

Abstract
Background
The β3-adrenoceptor (β3-AR) is implicated in cardiac remodeling. Since metabolic dysfunction due to loss of mitochondria plays an important role in heart diseases, we examined the effects of β3-AR on mitochondrial biogenesis and energy metabolism in atrial fibrillation (AF).
Methods
Atrial fibrillation was created by rapid atrial pacing in adult rabbits. Rabbits were randomly divided into 4 groups: control, pacing (P7), β3-AR antagonist (L748337), and β3-AR agonist (BRL37344) groups. Atrial effective refractory period (AERP) and AF induction rate were measured. Atrial concentrations of adenine nucleotides and phosphocreatine were quantified through high-performance liquid chromatography. Mitochondrial DNA content was determined. Real-time polymerase chain reaction and Western blot were used to examine the expression levels of signaling intermediates related to mitochondrial biogenesis.
Results
After pacing for 7 days, β3-AR was significantly upregulated, AERP was reduced, and the AF induction rate was increased. The total adenine nucleotides pool was significantly reduced due to the decrease in adenosine triphosphate (ATP). The P7 group showed decreased activity of F0F1-ATPase. Mitochondrial DNA content was decreased and mitochondrial respiratory chain subunits were downregulated after pacing. Furthermore, expression of transcription factors involved in mitochondrial biogenesis, including peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), nuclear respiratory factor 1 (NRF-1), and mitochondrial transcription factor A (Tfam), was lower in the P7 group in response to β3-AR activation. Further stimulation of β3-AR with BRL37344 exacerbated these effects, together with a significant decrease in the levels of phosphocreatine. In contrast, inhibition of β3-AR with L748337 partially restored mitochondrial biogenesis and energy metabolism of atria in the paced rabbits.
Conclusion
The activation of β3-AR contributes to atrial metabolic remodeling via transcriptional downregulation of PGC-1α/NRF-1/Tfam pathway that are involved in mitochondrial biogenesis, which ultimately perturbs mitochondrial function in rapid pacing-induced AF. The β3-AR is therefore a potential novel therapeutic target for the treatment or prevention of AF.
Keywords: atrial fibrillation, β3-adrenoceptor, metabolic remodeling, mitochondrial biogenesis
Introduction
Atrial fibrillation (AF), the most common tachyarrhythmia in clinical practice, is associated with increased risk of heart failure (HF) and stroke. Atrial fibrillation is characterized by a series of striking alterations in atrial electrical, structural, and functional properties,1,2 which in turn promote maintenance and self-perpetuation of AF; that is, “AF begets AF.” A number of studies have emphasized the role of perturbed cardiac metabolism in HF. Metabolic alterations are thought to precede and trigger the sustained functional and structural remodeling found in the stressed heart.3 Elevated release of adenine nucleotide metabolites was also found following glycoside-induced ventricular fibrillation.4 Recently, human and animal models of AF have documented dramatic modifications in cardiac energy metabolism.5,6 Therefore, understanding the mechanisms that contribute to metabolic remodeling during AF is of great importance. Mitochondria are the canonical organelles associated with energy metabolism. A study involving patients with AF showed increased mitochondrial DNA (mtDNA) deletion mutation.7 Moreover, mitochondria are considered to be the sources of metabolic stress and lethal arrhythmias.8,9 Although compelling studies have shown marked changes of myocardial mitochondrial morphology and function in AF,10–12 limited data are available regarding the underlying impact of cardiac mitochondrial function and energy metabolism upon the initiation and persistence of AF.
The β-ARs are primary regulators of cardiac performance, and the current family contains at least 3 β-ARs (including β1, β2, and β3 subtypes). Chronic stimulation of β-ARs plays a vital role in physiological and pathological cardiac remodeling. The roles of β1- and β2-AR in regulating cardiac structure and function in humans and other mammals are now well established. The β3-AR differs from β1- and β2-AR with respect to its molecular structure and pharmacological profile. Considerable attention has been focused on β3-AR with respect to cardiovascular pathologies such as HF, hypertension, and diabetes mellitus, whereas the precise mechanisms of β3-AR in cardiac regulation during AF are not yet completely understood. Sheng et al showed that β3-AR was elevated in paced atria and contributed to atrial oxidative stress and structural remodeling in a canine model.13 Additionally, Liu et al found that stimulation of β3-AR resulted in reduced fatty acid metabolism, which exacerbated atrial remodeling in pacing induced AF.14 These studies imply a close relationship between AF and β3-AR; therefore, a better knowledge regarding the regulatory role of β3-AR in AF is obligatory. Meanwhile, nebivolol, a third-generation β-blocker, was shown to increase mtDNA copy number and upregulate the mitochondrial protein levels by activation of β3-AR in 3T3-L1 adipocytes.15 In addition, a study from differentiated H9c2 cardiomyoblasts treated with isoproterenol showed loss of the mitochondrial respiratory chain.16 Nevertheless, the role of β3-AR in the regulation of cardiac mitochondrial function and energy metabolism in AF has not been reported yet.
Peroxisome proliferator-activated receptor coactivator 1α (PGC-1α), which is abundantly expressed in cardiac muscle, is a central promoter of mitochondrial biogenesis and energy metabolism through nuclear respiratory factor (NRF-1 and NRF-2).17–19 Mitochondrial transcription factor A (Tfam), a nuclear encoded transcription factor that is responsible for both the replication and transcription of mtDNA, is induced by the concerted activities of PGC-1α and NRF-1. Consistent with their key roles in the maintenance of mitochondrial health, cardiac-specific deletion of NRF-1 and Tfam resulted in decreased mitochondrial function.20,21 Following adrenergic signaling via G-protein coupled receptors, PGC-1α can be activated by p38 mitogen-activated protein kinase (MAPK) and cyclic adenosine monophosphate (cAMP).22 Rabbit cardiac function is similar to that of the human heart,23 and it has been shown that rabbit endogenously expresses β3-AR in ventricular cardiomyocytes, regulating mechanical and electrical cardiac function.24 Therefore, we designed the current study to investigate the potential significance of β3-AR in the development of AF in a rabbit model of rapid atrial pacing (RAP) and to test the hypothesis that changes in cardiac mitochondrial biogenesis and energy metabolism during AF are β3-AR activation dependent.
Materials and Methods
Animals
Thirty-eight adult New Zealand white rabbits (either sex) weighing 2 to 2.5 kg were purchased from the Experimental Animal Center of the First Affiliated Hospital of Harbin Medical University (Harbin, China) and randomly divided into 4 groups: the control (CON) group (n = 8), pacing (P7) group (n = 10), β3-AR antagonist (L748337) group (n = 10), and β3-AR agonist (BRL37344) group (n = 10). All surgeries and experiments were approved by the Institutional Animal Care and Use Committee and Animal Experimentation Ethics Committee of Harbin Medical University and animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals from the Institute for Laboratory Animal Research (NIH Publication No. 85–23, Revised 1996).
RAP Model
Light thoracotomy was performed in rabbits under anesthesia with ketamine (35 mg kg−1; Sigma Aldrich, St Louis, Missouri) and xylazine (5 mg kg−1, Sigma Aldrich). A unipolar lead was then sutured onto the right atrium and a pacemaker (Harbin Polytechnic University, Harbin, China) was implanted subcutaneously in the chest. After recovery from surgery for 1 week, pacing was started at 600 beats per minute (bpm) for 7 days. Rabbits in the CON group were operated on with an identical surgical procedure but were not subjected to RAP. After pacing was initiated, animals in the L748337 group and BRL37344 group received daily infusions of the β3-AR antagonist, L748337 (76 μg kg−1; Cat. No. L7045; Sigma Aldrich)13,25 or the β3-AR agonist, BRL37344 (9 μg kg−1; Cat. No. B169; Sigma Aldrich)14 by infusion pump for 30 minutes, respectively. The rabbits were injected with nadolol (1 mg kg−1; Cat. No. N1892; Sigma-Aldrich)14,26 20 minutes prior to intervention with either β3-AR agonist or antagonist. In the CON and P7 groups, the rabbits only received infusion of nadolol (1 mg kg−1) daily for 7 days. An echocardiogram was conducted both before pacing and at the end of the experimental period. A surface electrocardiography was daily recorded to confirm maintenance of 1:1 atrial pacing at 600 bpm.
