Life Technologies), Fibronectin, Collagen type IV (both from Sigma-Aldrich) at indicated concentrations, or 1% bovine serum albumin (BSA) in phosphate buffered saline (PBS) (negative control), or left uncoated. Cells were starved for 24 h without serum, trypsinized, washed twice with serum-free medium, and 2.5x104 cells/well were resuspended in a serum-free medium and plated in 4 replicas for each condition. Cells were allowed to attach for 30 min (unless otherwise stated) at 37°C, then unattached cells were removed by washing 3 times with a serum-free medium. The remaining cells were fixed with 4% PFA for 20 min at room temperature. The cells were stained with 0.1% Crystal Violet for 30 min at room temperature, and then the plates were washed 10 times with PBS. Plates were air dried overnight, and the crystal violet was then extracted using 0.5% Triton-X100 in PBS for 30 min on a shaker. Absorbance at 540 nm was measured on PheraStar instrument (BioTek). Background absorbance (from blank wells) was subtracted from all test wells and the signal was normalized to the input (cells fixed 6 h after plating without washing). The assay was always performed in 5 technical and 3 biological replicas.
Cell spreading assay
MDA-MB-231 control and GPRC5A knock-out cells carrying mCherry fluorescent protein-coding construct were starved 24 h without serum, trypsinized and washed twice with serum-free medium 7.5x104 cells per 1 mL of a serum-free medium were added onto Collagen I – coated glass coverslips. Fifteen or 30 minutes after plating, cells were fixed with 2% paraformaldehyde at 37°C for 10 minutes. The fixative was washed away with PBS containing 20 mM Glycine, and the cells were counterstained with DAPI to visualize nuclei. After a brief wash with PBS, the coverslips were mounted in ProLong Gold Antifade mounting medium (Invitrogen), and imaged using Nikon Eclipse 90i epifluorescence microscope (Nikon) equipped with DS-Fi1 camera. Images were processed with the Nikon NIS Elements AR software using automatic object mask detection for mCherry and DAPI channels to measure cell area and nucleus area, respectively. Over sixty cells were quantified for each sample. The experiment was performed in 2 technical and 2 biological replicas.
Migration assay
A gradient-directed cell migration assay was performed according to the Incucyte™ chemotaxis cell migration assay protocol for adherent cells” using IncuCyte™ ClearView 96-Well Cell Migration Plates (Essen BioSciences) according to manufacturer's instructions. Briefly, cells were starved for 24 h without serum, trypsinized, washed, and resuspended in a serum-free DMEM. 103 cells resuspended in 60 µl serum-free DMEM were placed in the upper chambers of a ClearView plate, while the lower chambers were filled with 0.75 ml of either serum-free DMEM (control), or DMEM containing 5% FBS. The cells were allowed to migrate toward the serum gradient for 72 hours at 37°C, and imaged and automatically quantified every 4 h using IncuCyte ZOOM scanner (Essen BioSciences). Each experiment was performed in 4 replicas.
