

 Alkaloid catalysed additions to triketopiperazines gives products in high yield and er (88 : 12 to 99 : 1), including bridged hydroxy-DKPs via Michael-addition–ring closure.
Alkaloid catalysed additions to triketopiperazines gives products in high yield and er (88 : 12 to 99 : 1), including bridged hydroxy-DKPs via Michael-addition–ring closure.
Abstract
Michael addition reactions of triketopiperazine (TKP) derivatives to enones, mediated by a cinchona alkaloid-derived catalyst, deliver products in high yield and enantiomeric ratio (er). Use of unsaturated ester, nitrile or sulfone partners gives bridged hydroxy-diketopiperazine (DKP) products resulting from a novel Michael addition–ring closure.
Introduction
Diketopiperazines (DKPs) have acquired the status of privileged structures for medicinal chemistry, for example tadalafil (or cialis®, 1) is a potent PDE5 inhibitor used for treating male erectile dysfunction, Fig. 1.1
Fig. 1. Structures of significant bioactive DKPs.

The DKP structure is also prevalent in many types of peptide antibiotics, including the clinically significant compound bicyclomycin (2), which has an interesting [4.2.2] ether-bridged core.2
Many other important natural product families incorporate DKP motifs, for example the bicyclo[2.2.2]diazaoctane core structure features in the fungal metabolite stephacidin A (3), which displays potent and selective antitumour activity.3 The extensive studies of the synthesis and medicinal chemistry of DKPs and their occurrence in bioactive natural products have been described in a comprehensive review by Borthwick.4
Despite this background, access to enantiomerically pure DKPs has relied very heavily on chiral pool materials and, with the exception of studies aimed at stephacidins and related natural products, reports of stereocontrolled access to α,α-disubstituted systems are scarce.5
Herein, we describe highly enantioselective access to α,α-disubstituted DKPs, including bridged products related to 2 and 3, by means of asymmetric Michael additions mediated by cinchona alkaloid catalysts.6 To date this field of organocatalysis has largely been limited to doubly activated Michael donors, such as malonates, β-diketones, and similarly acidic substrates,7 and the use of amidic donors has been scarcely explored.8 Key to our new chemistry is the finding that the under-explored triketopiperazine (TKP) motif provides exceptional activation for deprotonation, whilst also enabling facile access to DKP products through nucleophilic addition.9
Results and discussion
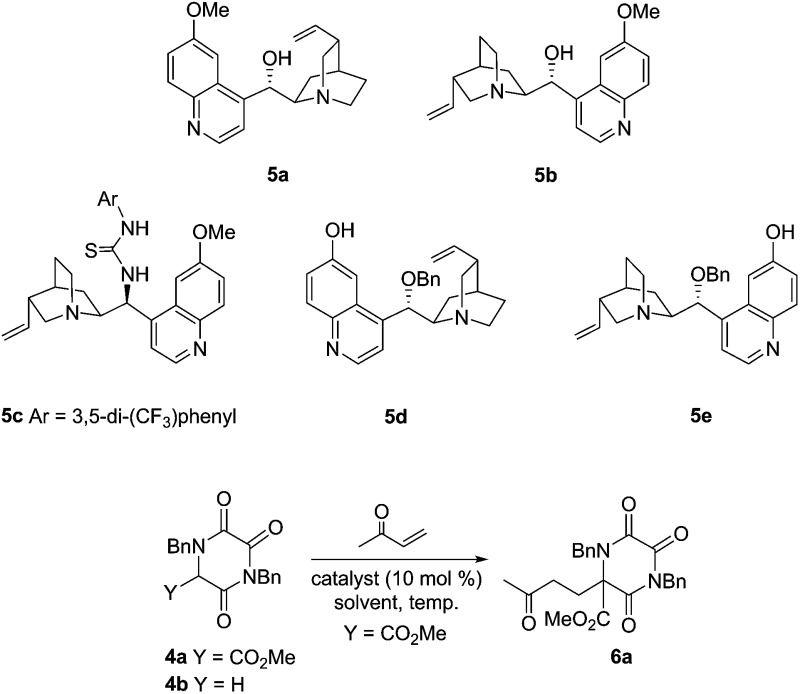
Both the doubly activated α-carboxymethyl TKP 4a, and the parent system 4b, were readily prepared using an established approach.9b,10 Mindful of the challenge of engaging masked glycine 4b in this type of chemistry, initial screening focused on the doubly activated TKP 4a, using a limited range of alkaloid catalysts 5a–e, and with MVK as Michael acceptor, Table 1.
