

Abstract
Uncontrolled inflammatory responses underlie the etiology of acute lung injury and acute distress respiratory syndrome, the most common late complications in trauma, the leading cause of death under the age of 59. Treatment with HTS decreases lung injury in clinical trials, rat models of trauma and hemorrhagic shock and inflammation in lung cell lines, although the mechanisms underlying these responses are still incompletely understood. Transcriptomics (RNaseq), proteomics, and U-13C-glucose tracing metabolomics experiments were performed to investigate the mechanisms of cellular responses to HTS treatment in primary small airway epithelial cells in the presence or absence of inflammatory injury mediated by a cocktail of cytokines (10 ng/mL of IFNγ, IL-1β, and TNFα). Modestly hyperosmolar HTS has an anti-inflammatory effect, triggers the p53-p21 signaling axis, and deregulates mitochondrial metabolism while inducing minimal apoptosis in response to a second hit by cytokines. Decreased transcription of pro-inflammatory cytokines suggested a role for the tumor suppressor protein p53 in mediating the beneficial effects of the HTS treatment. The anti-inflammatory mechanisms induced by HTS involves p53 gene regulation, promotes cell cycle arrest, and prevents ROS formation and mitochondria depolarization. Pharmaceutical targeting of the p53-p21 axis may mimic or reinforce the beneficial effects mediated by HTS when sustained hypertonicity cannot be maintained.
Keywords: metabolomics, proteomics, RNaseq, mitochondria, osmotic stress
Graphical Abstract
INTRODUCTION
Trauma and hemorrhagic shock is the global leading cause of death in the age window of 0–59 years and accounts for ~30% of total life years lost in 2014.1 Approximately 40% of deaths by trauma occur hours or days after injury, when sequelae to severe trauma and hemorrhage trigger multiple organ dysfunction, such as acute respiratory distress syndrome (ARDS), acute kidney failure, cardiac dysfunction, infections, and sepsis.2 In particular, lung inflammation and dysfunction typically precede other organ failure.3 Despite a decrease in mortality due to multiple organ failure, morbidity and prolonged hospital stays remain high (~44% of survivors) after trauma and hemorrhagic shock,3 mostly due to post-traumatic acute lung injury (ALI) and ARDS.4
Clinical trials have been performed to test the effectiveness of hypertonic saline (HTS) in the treatment of ALI5 and ARDS,6 showing promise for resuscitation in early trials for trauma patients.5–8 These clinical studies were recently supported by in vitro evidence, indicating a significant anti-inflammatory benefit of HTS.9–11 By the alteration of ionicity and osmolarity, thus causing fluid shifts,12 HTS could be leveraged for clinical applications such as a mucolytic agent in cystic fibrosis in children13 or proposed as a treatment to decrease brain swelling after traumatic brain injury. However, the mechanisms underlying the beneficial effects observed in HTS-treated patients remain elusive. A recent study showed that the efficacy of HTS therapy in the cystic fibrosis lung may be due to under-investigated metabolic mechanisms rather than to the osmotic sensitivity of bacteria such as Pseudomonas aeruginosa.14 Despite discouraging results from clinical trials on traumatic brain injury patients,15,16 it has been proposed that a single early small dose of HTS could influence the long-term survival of this population.16
Resuscitative strategies based upon systemic or nebulized administration of HTS have been questioned, owing to the rapid normalization of tonicity, especially in plasma.13 However, further not-yet-elucidated mechanisms other than tonicity may underlie the clinical benefits observed in patients. In vivo and in vitro studies performed by our group suggested that some of the beneficial anti-inflammatory effects mediated by HTS could be due to hyperosmolarity rather than hypertonicity.17,18 Following hemorrhagic shock in rats, nebulized HTS reduced lung inflammation, alveolar macrophage activation, and neutrophil recruitment into the lung.11 Besides this, inhaled HTS decreased lung leak and improved lung extracellular matrix homeostasis by promoting CINC1 production, MMP13 activation, and preservation of the overall lung architecture.11
In vitro, HTS directly modulates human neutrophil,19 T-cell,20 and macrophage8 responses. Additional in vitro evidence has suggested that HTS has an anti-inflammatory activity on lung epithelial cells by deregulating the pro-inflammatory signaling of ubiquitous cytokines such as tumor necrosis factor-a (TNFα) and interleukin 1b (IL-1β), key players in the etiology of pulmonary inflammation and post-traumatic ARDS and ALI.21,22