Immunoblotting
For Western blotting analysis, cells were lysed on ice in the RIPA buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1% sodium dodecylsulfate, 4 mM EDTA, 50 mM NaF, 1 mM sodium orthovanadate, Pierce protease inhibitor cocktail (Thermo Fisher Scientific). Lysates were centrifuged for 10 min at 16000 g at 4C to eliminate cell debris. Proteins were separated by SDS-PAGE and transferred to a Hybond-C nitrocellulose membrane (GE Healthcare). Membranes were blocked with 5% non-fat milk in Tris buffered saline (TBS) buffer containing 0.05% Tween-20, and incubated with appropriate primary antibodies overnight at +4°C. The following primary antibodies have been used: rabbit anti-integrin β1 (#EP1041Y, Abcam, diluted 1:2500); rabbit anti-GPRC5A (#HPA007928, Sigma Aldrich, diluted 1:1000); rabbit anti-FAK antibody sampler kit (#9330, Cell Signaling Technologies, 1:1000); rabbit anti-Rho GTPases antibody sampler kit (#9968, Cell Signaling Technologies, 1:1000); mouse anti-β actin (A2228, Sigma-Aldrich, 1:2000); goat anti-EphA2 (AF3035, R&D Systems, 1:2000); and mouse anti-GAPDH (#NB300-328, Novus Biologicals, 1:5000). Secondary antibodies were: goat anti-rabbit IRDye680RD, donkey anti-goat IRDye800RD, and goat anti-mouse IRDye800RD conjugates were from LI-COR Biosciences; goat anti-mouse horserad ish peroxidase (HRP) (ab6789) and goat anti-rabbit HRP (ab6721) conjugates were from Abcam. Incubation with secondary antibodies diluted 1:10000 was carried out for 1 h at room temperature. Bound immunocomplexes were detected using LI-COR Odyssey infrared fluorescence and chemiluminescence scanner (LI-COR Biosciences). Band intensities were quantified using Image Studio software (version 5.2., LI-COR Biosciences).
Immunoprecipitation assay
For immunoprecipitation assays, cells were lysed using non-denaturing lysis buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% sodium deoxycholate, 4 mM EDTA, 5 mM β-glycerophosphate, 50 mM NaF, 10 mM sodium pervanadate, Pierce protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). 500 µg of total protein lysate was incubated with 5 µg of either rabbit anti-GPRC5A (#HPA007928, Sigma Aldrich), or non-immune rabbit IgG, or 1µg of Anti-Phosphotyrosine Antibody, clone 4G10 (#05-321, EMD Millipore) for 3 h at 4°C, followed by precipitation using 100 µl of SureBeads protein G magnetic beads (BioRad) at 4°C overnight. Beads-bound immunocomplexes were washed 3 times with TBS-0.05% tween. Bound proteins were then eluted using Glycine-HCl buffer, pH 2.0 and analyzed by immunoblotting as described above.
Rho GTPase activation assay
MDA-MB-231 cells were starved overnight, trypsinized for 5 min, washed twice in serum-free DMEM, and 107 cells were plated on 15 cm dishes coated with 0.1 mg/mL Collagen I (Corning). In 30 min, the cells were lysed and 400 μg total protein per sample was processed using the RhoA / Rac1 / Cdc42 Activation Assay Combo Biochem Kit (Cytoskeleton Inc.) according to manufacturer's instructions. Twenty ng His-tagged recombinant RhoA, Rac1, or Cdc42 proteins were used as positive controls. GTP-bound RhoA, Rac1, and cdc42 were detected using primary antibodies provided with the kit, followed by 1 h incubation with secondary HRP-conjugated goat anti-mouse IgG (ab6789, Abcam) diluted 1:5000. Detection was performed using the SuperSignal Pierce ECL substrate (Thermo Fischer Scientific). Blot images were quantified using Image Studio software (version 5.2., LI-COR Biosciences). The assay was performed in 3 independent biological replicas.
Liquid chromatography–mass spectrometry (LC-MS/MS) proteomics
Eluted immunocomplexes were separated on Criterion BisTris 4-12% gel with MOPS running buffer (Bio-Rad) and silver stained. Selected protein bands were cut out from the gel and in-gel digested as described earlier.60 The LC-ESI-MS/MS analyses were performed on a nanoflow HPLC system coupled to the QExactive mass spectrometer (Thermo Fisher Scientific). Peptides were first loaded on a trapping column and subsequently separated inline on a 15 cm C18 column. The mobile phase consisted of water with 0.1% formic acid (solvent A) and acetonitrile/water (80:20 (v/v)) with 0.1% formic acid (solvent B). A linear 10 min gradient from 5% to 48% B was used to elute peptides. Proteome Discoverer 1.4 software (Thermo Fisher Scientific) connected to an in-house server running the Mascot 2.4.1 software (Matrix Science) was used for protein database searches against the SwissProt database (release 2016_01). The database search settings included a taxonomy filer ‘human’ and trypsin as an enzyme. One missed cleavage was allowed. A significance threshold of p < 0 .05 was used.
