

Abstract
We report detailed characterizations of stochastic fluorescence switching of unmodified nucleic acids under visible light illumination. Although the fluorescent emission from nucleic acids under the visible light illumination has long been overlooked due to their apparent low absorption cross section, our quantitative characterizations reveal the high quantum yield and high photon count in individual fluorescence emission events of nucleic acids at physiological concentrations. Owing to these characteristics, the stochastic fluorescence switching of nucleic acids could be comparable to that of some of the most potent exogenous fluorescence probes for localization-based super-resolution imaging. Therefore, utilizing the principle of single-molecule photon-localization microscopy, native nucleic acids could be ideal candidates for optical label-free super-resolution imaging.
OCIS codes: (300.2530) Fluorescence, laser-induced; (260.5130) Photochemistry; (180.2520) Fluorescence microscopy; (100.6640) Superresolution
1. Introduction
Since being discovered, nucleic acids have been a central focus of studies in biological, physical, and chemical sciences. In biological systems, nucleic acids form highly complex and intricate structures that house, maintain, and regulate access to the genetic information critical for life. Increasingly, it has become apparent that the nanoscale topology of these structures has a prominent role in the regulation of essential cellular functions [1], such as gene transcription and replication. As such, directly visualizing these complex cellular systems will help expand our understanding of biological interactions, providing insight on gene regulation and cellular behavior. Recently, super-resolution fluorescence microscopy techniques, including stimulated emission depletion microscopy (STED), structured illumination microscopy (SIM), and photon localization microscopy (PLM), such as photoactivated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM), have extended the ultimate resolving power of optical microscopy far beyond the diffraction limit [1–7], facilitating access to the nanoscale organization of chromatin [8–11]. However, the majority of strategies used to image structures formed by nucleic acids require methods that label DNA-associated proteins instead of DNA itself or utilize small molecule dyes that may alter the structure and function of the native structures and affect cell viability [12,13].
Owing to these limitations, developing label-free optical super-resolution imaging methods to visualize DNA topology under native, non-perturbing conditions becomes attractive. It is well-known that nucleotides fluoresce under UV illumination and have significantly weaker absorption in the visible range. However, we have observed visible light-excited fluorescence of unmodified nucleic acids when their concentration approached the value of physiological conditions [14]. This intrinsic fluorescence has likely been overlooked previously because most photochemical studies of nucleotides were performed in dilute solutions with concentrations ranging between 10-100 μM [15–18], which is significantly lower than that in nuclei and chromosomes (0.1-1 M) [19–21]. More importantly, we further observed the stochastic fluorescence switching of nucleotides under visible illumination, which sets the foundation of using unmodified nucleic acids as endogenous contrast agents for nanoscopic imaging with photon localization microscopy (PLM) [14]. This phenomenon sets the stage for developing new label-free super-resolution optical imaging methods to resolve macromolecular structures with nucleotide topologies.
Here, we present the detailed photochemical characteristics of nucleotides at physiological concentrations under visible light illumination. To explain the mechanism of the observed stochastic fluorescence switching, we examined the fluorescence recovery of nucleotides under varying depletion conditions and demonstrated that these results fit well with the theory of ground state depletion (GSD) [6,7]. Furthermore, due to the relatively high quantum yield and low intersystem crossing probability of nucleotides, the photon count and blinking duration of individual emission events were found to be comparable to those of some of the most potent exogenous dyes used in PLM, making DNA molecules themselves ideal candidates as imaging contrasts in PLM.
2. Materials and methods
2.1. Chemicals and materials
All chemicals used in the photochemical measurements are commercially available for independent reproduction. Mononucleotide samples used in the measurements are HPLC grade (G8377, A1752, C1006, T7004) and were used as purchased from Sigma Aldrich. 2- and 4-base oligonucleotides were synthesized by Midland Science while 8-, 16- and 20-base oligonucleotides were synthesized by IDT. All synthetic oligonucleotides were used as purchased without further purification.
All samples used were derived both by extraction from natural synthesis (yeast, salmon) and through chemical synthesis (oligonucleotides from IDT and Midland). There is not a common denominator for their manufacture, other than their nucleic acid origin. Critically, these samples are for use as purity standards in analytical studies. For instance, the mononucleotides purchased from Sigma Aldrich are isolated from yeast and are used as the analytical standard for assessment of mononucleotide synthesis by mass spectrometry. In comparison, 20-base oligonucleotides are synthetically produced by phosphoramidite synthesis. The use of these synthetic oligonucleotides is for polymerase chain reactions. As a result, all of these molecules are of very high purity and produced or isolated by independent means for use as scientifically accepted standards.
For absorbance and fluorescence measurements, samples were dissolved in molecular biology grade nuclease-free distilled water (AM9938, Ambion, Invitrogen). Photochemistry measurements, including absorbance and fluorescence, were performed with commercial instruments in a 1-cm quartz cuvette (Z600717, Sigma). Baselines have been carefully measured with pure nuclease-free distilled water in the same cuvette following exactly the same preparation. Between each measurement, the cuvette was washed by acetone and methanol sequentially, triple rinsed with water, and then air dried. Finally, to account for any incidental contamination during the aliquoting of samples, all pipette tips [200 μL (37001-528, VWR) and 2,000 μL (83007-378, VWR)] used were from the same box without reuse.
2.2. LC-TOF analysis of mononucleotide samples
Mononucleotide samples used in the measurements are HPLC grade (the highest grade commercially available) and were used as purchased from Sigma Aldrich. While these materials have a certified purity greater than 99.9%, we understand the concerns that the observed fluorescent properties could be from organic impurities. To further exclude this possibility, we have performed liquid chromatography time-of-flight (LC-TOF) mass spectrometry on these mononucleotide samples. The mass spectra of the mononucleotides were measured using a high resolution electrospray ionization (HR-ESI) Agilent 6210 LC-TOF mass spectrometer with Agilent 1200 HPLC introduction. The results from these measurements show no indication of impurities compared to blank injections according to both 280nm UV absorbance and mass analysis for all nucleotides.
