

 The room temperature sp2–sp3 cross-coupling of flow-generated diazo compounds with boronic acids is reported.
The room temperature sp2–sp3 cross-coupling of flow-generated diazo compounds with boronic acids is reported.
Abstract
The work takes advantage of an important feature of flow chemistry, whereby the generation of a transient species (or reactive intermediate) can be followed by a transfer step into another chemical environment, before the intermediate is reacted with a coupling partner. This concept is successfully applied to achieve a room temperature sp2–sp3 cross coupling of boronic acids with diazo compounds, these latter species being generated from hydrazones under flow conditions using MnO2 as the oxidant.
Introduction
Chemical processes define our modern world in terms of providing society's functional materials.1 Many of these processes involve complex reaction pathways and proceed via transient reactive intermediates. The lifetime of these reactive species can vary enormously and their selectivity can often be difficult to exploit. Their high level of reactivity, when generated in batch mode, can be problematic because of incompatibilities with their chemical environment or functional groups, or because of associated exothermic properties. A case in point relates to diazo compounds which are valuable reactive intermediates.2 For example, isolated stabilised diazo compounds, bearing one or more electron withdrawing groups, have been extensively investigated in a wide range of applications such as dipolar cycloadditions,3 carbonyl homologation4 and cyclopropanation.5 However, their unstabilised alkyl and aryl diazo counterparts are much more difficult to harness.2,6 Their chemical preparation is limited due to the often tedious, corrosive or toxic synthesis protocols.6a,7 In situ generation of diazo compounds therefore represents an interesting opportunity for synthesis,8 although any new procedure must be compatible with both the generation of the diazo compound and with its subsequent chemical transformation.
Manganese dioxide (MnO2) has been used previously in batch mode to generate diazo compounds from hydrazones, although with low efficiency and with very specific substrates.9 Following on from the advantages that accrue when using MnO2 under continuous flow reactions,10 we believed that we could devise a system capable of delivering reactive unstable diazo compounds on demand. Also, with the aim of better exploiting the potential of the flow chemistry,11,12 we anticipated that these flow-generated transient species could be rapidly transferred into another chemical environment, in order to react the aforementioned species with a suitable partner, hence avoiding interference by spent reagents (Scheme 1). This generation/translocation/reaction sequence should immediately provide access to new chemical space for the discovery of new reactions for these reactive intermediates.
Scheme 1. Flowing scheme for the generation, translocation and reaction of the diazo species.

Metal free sp2–sp3 cross-coupling reactions have recently become a new focus of the chemical community.13 In 2009, Barluenga et al. reported an efficient coupling between tosylhydrazones and boronic acids.13a Further studies with saturated heterocyclic sulfonylhydrazones by Nakagawa,13c Kirschning,13e and ourselves,13f demonstrated a wider scope for the reaction (Scheme 2). Recently, Wang and co-workers reported an efficient synthesis of α-aryl esters and nitriles via a diazotization method for the generation of stabilised diazo compounds.13g However, the relatively harsh reaction conditions (up to 110 °C), large amount of inorganic additives (bases, NaNO2, NH4Cl) and low atom economy represent potential drawbacks of these transformations. We envisaged that we could access new reactivities of unstabilised diazo compounds, in particular their coupling with boronic acids at room temperature.
Scheme 2. Selected metal-free sp2–sp3 cross-coupling reactions.

Results and discussion
Generation of diazo compound
Initial investigations focused on 4-bromophenyl hydrazone (1a) as model substrate for the generation of the diazo compound 2a. To evaluate the generation of the transient diazo species, the material processed was quenched with acetic acid to yield the corresponding acetic acid ester 3a (Scheme 3).6 Amorphous MnO2 packed into the flow cartridge gave very poor conversion to 2a with mainly starting material being recovered. By contrast, a column packed with activated MnO2 gave full conversion to various products but with aldehyde 4a being the main product observed. This result was also consistent with batch studies where aldehyde or azine by-products were generally observed at ca. 30% yield, even under carefully controlled reaction conditions.9b,c,f However, conditioning of the packed column with a small amount of sacrificial hydrazone14 effectively reduced the formation of aldehyde and increased the production of diazo compound 2a as determined via the formation of ester 3a. Furthermore, addition of a base (Hünig's base, 2 equiv.) to the feedstock solution provided a reliable system with greatly reduced amounts of by-products generated.15
Scheme 3. Flow generation and quenching of 1-bromo-4-(diazomethyl)benzene.