Immunofluorescence
Cells were allowed to attach to coverslips coated with 0.1 mg/mL Collagen I or 5% Matrigel (BD Bioscience) for 30 min or 6 h and then fixed with 4% paraformaldehyde (Sigma Aldrich), washed with PBS and parmeabilized with 0.25% Triton-X100 in PBS. Antibody-blocking buffer contained PBS, 0.5% BSA, 0.15% glycine and 0.1% Triton X-100. Primary antibodies purchased from Sigma Aldrich were: rabbit anti-GPRC5A (cat# HPA007928, diluted 1:1000), and mouse anti-vinculin (cat# V9131, 1:500). Secondary antibodies were goat anti-rabbit Alexa Fluor 555 (cat# A-21428) and goat anti-mouse Alexa Fluor 647 (cat# A-21235), diluted 1:1000 (Life Technologies). Samples were incubated for 5 min with Alexa Fluor 488-phalloidin (cat# A-12379, Life Technologies) diluted 1:50 in PBS, 0.05% Triton X-100 to visualize F-actin. Nuclei were counterstained with Hoechst at 0.5 μg/ml. Images were taken using Nikon Eclipse 90i epifluorescence microscope (Nikon) equipped with DS-Fi1 camera and processed using the Nikon NIS Elements AR software.
Statistical methods
Statistical analysis was performed using the GraphPad Prism software (version 6.0.7., GraphPad Software Inc.). Non-parametric Mann-Whitney test and one-way ANOVA were used to compare only 2 data series. Analysis of 3 and more data series was carried out using one- or 2-way ANOVA with the Tukey post-hoc testing for multiple comparisons. P value ≤ 0.05 was considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr. Jyrki Kukkonen (University of Helsinki) for helpful discussions; to Dr. Johanna Ivaska (University of Turku) for providing critical comments on the manuscript.
Funding
Financial support for the project was provided to S.G.K. by the Academy of Finland (grant 253862), Finnish Medical Foundation, Sigrid Juselius Foundation, Cancer Society of Finland, and Biocenter Finland. D.R.B. was supported by a fellowship from the Integrative Life Science Doctoral Program of the University of Helsinki and personal grants from the K. Albin Johanssons Stiftelse and Biomedicum Helsinki Foundation. D.L. was supported by the Drug Research Doctoral Program (DRDP, University of Turku) and the Finnish Cultural Foundation. T.A. was supported by the Academy of Finland (grants 272437, 269862, 279163) and Cancer Society of Finland
Author contributions
D.R.B. and S.G.K. designed the study. D.R.B. performed the experiments. Y.A.A. generated CRISPR/Cas9 knock out cell lines. A.R. and P.K. performed Mass Spectrometry analysis. T.D.L. performed and T.A. supervised the data analysis. T.P. supervised adhesion experiments and shared key reagents. D.B. and S.G.K. wrote the manuscript. S.G.K. supervised the project. All authors reviewed the manuscript.