A similar, independent analysis was performed for thymidine monophosphate (T7004) by the Scripps Center for Metabolomics and can be found online at the METLIN metabolite database (https://metlin.scripps.edu/metabo_info.php?molid=3451). These results demonstrate that the mononucleotides have a purity exceeding 99.9%. Chemical analysis by mass spectrometry indicates the remaining molecules <0.1% are nucleotide metabolites (the fragments of nucleotides and the pentose monosaccharide ribose) that unavoidably appear in any nucleotide samples due to dissociation.
2.3. Size exclusion HPLC analysis of polynucleotide samples
The main impurities present in the synthetic oligonucleic acids are incompletely elongated strands (shorter sequences) and trace quantities of organic salts that are removed by desalting. Using HPLC, the nucleic acids account for 99.06% of the sample. The purity of these samples is measured by IDT and was confirmed using size exclusion high-performance liquid chromatography (HPLC) performed independently by the Northwestern University Keck Biophysics core. The size exclusion HPLC was coupled to a photo-diode array detectors (DAD) absorbance fluorimeter set to measure absorbance at 230 nm and 532 nm. HPLC grade Water (ThermoFisher) and molecular or HPLC grade methanol (>99.9%), ethanol (>99.45%), and acetic acid (>99.85%) were used during sample preparation as needed. Measurements were performed at high concentrations (4-5 mg/ml) to match our photochemistry experiments.
2.4. Absorbance spectra
In order to satisfy the sample quantity needed for fluorescence spectrophotometry, we chose commercially available mononucleotides (Sigma), including adenine (A), guanine (G), cytosine (C), and thymine (T), that were dissolved in nuclease free distilled water at a concentration of 0.1 M. Absorbance (A = − log10 T, where T is transmittance) were measured in 1-cm quartz cuvettes using a dual-beam UV-VIS Spectrophotometer (UV-1800, Shimadzu). A control (nuclease-free distilled water, Ambion, Invitrogen) prepared by exactly same procedure was placed in the reference beam to compensate for any environmental variation during the measurement. According to the instrument specification, the measurement accuracy is ±0.001 absorbance units (A.U.) at 0.5 A.U.
Due to the difficulty of acquiring large quantity of polynucleotide samples, the absorbance spectra of 100-μM poly-G nucleotide were measured in a 45-μL cuvette with path length of 3-mm (Z802336, Sigma) by using the same protocol used for mononucleotide solutions.
2.5. Fluorescence spectra and lifetimes
Fluorescence spectra of polynucleotides were measured by using a spectrofluorimeter (Nanolog, Horiba Jobin-Yvon) equipped with an iHR 320 spectrometer with a 100 g/mm grating at 600 nm, a 150-W xenon arc lamp and a picosecond photon-detection module (PPD-850). Due to the difficulty of acquiring large quantity of polynucleotide samples, fluorescence spectra of polynucleotides were measured in a 45-μL cuvette with path length of 3-mm.
Fluorescence lifetimes of mononucleotides were measured by the time-correlated single photon counting method as perviously reported in [22]. Mononucleotides illuminated at 532 nm have exceptionally longer fluorescence decays with respective lifetimes of 1.98±0.08 ns, 2.00±0.03 ns, 2.48±0.05 ns, and 2.56±0.07 ns for A, G, C, and T.
2.6. Absolute quantum yield measurement
To measure the absolute quantum yields (QYs) of mononucleotides, we used a Horiba Quanta-Phi F-3029 integrating sphere mounted in the sample compartment of the Nanolog (Horiba Jobin-Yvon) spectrofluorimeter. The measurements were performed at an illumination wavelength of 532 nm at room temperature. Data was processed by software supplied by Horiba-Jobin-Yvon, from which G and C have QYs of 0.0559±0.0072 and 0.1057±0.0107, respectively; A and T have somewhat lower QYs of 0.0194±0.0024 and 0.0199±0.0025, respectively.
2.7. Experimental validation of GSD mechanism
10 μL polynucleotides solution (100 μM, IDT) were dropped on plasma cleaned coverslips (#1.5, Fisher Scientific) and dried at 20°C overnight to form hydrogel thin films. We used an inverted microscope (Nikon, Eclipse Ti-U) with an objective lens (Nikon, TIRF 100×, 1.49 NA) and used a 532 nm diode-pumped solid-state laser with 1-W maximum output for illumination. We determined the fraction of residual singlet state molecules using a pump-probe mode with a constant probe (0.3 kWcm−2) and pump pulses of varying intensity (100 ms, 1-25 kWcm−2) for shelving the molecules into dark states. The fluorescence recovery was monitored for calculating the recovery lifetime by applying an exponential fitting.
2.8. Single-molecule photon localization miroscopy
To examine the properties of nucleic acid blinking, we built a single molecule optical imaging system based on an inverted microscope. As shown in Fig. 1 , a 532 nm diode-pumped solid-state laser with 300-mW maximum output was passed through the microscope body (Nikon, Eclipse Ti-U) and was focused at the back focal plane of an objective lens (Nikon, TIRF 100×, 1.49 NA). The intensity of the illumination beam fluence was adjusted by an achromatic half-wave plate (HWP, AHWP05M-600, Thorlabs) and a Glan-Taylor polarizer (GTP, GT10, Thorlabs). The illumination beam size was controlled by a dual lens assembly. A long-pass filter (BLP01-532R-25, Semrock) was used to reject the reflected laser beam. The fluorescence image was collected through a 550-nm long-pass filter before video acquisition by an EMCCD (Andor, iXon 897 Ultra). We performed single molecule imaging of 20-base Poly-G DNA with Iex=7.14 kWcm−2 by acquiring movies consisting of 1,000 frames at exposure times of 10-ms per frame.