Under the optimised conditions, a 0.1 M solution of hydrazone 1 was passed through a conditioned MnO2 column (0.86 g, activated MnO2) at 0.5 mL min–1 with 2 equiv. of Hünig's base in CH2Cl2. In-line IR data (collected using an in-line infrared spectrometer) suggested that a typical load of activated MnO2 (10 mmol) could cleanly generate 2 mmol of diazo compound over 40 minutes. The generation of diazo compound, which proved to be highly practical as the whole process was carried out at room temperature, was followed by an esterification step of the transient species produced.
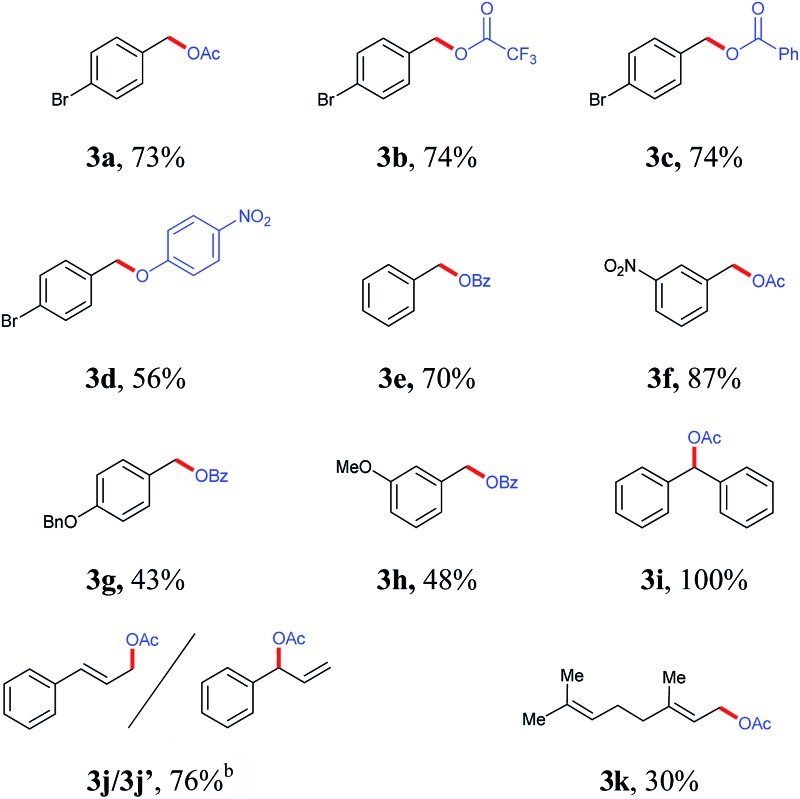
To explore the general application of this process, different hydrazone derivatives were examined (Table 1). The generated diazo compound was quenched with different carboxylic acids (3a–3c) and one phenol substrate (3d). The reaction was compatible with both electron poor and electron rich aromatic hydrazones. Electron poor aryl diazo compound gave in general better yields as they are more stabilised. Investigation of the reactivity of a vinylic diazo compound revealed the formation of two regioisomers (3j and 3j′) after reaction with excess AcOH. However, when geranyl hydrazone was processed via our protocol only one isomer was obtained, though only in moderate yield (3k). On the other hand, diphenyl ketone hydrazone gave ester 3i in excellent yield.
Table 1. Titration of generated diazo compound to the corresponding esters a .
|
|
aIsolated yield over 3 steps based on aldehyde precursor.
bCombined yield (r.s. = 1 : 1).