References
- [1].Mescher A, Mescher A. Junqueira's Basic Histology: Text and Atlas. 12th ed. 2009 [Google Scholar]
- [2].Makrilia N, Kollias A, Manolopoulos L, Syrigos K. Cell adhesion molecules: role and clinical significance in cancer. Cancer Invest 2009; 27:1023-37; PMID:19909018; https://doi.org/ 10.3109/07357900902769749 [DOI] [PubMed] [Google Scholar]
- [3].Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion – and cell survival. Cancer Metastasis Rev 2005; 24:425-39; PMID:16258730; https://doi.org/ 10.1007/s10555-005-5134-3 [DOI] [PubMed] [Google Scholar]
- [4].Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010; 10:9-22; PMID:20029421; https://doi.org/ 10.1038/nrc2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol 2007; 213:565-73; PMID:17680633; https://doi.org/ 10.1002/jcp.21237 [DOI] [PubMed] [Google Scholar]
- [6].Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002; 110:673-87; PMID:12297042; https://doi.org/ 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- [7].Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol 2009; 10:21-33; PMID:19197329; https://doi.org/ 10.1038/nrm2593 [DOI] [PubMed] [Google Scholar]
- [8].Hamadi A, Bouali M, Dontenwill M, Stoeckel H, Takeda K, Ronde P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J Cell Sci 2005; 118:4415-25; PMID:16159962; https://doi.org/ 10.1242/jcs.02565 [DOI] [PubMed] [Google Scholar]
- [9].Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 2006; 18:516-23; PMID:16919435; https://doi.org/ 10.1016/j.ceb.2006.08.011 [DOI] [PubMed] [Google Scholar]
- [10].Hamadi A, Deramaudt TB, Takeda K, Ronde P. Hyperphosphorylated FAK Delocalizes from Focal Adhesions to Membrane Ruffles. J Oncol 2010; 2010:pii: 932803; PMID:20847951; https://doi.org/ 10.1155/2010/932803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol 1995; 15:954-63; PMID:7529876; https://doi.org/ 10.1128/MCB.15.2.954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tomakidi P, Schulz S, Proksch S, Weber W, Steinberg T. Focal adhesion kinase (FAK) perspectives in mechanobiology: implications for cell behaviour. Cell Tissue Res 2014; 357:515-26; PMID:24988914; https://doi.org/ 10.1007/s00441-014-1945-2 [DOI] [PubMed] [Google Scholar]
- [13].Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 2010; 11:633-43; PMID:20729930; https://doi.org/ 10.1038/nrm2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci 2009; 122:1059-69; PMID:19339545; https://doi.org/ 10.1242/jcs.039446 [DOI] [PubMed] [Google Scholar]
- [15].Marcoux N, Vuori K. EGF receptor mediates adhesion-dependent activation of the Rac GTPase: a role for phosphatidylinositol 3-kinase and Vav2. Oncogene 2003; 22:6100-6; PMID:12955089; https://doi.org/ 10.1038/sj.onc.1206712 [DOI] [PubMed] [Google Scholar]
- [16].Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol 2000; 2:62-9; PMID:10655584; https://doi.org/ 10.1038/35000008 [DOI] [PubMed] [Google Scholar]
- [17].Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G, Ramponi G, Chiarugi P. EphrinA1 activates a Src/focal adhesion kinase-mediated motility response leading to rho-dependent actino/myosin contractility. J Biol Chem 2007; 282:19619-28; PMID:17449913; https://doi.org/ 10.1074/jbc.M701319200 [DOI] [PubMed] [Google Scholar]