Fig. 1.

Schematic of the experimental setup for single-molecule photon localization microscopy. LF: laser clean-up filter; SF: spatial filter; HWP: achromatic half-wave plate; GTP: Glan-Taylor polarizer; DM: dichroic mirror; OBJ: objective lens; LPF: long-pass filter; RM: reflecting mirror; TL: tube lens; EMCCD: electron multiplying charge coupled device.
2.9. Control experiments on glass coverslips
In order to verify the fluorescence blinking events we observed from the polynucleotide are not introduced from contamination on the glass coverslip, we performed control experiments with using cleaned and water-covered microscope coverslips. Cleaned coverslips (Fisherfinest™ Premium Cover Glasses, Fisher Scientific) were treated by a plasma cleaner (SBT PC-2000) to remove organic contaminants. Water-coated coverslips were prepared by spin coating nuclease free distill water at 3,000 rpm on the cleaned coverslip and then air dried. We recorded blinking events from these samples over 50 seconds under the 532-nm illumination (7.14 kWcm−2). As shown in Fig. 2 , the results indicate the cleaned and water-coated coverslips have far fewer (two orders of magnitude fewer) fluorescence blinking events compared with polynucleotide deposited coverslips, which exclusively demonstrates the blinking is not from contamination on the glass coverslip or in the water. Moreover, the result shown in Fig. 2(c) indicates that the number of blinking events is reasonably invariant within the acquisition period. This suggests photodamaging of DNA can be neglected during blinking measurements in the hydrogel samples under the excitation wavelength and power used in our experiments.
Fig. 2.

Max projection images and number of blinking events over 50 seconds under various conditions: (a) coverslip surface treated by plasma cleanering; (b) water-coated coverslip; and (c) polynucleotides deposited coverslip. The results show at least 99% of the blinking is from polynucleotides.
3. Results and discussion
3.1. Photochemical characteristics of nucleotides at physiological concentration
The physiological concentration of DNA in interphase nuclei is ∼0.10-0.40 g/mL and even higher in metaphase chromosomes [19–21], which correspond to concentrations about 0.26-1.04 M in solution, respectively. These physiological concentrations are significantly higher than the concentrations (10-100 μM) used in most photochemical studies of nucleic acids [15–18]. To establish the photochemistry of DNA for conditions approaching those observed in chromatin, we examined ultra-pure nucleotide solutions with various concentrations. At these higher concentrations, it is critical to first demonstrate that all observations are produced intrinsically from the nucleic acids and are not consistent with results from trace contaminants or impurities. Therefore, throughout our experiments we utilized the highest grade reagents available (Molecular or HPLC grade) and performed rigorous measurements of negative controls to rule out the possibility of introduced impurities (see Materials and methods). In order to satisfy the sample quantity and quality needed for fluorescence spectrophotometry, we chose commercially available, HPLC grade (>99.9% pure for all samples and 100% pure for adenine) mononucleotides (Sigma-Aldrich), including adenine (A), guanine (G), cytosine (C), and thymine (T), that were dissolved in nuclease free distilled water. Further, we verified the purity of the mononucleotides using LC-TOF mass spectrometry on all of the mononucleotides, confirming that all of the samples are analytically pure, with the remaining components consisting of nucleic acid metabolites (e.g. the naturally disassociated ribose and phosphate groups).
Figure 3(a) illustrates absorbance spectra of guanine monophosphate as a function of concentration. Although dilute aqueous solutions of the mononucleotide do not show absorption in the visible range, the UV-visible absorption spectra of concentrated solutions of nucleic acids reveal a broad absorption band in visible range, growing in intensity with increasing concentration. This is particlarly clear in mononucleotide solutions at physiological concentration (0.1 M-1 M). Figure 3(b) further shows the absorbance spectra of mononucleotides solution at a concentration of 0.1 M. The molar extinction of mononucleotides E = A/c [M−1cm−1], where c [M] is the solution concentration, indicates E is ∼ 3-10 M−1cm−1 for the four mononucleotides at 532 nm. Such photochemistry has not been reported previously, and the absorption further produces noticeable fluorescence under visible light illumination.
Fig. 3.

(a) Absorbance spectra of guanine monophosphate as a function of concentration. (b) Absorbance spectra of 0.1-M mononucleotide solutions.
It is an established fact that the fluorescence spectrum of DNA extends into the visible range even under UV excitation at diluted condition. This has been frequently observed and reported elsewhere [15, 17]. However, this phenomenon has long been overlooked due to DNA’s weak absorption coefficient in visible. One important observation is that mononucleotides illuminated at 532 nm have exceptionally longer fluorescence decays with respective lifetimes of ∼2 ns, which are significantly longer than the reported fluorescence lifetimes of mononucleotides (∼1 ps) at 267-nm illumination [22], and comparable to fluorescence lifetimes of high quantum yield fluorophores. This is also consisitent with the relatively high quantum yields (QYs) of these nucleotides measured under visible illumination (QY ∼ 0.02-0.11, see Materials and methods), which are significantly (two to three orders of magnitude) higher than the reported QY measured at 267-nm illumination (3×10−4) [22]. Particularly significant is the observation that these properties are reproducibly distinct for each mononucleotide.
Furthermore, visible fluorescence has also been previously observed in ex vivo nucleic acid studies [23]. Incidentally, the observation of this autofluorescence within the visible range (473-632 nm) was in a study designed for the elimination of autofluorescence in nucleic acid samples deposited on glass for microarray measurements. While the authors report similar visible fluorescent findings in nucleic acids deposited on microarray glass slides, they dismiss the utility of exploring the observed phenomenon. Critically, this observation confirms that visible light fluorescence has been measured in unmodified nucleic acids in earlier and independent studies.