Electrophysiological Measurements
After RAP for 7 days, atrial effective refractory period (AERP) and AF induction rate were measured using a Prucka CardioLab multi-channels cardiac electrophysiological polygraph (GE Co Ltd; Fairfield, Connecticut). Electrical stimulation was delivered using a bipolar epicardial electrode attached to the right atrial appendage, and AERP was determined at a representative basic cycle length of 200 milliseconds. The AERP200ms was measured with a train of 8 basic stimuli (S1) followed by a premature stimulus (S2) at an interval that was progressively shortened from 180 milliseconds in 10-millisecond decrements until the S2 failed to generate a propagated response. The S1 to S2 interval was then increased by 5-milliseconds and decreased by 2-milliseconds in steps until S2 capture failure. The longest S1 to S2 interval that failed to generate a response was considered the AERP200ms value. The AF duration was induced by a burst of stimuli (10 Hz, 2 milliseconds) to the right atrium at 4 times the threshold current. AF was registered as a rapid and irregular atrial rhythm with varied atrial electrogram morphology. The hearts were then removed immediately and the atrial tissues were rapidly separated. A portion of free atrial tissue was fixed in 2.5% glutaraldehyde (4°C), and the other portions were either used for mitochondrion isolation or quickly frozen in liquid nitrogen and stored at −80°C for future research.
Isolation of Mitochondria From Atrial Tissues
Mitochondria isolation kit (Beyotime Institute of Biotechnology, Shanghai, China) was purchased. All procedures were performed per the manufacturer’s instructions at 4°C in 1 hour.
Measurement of Mitochondrial Cytochrome c Oxidase and F0F1-ATPase Activities
Mitochondrial cytochrome c oxidase and F0F1-ATPase activity measurement kits (Genmed Gene Pharmaceutical Technology Co, Shanghai, China) were purchased. All experiments were conducted following the manufacturer’s instructions. Mitochondrial protein was quantified using a bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China).
Electron Microscopy
After fixation for at least 2 hours, atrial tissues were postfixed in 1% osmium tetroxide, dehydrated in ethanol, and embedded in Epon. Ultrathin sections were cut from each sample, counterstained with uranium acetate and lead citrate, and evaluated under a JEM-7650 transmission electron microscope (Hitachi, Tokyo, Japan).
Determination of Adenine Nucleotides and Phosphocreatine Content
The atrial tissue concentrations of adenine nucleotides, including adenosine triphosphate (ATP), adenosine diphosphate (ADP), AMP, and phosphocreatine (Pcr), were measured by means of high-performance liquid chromatography (HPLC) as described previously.27 Briefly, liquid nitrogen frozen atria tissues (100 mg) were homogenized with 0.4 mol/L perchloric acid and centrifuged (10000 × g for 10 minutes at 0°C). The supernatant was then neutralized with 0.2 mol/L potassium hydrate and clarified with a second centrifugation (3000 × g for 10 minutes at 0°C). The supernatant was again collected and filtrated, and an aliquot of 20 μL was applied to the HPLC system (Shimadzu, Japan) with a Hypersil GOLD aQ column. All steps were conducted on ice. Standards of ATP, AMP, ADP, and Pcr were purchased (Sigma-Aldrich). The amount of total adenine nucleotides (TANs) was calculated as ATP + ADP + AMP.
Determination of mtDNA Content
Total DNA was extracted from atrial tissues using a genomic DNA extraction kit (Tiangen, Beijing, China) according to the manufacturer’s instructions. A 5-μL sample DNA was used for agarose gel electrophoresis.
The mtDNA content was determined according to the ratio between the mitochondrial D-loop gene and the nuclear encoded β-actin gene. Quantitative real-time polymerase chain reaction (qPCR) amplification was performed using an ABI 7500 real-time PCR system (Applied Biosystems, Foster, California) in a 20-μL reaction volume containing 10 μL of 2 × UltraSYBR Mixture (TaKaRa, Dalian, China), 0.4 μL of forward primers (10 mmol/L), 0.4 μL of reverse primers (10 mmol/L), and 2 μL of sample DNA. Data analysis was based on measurement of the cycle threshold (CT). The CT value differences, that is, CT (D-loop)-CT (β-actin), were used to quantify mtDNA copy number.28 The primers used are listed in Table 1.
Table 1.
Primers Used to Estimate mtDNA Content.
| Genes | Sequence |
|---|---|
| D-loop | Forward primer 5′-GGTTCTTACCTCAGGGCCATGA-3′ |
| Reverse primer 5′-GATTAGTCATTAGTCCATCGAGAT-3′ | |
| β-Actin | Forward primer 5′-ATCGTGCGCGACATCAAGGAGAAG-3′ |
| Reverse primer 5′-GCAGCTCGTAGCTCTTCTCCAG-3′ |
Abbreviation: mtDNA, mitochondrial DNA.
Western Blot
After homogenization, total protein was extracted and quantified using a BCA protein assay kit (Beyotime Institute of Biotechnology, China). Total protein was fractionated by electrophoresis and transferred onto polyvinylidene difluoride membranes. The membranes were then, respectively, incubated overnight at 4°C with a specific antibody targeting β3-AR (1:100; Santa Cruz, Santa Cruz, California, Tfam (1:100; Santa Cruz), PGC-1α (1:500; Abcam, Cambridge, United Kingdom), NRF-1 (1:900; Abcam), MitoProfile Total OXPHOS Rodent WB Antibody Cocktail (1:1000; Abcam), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000; Bioss, Beijing, China). After conjugation with the second antibody for 1 hour and washing 4 times with TBST, membranes were exposed to ECL buffer and the signals were captured by ChemiDoc XRS (Bio-Rad, Hercules, California). Bands were quantified using Bio-Rad image analysis software. GAPDH was evaluated as a loading control.
Primer Design and Real-Time PCR
To measure the expression levels of target genes, total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, California). The RNA sample was quantified using fluorescence spectrophotometry and then reverse transcribed via a Prime-Script RT reagent kit with gDNA Eraser (Takara, Japan). A 20-μL reaction mixture consisting of 10 μL of FastStart Universal SYBR Green Master (ROX; Roche, Basel, Switzerland), 0.5 μL of forward primers (10 mmol/L), 0.5 μL of reverse primers (10 mmol/L), and 2 μL of complementary DNA was applied to the ABI 7500 Real Time PCR system (Applied Biosystems). The relative quantification was calculated as the 2−ΔΔCT, and β-actin was used as an internal control. The primers used are shown in Table 2.
Table 2.
Primers Used for Real-Time PCR.
| Genes | Sequence |
|---|---|
| β3-AR | Forward primer 5′-CACGCTGGGGCTCATTAT-3′ |
| Reverse primer 5′-TGAAGGCAGAGTTGGCAT-3′ | |
| PGC-1 | Forward primer 5′-GAGAAGCGGGAATCGGAAAG-3′ |
| Reverse primer 5′-GCATCACAGGTGTATCGGTAGGT-3′ | |
| Tfam | Forward primer 5′-TTCCGAGATGGTGTTCATCC-3′ |
| Reverse primer 5′-TGCCTCTGGGTTCTTAGCTT-3′ | |
| β-Actin | Forward primer 5′-GTCAGGTCATCACTATCGGCAAT-3′ |
| Reverse primer 5′-AGAGGTCTTTACGGATGTCAACGT-3′ |
Abbreviations: PCR, polymerase chain reaction; β3-AR, β3-adrenoceptor; PGC-1, peroxisome proliferator-activated receptor γ coactivator 1; Tfam, mitochondrial transcription factor A.
