

Abstract
Essentials.
Part 1 of this review summarizes recent findings on the exquisite balance between activating and inhibitory mechanisms that control platelet adhesiveness in the circulation and at sites of vascular injury.
Part 2 of this review discusses molecular differences and similarities for how platelets secure vascular integrity at sites of inflammation and at sites of mechanical trauma.
In this brief review paper, we will summarize the State‐of‐the‐Art on how platelet reactivity is regulated in circulation and at sites of vascular injury. Our review discusses recent and ongoing work presented at this year's International Society on Thrombosis and Haemostasis (ISTH) meeting, on the role of platelets in (i) classical hemostasis at sites of mechanical injury, and (ii) the maintenance of vascular integrity at sites of inflammation.
Keywords: adhesion, hemostasis, platelets, signaling, vascular integrity
1. CLASSICAL HEMOSTASIS—BALANCING PLATELET ADHESIVENESS IN CIRCULATION AND AT THE VASCULAR INTERFACE
Platelets are critical for the body's response to vascular injury. To form a hemostatic plug, they rely on a signal transduction machinery, which is optimized to sense and respond to minor changes in the environment, and to mediate a near‐immediate conversion of cell‐surface integrins from an anti‐adhesive to a pro‐adhesive state. While patrolling the vasculature, however, platelet activation has to be prevented in order to avoid thrombocytopenia and/or thrombosis. In this first section of our review, we will discuss the exquisite balance between activating and inhibitory mechanisms that control platelet adhesiveness in the circulation and at sites of vascular injury.
To efficiently plug a vascular lesion and prevent blood loss into the surrounding tissue, platelets have evolved highly specialized adhesion mechanisms, which enable cell‐matrix and cell‐cell interactions in the presence of fluid shear stress. Initial platelet recruitment at sites of vascular injury is mediated by glycoprotein (GP) Ib‐V‐IX complex binding to von Willebrand factor (VWF). This interaction has a sufficiently fast association rate to enable platelet capture even at very high shear rates, but it supports only transient adhesion. Thus, it serves the purpose of decelerating platelets in the proximity of the lesion, and allow enough time for the platelets to become activated and engage more stable adhesive interactions.1 Firm adhesion is mediated by receptors that belong to the integrin superfamily. Integrins are transmembrane αβ heterodimers that are expressed on the surface of resting platelets in a low‐affinity binding state. Stimulation of platelets, via extracellular matrix (ECM) molecules exposed at the site of injury (eg, collagen), and/or locally generated soluble agonists (eg, thrombin), triggers intracellular signaling cascades that ultimately lead to the inside‐out activation of integrins. Important in this process is the recruitment of the FERM‐domain containing proteins, TALIN2, 3 and KINDLIN3,4 to the integrin cytoplasmic tail.5 Once bound, TALIN triggers conformational changes that increase integrin ligand‐binding affinity,6 while KINDLIN3 promotes multivalent ligand binding by increasing the local density (avidity) of TALIN‐activated integrins.7 TALIN and KINDLIN3 are also directly linked to the cytoskeleton and increasing evidence presented at the 2017 ISTH conference suggests that altered cytoskeletal dynamics may result in dysregulated integrin activation.8, 9, 10 Active β1 integrins (α2β1, α6β1, α5β1) support adhesion to the ECM for the first platelet layer, which then becomes a new reactive surface for further platelet deposition. Active αIIbβ3 integrins, which are extremely abundant on the platelet surface, bind multivalent plasma proteins (eg, fibrinogen or VWF) that bridge adjacent activated platelets and thus support platelet‐platelet cohesion.11, 12
The main signals that drive the three‐dimensional growth of the hemostatic plug are soluble agonists (Figure 1), such as the product of the coagulation cascade, thrombin, or the second wave mediators released from activated platelets, adenosine diphosphate (ADP) and thromboxane (Tx)A2. Being soluble, these agonists can recruit free‐flowing platelets into the growing thrombus. Soluble agonists bind to G protein coupled receptors (GPCRs), which induce a very rapid and robust signaling response.13, 14, 15 The key to GPCR signaling is the activation (GTP loading) of heterotrimeric G proteins. The GPCR hereby serves as a guanine nucleotide exchange factor (GEF), which induces the release of GDP from the alpha subunit of the heterotrimeric G protein. Since GTP loading of the nucleotide‐free G protein happens very rapidly, this on‐switch mechanism provides a perfect system to transduce signals on a millisecond scale required for platelet adhesion under high shear stress conditions.