Table 1. Initial catalyst screening using TKP 4a.
| ||||||
| Entry | Catalyst | Temp | Solvent | Time a | 6a b [%] | er c |
| 1 | 5a | RT | CH2Cl2 | 20 | 94 | 82 : 18 |
| 2 | 5b | RT | CH2Cl2 | 20 | 91 | 25 : 75 |
| 3 | 5c | RT | CH2Cl2 | 20 | 98 | 31 : 69 |
| 4 | 5d | RT | CH2Cl2 | 20 | 99 | 96 : 4 |
| 5 | 5d | RT | THF | 20 | 98 | 95 : 5 |
| 6 | 5d | RT | Toluene | 20 | 98 | 94 : 6 |
| 7 | 5d | RT | MeCN | 20 | 99 | 94 : 6 |
| 8 | 5d | 0 °C | CH2Cl2 | 60 | 99 | 98 : 2 |
| 9 | 5d | 0 °C | CH2Cl2 | 120 | 91 d | 97 : 3 |
| 10 | 5e | 0 °C | CH2Cl2 | 60 | 82 | 2 : 98 |
aMinutes.
bIsolated yield after chromatography.
cDetermined by HPLC analysis.
dReaction conducted using 1 mol% catalyst.
All of the catalysts explored gave good levels of conversion to the desired Michael adduct 6a within only 20 minutes at room temperature, with isolated yields being very high. Natural alkaloids quinidine (5a) and quinine (5b) gave interesting levels of enantioselection, with the modified thiourea system (5c) being slightly less selective.11 The catalyst systems 5d and 5e, developed by the Deng group, having a free quinoline phenol and an alkylated C-9 secondary alcohol, immediately displayed exciting levels of selectivity, especially in CH2Cl2 at 0 °C.12 As anticipated, pseudoenantiomeric catalysts 5d and 5e gave opposite enantiomers of the product 6a, in this case with very high selectivity. The use of only 1 mol% of catalyst 5d was still adequate to effect smooth conversion to product 6a without erosion of enantioselectivity (entry 9).13
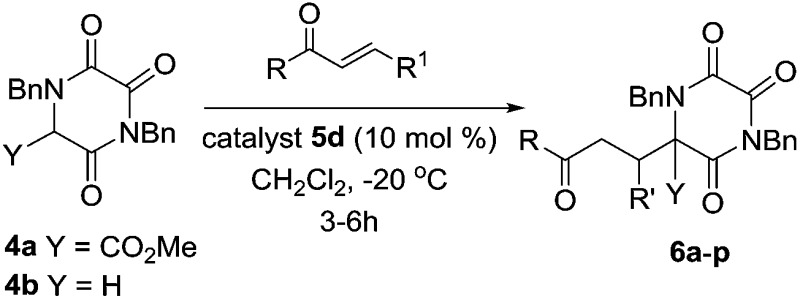
When these partly optimised conditions were explored with alternative Michael acceptors, and then with the parent TKP 4b, we found some slight further tailoring of the conditions was required. In particular, TKP 4b showed a tendency towards double Michael addition, which could be controlled simply by reducing the reaction temperature to –20 °C. The use of this lower temperature in turn extended reaction times, particularly with β-substituted Michael acceptors, and required us to maintain catalyst loading at 10 mol%. Table 2 shows the results obtained with TKPs 4a and 4b with a range of acceptors, including MVK, acrolein and various enones.