Statistical Analysis
Data are presented as mean ± standard deviation. For comparisons of all experimental data, 1-way analysis of variance followed by Dunnett T3 or Tukey post hoc test was used. A P value < 0.05 was considered to be significant.
Results
Effect of β3-AR on AERP200ms and AF Inducibility in RAP Rabbits
After pacing, the level of β3-AR mRNA and protein in atrial tissue was increased compared with the CON group (Figure 1A-C). Additionally, when compared to the P7 group, the expression of β3-AR was further elevated following treatment with the β3-AR agonist, BRL37344, but was reduced upon addition of the β3-AR antagonist L748337. This finding is consistent with our previous results in this model, in which expression of β3-AR protein was barely detectable after treatment with the β3-AR antagonist, SR59230A.14
Figure 1.
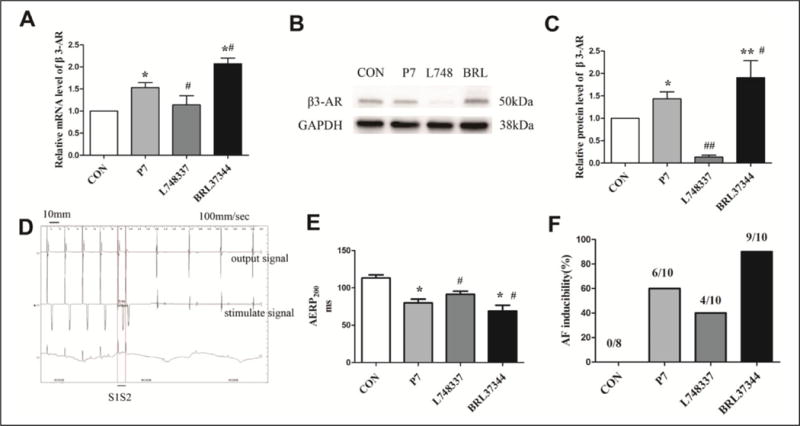
Expression levels of β3-adrenoceptor (β3-AR) and electrophysiological measurements in rapid atrial pacing (RAP) rabbits. A-C, Expression levels of β3-AR messenger RNA (mRNA) and protein were determined by real-time polymerase chain reaction (PCR and Western blot. D, A representative bipolar recording of atrial effective refractory period (AERP)200ms in the β3-AR antagonist (L748337) group: the premature stimulus (S2) failed to generate a response after a train of 8 basic stimuli (S1); the S1S2 value was 92 milliseconds. E, AERP200ms was determined at a representative basic cycle length of 200 milliseconds. F, Atrial fibrillation (AF) was registered as a rapid and irregular atrial rhythm with varied atrial electrogram morphology. A, C, and E, Data are presented as mean ± standard deviation (SD). *P < .05 versus control (CON) group; **P < .01 versus CON group; #P < .05 versus pacing (P7) group; ##P < .01 versus P7 group.
The present study investigated the effect of the β3-AR agonist and antagonist on AERP200ms (Figure 1E) and AF inducibility (Figure 1F) in RAP rabbits. In comparison with the CON group, AERP200 ms was significantly reduced in the P7 group (from 113.25 ± 4.13 milliseconds to 80.00 ± 4.99 milliseconds, P < .05). Application of BRL37344 enhanced RAP-induced AERP200ms reduction (from 80.00 ± 4.99 milliseconds to 67.20 ± 5.00 milliseconds, P < .05). The AERP200ms was increased by L748337 compared with the P7 group (from 80.00 ± 4.99 milliseconds to 91.60 ± 3.98 milliseconds, P < .05). In the CON group, no episode of AF was captured. RAP resulted in an increased AF induction rate (from 0% to 60%). The AF induction rate was further increased by treatment with BRL37344 compared with the P7 group (from 60% to 90%). By contrast, the group treated with the β3-AR antagonist L748337 decreased the RAP-induced AF induction rate (from 60% to 40%).
Transmission Electron Microscopy
Atrial myocytes from the CON group showed a regular sarcomere structure with rows of uniformly sized mitochondria (Figure 2A). However, after RAP for 7 days, partial disintegration of myofilaments, swelling of mitochondria, accumulation of glycogen (Figure 2B), and formation of contraction bands (Figure 2C) were present. After treatment with BRL37344, most of the myofilaments were disintegrated and replaced by fibrous tissues, and mitochondrial swelling was aggravated (Figure 2D). We also observed nuclear swelling in the BRL37344 group (Figure 2E), and these changes were somewhat attenuated by treatment with L748337 (Figure 2F).
Figure 2.
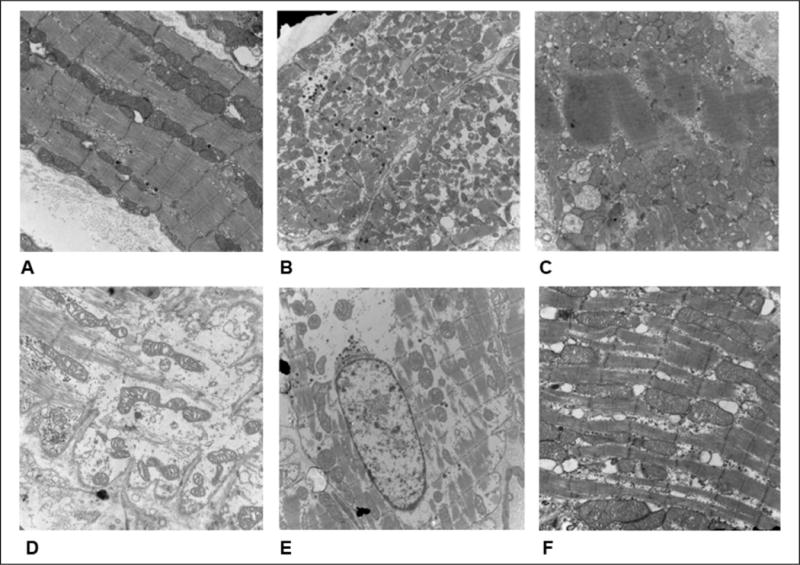
Comparison of atrial ultrastructure among groups. A, The control group showed regular sarcomere organization and uniformly sized mitochondria between sarcomeres. B, In atrial tissues from the pacing (P7) group, partial myofilaments were disintegrated and replaced by fibrous tissues, and mitochondria were slightly swollen. C, In the P7 group, formation of contraction bands was observed in the atrial myocardium. D, After treatment with β3-AR agonist (BRL37344), most of the myofilaments were disintegrated and replaced by fibrous tissues, and mitochondria showed severe swelling that was accompanied by fractured cristae. E, Nuclear swelling in the BRL37344 group. F, Reversion of the effects following β3-AR antagonist (L748337)-dependent β3-AR inhibition. (Magnification: A, D, and F ×20000; B, C, and E ×10000, n = 8 for control group, n = 10 for P7, L748337, and BRL37344 groups).
Effect of β3-AR on Adenine Nucleotides and Pcr Content in RAP Rabbits
To explore the energy status during AF, we quantified the content of ATP, ADP, AMP, and Pcr following RAP-induced AF. After 7 days of pacing, the ATP and ADP content, and the TAN pool decreased compared with the control atria (P < .05; Figure 3A, B, and D). Further activation of β3-AR with BRL3744 exacerbated the decrease in the levels of ATP, ADP, AMP, and TAN. In contrast, inhibition of β3-AR resulted in higher levels of ATP content and TAN pool compared with the P7 group (P < .01; Figure 3A and D). Moreover, stimulation of β3-AR with BRL37344 resulted in a significantly lower Pcr content than that in the CON group (P < .05; Figure 3E). Finally, in comparison to the CON group, the Pcr/ATP ratio was significantly higher in the P7 and BRL37344 groups (P < .05; Figure 3F).