Recently, we associated HTS treatment to pulmonary epithelial cancer cells with the disruption of transcriptional signaling axis involving nuclear factor kB (NF-kB),18 a transcription factor for several genes involved in the inflammation in both animals and humans,23 especially in response to post-traumatic ARDS.24 Although NF-kB-dependent inflammatory gene expression is well-established, inflammatory cascades often involve multiple transcription factors that vary by cell phenotype and stimulus. Previously, we have determined that HTS may also prevent NF-kB activation by decreasing signaling through IKK in A549 human lung carcinoma18 and human vascular smooth muscle cells.17 NF-kB-independent anti-inflammatory effects of HTS have been also attributed to differential attenuation in ERK1/2 versus p38MAPK pathways and transcriptional signaling by IRF-1,25 suggesting a complex role for HTS in mediating transcriptional cascades. Other than exerting a regulatory transcriptional effect on inflammatory cytokines, HTS treatment may also promote the endocytosis of inflammatory cytokine receptors26 and cell apoptosis.27
More recently, hyperosmotic stress has been associated with the transcriptional activation of the prototypic stress-associated transcription factor, p53,28 mostly known for its central role as a tumor suppressor guardian of the genome.29 Intriguingly, an emerging concept in cancer research and immunity posits that key tumor suppressors and oncogenes such as p53 may mediate systemic metabolic changes,30 which in turn affect not just cancer cell proliferation potential but also immune cell responses.31 Indeed, metabolic reprogramming is a key feature in immune system activation32 (e.g., succinate release as a mediator of inflammation through IL-1β and HIF1α33), and it underlies the pro-inflammatory phenotype associated with acute and chronic lung diseases, such as pulmonary hypertension.34,35
In this view, in the present study, we adopted an integrated omics approach, based upon transcriptomics (RNaseq), proteomics, and metabolomics (including U-13C-glucose tracing experiments) to further dissect the transcriptional mechanisms and signaling cascades through which HTS might affect cellular homeostasis and inflammation in primary nontumor lung epithelial cells as to disrupt inflammatory signaling triggered by pro-inflammatory cytokines such as TNFα, IL-1β, and IFNγ.
METHODS
Cell Culture and Treatments
Primary cells, media and additives, wash buffers, and trypsin/trypsin inhibitor were purchased from Lonza (Walkersville, MD).
Small airway epithelial cells (SAEC, no. CC-2547) were grown in full growth medium (SABM+singlequots) at 37 °C in the presence of 5% CO2 to 70–80% confluence and then trypsinized and reseeded for subsequent experiments. Treatments were performed as previously reported, consistent with the minimum increases in osmolarity necessary to promote anti-inflammatory activity in pulmonary epithelial cells in our previous studies.18 Cells were treated with isotonic (290 mOsm) medium (controls) or 400 mOsm NaCl (hypertonic saline, HTS) media in the presence or the absence of 10 ng/mL TNFα, 10 ng/mL IL-1β, and 10 ng/mL IFNγ (cytomix (CMX); Sigma-Aldrich, St. Louis, MO).
RNaseq
RNaseq analyses were performed as previously reported.36 Briefly, total RNA was extracted from three independent biological replicates of SAECs, either untreated or treated with either HTS or CMX or both using RNeasy kit (Qiagen Inc., Valencia, CA). cDNA libraries were constructed for each sample using the TruSeq mRNA sample preparation kit no. RS-122-9400 (Illumina). For each condition, independently and uniquely indexed libraries were pooled and loaded onto a single lane of an Illumina HiSeq2000 flow cell yielding single-pass 100 bp reads. The resulting sequence from the cDNA libraries were subjected to removal of low-quality bases (Phred score of <15) using a custom Python script. The remaining sequences were mapped to the human genome (hg19) using GSNAP, as reported.36 After strand alignment of the human sequences, the fragments per kilobase of exon per million mapped reads (FPKM; a relative expression level normalized to the sum of all sequences mapped as well as by specific gene length) were determined using CUFFLINKS. In R, FPKM values were used to determine significant differential gene expression using ANOVA. Differentially expressed genes upon false discovery rate correction (Q < 0.01) per cell line were analyzed with Gene Set Enrichment Analysis software (Broad Institute, Cambridge, MA) to identify pathways of interest that may have been affected by either HTS or Cytomix or combined treatments. Parameters used for the analysis are as follows: c2.all.v4.0.symbols.gmt database, 1000 permutations, and gene set permutation type.