Figure 1.
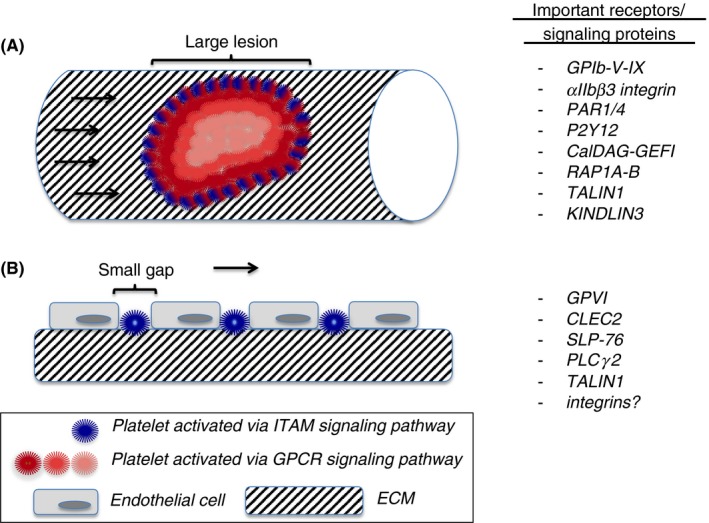
Platelets at the vascular interface. (A) Mechanical injury. Shown is a hemostatic plug with platelets activated via the ITAM signaling pathway (blue color) or the GPCR signaling pathway (red colors). Lighter shades of red indicate weaker cellular activation. ITAM signaling plays a minor role in hemostatic plug formation, as only few platelets (blue) are in direct contact with the extracellular matrix (ECM). In contrast, GPCRs like PAR1/4 and P2Y12, receptors that sense soluble agonists, play a critical role. The GPIb‐V‐IX complex and integrin αIIbβ3 are critical for transient and firm platelet adhesion, respectively. The CalDAG‐GEFI/RAP1A‐B signaling pathway is critical for rapid integrin activation following GPCR engagement. (B) Inflammation. Shown are small gaps between endothelial cells, induced by infiltrating leukocytes (not shown), and single platelets that occupy these gaps. Due to their direct contact with the ECM, platelet adhesion at sites of inflammation is strongly dependent on the ITAM receptors, GPVI and CLEC2, and downstream signaling molecules, such as SLP‐76 and PLCγ2
To keep these highly sensitive receptors in check, activation by soluble agonists is not an all‐or‐nothing process. In fact, it is a two‐step process, carried out by dual receptor systems. Thrombin stimulates human platelets through the protease‐activated receptors, PAR1 and PAR4, while ADP activation occurs through the purinergic receptors, P2Y1 and P2Y12. PAR1 is triggered by very low concentrations of thrombin, but it also rapidly desensitizes. Thus, it is critical for high sensitivity to thrombin and a fast but reversible integrin activation response. PAR4 binds thrombin with much lower affinity, but it also internalizes more slowly and thus generates a steady and sustained integrin activation signal.16, 17, 18 Similarly, the ADP receptor P2Y1, which is not very abundant on the platelet surface,19 is rapidly desensitized.20, 21 Thus, it can only cause a rapid but short‐lived rise of the cytosolic calcium concentration and needs co‐signaling through the P2Y12 receptor to support platelet aggregation.22, 23 So both PAR1 and P2Y1 are very sensitive and provide a fast but reversible activation response, while PAR4 and P2Y12 are required to sustain the signal. As only the stimulation of both receptors provides an integrated response that is both rapid and sustained, this provides an elegant mechanism to limit thrombus growth in the outer layers of the thrombus, where agonist concentrations diminish.
Duration and intensity of the GPCR signal are limited not only by receptor desensitization but also by Regulator of G‐protein Signaling (RGS) proteins that increase the rate of GTP hydrolysis and return G proteins to the inactive state. Consistently, it was previously shown that knockdown of RGS18,24, 25 RGS10,26 or mutation of the RGS binding site on Gαi27 results in increased platelet reactivity. At this ISTH conference, Gupta et al. confirmed that knockdown of RGS10 results in a gain of platelet function, in particular in response to stimulation by the second wave mediators, ADP and TxA2.28 Unexpectedly, the same group reported preliminary studies that defective RGS binding to Gαq causes a decrease in platelet activation.29 The reasons for this surprising platelet hypofunction are currently unclear.