Figure 3.
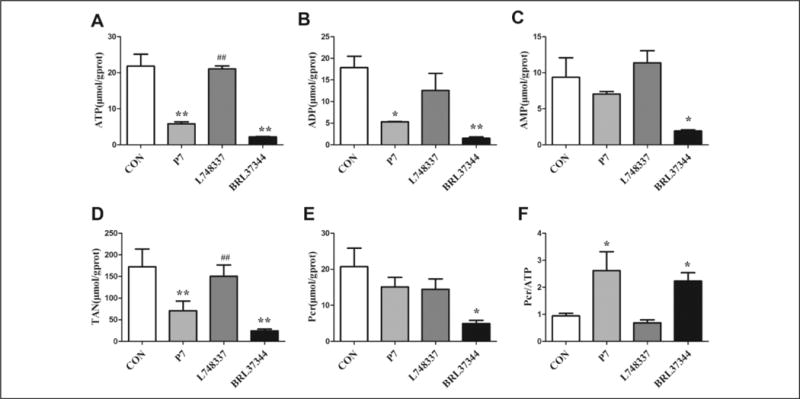
Adenine nucleotides and phosphocreatine (Pcr) content in rapid atrial pacing (RAP) rabbits. Atrial concentrations of adenine nucleotides and Pcr were measured by high-performance liquid chromatography (HPLC). Data are presented as mean ± standard deviation (SD). *P < .05 versus control (CON) group; **P < .01 versus CON group; ##P < .01 versus pacing (P7) group. ATP, adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; TAN, total adenine nucleotides; Pcr, phosphocreatine, respectively.
Effect of β3-AR on Cytochrome c Oxidase and F0F1-ATPase Activity in RAP Rabbits
The activity of mitochondrial cytochrome c oxidase and F0F1-ATPase is closely related to energy metabolism in the heart. We found that the activity of F0F1-ATPase was decreased in the P7 group compared with the CON group (P < .05; Figure 4A). Upon injection with BRL37344, the activity of F0F1-ATPase was further depressed (P < .05). The activity of F0F1-ATPase was higher in the L748337 group than that in the P7 group (P < .05). Although the activity of cytochrome c oxidase was not significantly different among the 4 groups, it tended to decrease in the P7 and BRL37344 groups compared with the CON group (Figure 4B).
Figure 4.
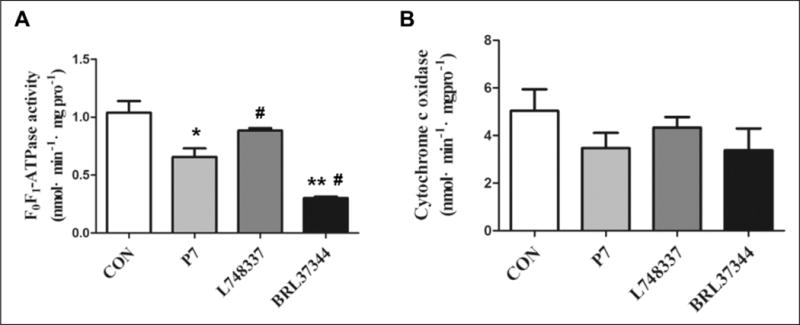
Mitochondrial F0F1-ATPase and cytochrome c oxidase activity in rapid atrial pacing (RAP) rabbits. Activity of atrial (A) F0F1-ATPase and (B) cytochrome c oxidase was determined by fluorescence spectrophotometry. Data are presented as mean ± standard deviation (SD). *P < .05 versus control (CON) group; **P < .01 versus CON group; #P < .05 versus pacing (P7) group.
Effect of β3-AR on mtDNA Content in RAP Rabbits
To determine whether the altered mitochondrial function and energy metabolism in atrial tissues are related to mitochondrial biogenesis, we also estimated the mtDNA content in the 4 groups (Figure 5). In our experiment, mtDNA copy number was significantly decreased after RAP for 7 days compared with the CON group (P < .01). The mtDNA copy number was further decreased by BRL37344 infusion. Conversely, the pacing-induced decrease in mtDNA copy number was significantly increased by inhibition of β3-AR with L748337 (P < .01).
Figure 5.
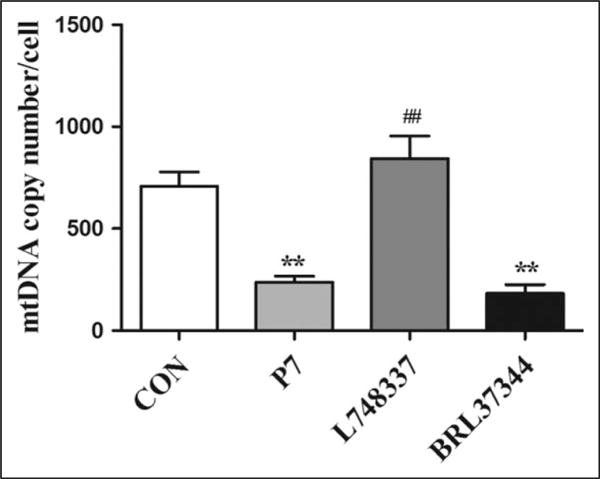
The mitochondrial DNA (mtDNA) content in rapid atrial pacing (RAP) rabbits. The mtDNA copy number was determined by real-time polymerase chain reaction (PCR). Data are presented as mean ± standard deviation (SD). **P < .01 versus control (CON) group; ##P < .01 versus pacing (P7) group.
Effect of β3-AR on Mitochondrial Respiratory Chain in RAP Rabbits
To complete our study, we examined the expression of mitochondrial respiratory chain complexes, which play a role in cardiac mitochondrial oxidative phosphorylation (OXPHOS) and consequent ATP production (Figure 6A and B). The expression of subunit of nicotinamide-adenine dinucleotide dehydrogenase (complex I; NDUFB8), subunit I of cytochrome c oxidase (complex IV; MTCO1), and the α subunit of the F0F1-ATP synthase (complex V; ATP5A) were lower in the P7 group than in the CON group (P < .05). In comparison with the CON group, the expression of the 30-kDa subunit of succinate dehydrogenase (complex II; SDHB) and core protein 2 of the ubiquinol–cytochrome c reductase complex (complex III; UQCRC2) showed a tendency to decrease in the P7 group. Furthermore, β3-AR antagonist treatment resulted in higher levels of NDUFB8, SDHB, UQCRC2, and MTCO1 (P < .05), suggesting that β3-AR inhibition promotes mitochondrial respiratory chain protein synthesis in rabbits following RAP-induced AF.
Figure 6.
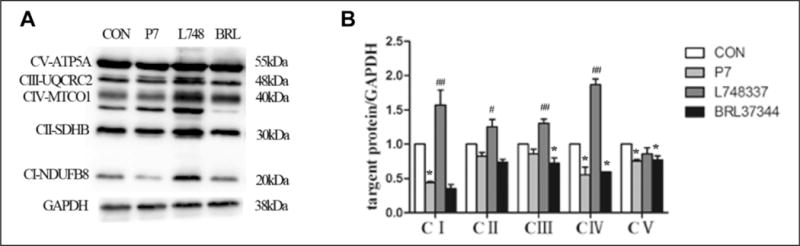
Protein levels of mitochondrial respiratory chain complexes (I, II, III, IV, and V) in rapid atrial pacing (RAP) rabbits. A, Western blot results for NDUFB8, SDHB, UQCRC2, MTCO1, and ATP5A protein expression. B, Quantification of NDUFB8, SDHB, UQCRC2, MTCO1, and ATP5A protein levels in the 4 groups. Data are presented as mean ± SD. *P < .05 versus control (CON) group; **P < .01 versus CON group; #P < .05 versus pacing (P7) group; ##P < .01 versus P7 group. NDUEB8 indicates subunit of nicotinamide-adenine dinucleotide dehydrogenase (complex I); SDHB, 30-kDa subunit of succinate dehydrogenase (complex II); UQCRC2, core protein 2 of ubiquinol-cytochrome c reductase complex (complex III); MTCO1, subunit I of cytochrome c oxidase (complex IV); ATP5A, α subunit of F0F1-ATP synthase (complex V); SD, standard deviation.