RT-PCR
Total RNA was isolated from SAECS using the RNeasy Mini Kit (Qiagen Inc.) and quantified spectrophotmetrically. A total of 1 μg of total RNA was used to prepare cDNA using the qScript cDNA supermix (Quanta Bioscience, Gaithesburg, MD; no. 95048-025). Real-time PCR (RT-PCR) was performed on an iQ5Multicolor Real-Time PCR Detection system (Bio-Rad). All primer-probe gene expression assays for real-time analyses were purchased from Applied Biosystems (Thermo Fisher) (tumor protein p53, TP53 assay ID Hs01034249_m1, p21 assay ID Hs00355782_m1, TIGAR assay ID Hs00608646_m1, and GAPDH assay ID Hs99999905_m1). A total of 2 μL of total cDNA was used to assay TP53, CDN1A, and TIGAR transcripts. A total of 1 ml of 1:10 dilution of the cDNA was to assay for GAPDH as a housekeeping control. Transcripts were amplified using the PerfeCTa FastMix II reagent (Quanta Bioscience catalog no. 95119-250) according to the manufacturer’s protocol.
Western Blots
SAECs were cultured in 100 mm tissue culture dishes to 80% confluence. After HTS pretreatment and CMX stimulation, as above, media were aspirated and cells lifted using 500 μL/dish of RIPA lysis and extraction buffer (Thermo Fisher Scientific, no. 89900) for 15 min at 4 °C. Cells were then lifted and collected, sonicated for three cycles of 20 s each, and spun at 17 000 relative centrifugal force for 5 min to separate debris. The supernatants were immediately aliquoted, flash-frozen, and stored at −80 °C for future analysis. A total of 15 μg of protein per sample were run by SDS PAGE in 15 well gels with 4–20% acrylamide (BioRad, Hercules, CA). The proteins were then transferred onto nitrocellulose membranes by semidry transfer (Bio Rad, Turbo blot) for 7 min. Even loading and transfer was verified by Ponceau S staining of the membranes (1% Ponceau S in 5% acetic acid, Sigma-Aldrich). After destaining, the membranes were incubated with blocking solution (5% BSA in PBS) for 1 h at room temperature. Primary antibodies were applied at a concentration of 2 μg/mL in 5% BSA in PBS overnight at 4 °C. After the washing of excess antibody, antispecies secondary antibodies conjugated to HRP (1:10 000; Pierce and Thermo Fisher Scientific, Grand Island, NY) were incubated with the membranes at room temperature for 1 h. Bands were visualized after the application of ECL substrate (Pierce, West Dura) using a Chemidoc MP system (Bio Rad). Band densitometry was normalized to densitometry of GAPDH bands on the same membrane (GAPDH antibody from Cell Signaling, Danver, MA).
Live-Cell Imaging
All live-cell reagents were purchased from Molecular Probes and Thermo Fisher Scientific (Pittsburgh, PA) and applied as per manufacturer’s protocols.
Cells were seeded at 100 000/mL in Delta-T dishes (Bioptechs Inc., Butler, PA), incubated for 24 h at 37 °C in a humidified chamber in the presence of 5% CO2 in resting medium (SABM+retinoic acid+ glutamine), and then treated with either isotonic or hypertonic growth medium for 30 min. Cells were then treated with 10 ng/mL each of TNFα, IL1β, and IFNγ (Cytomix) (Sigma/Aldrich) for 4 h. A total of 15–30 min before the end of the 4 h time, live-cell stains were added as per manufacturer’s suggestions.
The combinations of live-cell stains studied were: JC1 (red and green fluorescence) for mitochondrial potential measurements; Cell-ROX Orange, Hoechst B (blue), and Cell Mask Green for cellular ROS generation measurements; and Mito SOX orange, Hoechst B, Cell Mask Green for mitochondrial peroxide generation measurements. Excess reagents were washed off twice with warm PBS, and 10 μL/ml of Prolong-Live (1:100 final concentration) in growth medium was added to all dishes to decrease fluorescence quenching during imaging. Images were collected at 4 h of incubation using a Zeiss Observer.Z1 inverted microscope equipped with a Bioptechs Delta-T 5 μ-Environmental controller. Images were acquired and analyzed using Slidebook 6.0 (Intelligent Imaging Innovations, Denver, CO). Boolean mask operations were applied and fluorescent intensity measures analyzed by ANOVA and t tests.
Fixed-Cell Imaging
Cells were grown in four well culture slides (Falcon and Thermo Scientific) to a 70–80% confluence and treated as above. Media were aspirated and cells rinsed twice in 100 mm PBS, fixed and permeabilized in 70% acetone and 30%methanol at −20 °C for 10 min, air-dried, and incubated in blocking solution (10% Normal Donkey Serum in PBS) for 1 h at room temperature.