In addition to GPCRs, platelets sense the environment through the ITAM‐coupled receptors GPVI and CLEC‐2, which predominantly interact with the ECM protein collagen and the extravascular protein podoplanin, respectively. The ITAM signaling cascade evolved from immune cells that signal over much longer time scales than platelets (minutes vs seconds). And even though GPVI has adapted to meet the accelerated temporal needs of hemostasis,30 the ITAM signaling response is still slower compared to the GPCR response. Consistently, the mild bleeding phenotype of patients31, 32 or mice33, 34 lacking GPVI suggests that this receptor has a minor role in classical hemostasis when compared to the major GPCRs.15 Surprisingly, GPVI was shown to be more important for thrombus stabilization than for thrombus initiation,35 suggesting the existence of ligands other than collagen. Fibrin, a protein deposited throughout the hemostatic plug, could be this ligand.36, 37 At this ISTH meeting, Haining et al. reported that CLEC‐2 also appears to have a function in thrombus stabilization, independent of its signaling function and thus potentially by binding an unknown intravascular ligand.38, 39 While it is not the major player in classical hemostasis, ITAM signaling is critical for another form of hemostasis that is responsible for maintaining vascular integrity at sites of inflammation40 (see next section).
Most GPCRs and ITAM‐coupled receptors induce the activation of phospholipase C (PLC), a key enzyme in the generation of the second messengers, calcium and diacylglycerol. At this ISTH meeting, we presented definitive evidence that the small GTPase RAP1 is the main signaling node that integrates these second messenger signals with integrin activation.41 Previous studies in other cells42, 43, 44, 45 and in heterologous cell lines expressing αIIbβ346 suggested that RAP1 controls integrin conversion to a high affinity state by recruiting TALIN to the plasma membrane. However, platelets lacking the most abundant RAP isoform, RAP1B, only displayed a partial integrin activation defect.47 Since systemic deficiency of the closely related isoform, RAP1A was reported to have no effect on platelet aggregation (unpublished observation47), this led to the wrong belief that there is a powerful RAP1‐independent signaling pathway that can mediate integrin activation in platelets.48 Employing mice where Rap1a and/or Rap1b were conditionally deleted in the megakaryocyte/platelet lineage, we have produced preliminary data (presented at the ISTH meeting) supporting a role for both RAP1A and RAP1B in platelet integrin activation.41 When comparing the Rap1a/b‐double deficient mice to Talin1‐deficient mice, we found that there is very limited integrin activation, mediated by TALIN1, in platelets lacking both RAP1 isoforms. Importantly, this residual RAP1‐indendent integrin activation can only support slow and sub‐maximal aggregation in vitro, while it was not sufficient to support hemostatic plug formation in vivo.41 Thus, the RAP1/TALIN signaling axis is critical in platelet integrin activation and classical hemostasis.
There is a very rational explanation for why RAP1A and RAP1B are critical in hemostatic plug formation. RAP GTPases, like the heterotrimeric G proteins, are molecular switches that can be turned on rapidly by GEF‐mediated GTP for GDP exchange. The main RAP‐GEF in platelets is CalDAG‐GEFI, a multidomain protein predominantly found in blood cells.49, 50 Studies in mice51, 52 and humans53, 54, 55 lacking functional CalDAG‐GEFI strongly suggest that its nucleotide exchange activity towards RAP1 is directly controlled by the binding of calcium to a pair of EF hands in the regulatory domain. Thus, CalDAG‐GEFI can sense agonist‐induced changes in the cytosolic calcium levels.56 The result is a near‐immediate integration of second messenger generation and integrin activation. Lack of CalDAG‐GEFI leads to impaired thrombus formation and bleeding.51, 53, 54, 55, 57, 58, 59, 60
CalDAG‐GEFI‐mediated RAP1 activation is antagonized by the GTPase activating protein (GAP), RASA3.61 Like RGS proteins, GAPs are important to enhance GTP hydrolysis and to return small GTPases back to the inactive state. RASA3 is active in circulating platelets, where it is required to counteract any unwanted RAP activation and to keep the cells in a quiescent state. Mice lacking Rasa3 are characterized by platelet preactivation and severe thrombocytopenia due to increased platelet clearance. At sites of vascular injury, however, this negative feedback is detrimental to hemostatic plug formation. For firm platelet adhesion to occur, RASA3 activity has to be down‐modulated. Critical for the down‐modulation of Rasa3 activity is signaling via the Gi‐coupled receptor for ADP, P2Y12.61 Thus, similar to the GPCR dual receptor systems, RAP activation is a two‐step process. A rapid but reversible activation step, mediated by calcium/CalDAG‐GEFI signaling, is followed by a second step, RASA3 inactivation, that is required to sustain the signal. Again, this two‐step system is important to facilitate controlled platelet adhesion and thrombus growth under high shear stress conditions.