Additionally, work has recently been published showing changes in the absorption properties of organic molecules as a function of concentration [24]. In this work that the absorption properties of molecule widely considered to be ‘dark’, nonanoic acid, change as a function of concentration. Indeed, they demonstrate that while at low concentrations, nonanoic acid has one primary absorbance band at 200 nm, increasing the concentrations of nonanoic acid produces both a red-shift in absorption spectra and creates a secondary absorption peak centered at 270 nm. Moreover, a concentration dependent fluorescence red-shift has been previously identified in carboxylic acids and esters, but has not been studied extensively in more complex organic molecules [25]. The phenomena we found on nucleic acids may be a part of a similar, but under explored, phenomena inherent to organic molecules.
3.2. Photochemical characteristics of polynucleotides with different lengths
We examined photochemical properties of nucleic acids with different sequence lengths. In this study, we chose poly-G nucleotides as model systems of nucleic acid polymer sequences. Similarly, we first confirmed their purities with size exclusion HPLC equipped with DAD (see experiment details in Materials and methods). Figure 4 shows the normalized HPLC DAD absorption spectra of 20-base polynucleotide (IDT), in which the composition of elution bands were verified by UV absorbance at 230 nm as well as by using the time of elution. The first elution band at 20 min consists of the isolated 20-base polynucleotides and has appreciable absorbance at 532 nm, which further confirms the presence of visible light absorption of DNA. In addition to the main polynucleotide band, there is an elution band consistent with small molecules at 65 minutes, which has no measureable absorbance at 532 nm above the baseline of HPLC grade water. Since the small molecules band has no measureable visible absorbance relative to the nucleic acids and they exist at such low concentrations relative to the polynucleotides (<1%), it is unlikely that they are considerably contributing to the experimentally observed fluorescence.
Fig. 4.

Normalized HPLC DAD absorption spectra of 20-base polynucleotide synthesized by IDT. Impurity is observed at 65 minutes as measured by 230 nm absorbance and is demarked by the arrow. Under 532 nm, this band does not show detectable absorbance above the baseline measurements performed on HPLC grade water (H2O) at the same time. Additionally, the purified elusion band consisting of pure polynucleotide demonstrates significant measureable absorbance at 532 nm.
We measured the absorbance spectrum of 100-μM mononucleotide and 20-base poly-G nucleotide in the visible range (Fig. 5 ). The measured Molar extinction coefficient E is 1,200±200 M−1cm−1 at 532 nm for the 20-base poly-G nucleotide, indicating an order of magnitude increase in absorption with longer oligomer length when compare to the mononucleotide. Further, we found sequence length has a distinct effect on the visible fluorescence properties. As shown in Fig. 6 , we investigated the effect of nucleotide length by acquiring fluorescence Intensity of poly-G nucleotides excited at 500 nm with different oligomer length (1-, 2-, 4-, 8-, 16- and 20-base). Critically, a strong increase in the fluorescence intensity is noted as a function of increasing base length [Fig. 6(a)]. As shown in Fig. 6(b), the fluorescence intensity per nucleotide (FL/N) increase by nearly an order of magnitude from mononucleotides to 20-base polynucleotide. This is consistent with the increasing of Molar extinction coefficient per nucleotide (E/N) shown in Fig. 6(c). For sequence longer than 4 bases, the fluorescence intensities per nucleotide appear no significant variation beyond the experimental error. This is in agreement with the previously measured delocalization length of 3.3±0.5 bp for UV light excitation [26].
Fig. 5.
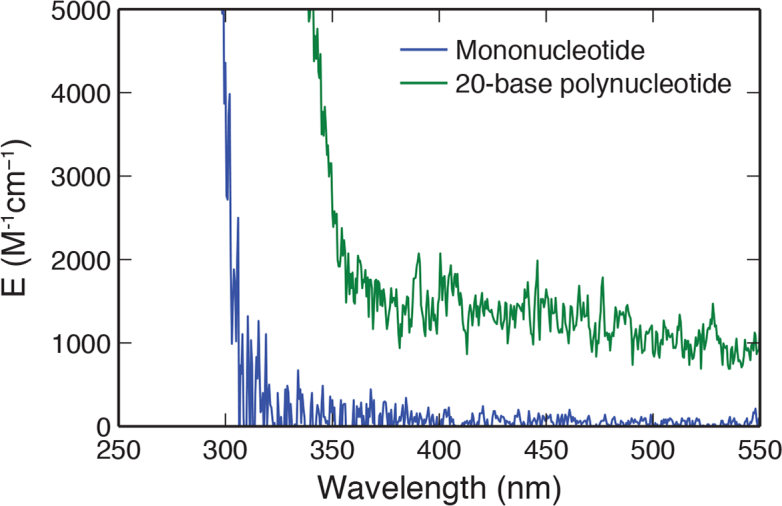
Molar extinction coefficient of 20-base poly-G nucleotide was measured using its aqueous solution with concentration of 100-μM. Measurement was performed in a 45-μL cuvette with a path length of 3-mm using a UV-VIS Spectrophotometer (UV-1800, Shimadzu). The Molar extinction coefficient of mononucleotide was measured under same condition for comparison.
Fig. 6.

(a) Fluorescence intensity per nucleotide with respect to oligomer lengths. Fluorescence of polynucleotides with 1-, 2-, 4-, 8-, 16- and 20-base were excited at 500 nm. Comparison of (b) fluorescence intensity per nucleotide and (c) Molar extinction coefficient per nucleotide for mononucleotide and 20-base polynucleotide. Critically, this indicates a non-linear relation between sequence length, fluorescence intensity, and excitation coefficient.