Table 2. Michael additions of TKPs 4 using catalyst 5d.
| |||||
| Entry | TKP | R′ | R | 6 a [%] | er b |
| 1 | 4a | H | Me | 6a 99 | 99 : 1 |
| 2 | 4a | H | H | 6b 99 | 96 : 4 c |
| 3 | 4a | H | Et | 6c 90 | 97 : 3 |
| 4 | 4a | H | cy-C6H11 | 6d 99 | 97 : 3 |
| 5 | 4a | H | Ph | 6e 98 | 99 : 1 |
| 6 | 4a | H | p-C6H4Br | 6f 98 | 99 : 1 |
| 7 | 4a | H | p-C6H4OMe | 6g 87 | 99 : 1 |
| 8 | 4b | H | Me | 6h 80 | 93 : 7 |
| 9 | 4b | H | Et | 6i 86 | 96 : 4 |
| 10 | 4b | H | cy-C6H11 | 6j 99 | 90 : 10 |
| 11 | 4b | H | p-C6H4OMe | 6k 93 | 88 : 12 |
| 12 | 4b | Ph | Ph | 6l 98 | 99 : 1 d |
| 13 | 4b | Ph | o-C6H4Br | 6m 91 | 88 : 12 d |
aIsolated yield after chromatography.
bDetermined by HPLC analysis.
cHPLC carried out on derived compound 8. See Scheme 1.
dIsolated as a single diastereomer.
Addition reactions with TKP 4a gave excellent levels of asymmetric induction, with er ≥ 97 : 3 (entries 1–7). The high selectivity observed for both aromatic and aliphatic enones, and for acrolein, is particularly notable. High yields were also obtained in reactions of TKP 4b, although levels of enantioselection proved more mixed. The lower figures appear to be due to variable erosion of er in the configurationally unstable TKP products rather than inherently lower levels of asymmetric induction. Thus, re-exposure of isolated TKP 6i to the catalyst 5d, under the usual conditions, led to erosion of er from 96 : 4 to 89 : 11. This problem should be overcome by more closely monitoring individual reactions, or perhaps by use of more hindered catalysts.
Notwithstanding these issues, the er values for MVK and EVK (entries 8 and 9) are good, and entry 12, involving chalcone as acceptor, demonstrates that excellent selectivities are still possible from the simple TKP 4b. Notably, both chalcone products 6l and 6m were isolated as single diastereoisomers, and no minor isomers could be detected in the 1H NMR spectra of the crude materials.
To our knowledge, these are the first enone Michael additions using a simple, non-benzylic, amidic donor,14 and are unusual in giving masked α-amino acid products retaining an α-hydrogen.
Following crystallisation, the absolute and relative configuration of adduct 6m, generated from reaction of TKP 4b and ortho-bromo chalcone, was determined by X-ray crystallography, Fig. 2.15
Fig. 2. The structure of one of the two crystallographically independent molecules of 6m, with ellipsoids drawn at the 50% probability level.

Interestingly, in the crystal there are two independent molecules of 6m having different conformations around the newly formed C–C bond (only one is shown, see ESI† for the other one).
From the data available to date, we confidently expect that the absolute configuration of the other products 6 is analogous, i.e. (S) at the newly formed asymmetric centre at C-6 on the TKP ring.16
This outcome is in accord with the stereochemical model proposed by Deng, in which the catalyst serves to organize both of the Michael addition partners in an extended array.12 This involves activation of the donor by enolate formation through the quinuclidine amine function, and a developing hydrogen-bonded association between the Michael acceptor and the quinoline phenol, Fig. 3.
Fig. 3. Model for 5d-catalyzed Michael addition leading to 6m.