Coupling reaction with boronic acids
Diazo compounds are commonly proposed as intermediates for several useful transformations, in particular carbon–carbon sp2–sp3 coupling between tosylhydrazone and boronic acids.13 High temperature and an inorganic base are typically required as the generation of the diazo compound is reported to be the limiting step of these reactions. We speculated that with our new protocol the chemical reactivity of diazo compounds could be studied separately without interference from the generation step. Pleasingly, after its generation, the stream of the diazo compound 2a reacted quickly at room temperature with boronic acid 7a in the first attempt, giving the coupled product 8a in very good yield (Scheme 4). This extremely mild and fast reaction conditions demonstrated the high chemical reactivity of diazo compounds and highlighted the advantages of the flow protocol for these reactive intermediates where the equivalent batch mode reaction was found to be unsuccessful.
Scheme 4. Flow generation, translocation and reaction of 2a with 7a.

Different coupling combinations were investigated using the flow procedure for the generation of clean diazo compounds (Table 2). With much milder reaction conditions compared to the original Barluenga coupling, the electronic properties exerted a strong influence on the reaction time. For electron rich or neutral boronic acids the transformation was complete within 1 hour (compounds 8a–c). By contrast, electron poor aryl boronic acids required longer reaction time or harsher reaction conditions (8d–e). Similar trends were observed with other diazo compounds. Electron poor (more stabilised) diazo compounds required, in general, longer reaction times when reacted with the same boronic acid (8j vs. 8b). Interestingly, vinyl diazo compounds are also compatible with this protocol giving the desired products in good yields (8n–q). Notably, in these cases a mixture of products was obtained resulting from a 1,3-borotropic rearrangement and the regioselectivity of the products mixtures was strongly influenced by the electronic properties of the two aromatic rings, ranging from 2 : 1 (8n) to 15 : 1 (8q). Different functionalities are tolerated such as halogens, nitro groups and ethers. Heterocycles such as pyridine (8e) are also compatible with this protocol. The highly reactive geranyl diazo gave the geranylated product (8l) in good yield, based on the titration of the corresponding diazo compound. This process is remarkable as it provides a complementary method for geranylation, where the normal Friedel–Crafts alkylation fails. Additionally, we were able to access sp2–sp3 coupling transformations for aryl–alkyl diazomethane derivatives in quite good yield (8m).
Table 2. Coupling products prepared by reaction of diazo compounds with boronic acids a .
|

|
aIsolated yield based on titration of diazo compound with acetic acid.
bMicrowave 100 °C in 15 min.
cCombined yields.
Since the boronic acids are often the more functionalised of the two starting materials, the flow-generated diazo partner was used in excess to fully convert the boronic acid 7b to the desired coupling product (Scheme 5). Pleasingly, the diaryl compound 8f was obtained in 96% isolated yield using only a slight excess of diazo compound (1.5 equiv.). In this case, the excess diazo was subsequently quenched via an acidic aqueous work-up protocol. The main by-products detected were both the corresponding azine and alcohol.
Scheme 5. Boronic acid 7b used as limiting reagent.

The strong dependence of the reaction kinetics on the electronic properties of both starting materials reveals some insight about the reaction mechanism. It is generally assumed that after nucleophilic attack of the diazo compound to the boronic acid, a first ate complex 9 is formed. This intermediate rapidly undergoes a 1,2-carbon migration forming the boronic species 10 which isomerises to the most stable isomer prior to a proto-deboronation to give the coupling product 8 (Scheme 6). Although this mechanism is assumed to describe the actual chemical combinations of the intermediates involved, no report has either identified the kinetics of this kind of transformation or the nature of the species involved. The reasons behind this are linked to the harsh conditions employed to generate the reactive intermediate.13 We started by investigating the kinetics involved in the sequence of events. Initial NMR studies focused on a model reaction between diazo compound 2b and boronic acid 7b and the analysis showed a two-step mechanism (Scheme 6), where the first step happens at a much faster rate than the second one. Using the data collected over a large series of dynamic experiments, we were able to define that the reaction is first order with respect to the diazo compound.