3.3. Ground state depletion of nucleotides
While the photochemical measurements demonstrate that unmodified nucleic acids have absorption and intrinsic fluorescence under visible light illumination, super-resolution imaging based on single-molecule photon localization requires the ability to detect blinking single-molecule emissions. Given these observed measured relatively high QY and considerable fluorescence emission under visible light illumination, we hypothesized that super-resolution imaging could be accomplished by leveraging GSD with dark-state shelving and stochastic return. GSD has previously been explored for super-resolution imaging using a typical three-level molecular system [7]. When excited by light with intensity Iex [Wcm−2], a molecule transits from its ground state (S0) to an excited state (S1) with the average rate kex = Iexσ/hν, where σ [cm2] is the absorption cross section; h [J·s] is the Planck constant (6.626×10−34 J·s); and ν [s−1]=c/λ is the frequency of the transition. From this state, the molecule can relax non-radiatively, emit a fluorescence photon with a probability equal to QY, or transit to a dark (e.g. triplet) state (T) via intersystem crossing (ISC) with a probability Φ. Φ is a key characteristic of the intersystem crossing from singlet state to a triplet state. If Φ ≪1, the dark states have a lifetime τ much longer than that of fluorescence (i.e. τ ∼ several hundreds of milliseconds ≫ τfl ∼ nanoseconds). Therefore, molecules increasingly shelve in a long-lived dark state with each illumination and no longer fluoresce. However, the excited molecules may return to their ground state with the average rate k = 1/τ, after which they again become excitable. This process creates the required “on” and “off” periods, or blinking for PLM. It can be described by a system of three differential equations:
| (1) |
where n0,1,2 [dimensionless] are the population probabilities of the molecule and . In this model, the parameters governing the behavior of the system are kfl and kisc, which are respectively the rate constants of fluorescence and internal conversion and follow the relationship of kfl/kisc = 1/Φ − 1. They can be further defined as kfl = 1/τfl and kisc = 1/τisc, where τfl and τisc are the fluorescence lifetime and the intersystem crossing lifetime, respectively. To estimate kex, the absorption cross section was calculated using .
For experimental validation of the proposed GSD mechanism, we used polynucleotides (20-base poly-A, G, C, and T, IDT) as model systems. We deposited polynucleotides solution (100 μM, IDT) on a plasma cleaned coverslip surface and dried at 20 °C overnight to form hydrogel thin films (with its concentration approaching the physiological range). At steady state, the population probabilities ni of molecules do not change: dni/dt = 0, giving
| (2) |
Since kfl + kisc = kisc/Φ and kisc ≫ k,
| (3) |
The measured fluorescence intensity F at the steady state is proportional to population probability of the ground state: F ∼ an0, where a is a proportionality constant depending on the quantum yield and the detection efficiency. If we define ε as the ratio of fluorescence intensities after and before GSD, ε is reduced by increasing kex as
| (4) |
where F0 is the fluorescence intensity before GSD (when kex →0). We measured fluorescence intensities of nucleotides at the steady state under various illumination intensities and estimated ε by normalizing them with respect to the fluorescence intensity under a moderate illumination (∼0.3 kWcm−2). As expected, 1/ε was linearly related to Iex as shown in Fig. 7 . This result agrees with the GSD model. Notably, while all polynucleotides were somewhat distinct, polynucleotides containing purines (A and G) and pyrimidines (C and T) share similar features, likely due to the similarity in their molecular structures.
Fig. 7.

Population of the ground state ε of four types of polynucleotides as function of Iex and their linear regression (solid lines). Inset: Jablonski diagram of a three level system.
Furthermore, the theory of GSD also predicts that once GSD has been induced by a strong pump illumination (Ipump for td=100 ms), the fluorescence induced by a weaker probe beam (Iprobe) will follow the exponential time course of the repopulation of the ground state with recovery timescale τ [6,7]. As given by a solution of Eq. (1):
| (5) |
where .
To validate the role of the long-lived dark state in the observed stochastic emission of nucleic acids, we performed the pump-probe measurements for all four types of polynucleotides and chose poly-G DNA to compare against the predictions of the GSD model [Eq. (5)]. As shown in Fig. 8 , the experimental recovery data under pump illuminations up to 24 kWcm−2 fit accurately with the GSD model with the measured E of 1,200 M−1cm−1 for the 20-base poly-G DNA. The fitting gives a recovery time τ of 220 ms, which is comparable to the lifetime of the triplet states in most exogenous dyes, and Φ ∼ 1.67 × 10−4. These long recovery time τ and a low value of Φ facilitate the detection of single-molecule signal and, along with a high quantum yield, produce a strong photon count of emission during a blinking event, as discussed below. The recovery lifetimes of all four types of polynucleotides were summarized in Table 1. Notably, although different polynucleotides have distinct τ, similar values were observed for purines and pyrimidines. In addition, we tested the influence of an additional triplet-specific quencher, β-mercaptoethanol, on the rate of fluorescence recovery. As expected for a GSD system, adding the triplet-specific quencher β-mercaptoethanol reduced τ by 36%, thus confirming the shelving of excited electrons in the dark, and most likely, triplet state.
Fig. 8.

Fluorescence recovery of poly-G DNA after 100 ms dark state shelving with Ipump up to 24 kWcm−2. The recovered signal (black line) was read out with Iprobe (532 nm, 0.3 kWcm−2), which can be accurately explained by the GSD model (red line). The recovery time τ and Φ were obtained by fitting the GSD model to the data.
Table 1.