Application of the Deng model for chalcone systems requires a seemingly counter-intuitive placement of the β-phenyl substituent (R′) endo with respect to the TKP ring (albeit a planar, all-sp2 ring). That this arrangement is plausible is supported by the finding that one of the crystal structure conformations of 6m (Fig. 1) exhibits precisely the local conformation about the newly formed C–C bond, as shown in Fig. 2. This arrangement apparently tolerates substitution of the α-hydrogen, Y = H, with an α-CO2Me function, this possibly further organising the transition state by an additional hydrogen bond to the quinoline phenol.12e
The TKP ring in these chiral products is expected to be electrophilic at C-3, this carbonyl function being both part of an imide, and part of an α-dicarbonyl.9 Thus, we anticipate that varied manipulations of these compounds will be possible to give DKPs of various kinds, and by way of preliminary demonstration of this potential, we have transformed 6b into the ether bridged DKP 8, via diol 7, Scheme 1.
Scheme 1. Conversion of Michael adduct 6b into bridged DKP 8.

The use of NaBH4 enabled reduction of the aldehyde in adduct 6b with concomitant regioselective TKP reduction, to give 7, which then underwent N-acyliminium cyclisation using TMSOTf to give 8.17 This sequence was used to convert 6b into an adduct more suitable for er determination, and the product 8 proved to be highly enantiomerically enriched (>96 : 4 er). This compound has a distinctive 4-atom ether-bridged DKP reminiscent of that present in the antibiotic bicyclomycin (2).2
We also established that reduction of TKPs to the corresponding DKPs could be achieved in very high overall yield by a two-step process involving initial reduction by l-selectride, then treatment with Et3SiH BF3–OEt2, Scheme 2.
Scheme 2. Regioselective reduction of TKP 6l to give DKP 10.

The initial reduction proved highly regioselective for the C-3 TKP C O function, even in the presence of a side-chain ketone. The hydroxy DKP 9 is an obvious precursor to an N-acyliminium type of intermediate, with many possibilities for further transformation.
In this case reduction under standard conditions gave DKP 10, the er of which (99 : 1) was found to be essentially the same as the starting TKP. This result is particularly significant for manipulation of these systems, since it demonstrates the possibility of converting a configurationally labile TKP into a less acidic, and so more configurationally stable, DKP product.
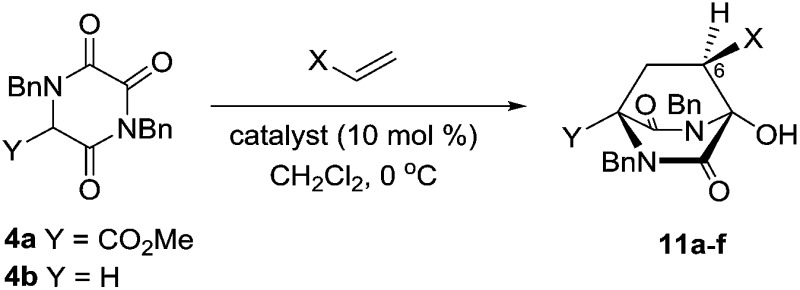
We subsequently extended our Michael addition studies to include other types of acceptor, including vinyl sulfone, acrylonitrile and acrylate esters. Reactions conducted using TKPs 4a and 4b with these partners provided bicyclic hydroxy DKP products 11 directly, via a novel Michael–ring-closure process,18Table 3.
Table 3. Enantioselective Michael–ring-closure of TKPs 4.
| |||||
| Entry | TKP | Cat. | X | 11 a [%] | er b |
| 1 | 4a | 5d | CO2Me | 11a 98 | 87 : 13 |
| 2 | 4a | 5d | CN | 11b 99 | 95 : 5 |
| 3 | 4a | 5d | SO2Ph | 11c 97 | 97 : 3 |
| 4 | 4b | 5d | CO2Me | 11d 89 | 83 : 17 |
| 5 | 4b | 5d | CN | 11e 98 | 91 : 9 |
| 6 | 4b | 5d | SO2Ph | 11f 82 | 93 : 7 |
| 7 | 4a | 5c | CN | 11b 88 | 7 : 93 c |
| 8 | 4b | 5c | CN | 11e 82 | 1 : 99 c |
aIsolated yield of single diastereomer after chromatography.
bDetermined by HPLC analysis.
cEnantiomeric structure to that shown.