Stimulation of β3-AR Resulted in Downregulation of PGC-1α/NRF-1/Tfam in RAP Rabbits
We next investigated the underlying mechanisms of β3-AR–mediated regulation of mitochondrial function and energy metabolism via profiling of PGC-1α, NRF-1, and Tfam expression in the atrial tissues following RAP-induced AF. The levels of PGC-1α and Tfam mRNA were reduced in the P7 and BRL37344 groups compared with the CON group but increased by L748337 when compared with the P7 group (Figure 7A and B). In comparison with the CON group, the protein levels of PGC-1α, NRF-1, and Tfam were significantly reduced in the P7 group (Figure 7C-H). Treatment with L748337 for 7 days triggered the expression of PGC-1α, NRF-1, and Tfam, suggesting that the effect was indeed caused by β3-AR inhibition during RAP-induced AF.
Figure 7.
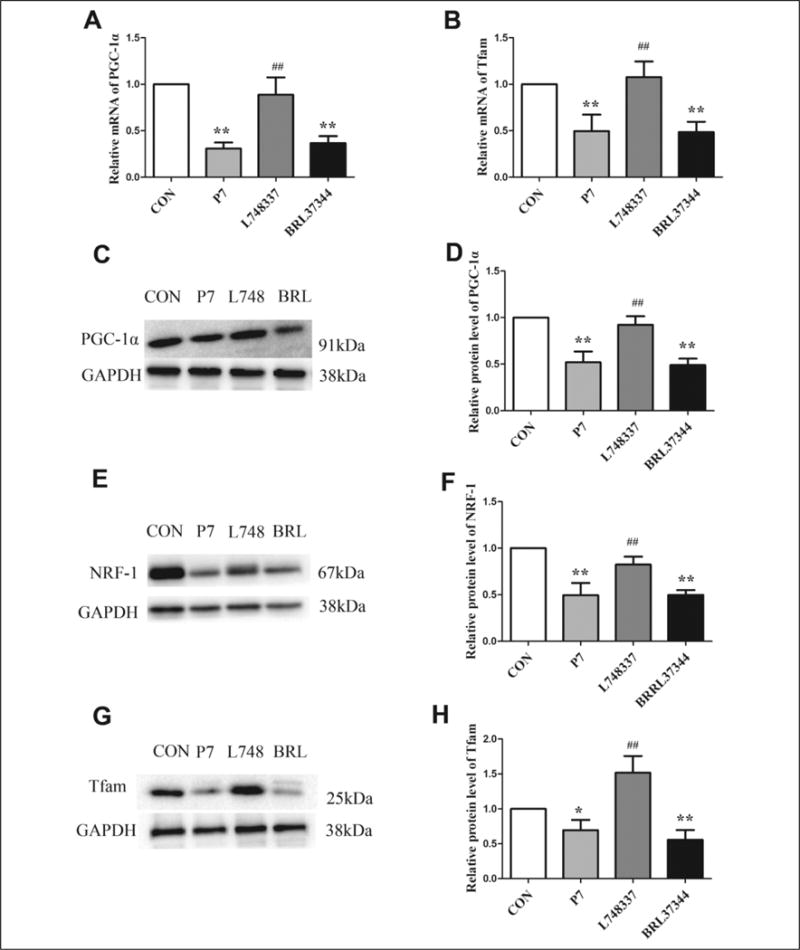
Gene and protein levels of mitochondrial biogenesis-related proteins in rapid atrial pacing (RAP) rabbits. A and B, The gene expression levels of PGC-1α and Tfam among the 4 groups. C, E, and G, Western blot results for PGC-1α, NRF-1, and Tfam protein expression. D, F, and H, Quantification of PGC-1α, NRF-1, and Tfam protein levels in the 4 groups. Data are presented as mean ± SD. *P < .05 versus control (CON) group; **P < .01 versus CON group; #P < .05 versus pacing (P7) group; ##P < .01 versus P7 group. PGC-1α indicates peroxisome proliferator-activated receptor γ coactivator 1α; NRF-1, nuclear respiratory factor 1; Tfam, mitochondrial transcription factor A; SD, standard deviation.
Discussion
This article provides, to the best of our knowledge, the first evidence for a relationship among β3-AR–mediated mitochondrial biogenesis, function, and AF. Rapid pacing for 7 days led to a marked upregulation of β3-AR, followed by a reduction in AERP200ms and an increase in AF induction. The paced atria showed reduced mtDNA content and downregulation of mitochondrial respiratory chain complexes and PGC-1α, NRF-1, and Tfam expression. These effects ultimately hinder the progression of atrial mitochondrial respiratory function and thus affect ATP generation and disturb cardiac energy balance. Considering that SR59230A displays higher affinity at human β1-AR and β2-AR, we administrated L748337 to inhibit β3-AR. In addition, BRL37344 and L748337 were still shown with low affinity for β1-AR and β2-AR, and all experiments were performed in the presence of nadolol, a selective β1- and β2-AR antagonist. The observation that the effects were stimulated by BRL37344 and blocked by L748337 led us to confirm that these responses were β3-AR dependent. Together, these data suggest the importance of β3-AR in regulation of mitochondrial biogenesis and function in a rabbit model of RAP-induced AF.
Activation of β3-AR Leads to AERP Reduction and an Increase in the Rate of AF
Stimulation of β-ARs by the sympathetic nervous system (SNS) is generally involved in regulation of cardiac function in response to acute or chronic stress, while the functional roles of β3-AR in cardiac performance have remained largely unclear and controversial. The expression level of β3-AR is extremely low in normal heart. The β3-AR is activated at higher concentrations of catecholamines, which is relatively resistant to desensitization and acts as a brake for excessive SNS activation.29 Morimoto A et al observed that block of β3-AR with L748337 did not affect heart rate, left ventricle end-systolic pressure or end-diastolic volume in normal dogs, nor was there any effect on cell contractile performance in normal myocytes.25 The expression level of β3-AR is upregulated markedly in HF and AF, suggesting its important role in cardiac pathophysiological disorders rather than physiological function.
Previously, β3-AR stimulation produced negative inotropic effect in human ventricle through Gi/o and endothelial nitric oxide synthase (NOs) signaling.30–33 Beyond that, a study from hypertrophic ventricular cardiomyocytes indicated that β3-AR activation mediated antioxidant and antihypertrophic effects via a neuronal NOs dependent mechanism.34 However, another study from paced canine atria showed increased levels of oxidative stress markers and decreased glutathione peroxidase by regulating p38 MAPK pathway.13 Meanwhile, it was reported that activation of β3-AR increased L-type Ga2+ channels and produced positive inotropic effect in human atrial tissues via the cAMP-dependent pathway.35,36 Similarly, there are controversial reports concerning the effect of β3-AR on effective refractory periods. The β1/2/3-AR knockout resulted in significantly prolonged cardiac conduction times and ERP, which were accompanied by a highly significant reduction of atrial and ventricular arrhythmias.37 In contrast, however, continuous BRL37344 infusion in a canine model significantly reduced the occurrence of ventricular tachycardia,38 and the same result was observed in rats with myocardial infarction.39 One possible reason for these findings might be that β3-AR performs diverse roles in atria compared to its function in ventricles by mediating different signaling pathway. Besides, the functional performance of β3-AR may depend on its expression level, since the expression level of β3-AR is much lower in atria than that in ventricle.30 In our study, β3-AR was significantly upregulated in the paced atria. BRL37344 exposure further increased the level of β3-AR expression, and this was along with a reduction in AEPR200ms in our RAP model, accounting for the higher vulnerability to AF induction. The finding that L748337 reversed the reduction in AEPR200 ms and increase in the AF rate supports our hypothesis that β3-AR inhibition decreased AF susceptibility. In view of the AF induction rate which decreased from 60% to 40% after β3-AR inhibition, persistent atrial remodeling induced by RAP, especially ultrastructural anatomic changes, regional myocardial ischemia, and inflammation, which would not be blocked completely by β3-AR inhibition, might account for this matter.