Comparison of recovery lifetime and averaged photon count and blinking time at Iex=7.14 kWcm−2 between four polynuceotides.
| Poly-A | Poly-G | Poly-C | Poly-T | |
|---|---|---|---|---|
| τ (ms) | 200 ± 50 | 220 ± 30 | 320 ± 50 | 380 ± 40 |
| 〈NB〉 (photon) | 264 ± 17 | 381 ± 23 | 441 ± 26 | 279 ± 19 |
| 〈τB〉 (ms) | 33 ± 20 | 32 ± 20 | 12 ± 10 | 15 ± 10 |
3.4. Fluorescence switching of polynucleotides
To examine the properties of nucleic acid blinking, we performed PLM single molecule imaging of 20-base Poly-G DNA with Iex=7.14 kWcm−2 (see details in Materials and methods). We acquired images consisting of 1,000 frames at exposure times of 10-ms per frame, as illustrated in Fig. 9(a) . Because the lifetime τ of the triplet state is much longer than that of fluorescence, the majority of molecules are ‘shelved’ to their long-lived triplet states under this illumination. As previously described for a GSD system, only a few molecules may return to their ground state at any given time, with the average rate of k = 1/τ, where they can then be repeatedly excited to the fluorescent state [Fig. 9(b)]. This creates the “on” and “off” periods, or blinking described above, yielding the required stochastic activity for precisely locating molecules with PLM. We measured the detected photon counts of each blinking event, NB, and the corresponding duration of the “on” time, τB. Figure 9(c) is a scatter plot of NB versus τB, which indicates NB is linearly proportional to τB. This demonstrated the fluorescence photon arrival rate during the “on” period Γ = NB/τB is nearly identical among each blinking event, and thus 〈Γ〉 can be fitted from the plot. Figures 9(d) and 9(e) show histograms of NB and τB. Critically, NB and τB follow an exponential distribution as predicted by the theory involving quantum jumps between weak and strong transitions [27,28].
Fig. 9.

(a) Consecutive images acquired from poly-G DNA sample with time interval of 10 ms, showing blinking events. (b) The illustration shows a typical fluorescence switching process. (c) Fluorescent photon count NB versus blinking duration τB at Iex=7.14 kWcm−2. Color was used to denote the density of data spots. Histograms of (d) τB and (e) NB follow exponential distribution.
To further examine the blinking dynamics with respect to illumination intensity, we performed single molecule imaging with varying Iex up to 11.0 kWcm−2 in their steady state. Prior to characterizing the blinking properties of the nucleic acids, baseline measurements of the plasma cleaned glass coverslip and water were performed to account for the influence of trace organic residues during sample preparation. The result exclusively demonstrates the blinking event we observed is from the nucleic acids. Based on fundamental photophysics, 〈Γ〉 is given by
| (6) |
As shown in Fig. 10(a) , we found that the experimentally observed Γ is linearly proportional to the illumination power, which is in strong agreement with the Eq. (6).
Fig. 10.

(a) Fluorescent photon emission rate Γ during “on” times, (b) mean blinking time τB and (c) mean photon count NB as a function of Iex, respectively. In steady state the model accurately explained the Γ, τB and NB as a function of Iex.
Likewise, by assuming that during the “on” period the triplet transition takes place at a constant rate, this produces the average “on” time 〈τB〉 as a function of Iex that is given by [28]
| (7) |
Figure 10(b) shows that in the steady state, the experimental measured blinking times as a function of Iex agrees reasonably well with the theoretical model. The deviation at low Iex is likely due to the low signal-to-background ratio of the recorded blinking which resulted in relatively relatively smaller 〈τB〉 values.
As 〈NB〉 is given by
| (8) |
〈NB〉 is independent of the absorption cross section and is in principle identical under various illumination intensities. As shown in Fig. 10(c), the measured 〈NB〉 grew asymptotically to reach a constant value with increasing Iex. Notably, 〈NB〉 is lower than the model at low Iex. This is likely due to an excessive background subtraction when the signal-to-background ratio of the recorded blinking is low.
Significantly, although bulk nucleotides absorb weakly in the visible range, the number of photons emitted from each blinking event is large enough for single-molecule-localization based imaging. This is due to the high QY of their visible-light fluorescence, which is much greater than that for the UV transitions and thereby compensates for the relatively weak absorption. Furthermore, the total photon count during an “on” time is independent of the absorption cross section and depends only on QY/Φ. A high QY (e.g. QY=0.0559 of poly-G DNA) and a small Φ for the transitions in the visible spectral range result in a high photon count, which in turn translates into a high spatial resolution of PLM imaging, given by where s is the full width at half maximum of the diffraction point spread function [29]. As shown in Table 1, although the four polynucleotides generate different numbers of collected photon in individual emission events, they are all capable of providing a sub-20-nm localization precision in PLM.
4. Conclusion
Generally, the unlabeled products usually have much lower extinction coefficient and likewise fluorescence emission if compared with fluorescence targets. As a result, they are normally neglected when mixed with fluorescent targets. Therefore, the observed anomalous signals from these samples are frequently classified as impurities or contaminations without further analysis. In fact, numerous methods have been developed and employed to suppress or eliminate signals that emanate from endogenous molecules. These methods include chemical means for suppression of endogenous emissions and mathematical analysis that eliminates events deemed unlikely to originate from the fluorescent tag [30–32]. The ubiquitous use of these methods has created a widely accepted notion that endogenous molecules lack the capacity for usable fluorescence emission. In contrast, we discovered the fluorescence emission of nucleotides in visible range, and more importantly, we explored its blinking mechanism, which facilitates super-resolution imaging by using the principle of single molecule localization microscopy.