The greater nucleophilicity of the intermediate anions or enolates involved in these reactions, compared to those involving enones, is presumably the origin of the observed aldol-like ring-closure. Remarkably, this process delivers bridged DKP products 11 as single diastereomers, with the creation of three new stereogenic centres, and with extraordinary efficiency and very good selectivity, especially in the cases involving vinyl sulfone.19 In the case of the nitriles 11b and 11e it was found that the thiourea catalyst 5c also gives excellent results (entries 7 and 8).
These DKPs possess the bicyclo[2.2.2]diazaoctane core structure of stephacidin and related natural products, and the relative configuration at C-6 (stephacidin numbering – see 3) is also correct for the majority of this family of alkaloids. The potential of this new process to deliver intermediates capable of further elaboration towards bioactive relatives of naturally occurring alkaloids is clear.
Conclusions
In conclusion, we have demonstrated the utility of the TKP motif in asymmetric cinchona alkaloid-catalyzed Michael additions to access various significant types of chiral DKP with high selectivities. We anticipate that this approach will enable the synthesis of many types of non-racemic TKPs, DKPs and derived structures, particularly those with fully substituted asymmetric centres, and further study of these systems is the subject of ongoing research in our laboratories.
Supplementary Material
Acknowledgments
We acknowledge the University of Birmingham, and AstraZeneca for support of AC. The NMR and HPLC instruments used in this research were obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). We also thank Neil Sumner of AstraZeneca for assistance with chromatographic er determination.
Footnotes
†Electronic supplementary information (ESI) available: Experimental procedures, analytical data and NMR spectra, X-ray data. CCDC 977363, 977364, 977365 and 1017592. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03218g
References
- (a).Daugan A., Grondin P., Ruault C., Le Monnier de Gouville A.-C., Coste H., Linget J. M., Kirilovsky J., Hyafil F., Labaudinière R. J. Med. Chem. 2003;46:4533. doi: 10.1021/jm0300577. [DOI] [PubMed] [Google Scholar]
- Williams R. M., Durham C. A. Chem. Rev. 1988;88:511. [Google Scholar]
- (a) Qian-Cutrone J. F., Huang S., Shu Y. Z., Vyas D., Fairchild C., Menendez A., Krampitz K., Dalterio R., Klohr S. E., Gao Q. J. Am. Chem. Soc. 2002;124:14556. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]; (b) Nising C. F. Chem. Soc. Rev. 2010;39:591. doi: 10.1039/b900407f. [DOI] [PubMed] [Google Scholar]; (c) Miller K. A., Williams R. M. Chem. Soc. Rev. 2009;38:3160. doi: 10.1039/b816705m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Borthwick A. D. Chem. Rev. 2012;112:3641. doi: 10.1021/cr200398y. [DOI] [PubMed] [Google Scholar]; (b) See also González J. F., Ortín I., de la Cuesta E., Menéndez J. C., Chem. Soc. Rev., 2012, 41 , 6902 . [DOI] [PubMed] [Google Scholar]
- Pichowicz M., Simpkins N. S., Blake A. J., Wilson C., Tetrahedron, 2008, 64 , 3713 , , and references therein . [Google Scholar]