Primary antibodies against p53, P21, and respective phosphorylated forms were purchased from Santa Cruz Biotech, Inc.(Dallas, TX) (p53 FL-393 sc-6243, p21 M-19 sc-471, p-p53 thr155 sc-17105, and p-p21 thr145 sc 20220). Antibodies against the specific proteins, and respective isotype controls, were applied at concentrations of 4 μg/mL overnight at 4 °C. Antispecies donkey secondary antibodies conjugated to Alexa Fluor 555, Alxa Fluor 647 WGA (wheat germ agglutinin, to visualize all cell membranes), and Prolong+DAPI anti-quenching and mounting medium were purchased from Molecular Probes and Thermo Fisher Scientific. Slides were washed three times for 10 min each in PBS to eliminate excess antibody, and secondary antibodies were applied at a 1:100 dilution, together with Alexa Fluor 647 WGA, for 1 h at room temperature. After the washing steps were repeated, the slides were mounted with Prolong+DAPI, sealed, and imaged.
Confocal images were collected using a Zeiss Observer.Z1 inverted microscope equipped with Semrock wheel filters with ec/em spectra for the Alexa Fluor probes used. Images were acquired and analyzed using Slidebook 6.0 (Intelligent Imaging Innovations, Denver, CO). Boolean mask operations were applied, and fluorescent intensity measures, minus nonspecific intensities calculated in slides incubated with isotytpes, were analyzed by ANOVA and t tests.
Proliferation and Caspase 3–7 Activation Assay
Cells were seeded in wells of a 96-well plate at 5000 cells per well and grown for 24 h. Cells were then preincubated with or without HTS for 30 min and stimulated with Cytomix, as above. The Incucyte kinetic caspase 3–7 activation reagent (#4440, 1:4000) (Essen Bioscience, Ann Arbor, MI) was added at time 0 (stimulation), and the cells were imaged in an Incucyte Zoom imaging system, both for green fluorescence (caspase activity) and in phase contrast (proliferation), at 10× magnification. Images were collected hourly for 24 h. In each well, green fluorescent and phase-contrast confluence were measured using the Basic analyzer function of the Incucyte ZOOM 2015A software. Data are expressed, both for Caspase activity and proliferation, as percent confluence at each time point, normalized to time 0 for each individual well.
Mass-Spectrometry-Based Metabolomics and Tracing Experiments
Metabolomics analyses were performed as previously reported.37 Briefly, 2 × 106 SAECs (n = 5), were either untreated or treated with HTS or CMX or both, as detailed above, in the presence or absence of 10 mM U-13C-glucose (for 0, 30, 120, and 240 min from the incubation with the labeled substrate). Cells were extracted in 1 mL of ice-cold lysis and extraction buffer (methanol/acetonitrile/water 5:3:2). After the protein pellets were discarded, water- and methanol-soluble fractions were run through a C18 reversed-phase column (250 μL/min, 9 min gradient 5–95% B; phase A: water, 0.1% formic acid; B: acetonitrile, 0.1% formic acid; Phenomenex, Torrance, CA) through an ultrahigh-performance chromatographic system (UHPLC; Vanquish, Themo Fisher). UHPLC was coupled on line with a high-resolution quadrupole Orbitrap instrument run in both polarity modes (QExactive, Thermo Fisher) at a 70 000 resolution (at 200 m/z). Metabolite assignment, isotopologue distributions upon correction for natural abundance of 13C, and peak integration for relative quantitation were performed through the software Maven (Princeton) against the KEGG pathway database and an in-house validated standard library (>650 compounds; SIGMA Aldrich; IROA-Tech). Integrated peak areas were exported into Excel (Microsoft, Redmond, CA) and elaborated for statistical analysis (t-Test and ANOVA) and hierarchical clustering analysis (HCA) through the software GraphPad Prism (GraphPad Software Inc., La Jolla, CA) and GENE E (Broad Institute), respectively.