In conclusion, we characterized the fluorescence of nucleic acids under visible light illumination as well as their fluorescence depletion and recovery that accurately matches a GSD model. In the photoswitching process, experimentally measured Γ, τB and NB as a function of Iex all satisfy the fundamental photophysics of single molecule fluorescence blinking, which in addition to the retraced parameter, Φ, are consistent with that obtained by the GSD model. Remarkably, the fluorescence characteristics of nucleic acids under visible light illumination make them ideal candidates for use as blinking fluorophores for super resolution imaging in biological systems. (1) They exhibit a long shelving lifetime τ at the range of hundreds of milliseconds, which is ideal for efficient depletion based on the GSD model. Consequently, only a small number of molecules fluoresce at the same time upon GSD, which is an essential requirement for beating the diffraction-limit in PLM. (2) Although nucleic acids have weak fluorescence under visible illumination due to their low absorption in bulk, the photon counts of individual emission events are comparable to those of some of some of the most potent exogenous dyes used in PLM due to their high QY and low intersystem crossing probability Φ. (3) Finally, the fluorescence properties of nucleic acids allows detection at lower illumination intensity (<10 kWcm−2), which is highly advantageous compared to the cell-damaging high light intensities typically used in some other super-resolution methods (e.g. up to 105 kWcm−2 in STED). In all, this discovery paves a new way to realize label-free optical super-resolution imaging of nucleic acids [14], which may provide an ideal technique to visualize the spatial organization of single or groups of nucleosomes and quantitatively estimates the nucleosome occupancy level of DNA in unstained chromosomes and nuclei.
Conflicts of interest
HFZ and CS have financial interests in Opticent Health. All other authors declare no competing financial interests.
Acknowledgments
We thank Dr. Michael R. Wasielewski for the support in photochemistry studies and Dr. Ryan M. Young and Mr. Yilei Wu for their assistance in spectroscopic measurements. We thank the Integrated Molecular Structure Education and Research Center at Northwestern University for mass spectrometry measurements. We thank Dr. Arabela Grigorescu and Theint Aung from the Northwestern University Keck Biophysics Core Facility for their assistance in size exclusion chromatography of polynucleotides.
Funding
National Institutes of Health (NIH) (R24EY022883, DP3DK108248, F30EY026472, U54CA193419, R01CA165309, T32GM008152, T32HL076139); National Science Foundation (NSF) (DBI-1353952, DGE-0824162, CBET-1055379, CBET-1240416, EEC-1530734); Research Catalyst Award by Northwestern McCormick School of Engineering.
References and links
- 1.Ellis R. J., Minton A. P., “Cell biology - Join the crowd,” Nature 425, 27–28 (2003). 10.1038/425027a [DOI] [PubMed] [Google Scholar]
- 2.Hell S. W., Wichmann J., “Breaking the diffraction resolution limit by stimulated-emission - stimulated-emission-depletion fluorescence microscopy,” Opt. Lett. 19, 780–782 (1994). 10.1364/OL.19.000780 [DOI] [PubMed] [Google Scholar]
- 3.Gustafsson M. G. L., “Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution,” P. Natl. Acad. Sci. USA 102, 13081–13086 (2005). 10.1073/pnas.0406877102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., “Imaging intracellular fluorescent proteins at nanometer resolution,” Science 313, 1642–1645 (2006). 10.1126/science.1127344 [DOI] [PubMed] [Google Scholar]
- 5.Rust M. J., Bates M., Zhuang X. W., “Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM),” Nat. Methods 3, 793–795 (2006). 10.1038/nmeth929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hell S. W., Kroug M., “Ground-State-Depletion Fluorescence Microscopy - A concept for breaking the diffraction resolution limit,” Appl. Phys. B 60, 495–497 (1995). 10.1007/BF01081333 [DOI] [Google Scholar]
- 7.Folling J., Bossi M., Bock H., Medda R., Wurm C. A., Hein B., Jakobs S., Eggeling C., Hell S. W., “Fluorescence nanoscopy by ground-state depletion and single-molecule return,” Nat. Methods 5, 943–945 (2008). 10.1038/nmeth.1257 [DOI] [PubMed] [Google Scholar]
- 8.Ricci M. A., Manzo C., Garcia-Parajo M. F., Lakadamyali M., Cosma M. P., “Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo,” Cell 160, 1145–1158 (2015). 10.1016/j.cell.2015.01.054 [DOI] [PubMed] [Google Scholar]
- 9.Boettiger A. N., Bintu B., Moffitt J. R., Wang S., Beliveau B. J., Fudenberg G., Imakaev M., Mirny L. A., Wu C. T., Zhuang X., “Super-resolution imaging reveals distinct chromatin folding for different epigenetic states,” Nature 529, 418–422 (2016). 10.1038/nature16496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuda A., Shao L., Boulanger J., Kervrann C., Carlton P. M., Kner P., Agard D., Sedat J. W., “Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP-histones,” Plos One 5, e12768 (2010). 10.1371/journal.pone.0012768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohn M., Diesinger P., Kaufmann R., Weiland Y., Muller P., Gunkel M., von Ketteler A., Lemmer P., Hausmann M., Heermann D. W., Cremer C., “Localization microscopy reveals expression-dependent parameters of chromatin nanostructure,” Biophys. J. 99, 1358–1367 (2010). 10.1016/j.bpj.2010.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beerman T. A., Mchugh M. M., Sigmund R., Lown J. W., Rao K. E., Bathini Y., “Effects of Analogs of the DNA minor groove binder Hoechst-33258 on topoisomerase II and I mediated activities,” Biochim. Biophys. Acta 1131, 53–61 (1992). 10.1016/0167-4781(92)90098-K [DOI] [PubMed] [Google Scholar]