- (a) Scheffler U., Mahrwald R. Chem.–Eur. J. 2013;19:14346. doi: 10.1002/chem.201301996. [DOI] [PubMed] [Google Scholar]; (b) Palomo C., Oiarbide M., López R. Chem. Soc. Rev. 2009;38:632. doi: 10.1039/b708453f. [DOI] [PubMed] [Google Scholar]; (c) Pellissier H. Adv. Synth. Catal. 2012;354:237. [Google Scholar]; (d) Marcelli T., Hiemstra H. Synthesis. 2010:1229. [Google Scholar]; (e) Connon S. J. Synlett. 2009:354. [Google Scholar]
- (a) Hong L., Wang R. Adv. Synth. Catal. 2013;355:1023. [Google Scholar]; (b) Qiao B., An Y., Liu Q., Yang W., Liu H., Shen J., Yan L., Jiang Z. Org. Lett. 2013;15:2358. doi: 10.1021/ol401062z. [DOI] [PubMed] [Google Scholar]; (c) Alba A.-N. R., Company X., Valero G., Moyano A., Rios R. Chem.–Eur. J. 2010;16:5354. doi: 10.1002/chem.200903025. [DOI] [PubMed] [Google Scholar]; (d) Alemán J., Milelli A., Cabrera S., Reyes E., Jørgensen K. A. Chem.–Eur. J. 2008;14:10958. doi: 10.1002/chem.200802030. [DOI] [PubMed] [Google Scholar]; (e) Mosey R. A., Fisk J. S., Tepe J. J., Tetrahedron: Asymmetry, 2008, 19 , 2755 , . See also vinylogous Michael additions of unsaturated butyrolactams . [Google Scholar]; (f) Choudhury A. R., Mukerjee S. Org. Biomol. Chem. 2012;10:7313. doi: 10.1039/c2ob25832c. [DOI] [PubMed] [Google Scholar]; (g) Yang Y., Dong S., Liu X., Lin L., Feng X. Chem. Commun. 2012;48:5040. doi: 10.1039/c2cc31470c. [DOI] [PubMed] [Google Scholar]; (h) Zhu J.-L., Zhang Y., Liu C., Zheng A.-M., Wang W. J. Org. Chem. 2012;77:9813. doi: 10.1021/jo302133n. [DOI] [PubMed] [Google Scholar]; (i) Alonso D. A., Kitagaki S., Utsumi N., Barbas III C. F. Angew. Chem., Int Ed. 2008;47:4588. doi: 10.1002/anie.200801088. [DOI] [PubMed] [Google Scholar]; (j) For additions of 5-aryl-1,3-dioxolan-4-ones, see Hynes P. S., Stranges D., Stupple P. A., Guarna A., Dixon D. J., Org. Lett., 2007, 9 , 2107 . [DOI] [PubMed] [Google Scholar]; (k) Polaske N. W., Dubey R., Nichol G. S., Olenyuk B. Tetrahedron: Asymmetry. 2009;20:2742. doi: 10.1016/j.tetasy.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B., Hernández-Torres G., Barbas III C. F. Angew. Chem., Int. Ed. 2012;51:5381. doi: 10.1002/anie.201200996. [DOI] [PubMed] [Google Scholar]
- TKP synthesis:; (a) Bailey P. D., Bannister N., Bernad M., Blanchard S., Boa A. N. J. Chem. Soc., Perkin Trans. 1. 2001:3245. [Google Scholar]; (b) Makino S., Nakanishi E., Tsuji T. Synlett. 2003:817. [Google Scholar]; (c) Safir S. R., Hlavka J. J., Williams J. H. J. Org. Chem. 1953;18:106. [Google Scholar]; (d) Mulliez M., Royer J., Tetrahedron, 1984, 40 , 5143 , . The only significant strand of research, of which we are aware, involves the unsaturated TKP motif as a feature in gliocladin C, and in intermediates towards epipolythio-dioxopiperazine alkaloids, see . [Google Scholar]; (e) DeLorbe J. E., Horne D., Jove R., Mennen S. M., Nam S., Zhang F.-L., Overman L. E. J. Am. Chem. Soc. 2013;135:4117. doi: 10.1021/ja400315y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jabri S. Y., Overman L. E., J. Am. Chem. Soc., 2013, 135 , 4231 , See also: Coste A., Kim J., Adams T. C., Movassaghi M., Chem. Sci., 2013, 4 , 3191 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- We adopted oxalyl dibenzotriazole for TKP synthesis, as opposed to the less easily prepared diimidazole reagent employed in (ref. 9b), see Katritzky A. R., Levell J. R., Pleynet D. M. P., Synthesis, 1998. , 153 . [Google Scholar]