What is special about the RAP1 signaling pathway, however, is how tightly controlled it has to be in order to avoid serious complications for the organism. The affinity for calcium of the CalDAG‐GEFI EF hands is ~80 nmol L−1,56 a value that is only minimally higher than the cytosolic calcium concentration measured in resting platelets (~20‐50 nmol L−1). Given these numbers, it can be concluded that (i) only minimal changes in cytosolic calcium are required to mount a robust CalDAG‐GEFI response, and (ii) that some CalDAG‐GEFI protein exists in an active conformation even in circulating, quiescent platelets. Consistent with these conclusions, complete CalDAG‐GEFI deficiency is ill tolerated as it causes moderate to severe bleeding in humans and mice.51, 53, 54, 55, 58, 59, 60 At the same time, CalDAG‐GEFI/RAP1 signaling has to be restrained by RASA3 in circulating platelets to avoid severe thrombocytopenia and blood‐lymphatic mixing in the developing embryo. Together, these specialized mechanisms ensure the tight regulation of platelet adhesiveness, which is crucial for the platelets’ ability to efficiently form a stable and self‐limiting hemostatic plug at sites of vascular injury. However, platelets are also important for the maintenance of vascular integrity at sites of inflammation,62 where a different platelet response is required to seal small gaps in the endothelial lining.
2. VASCULAR INTEGRITY VS HEMOSTATIC PLUG FORMATION—LESIONS OF DIFFERENT SIZE REQUIRE UNIQUE PLATELET RESPONSES
As outlined above, platelet function during hemostatic plug formation depends strongly on G protein signaling. Hemostatic plugs are aggregates of platelets, where only few of these cells make direct contact with the ECM in the damaged vascular wall (Figure 1A). GPCRs sense soluble agonists that diffuse into the lesion, and they initiate a platelet signaling response characterized by the very rapid activation of PLC and the generation of second messengers. The on‐off nature of the small GTPase system provides a unique system to quickly transduce the signal to the integrin and thus facilitate platelet adhesion under shear stress conditions. Interestingly, recent studies suggest that platelet GTPase signaling is much less important in situations where vascular lesions are very small. At sites of inflammation, single platelets plug holes in the vascular lining (Figure 1B),63 tiny lesions induced by transmigrating inflammatory cells.64 Without platelets (severe thrombocytopenia), these vessels become leaky for RBCs, causing hemorrhage in the affected areas.62 We have recently demonstrated that GPCRs are not critical for platelet function in this setting.40, 65 Instead, vascular integrity at sites of inflammation depends strongly on platelet ITAM signaling, mediated by GPVI and CLEC‐2. This ISTH meeting provided important new information on the molecular mechanisms underlying this unique form of hemostasis. In our ongoing studies, we observed that mice lacking both RAP1 isoforms, RAP1A and RAP1B, do not bleed at sites of inflammation, even though classical hemostasis is strongly impaired in these mice.41 Our findings of significant bleeding at sites of inflammation in the skin (reverse passive Arthus reaction) of Talin1‐deficient mice, however, do suggest that platelet integrin signaling is critical in the prevention of inflammatory hemorrhage. There are several questions that arise from these observations. For example, why do we observe inflammatory bleeding in Talin1‐deficient mice while no such bleeding was observed in mice lacking αIIbβ362? Like GPCRs, αIIbβ3 is particularly important for platelet‐platelet aggregate formation. The adhesion to the ECM, however, can also be mediated by β1 integrins, such as α2β1 (collagen) and α6β1 (laminin). Thus, it is likely that a redundancy between β3 and β1 integrins in mediating platelet adhesion to the ECM protects β3‐deficient mice from inflammatory hemorrhage, a hypothesis that warrants further investigation. Another important question is why Rap1‐deficiency does not cause inflammatory hemorrhage like Talin1‐deficiency, while both RAP1 and TALIN1 are critical for classical hemostasis? Our preliminary studies presented at the ISTH conference show that platelets lacking RAP1A and RAP1B retain a weak but significant ability to activate integrins in response to agonist stimulation.41 The resulting slow aggregation response observed in vitro, however, is not sufficient for hemostatic plug formation at sites of mechanical injury.41 How RAP1‐deficient platelets manage to prevent inflammatory hemorrhage is currently not clear. A likely explanation is that shear stress is less of a factor in this situation as platelets may simply get trapped in the ECM (Figure 1B). If that is the case then the delay in integrin activation in Rap1‐deficient platelets would be negated by this trapping mechanism, which retains platelets in the EC gaps long enough for integrins to get engaged.