- 13.Almassalha L. M., Bauer G. M., Chandler J., Gladstein S., Cherkezyan L., Stypula-Cyrus Y., Weinberg S., Zhang D., Thusgaard Ruhoff P., Roy H., Subramanian H., Chandel N., Szleifer I., Backman V., “Label-free imaging of the native, living cellular nanoarchitecture using partial-wave spectroscopic microscopy,” P. Natl. Acad. Sci. USA 113 E6372–E6381 (2016). 10.1073/pnas.1608198113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong B., Almassalha L. M., Stypula-Cyrus Y., Urban B. E., Chandler J. E., Nguyen T. Q., Sun C., Zhang H. F., Backman V., “Superresolution intrinsic fluorescence imaging of chromatin utilizing native, unmodified nucleic acids for contrast,” P. Natl. Acad. Sci. USA 113, 9716–9721 (2016). 10.1073/pnas.1602202113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaya I., Gustavsson T., Miannay F. A., Douki T., Markovitsi D., “Fluorescence of natural DNA: From the femtosecond to the nanosecond time scales,” J. Am. Chem. Soc. 132, 11834–11835 (2010). 10.1021/ja102800r [DOI] [PubMed] [Google Scholar]
- 16.Anders A., “DNA Fluorescence at room-temperature excited by means of a dye-laser,” Chem. Phys. Lett. 81, 270–272 (1981). 10.1016/0009-2614(81)80250-3 [DOI] [Google Scholar]
- 17.Plessow R., Brockhinke A., Eimer W., Kohse-Hoinghaus K., “Intrinsic time- and wavelength-resolved fluorescence of oligonucleotides: A systematic investigation using a novel picosecond laser approach,” J. Phys. Chem. B 104, 3695–3704 (2000). 10.1021/jp994132u [DOI] [Google Scholar]
- 18.Takaya T., Su C., de La Harpe K., Crespo-Hernandez C. E., Kohler B., “UV excitation of single DNA and RNA strands produces high yields of exciplex states between two stacked bases,” P. Natl. Acad. Sci. USA 105, 10285–10290 (2008). 10.1073/pnas.0802079105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bancaud A., Lavelle C., Huet S., Ellenberg J., “A fractal model for nuclear organization: current evidence and biological implications,” Nucleic Acids Res. 40, 8783–8792 (2012). 10.1093/nar/gks586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daban J. R., “High concentration of DNA in condensed chromatin,” Biochem. Cell Biol. 81, 91–99 (2003). 10.1139/o03-037 [DOI] [PubMed] [Google Scholar]
- 21.Bohrmann B., Haider M., Kellenberger E., “Concentration evaluation of chromatin in unstained resin-embedded sections by means of low-dose ratio-contrast imaging in STEM,” Ultramicroscopy 49, 235–251 (1993). 10.1016/0304-3991(93)90230-U [DOI] [PubMed] [Google Scholar]
- 22.Callis P. R., “Polarized fluorescence and estimated lifetimes of the DNA bases at room-temperature,” Chem. Phys. Lett. 61, 563–567 (1979). 10.1016/0009-2614(79)87174-2 [DOI] [Google Scholar]
- 23.Raghavachari N., Bao Y. P., Li G. S., Xie X. Y., Muller U. R., “Reduction of autofluorescence on DNA microarrays and slide surfaces by treatment with sodium borohydride,” Anal. Biochem. 312, 101–105 (2003). 10.1016/S0003-2697(02)00440-2 [DOI] [PubMed] [Google Scholar]
- 24.Rossignol S., Tinel L., Bianco A., Passananti M., Brigante M., Donaldson D. J., George C., “Atmospheric photochemistry at a fatty acid–coated air-water interface,” Science 353, 699–702 (2016). 10.1126/science.aaf3617 [DOI] [PubMed] [Google Scholar]
- 25.Caswell L. R., Howard M. F., Onisto T. M., “Solvent and substituent effects upon n → π* transition of aliphatic carboxylic-acids and esters,” J. Org. Chem. 41, 3312–3316 (1976). 10.1021/jo00882a023 [DOI] [Google Scholar]
- 26.Buchvarov I., Wang Q., Raytchev M., Trifonov A., Fiebig T., “Electronic energy delocalization and dissipation in single- and double-stranded DNA,” P. Natl. Acad. Sci. USA 104, 4794–4797 (2007). 10.1073/pnas.0606757104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook R. J., Kimble H. J., “Possibility of direct observation of quantum jumps,” Phys. Rev. Lett. 54, 1023–1026 (1985). 10.1103/PhysRevLett.54.1023 [DOI] [PubMed] [Google Scholar]
- 28.Burzykowski T., Szubiakowski J., Ryden T., “Analysis of photon count data from single-molecule fluorescence experiments,” Chem. Phys. 288, 291–307 (2003). 10.1016/S0301-0104(03)00034-X [DOI] [Google Scholar]
- 29.Mortensen K. I., Churchman L. S., Spudich J. A., Flyvbjerg H., “Optimized localization analysis for single-molecule tracking and super-resolution microscopy,” Nat. Methods 7, 377–381 (2010). 10.1038/nmeth.1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rubin-Delanchy P., Burn G. L., Griffie J., Williamson D. J., Heard N. A., Cope A. P., Owen D. M., “Bayesian cluster identification in single-molecule localization microscopy data,” Nat. Methods 12, 1072–1076 (2015). 10.1038/nmeth.3612 [DOI] [PubMed] [Google Scholar]
- 31.Sage D., Kirshner H., Pengo T., Stuurman N., Min J., Manley S., Unser M., “Quantitative evaluation of software packages for single-molecule localization microscopy,” Nat. Methods 12, 717–724 (2015). 10.1038/nmeth.3442 [DOI] [PubMed] [Google Scholar]
- 32.Baumgart F., Arnold A. M., Leskovar K., Staszek K., Folser M., Weghuber J., Stockinger H., Schutz G. J., “Varying label density allows artifact-free analysis of membrane-protein nanoclusters,” Nat. Methods 13, 661–664 (2016). 10.1038/nmeth.3897 [DOI] [PMC free article] [PubMed] [Google Scholar]