- [18].Tharmalingam S, Daulat AM, Antflick JE, Ahmed SM, Nemeth EF, Angers S, Conigrave AD, Hampson DR. Calcium-sensing receptor modulates cell adhesion and migration via integrins. J Biol Chem 2011; 286:40922-33; PMID:21969374; https://doi.org/ 10.1074/jbc.M111.265454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol 2012; 24:600-6; PMID:22980731; https://doi.org/ 10.1016/j.ceb.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pham H, Chen M, Takahashi H, King J, Reber HA, Hines OJ, Pandol S, Eibl G. Apigenin inhibits NNK-induced focal adhesion kinase activation in pancreatic cancer cells. Pancreas 2012; 41:1306-15; PMID:22889981; https://doi.org/ 10.1097/MPA.0b013e31824d64d9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Slack BE. Tyrosine phosphorylation of paxillin and focal adhesion kinase by activation of muscarinic m3 receptors is dependent on integrin engagement by the extracellular matrix. Proc Natl Acad Sci U S A 1998; 95:7281-6; PMID:9636140; https://doi.org/ 10.1073/pnas.95.13.7281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Valtcheva N, Primorac A, Jurisic G, Hollmen M, Detmar M. The orphan adhesion G protein-coupled receptor GPR97 regulates migration of lymphatic endothelial cells via the small GTPases RhoA and Cdc42. J Biol Chem 2013; 288:35736-48; PMID:24178298; https://doi.org/ 10.1074/jbc.M113.512954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yago T, Tsukamoto H, Liu Z, Wang Y, Thompson LF, McEver RP. Multi-Inhibitory Effects of A2A Adenosine Receptor Signaling on Neutrophil Adhesion Under Flow. J Immunol 2015; 195:3880-9; PMID:26355151; https://doi.org/ 10.4049/jimmunol.1500775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li HY, Cui XY, Wu W, Yu FY, Yao HR, Liu Q, Song EW, Chen JQ. Pyk2 and Src mediate signaling to CCL18-induced breast cancer metastasis. J Cell Biochem 2014; 115:596-603; PMID:24142406; https://doi.org/ 10.1002/jcb.24697 [DOI] [PubMed] [Google Scholar]
- [25].Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 2004; 23:7969-78; PMID:15489914; https://doi.org/ 10.1038/sj.onc.1208162 [DOI] [PubMed] [Google Scholar]
- [26].Zhou H, Rigoutsos I. The emerging roles of GPRC5A in diseases. Oncoscience 2014; 1:765-76; PMID:25621293; https://doi.org/ 10.18632/oncoscience.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dairkee SH, Sayeed A, Luciani G, Champion S, Meng Z, Jakkula LR, Feiler HS, Gray JW, Moore DH. Immutable functional attributes of histologic grade revealed by context-independent gene expression in primary breast cancer cells. Cancer Res 2009; 69:7826-34; PMID:19789341; https://doi.org/ 10.1158/0008-5472.CAN-09-1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu H, Zhang Y, Hao X, Kong F, Li X, Yu J, Jia Y. GPRC5A overexpression predicted advanced biological behaviors and poor prognosis in patients with gastric cancer. Tumour Biol 2015; 37:503-10; [DOI] [PubMed] [Google Scholar]
- [29].Zheng J, Guo X, Gao X, Liu H, Tu Y, Zhang Y. Overexpression of retinoic acid-induced protein 3 predicts poor prognosis for hepatocellular carcinoma. Clin Transl Oncol 2014; 16:57-63; PMID:23632812; https://doi.org/ 10.1007/s12094-013-1040-2 [DOI] [PubMed] [Google Scholar]
- [30].Nagahata T, Sato T, Tomura A, Onda M, Nishikawa K, Emi M. Identification of RAI3 as a therapeutic target for breast cancer. Endocr Relat Cancer 2005; 12:65-73; PMID:15788639; https://doi.org/ 10.1677/erc.1.00890 [DOI] [PubMed] [Google Scholar]
- [31].Hirano M, Zang L, Oka T, Ito Y, Shimada Y, Nishimura Y, Tanaka T. Novel reciprocal regulation of cAMP signaling and apoptosis by orphan G-protein-coupled receptor GPRC5A gene expression. Biochem Biophys Res Commun 2006; 351:185-91; PMID:17055459; https://doi.org/ 10.1016/j.bbrc.2006.10.016 [DOI] [PubMed] [Google Scholar]
- [32].Hynes RO. The extracellular matrix: not just pretty fibrils. Science (80- ) 2009; 326:1216-9; https://doi.org/ 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol 2015; 25:234-40; PMID:25572304; https://doi.org/ 10.1016/j.tcb.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Popova SN, Lundgren-Akerlund E, Wiig H, Gullberg D. Physiology and pathology of collagen receptors. Acta Physiol 2007; 190:179-87; https://doi.org/ 10.1111/j.1748-1716.2007.01718.x [DOI] [PubMed] [Google Scholar]