Our preliminary studies also demonstrate that inflammatory hemorrhage is significantly less severe in Talin1‐deficient mice when compared to thrombocytopenic mice.41 This, of course, begs the question about the nature of this TALIN (maybe integrin)‐independent contribution of platelets to vascular integrity in inflammation. One potential explanation that was brought forward at the ISTH meeting is that CLEC2, the hemITAM signaling receptor critical in this response, may serve as an adhesion receptor in addition to its function in platelet activation. As outlined in the abstract by Haining et al., studies in mice expressing a signaling defective mutant of CLEC2 (Y7A KI) suggest a signaling‐independent contribution of the CLEC2 ectodomain to thrombus stability38, 39 A similar signaling‐independent, adhesive role was also reported for GPVI.36, 37
Another interesting study on this topic was presented at ISTH 2017 by Rayes et al., who demonstrated that podoplanin, the ligand for CLEC2, is found in the deeper layers of the vasculature.66 The authors suggest that the CLEC2‐podoplanin interaction is required as a fail‐safe in case GPVI is malfunctional, a hypothesis that is supported by the observation that megakaryocyte/platelet‐specific deletion of CLEC2 in mice does not cause increased inflammatory hemorrhage, unless the mice are crossed with GPVI‐deficient mice.66 Interestingly, the preliminary studies by Rayes et al. also suggest that platelets contribute to vascular integrity in LPS challenged lungs by a GPVI/CLEC2‐independent but GPIbα‐dependent mechanism, findings that are in conflict with previous findings.40 Additional studies are required to clarify whether there is indeed an organ and/or inflammatory trigger‐specific platelet response in this form of hemostasis.
Release of vasoactive substances from platelet granules has been proposed as another, TALIN‐independent, contribution of platelets to vascular integrity in inflammation.67, 68 However, elegant studies by Deppermann et al., presented at the 2017 ISTH meeting and recently published,69 question this hypothesis. Utilizing mice with a defect in both alpha and dense granule secretion (Unc13d −/− /Nbeal2 −/−), the authors investigated the contribution of platelet granule content on vascular integrity in the two most commonly used models of localized inflammation: the reverse passive Arthus reaction in the skin, and LPS‐induced inflammation in the lung. No increase in bleeding was observed in inflamed tissues of Unc13d −/− /Nbeal2 −/− mice when compared to controls. This result does not exclude a contribution of platelet granule secretion to vascular integrity in inflammation, but it does suggest that such a contribution would not be required during the acute phase of the hemostatic response. Interestingly, the authors did observe significant bleeding in Unc13d −/− /Nbeal2 −/− mice challenged in a model of thrombo‐inflammation in the brain. This finding is interesting as previous studies demonstrated that hemostasis in this model is dependent on integrin αIIbβ3, but not GPVI and GPIb‐V‐IX.70 These results could be explained by (i) brain ischemia‐reperfusion injury inducing an inflammatory injury to the vasculature that requires a unique platelet response, or (ii) intermediate size lesions being generated following brain ischemia‐reperfusion injury, which require platelets to release their granules and to aggregate.
3. CONCLUSIONS
Decades of research have uncovered many of the molecular mechanisms underlying platelet activation and adhesion at sites of vascular injury. In this review, we provide an overview of the main regulatory mechanisms controlling platelet reactivity, both in circulation and at the vascular interface. We also discuss the latest developments in this field, presented at this year's ISTH meeting. These studies provide critical new information on the platelet signaling machinery, filling gaps but also challenging our current thinking of how platelets work. We are excited to see whether the many important scientific questions raised by these studies can be answered in time for presentation at the next ISTH meeting.
RELATIONSHIP DISCLOSURE
None of the authors have any disclosures relevant to this paper.