- (a) Vakulya B., Varga S., Csámpai A., Soós T. Org. Lett. 2005;7:1967. doi: 10.1021/ol050431s. [DOI] [PubMed] [Google Scholar]; (b) Li B.-J., Jiang L., Liu M., Chen Y.-C., Ding L.-S., Wu Y. Synlett. 2005:603. [Google Scholar]
- (a) Wang B., Wu F., Wang Y., Liu X., Deng L. J. Am. Chem. Soc. 2007;129:768. doi: 10.1021/ja0670409. [DOI] [PubMed] [Google Scholar]; (b) Li H., Song J., Deng L. Tetrahedron. 2009;65:3139. doi: 10.1016/j.tet.2008.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu F., Hong R., Khan J., Liu X., Deng L. Angew. Chem., Int. Ed. 2006;45:4301. doi: 10.1002/anie.200600867. [DOI] [PubMed] [Google Scholar]; (d) Wu F., Li H., Hong R., Deng L. Angew. Chem., Int. Ed. 2006;45:947. doi: 10.1002/anie.200502658. [DOI] [PubMed] [Google Scholar]; (e) Li H., Wang Y., Tang L., Wu F., Liu X., Guo C., Foxman B. M., Deng L. Angew. Chem., Int. Ed. 2005;44:105. doi: 10.1002/anie.200461923. [DOI] [PubMed] [Google Scholar]; (f) Li H., Wang Y., Tang L., Deng L. J. Am. Chem. Soc. 2004;126:9906. doi: 10.1021/ja047281l. [DOI] [PubMed] [Google Scholar]
- Giacalone F., Gruttadauria M., Agrigento P., Noto R. Chem. Soc. Rev. 2012;41:2406. doi: 10.1039/c1cs15206h. [DOI] [PubMed] [Google Scholar]
- For additions of β-ketoamides, see ; (a) del Mar Sanchez Duque M., Baslé O., Isambert N., Gaudel-Siri A., Génisson Y., Plaquevent J.-C., Rodriguez J., Constantieux T. Org. Lett. 2011;13:3296. doi: 10.1021/ol200924e. [DOI] [PubMed] [Google Scholar]; (b) Dai X., Wu X., Fang H., Nie L., Chen J., Deng H., Cao W., Zhao G. Tetrahedron. 2011;67:3034. [Google Scholar]; (c) Raimondi W., del Mar Sanchez Duque M., Goudedranche S., Quintard A., Constantieux T., Bugaut X., Bonne D., Rodriguez J., Synthesis, 2013, 45 , 1659 , . For additions of succinimidyl esters, see . [Google Scholar]; (d) Jacubec P., Cockfield D. M., Hynes P. S., Cleator E., Dixon D. J. Tetrahedron: Asymmetry. 2011;22:1147. [Google Scholar]
- ESI.
- All of the products obtained using catalyst 5d show negative specific optical rotations. We have also been able to determine the absolute configurations of 6l and a dioxolane acetal derived from MVK adduct 6h by X-ray crystallography (see ESI)
- For analogous bridge formation, see; (a) Sato Y., Shin C., Sumiya S., Nakajima Y., Yoshimura J., Bull. Chem. Soc. Jpn., 1984, 57 , 1265 , . For use of TMSOTf in triggering cationic cyclisations from hydroxy-DKPs, see . [Google Scholar]; (b) Frebault F. C., Simpkins N. S. Tetrahedron. 2010;66:6585. [Google Scholar]; (c) Frebault F., Simpkins N. S., Fenwick A. J. Am. Chem. Soc. 2009;131:4214. doi: 10.1021/ja900688y. [DOI] [PubMed] [Google Scholar]
- (a) Review of organocatalytic cyclization and cycloaddition reactions, see Moyano A., Rios R., Chem. Rev., 2011, 111 , 4703 . [DOI] [PubMed] [Google Scholar]; (b) For a related process with anthrones, see Shen J., Nguyen T. T., Goh Y.-P., Ye W., Fu X., Xu J., Tan C.-H., J. Am. Chem. Soc., 2006, 128 , 13692 . [DOI] [PubMed] [Google Scholar]
- The absolute and relative stereochemistry shown is based on an X-ray determination of 11d (see ESI). The absolute stereochemistry is consistent with that of Michael adducts from enones in Table 2
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.