- [35].Brakebusch C, Fassler R. β 1 integrin function in vivo: adhesion, migration and more. Cancer Metastasis Rev 2005; 24:403-11; PMID:16258728; https://doi.org/ 10.1007/s10555-005-5132-5 [DOI] [PubMed] [Google Scholar]
- [36].Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008; 9:690-701; PMID:18719708; https://doi.org/ 10.1038/nrm2476 [DOI] [PubMed] [Google Scholar]
- [37].Faoro L, Singleton PA, Cervantes GM, Lennon FE, Choong NW, Kanteti R, Ferguson BD, Husain AN, Tretiakova MS, Ramnath N, et al.. EphA2 mutation in lung squamous cell carcinoma promotes increased cell survival, cell invasion, focal adhesions, and mammalian target of rapamycin activation. J Biol Chem 2010; 285:18575-85; PMID:20360610; https://doi.org/ 10.1074/jbc.M109.075085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sugiyama N, Gucciardo E, Lehti K. EphA2 bears plasticity to tumor invasion. Cell Cycle 2013; 12:2927-8; PMID:23974090; https://doi.org/ 10.4161/cc.26180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ye X, Tao Q, Wang Y, Cheng Y, Lotan R. Mechanisms underlying the induction of the putative human tumor suppressor GPRC5A by retinoic acid. Cancer Biol Ther 2009; 8:951-62; PMID:19279407; https://doi.org/ 10.4161/cbt.8.10.8244 [DOI] [PubMed] [Google Scholar]
- [40].Tao Q, Cheng Y, Clifford J, Lotan R. Characterization of the murine orphan G-protein-coupled receptor gene Rai3 and its regulation by retinoic acid. Genomics 2004; 83:270-80; PMID:14706456; https://doi.org/ 10.1016/S0888-7543(03)00237-4 [DOI] [PubMed] [Google Scholar]
- [41].Tao Q, Fujimoto J, Men T, Ye X, Deng J, Lacroix L, Clifford JL, Mao L, Van Pelt CS, Lee JJ, et al.. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl Cancer Inst 2007; 99:1668-82; PMID:18000218; https://doi.org/ 10.1093/jnci/djm208 [DOI] [PubMed] [Google Scholar]
- [42].Fujimoto J, Kadara H, Garcia MM, Kabbout M, Behrens C, Liu DD, Lee JJ, Solis LM, Kim ES, Kalhor N, et al.. G-protein coupled receptor family C, group 5, member A (GPRC5A) expression is decreased in the adjacent field and normal bronchial epithelia of patients with chronic obstructive pulmonary disease and non-small-cell lung cancer. J Thorac Oncol 2012; 7:1747-54; PMID:23154545; https://doi.org/ 10.1097/JTO.0b013e31826bb1ff [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kadara H, Fujimoto J, Men T, Ye X, Lotan D, Lee JS, Lotan R. A Gprc5a tumor suppressor loss of expression signature is conserved, prevalent, and associated with survival in human lung adenocarcinomas. Neoplasia 2010; 12:499-505; PMID:20563252; https://doi.org/ 10.1593/neo.10390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen Y, Deng J, Fujimoto J, Kadara H, Men T, Lotan D, Lotan R. Gprc5a deletion enhances the transformed phenotype in normal and malignant lung epithelial cells by eliciting persistent Stat3 signaling induced by autocrine leukemia inhibitory factor. Cancer Res 2010; 70:8917-26; PMID:20959490; https://doi.org/ 10.1158/0008-5472.CAN-10-0518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang J, Farris AB, Xu K, Wang P, Zhang X, Duong DM, Yi H, Shu HK, Sun SY, Wang Y. GPRC5A suppresses protein synthesis at the endoplasmic reticulum to prevent radiation-induced lung tumorigenesis. Nat Commun 2016; 7:11795; PMID:27273304; https://doi.org/ 10.1038/ncomms11795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Deng J, Fujimoto J, Ye XF, Men TY, Van Pelt CS, Chen YL, Lin XF, Kadara H, Tao Q, Lotan D, et al.. Knockout of the tumor suppressor gene Gprc5a in mice leads to NF-kappaB activation in airway epithelium and promotes lung inflammation and tumorigenesis. Cancer Prev Res 2010; 3:424-37; https://doi.org/ 10.1158/1940-6207.CAPR-10-0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Deng J, Zhong S, Yin H, Liao Y, Yao F, Li Q, Zhang J, Jiao H, Zhao Y, Xu D, et al.. Lung tumor suppressor GPRC5A binds EGFR and restrains its effector signaling. Cancer Res 2015. [DOI] [PubMed] [Google Scholar]