Scheme 6. Mechanistic studies of coupling reaction between diazo compound and boronic acid.

Remarkably, using our mild flow protocol we were also able for the first time to intercept the secondary boronic intermediate 10, proving the presence of this intermediate prior to proto-deboronation. Indeed, we envisaged that the generation of a relatively stable boronic intermediate 10 would reduce the rate of proto-deboronation and this new reactive intermediate could be intercepted via an alternative reaction pathway. This idea was realised with the use of acetophenone hydrazone (1b) as starting material, which was passed through the oxidant-column, packed with activated MnO2, to form the corresponding diazo compound (as detected by in-line IR); the transient diazo species was translocated to react with boronic acid 7a and then further reacted with a solution of hydrogen peroxide. The analysis of the material revealed the prevalence of alcohol 11 (68%) within the mixture, corroborating the presence of a boronic species 10c as an intermediate.
To increase the efficiency of the new protocol, the regeneration and re-use of MnO2 column was briefly investigated. The spent manganese oxide could be oxidised by passage of a solution of tert-butyl hydroperoxide (TBHP, 1 M in CH2Cl2) (Scheme 7). Interestingly, the regenerated column produced diazo compound without the need for a pre-conditioning phase. The heterogeneous oxidant was recycled over three different cycles with just a slight decrease in reactivity after each recycle as detected by in-line IR (Fig. 1).
Scheme 7. Flow regeneration of MnO2 reagent using TBHP.

Fig. 1. IR data for the four regeneration cycles of oxidation for a single MnO2 load (0.86 g).

Moreover, in a preliminary attempt with tube-in-tube technology,16 oxygen gas also proved to be a useful re-oxidant for a low cost regeneration of MnO2 but with lower efficiency.17
Conclusion
In summary, we have reported a practical flow generation of unstable diazo compounds as reactive intermediate using the cheap, recyclable and non-toxic oxidant, MnO2. The diazo flow stream can be accurately titrated or followed by in-line IR. Using our protocol, we were able to deliver sp2–sp3 coupling reactions of the freshly generated diazo species with boronic acids, under extremely mild and simple reaction conditions providing alkylated products in good to excellent yield. The mechanistic studies demonstrated for the first time the presence of a specific boronic intermediate. These results demonstrate that flow generation/translocation/reaction of transient species opens up opportunities for the exploitation and study of new chemical reactivities. Further studies in this area are underway in our laboratories.
Acknowledgments
We are grateful to the Swiss National Science Foundation (DNT), Pfizer Worldwide Research and Development (CB and JMH), and the EPSRC (SVL, grant no. EP/K0099494/1). We also thank the EPSRC (core capability grant no. EP/K039520/1) for improvements to analytical facilities.
Footnotes
References