PROTEOMICS
1D-SDS-PAGE
Proteomics analyses were performed through the in-gel tryptic digestion approach, as previously described.38 Briefly, protein concentrations of SAECs, either untreated or treated with HTS or CMX or both, were determined through bicinchoninic assay. A total of 30 μg of five pooled samples were loaded onto a 1.5 mm thick NuPAGE Bis-Tris 4–12% gradient gel (Invitrogen). The BenchMark Protein Ladder (Invitrogen) was used as a protein molecular mass marker. The gel was stained using SimplyBlue SafeStain (Invitrogen, Carlsbad, CA) stain and destained with water according to the manufacturer’s protocol. Each lane of the gel was divided into 12 equal-sized bands, and proteins in the gel were digested as follows. Gel pieces were destained in 200 μL of 25 mM ammonium bicarbonate in 50% v/v acetonitrile for 15 min and washed with 200 μL of 50% (v/v) acetonitrile. Disulfide bonds in proteins were reduced by incubation in 10 mM dithiothreitol (DTT) at 60 °C for 30 min, and cysteine residues were alkylated with 20 mM iodoaceta-mide (IAA) in the dark at room temperature for 45 min. Gel pieces were subsequently washed with 100 μL of distilled water followed by the addition of 100 μL of acetonitrile and dried on a SpeedVac (Savant ThermoFisher). Next, 100 ng of trypsin was added to each sample and allowed to rehydrate the gel plugs at 4 °C for 45 min, and the sample was then incubated at 37 °C overnight. The tryptic mixtures were acidified with formic acid up to a final concentration of 1%. Peptides were extracted two times from the gel plugs using 1% formic acid in 50% acetonitrile. The collected extractions were pooled with the initial digestion supernatant and dried on a SpeedVac.
Mass Spectrometry Analysis
Samples were analyzed on an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific) coupled to an Eksigent nanoLC-2D system through a nanoelectrospray liquid chromatography-mass spectrometry (LC-MS) interface. The source was operated at 2.25–2.5 kV, with no sheath gas flow and with the ion transfer tube at 275 °C. A volume of 8 μL of sample was injected into a 10 μL loop using the autosampler. To desalt the sample, material was flushed out of the loop and loaded onto a trapping column (ZORBAX 300SB-C18, dimensions of 5 × 0.3 mm 5 μm) and washed with 0.1% FA at a flow rate of 5 μL/min for 5 min. The analytical column was then switched online at 600 nL/min over an in-house-made 100 μm i.d. × 150 mm fused silica capillary packed with 4 μm 80 Å Synergi Hydro C18 resin (Phenomenex). After 10 min of sample loading, the flow rate was adjusted to 350 nL/min, and each sample was run on a 90 min linear gradient of 2–32% ACN with 0.1% formic acid to separate the peptides. LC mobile-phase solvents and sample dilutions used 0.1% formic acid in water (Buffer A) (Optima LC-MS grade; Fisher Scientific) and 0.1% formic acid in acetonitrile (Buffer B) (Optima LC-MS grade; Fisher Scientific). Data acquisition was performed using the instrument-supplied Xcalibur (version 2.1) software. The mass spectrometer was operated in the positive-ion mode. Full MS scans were acquired in the Orbitrap mass analyzer over the m/z 300–1800 range with a resolution of 60 000 (m/z 400). The target value was 5.00 × 1005. The 20 most-intense peaks with charge states of ≥2 were selected for sequencing and fragmented in the ion trap with normalized collision energy of 35%, activation q = 0.25, activation time of 30 ms, and one microscan.
Database Searching and Protein Identification
Tandem mass spectrometry (MS/MS) spectra were extracted from raw data files and converted into .mgf files using a PAVA script (UCSF, MSF, San Francisco, CA). These .mgf files were then independently searched against mouse SwissProt database using an in-house Mascot server (Version 2.2.06, Matrix Science). Mass tolerances were ±10 ppm for MS peaks and ±0.6 Da for MS/MS fragment ions. Trypsin specificity was used, allowing for one missed cleavage. Met oxidation, protein N-terminal acetylation, peptide N-terminal pyroglutamic acid formation, and phosphorylation of of serines, threonines, and tyrosines were allowed for variable modifications, while carbamidomethyl of Cys was set as a fixed modification.
Scaffold (version 4.4, Proteome Software, Portland, OR) was used to validate MS/MS-based peptide and protein identi-fications. Peptide identifications were accepted if they could be established at greater than 95.0% probability, as specified by the Peptide Prophet algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least two identified unique peptides.