AUTHOR CONTRIBUTIONS
L. Stefanini drafted and reviewed literature and ISTH abstracts for this manuscript. W. Bergmeier reviewed literature and ISTH abstracts, and drafted and edited the manuscript.
ACKNOWLEDGMENTS
We thank Dorsaf Ghalloussi, Robert Lee and David Paul for their feedback on exciting science presented at the 2017 ISTH meeting. This work was supported by the American Heart Association (14EIA18910004) and NIH grant P01 HL120846 (W.B.) and the Young Researchers Program Rita Levi Montalcini (L.S.).
Bergmeier W, Stefanini L. Platelets at the vascular interface. Res Pract Thromb Haemost. 2018;2:27–33. 10.1002/rth2.12061
REFERENCES
- 1. Ruggeri ZM. Platelet adhesion under flow. Microcirculation. 2009;16:58–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petrich BG, Marchese P, Ruggeri ZM, et al. Talin is required for integrin‐mediated platelet function in hemostasis and thrombosis. J Exp Med. 2007;204:3103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nieswandt B, Moser M, Pleines I, et al. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med. 2007;204:3113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fässler R. Kindlin‐3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–30. [DOI] [PubMed] [Google Scholar]
- 5. Lagarrigue F, Kim C, Ginsberg MH. The Rap1‐RIAM‐talin axis of integrin activation and blood cell function. Blood. 2016;128:479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–6. [DOI] [PubMed] [Google Scholar]
- 7. Ye F, Petrich BG, Anekal P, et al. The mechanism of kindlin‐mediated activation of integrin αIIbβ3. Curr Biol. 2013;23:2288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Becker I, Stritt S, Beck S, et al. Twinfilin 2a is a central regulator of platelet reactivity and turnover in mice. Res Pract Thromb Haemost. 2017;1(Suppl 1):232. [Google Scholar]
- 9. Stritt S, Beck S, Becker IC, et al. Twinfilin 2a regulates platelet reactivity and turnover in mice. Blood. 2017;130:1746–56. [DOI] [PubMed] [Google Scholar]
- 10. Beck S, Stritt S, Sorrentino S, et al. Profilin 1 is a central regulator of integrin turnover and function in mouse platelets. Res Pract Thromb Haemost. 2017;1(Suppl 1):245. [Google Scholar]
- 11. Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–85. [DOI] [PubMed] [Google Scholar]
- 12. Varga‐Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403–12. [DOI] [PubMed] [Google Scholar]
- 13. Stalker TJ, Welsh JD, Brass LF. Shaping the platelet response to vascular injury. Curr Opin Hematol. 2014;21:410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dubois C, Panicot‐Dubois L, Merrill‐Skoloff G, Furie B, Furie BC. Glycoprotein VI‐dependent and ‐independent pathways of thrombus formation in vivo. Blood. 2006;107:3902–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bynagari‐Settipalli YS, Cornelissen I, Palmer D, et al. Redundancy and interaction of thrombin‐ and collagen‐mediated platelet activation in tail bleeding and carotid thrombosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:2563–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kahn ML, Zheng YW, Huang W, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–4. [DOI] [PubMed] [Google Scholar]
- 17. Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39:5458–67. [DOI] [PubMed] [Google Scholar]
- 18. Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease‐activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. 2000;275:25216–21. [DOI] [PubMed] [Google Scholar]
- 19. Ohlmann P, de Castro S, Brown GG, Gachet C, Jacobson KA, Harden TK. Quantification of recombinant and platelet P2Y(1) receptors utilizing a [(125)I]‐labeled high‐affinity antagonist 2‐iodo‐N(6)‐methyl‐(N)‐methanocarba‐2′‐deoxyadenosine‐3′,5′‐bisphosphate ([(125)I]MRS2500). Pharmacol Res. 2010;62:344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baurand A, Eckly A, Bari N, et al. Desensitization of the platelet aggregation response to ADP: differential down‐regulation of the P2Y1 and P2cyc receptors. Thromb Haemost. 2000;84:484–91. [PubMed] [Google Scholar]
- 21. Hardy AR, Conley PB, Luo J, Benovic JL, Poole AW, Mundell SJ. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase‐dependent mechanisms. Blood. 2005;105:3552–60. [DOI] [PubMed] [Google Scholar]
- 22. Jin J, Kunapuli SP. Coactivation of two different G protein‐coupled receptors is essential for ADP‐induced platelet aggregation. Proc Natl Acad Sci USA. 1998;95:8070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hechler B, Léon C, Vial C, et al. The P2Y1 receptor is necessary for adenosine 5′‐diphosphate‐induced platelet aggregation. Blood. 1998;92:152–9. [PubMed] [Google Scholar]