- (a) Ley S. V. Chem. Rec. 2012;12:378–390. doi: 10.1002/tcr.201100041. [DOI] [PubMed] [Google Scholar]; (b) Myers R. M., Fitzpatrick D. E., Turner R. M., Ley S. V. Chem.–Eur. J. 2014;20:12348–12366. doi: 10.1002/chem.201402801. [DOI] [PubMed] [Google Scholar]
- (a) Doyle M. Acc. Chem. Res. 1986;19:348–356. [Google Scholar]; (b) Padwa A. J. Organomet. Chem. 2001;617:3–16. [Google Scholar]; (c) Regitz M. and Mass G., in Diazo Compounds Properties and Synthesis, Academic Press, Inc., Orlando, 1986. [Google Scholar]; (d) Doyle M. P., McKervey M. A. and Ye T., Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998. [Google Scholar]; (e) Johnston J. N., Muchalski H., Troyer T. L. Angew. Chem., Int. Ed. 2010;49:2290–2298. doi: 10.1002/anie.200904828. [DOI] [PubMed] [Google Scholar]
- (a) Huisgen R., in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984. [Google Scholar]; (b) Mass G., in Heterocyclic Compounds: 1,3-Dipolar Cycloaddition, ed. A. Padwa and W. H. Pearson, John Wiley and Sons, Inc., New York, 2002, vol. 59, pp. 541–621. [Google Scholar]
- Selected examples for carbonyl homologation: ; (a) Gutsche C. D. Org. React. 1954;8:364–429. [Google Scholar]; (b) Holmquist C. R., Roskamp E. J. J. Org. Chem. 1989;54:3258–3260. [Google Scholar]; (c) Moebius D. C., Kingsbury J. S. J. Am. Chem. Soc. 2009;131:878–879. doi: 10.1021/ja809220j. [DOI] [PubMed] [Google Scholar]; (d) Allwood D. M., Blakemore D. C., Ley S. V. Org. Lett. 2014;16:3064–3067. doi: 10.1021/ol5011714. [DOI] [PubMed] [Google Scholar]
- Selected examples for cyclopropanation: ; (a) Aggarwal V. K., de Vicente J., Bonnert R. V. Org. Lett. 2001;3:2785–2788. doi: 10.1021/ol0164177. [DOI] [PubMed] [Google Scholar]; (b) Lebel H., Marcoux J.-F., Molinaro C., Charette A. B. Chem. Rev. 2003;103:977–1050. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; (c) Maas G. Chem. Soc. Rev. 2004;33:183–190. doi: 10.1039/b309046a. [DOI] [PubMed] [Google Scholar]; (d) Goudreau S. R., Charette A. B. J. Am. Chem. Soc. 2009;131:15633–15635. doi: 10.1021/ja9074776. [DOI] [PubMed] [Google Scholar]
- (a) Maas G. Angew. Chem., Int. Ed. 2009;48:8186–8195. doi: 10.1002/anie.200902785. [DOI] [PubMed] [Google Scholar]; (b) Rendina V. L., Kingsbury J. S. J. Org. Chem. 2012;77:1181–1185. doi: 10.1021/jo202214e. [DOI] [PubMed] [Google Scholar]; (c) Wommack A. J., Kingsbury J. S. J. Org. Chem. 2013;78:10573–10587. doi: 10.1021/jo401377a. [DOI] [PubMed] [Google Scholar]; (d) Holton T. L., Shechter H. J. Org. Chem. 1995;60:4725–4729. [Google Scholar]
- (a) Smith L. I., Howard K. L. Org. Synth. 1944;24:53. [Google Scholar]; (b) Nakagawa K., Onoue H., Minami K. Chem. Commun. 1966:730. [Google Scholar]; (c) Javed M. I., Brewer M. Org. Lett. 2007;9:1789–1792. doi: 10.1021/ol070515w. [DOI] [PubMed] [Google Scholar]