RESULTS
RNaseq Data Confirmation of a Potential Anti-inflammatory Activity of HTS and Suggestion of a Role for HTS in Mediating the Activation of the p53 Signaling Axis
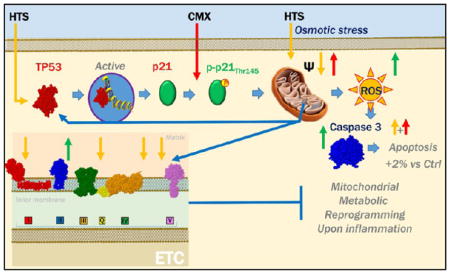
In the light of previous evidence showing a role for HTS in modulating transcriptional regulation in lung carcinoma A549 cells,18 RNaseq analyses were performed to investigate the role of HTS in the modulation of transcriptional regulation in primary human small airway epithelial cells (SAECs). Of the about 97 mRNA upregulated by >3 fold (p < 0.0001) by HTS (Supplementary Table 1), most are intracellular proteins, including transcription factors and phosphatases. However, among the ~100 mRNAs down-regulated by HTS, many are secreted cytokines (CCL5/RANTES,CXCL11, IL-36; Supplementary Table 1) as well as TNFRSF and IL-1 receptors (p < 0.001). Gene ontology term enrichment was performed to highlight top up- and down-regulated pathways following HTS treatment (Supplementary Table 1 and Figure 1A,B). Results indicated that HTS treatment deregulates transcriptional cascades and post-transcriptional regulation (RNA polymerase and degradation pathways) and nucleoside metabolism (purine and pyridimidine metabolism; Figure 1A). However, p53 signaling was the most up-regulated pathway following HTS treatment (Figure 1B). Specifically, significant up-regulation of the tumor protein p53 downstream targets, p21 and TP53AIP1, was observed in the RNaseq data set (Figure 1C). RT-PCR analyses were performed to validate RNaseq data while determining the effect on transcriptional regulation of TP53 and its target p21 after HTS treatment, either alone or in combination with a cocktail of pro-inflammatory cytokines (TNFα, IL-1β, and IFNγ, 10 ng/mL each). RT-PCR data suggested a transcriptional up-regulation of TP53 in response to cytomix treatment, alone or in combination with HTS, while HTS alone was sufficient to elicit transcriptional expression of p21 (Figure 1D). However, at the protein level, HTS treatment, but not CMX alone, promoted the expression of p53 and p21 (Figure 1E).
Figure 1.

RNaseq data suggestion of a role for HTS in mediating activation of the p53 signaling axis. Down-regulated (A) and up-regulated (B) pathways in the RNaseq screening of SAECs cells, either control or treated with hypertonic saline (HTS). Consistent with the tumor protein p53 signaling pathway ranking top in (B), significant up-regulation of TP53 and its downstream targets, p21 and TP53AIP1, was confirmed in the RNaseq data set (C). RT-PCR indicated transcriptional up-regulation of TP53 in response to cytomix (CMX) treatment, alone or in combination with HTS, while HTS alone was sufficient to elicit transcriptional expression of p21 (D). In (E), increased protein expression of TP53 and p21 was observed in response to HTS treatment.
HTS Treatment Priming of Increases of Total TP53 and Preservation of Stability of the TP53 Pool through Promotion of Degradative Phosphorylation at T155
Transcriptional activity of p53 is determined by its subcellular localization, which in turn is affected by the p53 phosphorylation state at threonine 155 (pT155).29 Fluorescence microscopy images confirmed Western blot data showing that total levels of p53 increased after treatment with HTS, either alone or in combination with CMX, while CMX alone decreased total levels of p53 (Figure 2A–D,E). These data are suggestive that p53 expression is up-regulated by HTS treatment, but, in the absence of the second hit by CMX, p53 turnover is promoted by increased pT155 and cytosolic localization (Figure 2J).
Figure 2.

HTS treatment priming of increases and nuclear translocation of p53 and preservation of the stability of the p53 pool through promotion of degradative phosphorylation at T155. Fluorescence microscopy images indicate that the total levels of cytosolic p53 and nuclear p53 increase after treatment with HTS and HTS+CMX (A–D,E). However, CMX treatment (either alone or in combination with HTS) decreased the percentage of cytosolic p53 phosphorylated at T155 (F–I), a post-translational modification that primes p53 degradation (J). Labeling: p53 (A–D) or pT155-p53 (F–I, red), cell nuclei (DAPI, blue), and membranes (WGA, green).
HTS Priming of p21 Expression and Cytosolic Localization by T145 Phosphorylation to Inhibit Cell Proliferation
As a downstream target of p53 family members, p21 expression was expected to increase in response to HTS treatment, as confirmed by fluorescence microscopy (Figure 3A–D). Also known as cyclin-dependent kinase inhibitor 1A protein, p21 is known to play a role on cell cycle arrest and induction of apoptosis dependently to its subcellular localization, which in turn is regulated by the phosphorylation state of threonine 145 (pT145).39 Fluorescence microscopy indicated that HTS treatment, especially in combination with CMX, promoted cytosolic pT145 of p21 (Figure 3E–H). These data are suggestive of a potential inhibitory effect on proliferation mediated by the HTS treatment, as confirmed by the Incucyte proliferation assay (Figure 1I). Inhibition of proliferation by the p53-p21 signaling axis may result in either cell-cycle arrest or induction of apoptosis (Figure 3J).