- 24. Ma P, Ou K, Sinnamon AJ, Jiang H, Siderovski DP, Brass LF. Modulating platelet reactivity through control of RGS18 availability. Blood. 2015;126:2611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delesque‐Touchard N, Pendaries C, Volle‐Challier C, et al. Regulator of G‐protein signaling 18 controls both platelet generation and function. PLoS ONE. 2014;9:e113215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hensch NR, Karim ZA, Druey KM, Tansey MG, Khasawneh FT. RGS10 negatively regulates platelet activation and thrombogenesis. PLoS ONE. 2016;11:e0165984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Signarvic RS, Cierniewska A, Stalker TJ, et al. RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood. 2010;116:6092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gupta S, Sampietro S, DeHelian D, et al. RGS proteins shape the hemostatic response by regulating the platelet signaling networks. Res Pract Thromb Haemost. 2017;1(Suppl 1):1248. [Google Scholar]
- 29. Gupta S, DeHelian D, Brass LF, Ma P. RGS‐insensitive G proteins as a model to study G protein‐dependent signaling. Res Pract Thromb Haemost. 2017;1(Suppl 1):1247. [Google Scholar]
- 30. Schmaier AA, Zou Z, Kazlauskas A, et al. Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc Natl Acad Sci USA. 2009;106:21167–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arthur JF, Dunkley S, Andrews RK. Platelet glycoprotein VI‐related clinical defects. Br J Haematol. 2007;139:363–72. [DOI] [PubMed] [Google Scholar]
- 32. Matus V, Valenzuela G, Sáez CG, et al. An adenine insertion in exon 6 of human GP6 generates a truncated protein associated with a bleeding disorder in four Chilean families. J Thromb Haemost. 2013;11:1751–9. [DOI] [PubMed] [Google Scholar]
- 33. Kato K, Kanaji T, Russell S, et al. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102:1701–7. [DOI] [PubMed] [Google Scholar]
- 34. Nieswandt B, Schulte V, Bergmeier W, et al. Long‐term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193:459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bender M, Hagedorn I, Nieswandt B. Genetic and antibody‐induced glycoprotein VI deficiency equally protects mice from mechanically and FeCl(3)‐induced thrombosis. J Thromb Haemost. 2011;9:1423–6. [DOI] [PubMed] [Google Scholar]
- 36. Mammadova‐Bach E, Ollivier V, Loyau S, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126:683–91. [DOI] [PubMed] [Google Scholar]
- 37. Alshehri OM, Hughes CE, Montague S, et al. Fibrin activates GPVI in human and mouse platelets. Blood. 2015;126:1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haining EJ, Stegner D, Cherpokova D, Wolf K, Watson SP, Nieswandt B. CLEC‐2 contributes to hemostasis independently of classical HemiTAM signaling in mice. Res Pract Thromb Haemost. 2017;1(Suppl 1):58. [DOI] [PubMed] [Google Scholar]
- 39. Haining EJ, Cherpokova D, Wolf K, et al. CLEC 2 contributes to hemostasis independently of classical hemITAM signaling in mice. Blood. 2017;130:2224–8. [DOI] [PubMed] [Google Scholar]
- 40. Boulaftali Y, Hess PR, Getz TM, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123:908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stefanini L, Paul DS, OShaughnessy E, et al. Rap1A and Rap1B functional redundancy in platelets and megakaryocytes. Res Pract Thromb Haemost. 2017;1(Suppl 1):226. [Google Scholar]
- 42. Katagiri K, Hattori M, Hattori M, et al. Rap1 is a potent activation signal for leukocyte function‐associated antigen 1 distinct from protein kinase C and phosphatidylinositol‐3‐OH kinase. Mol Cell Biol. 2000;20:1956–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reedquist KA, Ross E, Koop EA, et al. The small GTPase, Rap1, mediates CD31‐induced integrin adhesion. J Cell Biol. 2000;148:1151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Caron E, Self AJ, Hall A. The GTPase Rap1 controls functional activation of macrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Curr Biol. 2000;10:974–8. [DOI] [PubMed] [Google Scholar]
- 45. Bertoni A, Tadokoro S, Eto K, et al. Relationships between Rap1b, affinity modulation of integrin alpha IIbbeta 3, and the actin cytoskeleton. J Biol Chem. 2002;277:25715–21. [DOI] [PubMed] [Google Scholar]
- 46. Han J, Lim CJ, Watanabe N, et al. Reconstructing and deconstructing agonist‐induced activation of integrin alphaIIbbeta3. Curr Biol. 2006;16:1796–806. [DOI] [PubMed] [Google Scholar]