- [48].Jorissen H, Bektas N, Dahl E, Hartmann A, ten Haaf A, Di Fiore S, Kiefer H, Thess A, Barth S, Klockenbring T. Production and characterisation of monoclonal antibodies against RAI3 and its expression in human breast cancer. BMC Cancer 2009; 9:200; PMID:19552806; https://doi.org/ 10.1186/1471-2407-9-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Batson J, MacCarthy-Morrogh L, Archer A, Tanton H, Nobes CD. EphA receptors regulate prostate cancer cell dissemination through Vav2-RhoA mediated cell-cell repulsion. Biol Open 2014; 3:453-62; PMID:24795148; https://doi.org/ 10.1242/bio.20146601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wakayama Y, Miura K, Sabe H, Mochizuki N. EphrinA1-EphA2 signal induces compaction and polarization of Madin-Darby canine kidney cells by inactivating Ezrin through negative regulation of RhoA. J Biol Chem 2011; 286:44243-53; PMID:21979959; https://doi.org/ 10.1074/jbc.M111.267047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, Chen J. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J Cell Sci 2008; 121:358-68; PMID:18198190; https://doi.org/ 10.1242/jcs.017145 [DOI] [PubMed] [Google Scholar]
- [52].Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, Cates JM, Coffman K, Jackson D, Bruckheimer E, Muraoka-Cook RS, et al.. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest 2008; 118:64-78; PMID:18079969; https://doi.org/ 10.1172/JCI33154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yamada M, Mugnai G, Serada S, Yagi Y, Naka T, Sekiguchi K. Substrate-attached materials are enriched with tetraspanins and are analogous to the structures associated with rear-end retraction in migrating cells. Cell Adh Migr 2013; 7:304-14; PMID:23676281; https://doi.org/ 10.4161/cam.25041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lin X, Zhong S, Ye X, Liao Y, Yao F, Yang X, Sun B, Zhang J, Li Q, Gao Y, et al.. EGFR phosphorylates and inhibits lung tumor suppressor GPRC5A in lung cancer. Mol Cancer 2014; 13:233; PMID:25311788; https://doi.org/ 10.1186/1476-4598-13-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sugiyama N, Gucciardo E, Tatti O, Varjosalo M, Hyytiainen M, Gstaiger M, Lehti K. EphA2 cleavage by MT1-MMP triggers single cancer cell invasion via homotypic cell repulsion. J Cell Biol 2013; 201:467-84; PMID:23629968; https://doi.org/ 10.1083/jcb.201205176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014; 11:783-4; PMID:25075903; https://doi.org/ 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al.. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003; 9:493-501; PMID:12649500; https://doi.org/ 10.1261/rna.2192803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gu Y, Bouwman P, Greco D, Saarela J, Yadav B, Jonkers J, Kuznetsov SG. Suppression of BRCA1 sensitizes cells to proteasome inhibitors. Cell Death Dis 2014; 5:e1580; PMID:25522274; https://doi.org/ 10.1038/cddis.2014.537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Humphries MJ. Cell adhesion assays. Methods Mol Biol 2009. [cited 2016 Jul 25]; 522:203-10; PMID:19247616; https://doi.org/ 10.1007/978-1-59745-413-1_14 [DOI] [PubMed] [Google Scholar]
- [60].Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 1996. [cited 2016 Jul 25]; 68:850-8; PMID:8779443; https://doi.org/ 10.1021/ac950914h [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.