Figure 3.

Dependency of cytosolic and nuclear localization of p21 on T145 phosphorylation and correlation with HTS inhibitory effect on proliferation. Expression of p21 in SAECs was enhanced by the HTS treatment (A–D). HTS treatment, alone or in combination with CMX, promoted phosphorylation of p21 on T145, which prevented nuclear translocation and promoted cytosolic localization (E–H). Increased p21 expression and phosphorylation after treatment with HTS inhibited cell proliferation over 24 h (I). Inhibition of proliferation mediated by HTS effect on p21 may result in either cell-cycle arrest or apoptosis, depending on cytosolic vs nuclear localization of p21, which is in turn dependent on phosphorylation of T145 of p21.46 Labeling: p21 (A–D) or pT145-p21 (E–H, red), cell nuclei (DAPI, blue), membranes (WGA, green).
CMX Induction of Mitochondrial Hyperpolarization and Pretreatment with HTS Prior to Second Hit by CMX Promotion of Mitochondrial Uncoupling with Limited Apoptosis
Cell-cycle assays were performed on SAECs treated with HTS, either alone or in combination with CMX. Even though SAECs are characterized by heterogeneous primary cell populations, preliminary cell cycle screening suggested a minor effect of HTS treatment, resulting in the arrest of the cell cycle at the G1 phase (data not shown). In the light of the inhibitory effect on cell proliferation, because the observed effect on the cell cycle was not statistically significant, we hypothesized that the activation of the p53-p21 signaling axis induced by HTS treatment would promote apoptosis of SAECs. Caspase 3 and 7 activity in response to either or combined treatment was assayed through the Incucyte approach as well as through fluorescent microscopy (phosphatidylserine exposure, Annexin V, and propidium iodide; Figures 4A and S1), indicating significant albeit minimal increase (~2%) over baseline level of apoptosis in response to HTS only after a second hit by CMX in SAECs. Because p53 mediates apoptosis through the intrinsic mitochondrial pathway,40 which in turn is affected by total and mitochondrial reactive oxygen species (ROS),41 we wondered whether HTS’s effect on proliferation and the double-treatment effect on apoptosis were mediated by alterations of cellular and mitochondrial redox level and mitochondrial activity. Strikingly, total cellular ROS were decreased in response to HTS treatment alone but increased in response to CMX only when SAECs were pretreated with HTS (Figure 4B–E,K). However, superoxide radical generation in mitochondria was depressed by all the treatments and, in particular, by HTS alone (Figure 4F–I,K). Mitochondrial membrane potential was significantly increased in response to CMX (JC1 aggregates in red), while HTS pretreatment prevented CMX induced mitochondrial hyperpolarization (Figure 4J,K). Increased green fluorescence signaling (JC1 monomers) was observed in HTS-treated cells, especially after the second hit by CMX (Figure 4J,K). Overall, these data suggest that activation of the p53-p21 signaling axis by HTS promotes mitochondrial depolarization and inhibition of proliferation, which in turn results in increased ROS, although these effects only result in minimal increase in the percentage of apoptotic cells in response to a second hit by CMX (Figure 4L).
Figure 4.

Double treatment of HTS and CMX promotion of increased oxidative stress but only minor apoptosis. HTS treatment primes mitochondrial membrane potential uncoupling that prevents CMX-induced mitochondrial hyperpolarization. Minor apoptosis (~2% increase over untreated controls) as a function of caspase 3 and 7 activity upon HTS or CMX treatment or both in SAECs was observed at 24 h of combined treatment (A). No further increases were observed at 48 h (data not shown) Total reactive oxygen species (DCFDA, red) decreased in response to HTS treatment and increased in the combined treatment in comparison to controls (B–E,K). However, both CMX and HTS treatments decreased mitochondrial superoxide generation (F–I), suggestive of deregulated mitochondrial metabolism and mitochondrial dysfunction. Consistently, uncoupling of mitochondrial membrane potential was observed in response to all treatments, with CMX promoting membrane hyperpoplarization and HTS promoting mitochondrial membrane depolarization, especially in the combined treatment (J,K). A schematic representation of the p53-p21 cascade activation, mitochondrial membrane depolarization and minor induction of apoptosis is proposed in (L).