- 47. Chrzanowska‐Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115:680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang G, Xiang B, Ye S, et al. Distinct roles for Rap1b protein in platelet secretion and integrin αIIbβ3 outside‐in signaling. J Biol Chem. 2011;286:39466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burkhart JM, Vaudel M, Gambaryan S, et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120:e73–82. [DOI] [PubMed] [Google Scholar]
- 50. Stefanini L, Bergmeier W. RAP1‐GTPase signaling and platelet function. J Mol Med. 2016;94:13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crittenden JR, Bergmeier W, Zhang Y, et al. CalDAG‐GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–6. [DOI] [PubMed] [Google Scholar]
- 52. Stefanini L, Roden RC, Bergmeier W. CalDAG‐GEFI is at the nexus of calcium‐dependent platelet activation. Blood. 2009;114:2506–14. [DOI] [PubMed] [Google Scholar]
- 53. Canault M, Ghalloussi D, Grosdidier C, et al. Human CalDAG‐GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J Exp Med. 2014;211:1349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kato H, Nakazawa Y, Kurokawa Y, et al. Human CalDAG‐GEFI deficiency increases bleeding and delays αIIbβ3 activation. Blood. 2016;128:2729–27233. [DOI] [PubMed] [Google Scholar]
- 55. Bermejo E, Alberto MF, Paul DS, et al. Marked bleeding diathesis in patients with platelet dysfunction due to a novel mutation in RASGRP2, encoding CalDAG‐GEFI (p.Gly305Asp). Platelets. 2017;1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iwig JS, Vercoulen Y, Das R, et al. Structural analysis of autoinhibition in the Ras‐specific exchange factor RasGRP1. eLife. 2013;2:e00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stolla M, Stefanini L, André P, et al. CalDAG‐GEFI deficiency protects mice in a novel model of Fcγ RIIA‐mediated thrombosis and thrombocytopenia. Blood. 2011;118:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lozano ML, Cook A, Bastida JM, et al. Novel mutations in RASGRP2, which encodes CalDAG‐GEFI, abrogate Rap1 activation, causing platelet dysfunction. Blood. 2016;128:1282–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sevivas T, Bastida JM, Paul DS, et al. Identification of two novel mutations in RASGRP2 affecting platelet CalDAG‐GEFI expression and function in patients with bleeding diathesis. Platelets. 2017;1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Westbury SK, Canault M, Greene D, et al. Expanded repertoire of RASGRP2 variants responsible for platelet dysfunction and severe bleeding. Blood. 2017;130:1026–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stefanini L, Paul DS, Robledo RF, et al. RASA3 is a critical inhibitor of RAP1‐dependent platelet activation. J Clin Invest. 2015;125:1419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goerge T, Ho Tin Noé B, Carbo C, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111:4958–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gros A, Syvannarath V, Lamrani L, et al. Single platelets seal neutrophil‐induced vascular breaches via GPVI during immune‐complex‐mediated inflammation in mice. Blood. 2015;126:1017–26. [DOI] [PubMed] [Google Scholar]
- 64. Hillgruber C, Pöppelmann B, Weishaupt C, et al. Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J Exp Med. 2015;212:1255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boulaftali Y, Hess PR, Kahn ML, Bergmeier W. Platelet immunoreceptor tyrosine‐based activation motif (ITAM) signaling and vascular integrity. Circ Res. 2014;114:1174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rayes J, Lax S, Gros A, et al. The contribution of platelet adhesion receptors to vascular integrity during inflammation is stimulus and organ dependent. Res Pract Thromb Haemost. 2017;1(Suppl 1):234. [Google Scholar]
- 67. Ho Tin Noé B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68:6851–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ho Tin Noé B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. 2011;9(Suppl 1):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Deppermann C, Kraft P, Volz J, et al. Platelet secretion is crucial to prevent bleeding in the ischemic brain but not in the inflamed skin or lung in mice. Blood. 2017;129:1702–6. [DOI] [PubMed] [Google Scholar]
- 70. Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115:2323–30. [DOI] [PubMed] [Google Scholar]