Stimulation of β3-AR Exacerbates Mitochondrial Function and Energy Metabolism During RAP-Induced AF
Defects in energy metabolism have been implicated in the progression of HF. In 2004, Bilsen et al presented the concept of metabolic remodeling in the failing heart, which included alterations in high-energy phosphate metabolism, mitochondrial dysfunction, and substrate shift utilization.40 The authors posited that these changes would trigger metabolic disturbance and cardiac dysfunction. Reduced ATP levels have also been observed in the atria of hearts from human subjects with AF, but it remains unclear whether this resulted from mitochondrial dysfunction or increased ATP consumption in the cardiomyocytes. In present study, the paced atrial myocardium exhibited a decreased ATP and TAN content accompanied by a decrease in the activity of F0F1-ATPase, indicating a reduction of mitochondrial ATP synthesis. Decreased F0F1-ATPase activity has been reported in a canine model of pacing-induced cardiac failure.41 Others, however, observed no significant differences in F0F1-ATPase activity in rabbits with acute stretch-related AF or in goats subjected to chronic AF.5,42 In fact, short episodes of AF even resulted in increased F0F1-ATPase activity.43 Rather, the increased F0F1-ATPase activity may have resulted from a hypermetabolic state in the early stage of AF. After 7 days of RAP, the cytochrome c oxidase activity tended to decrease compared to the CON group, although the difference did not reach statistical significance. Previous investigations have shown that energy-sensing ATP-sensitive potassium channels was blocked by intracellular ATP,44 and their opening would abbreviate cardiac action potential duration (APD) and shortens the AERP.45,46 Moreover, F0F1-ATPase is tightly related to mitochondrial H+ electrochemical gradient and ATP generation. Mitochondria play an important role in regulation of intracellular calcium homeostasis through Ca2+ uptake with a mitochondrial Ca2+ uniporter and Ca2+ outward transport with a Na+/Ca2+ exchanger or a Na+-independent efflux mechanism, all of which are thought to be driven by the energy of H+ electrochemical gradient.47,48 In addition, mitochondria also provide ATP for activity of calcium pumps. Defects of calcium homeostasis in myocytes would induce a decrease in L-type Ca2+ channel, resulting in shortening of APD and AERP,49 facilitating the initiation and persistence of AF. Thus, reduced ATP content and F0F1-ATPase activity following β3-AR activation in atrial tissues would participate in cardiac electrical remodeling and enhance the propensity for arrhythmia.
Defects of mitochondrial respiratory function have been noted in failed hearts in both animal models and human myocardium.50,51 Similarly, Montaigne et al identified a general downregulation of the mitochondria/OXPHOS gene cluster in atrial tissues of patients in whom AF developed.52 The expression of mitochondrial respiratory chain complex subunits, including ATP5A, MTCO1, and NDUFB8, was downregulated after pacing in our model. The mtDNA encodes for 13 polypeptides of respiratory complexes I, III, IV, and V, which are important for mitochondrial respiratory chain function. Together with our current observation that mtDNA content was reduced by activation of β3-AR in the paced atria, these data suggest that mitochondrial gene transcription and replication are impaired in this model.
The decrease in levels of adenine nucleotides, in content and activity of mitochondrial respiratory chain, and in copy number of mtDNA was further exacerbated by β3-AR agonist, BRL37344. These findings support our notion that mitochondrial respiratory function and subsequent ATP generation are impaired in the atrial tissues following stimulation of β3-AR. The β3-AR activation-dependent decline in ATP was concomitant with a significant reduction in Pcr content. However, the relative reduction of ATP level was great than that of Pcr, which may explain the increased Pcr/ATP ratio in the P7 and BRL37344 groups. In agreement, our previous study showed that fatty acid and glucose oxidation are reduced in rabbits after treatment with the β3-AR agonist, SR59230A, implying a deficiency of intracellular energy.14 Sustained β3-AR activation also enhanced oxygen consumption and weakened mitochondrial efficiency by induction of mitochondrial uncoupling protein.53 Consistent with our notion were observations gained from atrial ultrastructure that indicated the occurrence of myocardial oxygen shortage and low energy metabolism in atria in response to β3-AR activation. Such abnormalities inevitably have a negative impact on global cardiac performance. Given that mtDNA content and mitochondrial respiratory subunit levels were increased by L748337, we suggest that inhibition of β3-AR induces mitochondrial gene transcription and replication, and thus restore cardiac mitochondrial respiratory function and subsequent energy production in AF.
β3-AR Activation-Dependent Downregulation of PGC-1α/NRF-1/Tfam Triggers Alterations in Atrial Tissue During RAP-Induced AF
Energy metabolism is controlled by activation of transcription factors and coactivators such as PGC-1α, which coordinately regulates cardiac long-term remodeling of entire ATP synthesis and utilizing pathways. In cultured cardiomyocytes, forced expression of PGC-1α resulted in increased mitochondrial content and coupled respiration, thereby promoting cardiac mitochondrial biogenesis.54 Peroxisome proliferator-activated receptor γ coactivator 1α is induced by a β3-AR agonist in brown adipose tissues of Wistar rats,55 and levels of this transcription factor are reduced in a rat model of cardiac failure56 as well as in patients who developed new-onset postoperative AF.57 Furthermore, NRF-1 expression is induced in neonatal cardiomyocytes receiving electrical stimulation.58 Chronic β3-AR activation has been reported to stimulate mitochondrial biogenesis and increase metabolic rate in white adipose tissue.59 Nevertheless, prior to the current study, evidence for a direct influence of β3-AR agonists on mitochondrial biogenesis in vivo during AF has been lacking. In contrast to the situation in white adipose tissue, we found that expression of NRF-1 and Tfam was decreased after RAP through β3-AR activation dependent downregulation of PGC-1α, which blocked the expression of nuclear and mitochondrial genes that encode mitochondrial proteins. This was true for both mtDNA content and mitochondrial respiratory subunit protein levels in our study. Regardless, the finding that L748337 upregulates PGC-1α/NRF-1/Tfam signaling supports the notion that this axis is the effective mediator of β3-AR responses during the changes that accompany mitochondrial biogenesis and energy metabolism in RAP-induced AF. Since transthoracic echocardiography showed that RAP-induced AF was not associated with overt cardiac dysfunction in this study (data not shown), it is unlikely that the observed changes of mitochondrial biogenesis were a part of a secondary response to hypertrophic growth signals.
Our work suggests that activation of β3-AR impairs cardiac mitochondrial biogenesis and function, contributing to atrial metabolic remodeling. Clearly, well-balanced metabolism is not only required in order to provide an efficient energy supply for cardiac contractile function and electrical stability but also for protection against oxidative stress, which is involved in the remodeling and progression of AF. Indeed, stimulation of β3-AR is known to promote electrical and structural remodeling in the atrial myocardium upon sustained AF.13,60 Taken together, these data show that β3-AR stimulation in the paced atrium is an important contributor to cardiac remodeling in AF and thus participates in the initiation and maintenance of the disease. We conclude that β3-AR should be further investigated as a promising therapeutic target for AF treatment. In particular, further work is necessary to characterize the interactions of β1-, β2-, and β3-AR in AF, as all 3 receptors are expressed in normal heart, and further research is also needed to understand the functional roles of β3-AR agonist/antagonist in normal heart. Furthermore, it remains to be established whether the β3-AR activation mediated downregulation of metabolic genes represents an early or late event in the clinical course of AF, as it has been reported that initial reduction in the Pcr content of atrial tissues is completely reversed between 8 and 16 weeks of AF. What’s more, it is necessary to establish a rapid-paced atrial myocyte model in which genetic upregulation/silencing of β3-AR is attempted to define the roles and mechanisms of β3-AR during AF at cellular and molecular levels.