- ; flow generation of diazo methane: ; (a) Maurya R. A., Park C. P., Lee J. H., Kim D.-P. Angew. Chem., Int. Ed. 2011;50:5952–5955. doi: 10.1002/anie.201101977. [DOI] [PubMed] [Google Scholar]; (b) Morandi B., Carreira E. M. Science. 2012;335:1471–1474. doi: 10.1126/science.1218781. [DOI] [PubMed] [Google Scholar]; (c) Rossi E., Woehl P., Maggini M. Org. Process Res. Dev. 2012;16:1146–1149. [Google Scholar]; (d) Proctor L. D., Warr A. J. Org. Process Res. Dev. 2002;6:884–892. [Google Scholar]; (e) Struempel M., Ondruschka B., Stark A. Org. Process Res. Dev. 2009;13:1014–1021. [Google Scholar]; (f) Mastronardi F., Gutmann B., Kappe C. O. Org. Lett. 2013;15:5590–5593. doi: 10.1021/ol4027914. [DOI] [PubMed] [Google Scholar]; (g) Martin L. J., Marzinzik A. L., Ley S. V., Baxendale I. R. Org. Lett. 2011;13:320–323. doi: 10.1021/ol1027927. [DOI] [PubMed] [Google Scholar]
- (a) Barakat M. Z., Abdel-Wahab M. F., El-Sadr M. M. J. Chem. Soc. 1956:4685–4687. [Google Scholar]; (b) Doyle M. P., Yan M. ARKIVOC. 2002:180–185. [Google Scholar]; (c) Doyle M. P., Yan M. J. Org. Chem. 2002;67:602–604. doi: 10.1021/jo016135k. [DOI] [PubMed] [Google Scholar]; (d) Denton J. R., Sukumaran D., Davies H. M. L. Org. Lett. 2007;9:2625–2628. doi: 10.1021/ol070714f. [DOI] [PubMed] [Google Scholar]; (e) Lee K.-H., Ko K.-Y. Bull. Korean Chem. Soc. 2006;27:185–186. [Google Scholar]; (f) Sharma T. C., Lal A., Saksena V. Bull. Chem. Soc. Jpn. 1976;49:2881–2882. [Google Scholar]
- (a) Battilocchio C., Hawkins J. M., Ley S. V. Org. Lett. 2014;16:1060–1063. doi: 10.1021/ol403591c. [DOI] [PubMed] [Google Scholar]; (b) Glöckner S., Tran D. N., Ingham R. J., Fenner S., Wilson Z. E., Battilocchio C., Ley S. V. Org. Biomol. Chem. doi: 10.1039/c4ob02105c. [DOI] [PubMed] [Google Scholar]
- Selected recent reviews for chemical flow systems: ; (a) Hartman R. L., McMullen J. P., Jensen K. F. Angew. Chem., Int. Ed. 2011;50:7502–7519. doi: 10.1002/anie.201004637. [DOI] [PubMed] [Google Scholar]; (b) Wegner J., Ceylan S., Kirschning A. Adv. Synth. Catal. 2012;354:17–57. [Google Scholar]; (c) Wiles C., Watts P. Green Chem. 2012;14:38–54. [Google Scholar]; (d) Malet-Sanz L., Susanne F. J. Med. Chem. 2012;55:4062–4098. doi: 10.1021/jm2006029. [DOI] [PubMed] [Google Scholar]; (e) Baxendale I. R. J. Chem. Technol. Biotechnol. 2013;88:519–552. [Google Scholar]; (f) Pastre J. C., Browne D. L., Ley S. V. Chem. Soc. Rev. 2013;42:8849–8869. doi: 10.1039/c3cs60246j. [DOI] [PubMed] [Google Scholar]
- Selected recent examples for chemical flow systems: ; (a) Kim H., Nagaki A., Yoshida J.-I. Nat. Commun. 2011;2:264. doi: 10.1038/ncomms1264. [DOI] [PubMed] [Google Scholar]; (b) Levesque F., Seeberger P. H. Angew. Chem., Int. Ed. 2012;51:1706–1709. doi: 10.1002/anie.201107446. [DOI] [PubMed] [Google Scholar]; (c) Deadman B. J., Battilocchio C., Sliwinski E., Ley S. V. Green Chem. 2013;15:2050–2055. [Google Scholar]; (d) He Z., Jamison T. F. Angew. Chem., Int. Ed. 2014;53:3353–3357. doi: 10.1002/anie.201310572. [DOI] [PubMed] [Google Scholar]; (e) Baumann M., Baxendale I. R. Beilstein J. Org. Chem. 2013;9:1613–1619. doi: 10.3762/bjoc.9.184. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Newton S., Carter C. F., Pearson C. M., Alves L. de C., Lange H., Thansandote P., Ley S. V. Angew. Chem., Int. Ed. 2014;53:4915–4920. doi: 10.1002/anie.201402056. [DOI] [PubMed] [Google Scholar]; (g) Hartwig J., Ceylan S., Kupracz L., Coutable L., Kirschning A. Angew. Chem., Int. Ed. 2013;52:9813–9817. doi: 10.1002/anie.201302239. [DOI] [PubMed] [Google Scholar]; (h) Nightingale A. M., Phillips T. W., Bannock J. H., de Mello J. C. Nat. Commun. 2014;5:3777. doi: 10.1038/ncomms4777. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fan X., Sans V., Yaseneva P., Plaza D. D., Williams J., Lapkin A. Org. Process Res. Dev. 2012;16:1039–1042. [Google Scholar]