Acknowledgments
This research was supported by grants from the “National Natural Science Foundation of China” (Grant 81270252, 81070160, 81100071 and 30971251), “Foundation for Innovative Research Groups of the National Natural Science Foundation of China” (Grant 81121003), and “Scientific Research Found of Heilongjiang Provincial Education Department” (Grant 12521206).
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Authors’ Contribution
Dong, J contributed to acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript; and agrees to be accountable for all aspects of work ensuring integrity and accuracy; Zhao, J contributed to acquisition and analysis; Zhang, M contributed to acquisition; Liu, G contributed to analysis; Wang, X contributed to acquisition; Liu, Y contributed to acquisition; Yang, N contributed to acquisition; Liu, Y contributed to acquisition; Zhao, G contributed to acquisition; Sun, J contributed to acquisition; Tian, J contributed to acquisition; Li, Y contributed to conception; Cheng, C contributed to conception and design; Wei, L contributed to conception and design; Li, W contributed to conception and design, critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54(2):230–246. doi: 10.1016/s0008-6363(02)00258-4. [DOI] [PubMed] [Google Scholar]
- 2.Krogh-Madsen T, Abbott GW, Christini DJ. Effects of electrical and structural remodeling on atrial fibrillation maintenance: a simulation study. PLoS Comput Biol. 2012;8(2):e1002390. doi: 10.1371/journal.pcbi.1002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taegtmeyer H, Wilson CR, Razeghi P, Sharma S. Metabolic energetics and genetics in the heart. Ann N Y Acad Sci. 2005;1047:208–218. doi: 10.1196/annals.1341.019. [DOI] [PubMed] [Google Scholar]
- 4.Bernauer W. Release of adenine nucleotide metabolites by toxic concentrations of cardiac glycosides. Basic Res Cardiol. 1994;89(4):308–321. doi: 10.1007/BF00795200. [DOI] [PubMed] [Google Scholar]
- 5.Kalifa J, Maixent JM, Chalvidan T, et al. Energetic metabolism during acute stretch-related atrial fibrillation. Mol Cell Biochem. 2008;317(1-2):69–75. doi: 10.1007/s11010-008-9832-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tu T, Zhou S, Liu Z, Li X, Liu Q. Quantitative proteomics of changes in energy metabolism-related proteins in atrial tissue from valvular disease patients with permanent atrial fibrillation. Circ J. 2014;78(4):993–1001. doi: 10.1253/circj.cj-13-1365. [DOI] [PubMed] [Google Scholar]
- 7.Lai LP, Tsai CC, Su MJ, et al. Atrial fibrillation is associated with accumulation of aging-related common type mitochondrial DNA deletion mutation in human atrial tissue. Chest. 2003;123(2):539–544. doi: 10.1378/chest.123.2.539. [DOI] [PubMed] [Google Scholar]
- 8.Akar FG, O’Rourke B. Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol Ther. 2011;131(3):287–294. doi: 10.1016/j.pharmthera.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown DA, O’Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res. 2010;88(2):241–249. doi: 10.1093/cvr/cvq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morillo CA, Klein GJ, Jones DL, Guiraudon CM. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation. 1995;91(5):1588–1595. doi: 10.1161/01.cir.91.5.1588. [DOI] [PubMed] [Google Scholar]
- 11.Montaigne D, Marechal X, Lefebvre P, et al. Mitochondrial dysfunction as an arrhythmogenic substratea translational proof-of-concept study in patients with metabolic syndrome in whom postoperative atrial fibrillation develops. J Am Coll Cardiol. 2013;62(16):1466–1473. doi: 10.1016/j.jacc.2013.03.061. [DOI] [PubMed] [Google Scholar]
- 12.Ausma J, Wijffels M, Thoné F, Wouters L, Allessie M, Borgers M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. 1997;96(9):3157–3163. doi: 10.1161/01.cir.96.9.3157. [DOI] [PubMed] [Google Scholar]
- 13.Sheng L, Shen Q, Huang K, et al. Upregulation of beta3-adrenergic receptors contributes to atrial structural remodeling in rapid pacing induced atrial fibrillation canines. Cell Physiol Biochem. 2012;30(2):372–381. doi: 10.1159/000339031. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Geng J, Li Y, et al. Beta3-adrenoceptor mediates metabolic protein remodeling in a rabbit model of tachypacing-induced atrial fibrillation. Cell Physiol Biochem. 2013;32(6):1631–1642. doi: 10.1159/000356599. [DOI] [PubMed] [Google Scholar]
- 15.Huang C, Chen D, Xie Q, Yang Y, Shen W. Nebivolol stimulates mitochondrial biogenesis in 3t3-l1 adipocytes. Biochem Biophys Res Commun. 2013;438(1):211–217. doi: 10.1016/j.bbrc.2013.07.055. [DOI] [PubMed] [Google Scholar]
- 16.Branco AF, Sampaio SF, Wieckowski MR, Sardao VA, Oliveira PJ. Mitochondrial disruption occurs downstream from beta-adrenergic overactivation by isoproterenol in differentiated, but not undifferentiated h9c2 cardiomyoblasts: Differential activation of stress and survival pathways. Int J Biochem Cell Biol. 2013;45(11):2379–2391. doi: 10.1016/j.biocel.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator pgc-1. Cell. 1999;98(1):115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 18.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18(4):357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 19.Shao D, Liu Y, Liu X, et al. Pgc-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by nrf-1 and err alpha. Mitochondrion. 2010;10(5):516–527. doi: 10.1016/j.mito.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 20.Huo L, Scarpulla RC. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol. 2001;21(2):644–654. doi: 10.1128/MCB.21.2.644-654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsson NG, Wang J, Wilhelmsson H, et al. Mitochondrial transcription factor a is necessary for mtdna maintenance and embryogenesis in mice. Nat Genet. 1998;18(3):231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 22.Andersson U, Scarpulla RC. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol. 2001;21(11):3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39(1):60–76. doi: 10.1016/s0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 24.Audigane L, Kerfant BG, El Harchi A, et al. Rabbit, a relevant model for the study of cardiac beta 3-adrenoceptors. Exp Physiol. 2009;94(4):400–411. doi: 10.1113/expphysiol.2008.045179. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto A, Hasegawa H, Cheng HJ, Little WC, Cheng CP. Endogenous beta3-adrenoreceptor activation contributes to left ventricular and cardiomyocyte dysfunction in heart failure. Am J Physiol Heart Circ Physiol. 2004;286(6):H2425–H2433. doi: 10.1152/ajpheart.01045.2003. [DOI] [PubMed] [Google Scholar]
- 26.Pelat M, Verwaerde P, Galitzky J, et al. High isoproterenol doses are required to activate beta3-adrenoceptor-mediated functions in dogs. J Pharmacol Exp Ther. 2003;304(1):246–253. doi: 10.1124/jpet.102.040691. [DOI] [PubMed] [Google Scholar]
- 27.Volonte MG, Yuln G, Quiroga P, Consolini AE. Development of an hplc method for determination of metabolic compounds in myocardial tissue. J Pharm Biomed Anal. 2004;35(3):647–653. doi: 10.1016/j.jpba.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Chen YH, Lin WW, Liu CS, Hsu LS, Lin YM, Su SL. Caveolin-1 provides palliation for adverse hepatic reactions in hypercholesterolemic rabbits. PLoS One. 2014;9(1):e71862. doi: 10.1371/journal.pone.0071862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Summers R, Kompa A, Roberts S. B-adrenoceptor subtypes and their desensitization mechanisms. J Auton Pharmacol. 1997;17(6):331–343. doi: 10.1046/j.1365-2680.1997.00055.x. [DOI] [PubMed] [Google Scholar]
- 30.Gauthier C, Tavernier G, Charpentier F, Langin D, Le Marec H. Functional beta3-adrenoceptor in the human heart. J Clin Invest. 1996;98(2):556–562. doi: 10.1172/JCI118823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozec B, Gauthier C. Beta3-adrenoceptors in the cardiovascular system: Putative roles in human pathologies. Pharmacol Ther. 2006;111(3):652–673. doi: 10.1016/j.pharmthera.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Gauthier C, Leblais V, Kobzik L, et al. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. 1998;102(7):1377–1384. doi: 10.1172/JCI2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL. Upregulation of beta(3)-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103(12):1649–1655. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- 34.Watts VL, Sepulveda FM, Cingolani OH, et al. Anti-hypertrophic and anti-oxidant effect of beta3-adrenergic stimulation in myocytes requires differential neuronal nos phosphorylation. J Mol Cell Cardiol. 2013;62:8–17. doi: 10.1016/j.yjmcc.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skeberdis VA, Gendviliene V, Zablockaite D, et al. Beta3-adrenergic receptor activation increases human atrial tissue contractility and stimulates the l-type ca2+ current. J Clin Invest. 2008;118(9):3219–3227. doi: 10.1172/JCI32519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sennitt MV, Kaumann AJ, Molenaar P, et al. The contribution of classical (beta1/2-) and atypical beta-adrenoceptors to the stimulation of human white adipocyte lipolysis and right atrial appendage contraction by novel beta3-adrenoceptor agonists of differing selectivities. J Pharmacol Exp Ther. 1998;285(3):1084–1095. [PubMed] [Google Scholar]
- 37.Stockigt F, Brixius K, Lickfett L, et al. Total beta-adrenoceptor knockout slows conduction and reduces inducible arrhythmias in the mouse heart. PLoS One. 2012;7(11):e49203. doi: 10.1371/journal.pone.0049203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou S, Tan AY, Paz O, et al. Antiarrhythmic effects of beta3-adrenergic receptor stimulation in a canine model of ventricular tachycardia. Heart Rhythm. 2008;5(2):289–297. doi: 10.1016/j.hrthm.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou L, Zhang P, Cheng Z, et al. Altered circadian rhythm of cardiac beta3-adrenoceptor activity following myocardial infarction in the rat. Basic Res Cardiol. 2011;106(1):37–50. doi: 10.1007/s00395-010-0110-7. [DOI] [PubMed] [Google Scholar]
- 40.van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: The cardiac burn-out syndrome? Cardiovasc Res. 2004;61(2):218–226. doi: 10.1016/j.cardiores.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 41.Marin-Garcia J, Goldenthal MJ, Moe GW. Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc Res. 2001;52(1):103–110. doi: 10.1016/s0008-6363(01)00368-6. [DOI] [PubMed] [Google Scholar]
- 42.Ausma J, Coumans WA, Duimel H, Van der Vusse GJ, Allessie MA, Borgers M. Atrial high energy phosphate content and mitochondrial enzyme activity during chronic atrial fibrillation. Cardiovasc Res. 2000;47(4):788–796. doi: 10.1016/s0008-6363(00)00139-5. [DOI] [PubMed] [Google Scholar]
- 43.Barbey O, Pierre S, Duran MJ, Sennoune S, Levy S, Maixent JM. Specific up-regulation of mitochondrial f0f1-atpase activity after short episodes of atrial fibrillation in sheep. J Cardiovasc Electrophysiol. 2000;11(4):432–438. doi: 10.1111/j.1540-8167.2000.tb00339.x. [DOI] [PubMed] [Google Scholar]
- 44.Sasaki N, Sato T, Marbán E, O’Rourke B. Atp consumption by uncoupled mitochondria activates sarcolemmal katp channels in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2001;280(4):H1882–H1888. doi: 10.1152/ajpheart.2001.280.4.H1882. [DOI] [PubMed] [Google Scholar]
- 45.Le Grand B, Hatem S, Le Heuzey JY, Deroubaix E, Benitah JP, Coraboeuf E. Pro-arrhythmic effect of nicorandil in isolated rabbit atria and its suppression by tolbutamide and quinidine. Eur J Pharmacol. 1992;229:91–96. doi: 10.1016/0014-2999(92)90290-k. [DOI] [PubMed] [Google Scholar]
- 46.Faivre JF, Findlay I. Action potential duration and activation of atp-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim Biophys Acta. 1990;1029(1):167–172. doi: 10.1016/0005-2736(90)90450-3. [DOI] [PubMed] [Google Scholar]
- 47.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol Cell Physiol. 1990;258(5 pt 1):C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 48.Poburko D, Demaurex N. Regulation of the mitochondrial proton gradient by cytosolic ca(2)(+) signals. Pflugers Arch. 2012;464(1):19–26. doi: 10.1007/s00424-012-1106-y. [DOI] [PubMed] [Google Scholar]
- 49.Yue L, Feng J, Gaspo R, Li G-R, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res. 1997;81(4):512–525. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]
- 50.Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88(5):529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 51.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32(12):2361–2367. doi: 10.1006/jmcc.2000.1266. [DOI] [PubMed] [Google Scholar]
- 52.Montaigne D, Marechal X, Lefebvre P, et al. Mitochondrial dysfunction as an arrhythmogenic substrate: A translational proof-of-concept study in patients with metabolic syndrome in whom post-operative atrial fibrillation develops. J Am Coll Cardiol. 2013;62(16):1466–1473. doi: 10.1016/j.jacc.2013.03.061. [DOI] [PubMed] [Google Scholar]
- 53.Nakamura Y, Nagase I, Asano A, et al. Beta 3-adrenergic agonist up-regulates uncoupling proteins 2 and 3 in skeletal muscle of the mouse. J Vet Med Sci. 2001;63(3):309–314. doi: 10.1292/jvms.63.309. [DOI] [PubMed] [Google Scholar]
- 54.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106(7):847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gomez-Ambrosi J, Fruhbeck G, Martinez JA. Rapid in vivo pgc-1 mrna upregulation in brown adipose tissue of wistar rats by a beta(3)-adrenergic agonist and lack of effect of leptin. Mol Cell Endocrinol. 2001;176(1-2):85–90. doi: 10.1016/s0303-7207(01)00451-8. [DOI] [PubMed] [Google Scholar]
- 56.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003;551(pt 2):491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun DM, Yuan X, Wei H, et al. Impaired myocardium energetics associated with the risk for new-onset atrial fibrillation after isolated coronary artery bypass graft surgery. Coron Artery Dis. 2014;25(3):224–229. doi: 10.1097/MCA.0000000000000081. [DOI] [PubMed] [Google Scholar]
- 58.Xia Y, Buja LM, Scarpulla RC, McMillin JB. Electrical stimulation of neonatal cardiomyocytes results in the sequential activation of nuclear genes governing mitochondrial proliferation and differentiation. Proc Natl Acad Sci U S A. 1997;94(21):11399–11404. doi: 10.1073/pnas.94.21.11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Granneman JG, Li P, Zhu Z, Lu Y. Metabolic and cellular plasticity in white adipose tissue i: Effects of beta3-adrenergic receptor activation. Am J Physiol Endocrinol Metab. 2005;289(4):E608–E616. doi: 10.1152/ajpendo.00009.2005. [DOI] [PubMed] [Google Scholar]
- 60.Yu J, Li W, Li Y, et al. Activation of β3-adrenoceptor promotes rapid pacing-induced atrial electrical remodeling in rabbits. Cell Physiol Biochem. 2011;28(1):87–96. doi: 10.1159/000331717. [DOI] [PubMed] [Google Scholar]