- Selected recent examples for metal free sp2–sp3 cross-coupling: ; (a) Barluenga J., Tomás-Gamasa M., Aznar F., Valdés C. Nat. Chem. 2009;1:494–499. doi: 10.1038/nchem.328. [DOI] [PubMed] [Google Scholar]; (b) Peng C., Zhang W., Yan G., Wang J. Org. Lett. 2009;11:1667–1670. doi: 10.1021/ol900362d. [DOI] [PubMed] [Google Scholar]; (c) Nakagawa S., Bainbridge K. A., Butcher K., Ellis D., Klute W., Ryckmans T. ChemMedChem. 2012;7:233–236. doi: 10.1002/cmdc.201100339. [DOI] [PubMed] [Google Scholar]; (d) Pérez-Aguilar M. C., Valdés C. Angew. Chem., Int. Ed. 2012;51:5953–5957. doi: 10.1002/anie.201200683. [DOI] [PubMed] [Google Scholar]; (e) Kupracz L., Kirschning A. J. Flow Chem. 2013;3:11–16. [Google Scholar]; (f) Allwood D. M., Blakemore D. C., Brown A. D., Ley S. V. J. Org. Chem. 2014;79:328–338. doi: 10.1021/jo402526z. [DOI] [PubMed] [Google Scholar]; (g) Wu G., Deng Y., Wu C., Zhang Y., Wang J. Angew. Chem., Int. Ed. 2014;53:10510–10514. doi: 10.1002/anie.201406765. [DOI] [PubMed] [Google Scholar]; (h) Wu G., Deng Y., Wu C., Wang X., Zhang Y., Wang J. Eur. J. Org. Chem. 2014:4477–4481. [Google Scholar]; (i) Metal Free C–H functionalization of aromatics, (Top. Heterocycl. Chem., 37, 2014), ed. V. Charushin and O. Chupakhin, Springer, Switzerland, 2014. [Google Scholar]
- Any aryl hydrazone can be used for conditioning the column
- We are currently investigating the mechanism of oxidation. We speculate that the oxidative process proceeds via either ionic or radical pathways
- Selected examples for tube-in-tube gas reactions: ; (a) Newton S., Ley S. V., Arce E. C., Grainger D. Adv. Synth. Catal. 2012;354:1805–1812. [Google Scholar]; (b) Cranwell P. B., O’Brien M., Browne D. L., Koos P., Polyzos A., Peña-López M., Ley S. V. Org. Biomol. Chem. 2012;10:5774–5779. doi: 10.1039/c2ob25407g. [DOI] [PubMed] [Google Scholar]; (c) Peterson T. P., Polyzos A., O’Brien M., Ulven T., Baxendale I. R., Ley S. V. ChemSusChem. 2012;5:274–277. doi: 10.1002/cssc.201100339. [DOI] [PubMed] [Google Scholar]; (d) Bourne S. L., O'Brien M., Kasinathan S., Koos P., Tolstoy P., Hu D. X., Bates R. W., Martin B., Schenkel B., Ley S. V. ChemCatChem. 2013;5:159–172. [Google Scholar]; (e) Mastronardi F., Gutmann B., Kappe C. O. Org. Lett. 2013;15:5590–5593. doi: 10.1021/ol4027914. [DOI] [PubMed] [Google Scholar]; (f) Tomaszewski B., Schmid A., Buehler K. Org. Process Res. Dev. doi: 10.1021/op5002116. [DOI] [Google Scholar]; (g) Gross U., Koos P., O'Brien M., Polyzos A., Ley S. V. Eur. J. Org. Chem. 2014:6418–6430. [Google Scholar]
- Depleted oxidant-activity column was flushed with a tube-in-tube system using oxygen 6% during 5 h. This column showed enhanced activity by producing diazo compound at ca. 25% yield (evaluated by IR)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.