Mediation of Depolarized Mitochondria in Nonapoptotic HTS-Treated SAECs by Decreased Levels of Electron Transport Chain Complexes I, III, IV, and V
Proteomics analyses were performed to confirm and expand on transcriptomics data (Figure 5A and Supplementary Table 2). Treatment with HTS and CMX increased the expression of several p53 targets involved in apoptotic signaling (Supplementary Figure 2), including the p53-induced glycolysis and apoptosis regulator (TIGAR)42 enzyme (Figure 5B). HTS treatment induced the up-regulation of several transporters (solute carriers, SLC44A2 and SLC38A2, and large amino acid transporter 1, LAT1), Krebs cycle enzymes involved in mitochondrial electron transport chain complex II (succinate dehydrogenase components) and glycolytic enzymes (glucose and lactate transporter, GLUT1 and MCT4; biphosphoglycerate mutae, BPGM; TIGAR and hexokinase 1; Figure 5C). However, HTS treatment decreased the levels of Krebs cycle enzymes (especially citrate synthase and oxoglutarate dehydrogenase) and glutathione biosynthesis enzymes (glutathione synthase and γ glutamyl cysteine lyase; Figure 5C).
Figure 5.

HTS-treated SAECs and decreased levels of electron transport chain complexes. Proteomics analyses were performed on SAECs by nanoUHPLC–MS/MS (A). Results confirmed Western blot data on TIGAR (B) and revealed that HTS treatment promoted the up-regulation of a wide series of membrane transporters and electron transport chain complex II components and down-regulation of enzymes involved in glutathione biosynthesis (C). In panels D and E, top pathways were down- or up-regulated upon treatment with HTS, as gleaned through GO term enrichment of >2-fold down- or up-regulated proteins with DAVID. These pathways included components of electron transport chain complexes and diseases, in which these components are altered (down, panel D), or proteasome, amino acid, and nucleoside (purine and pyrimidine) metabolism and succinate dehydrogenase complex components (up, panel E). The majority of the down-regulated proteins were part of mitochondrial electron transport chain complexes I, III, IV, and V, where the majority of the components decreased at least 2-fold (asterisks in panel F), as detailed in the panel.
In light of the data suggesting a role for HTS in inducing the inhibition of cell proliferation concomitantly to the deregulation of redox and mitochondrial homeostasis, consistent with the role of p53 in the regulation of mitochondrial respiration,43 we performed gene ontology term enrichment for metabolic function of 2-fold up- and down-regulated proteins in response to HTS treatment. Results revealed that HTS deregulated proteins involved in the electron transport chain complexes (and diseases in which ETC is uncoupled, such as Parkinson’s, Alzheimer’s, and Huntington’s diseases; Figure 5D). In Figure 5F, we schematically summarized ETC subunits (complexes I, III, IV, and V) deregulated by HTS alone or in combination with CMX. The top (and the only significantly regulated; p < 0.05) pathway up-regulated by the HTS treatment was referred to spliceosome and spliceosome components (Figures 5E and S3). Although not reaching significance, proteasome components and enzymes involved in amino acid and nucleotide metabolism were up-regulated (Figures 5E and S3). These results are consistent with a significant transcriptional down-regulation of components of these pathways by HTS (Supplementary Table 1 and Figure 1A).
HTS-Dependent Alteration of Mitochondrial and Redox Homeostasis Promotion of Metabolic Reprogramming of SAECs, Resulting in the Decreased Accumulation of TCA Cycle Intermediates, Increased Oxidative Stress, and Amino Acid Uptake but Not Catabolism
Cell-cycle regulation and induction of intrinsic mitochondrial pathway apoptosis by the p53-p21 signaling axis is known to promote significant metabolic reprogramming in cancer cells.30 UHPLC-MS metabolomics analyses were performed on SAECs cells and supernatants (n = 5 - Figure 6A and Supplementary Tables 3 and 4, respectively) to reveal a significant effect on metabolic phenotypes in response to HTS and CMX treatments, as gleaned through partial least-squares-discriminant analysis (Figure 6B). Heat map overviews of supernatants (Figures 6C and S4) and cells (Figures 6D and S5) show at a glance the effect of HTS treatment (alone or in combination with CMX) on the depression of nucleotide metabolism and increased amino acid uptake and decreased amino acid catabolism in HTS-treated cells. HTS-dependent TIGAR expression observed by proteomics (Figure 5B) was confirmed by Western blot (Figure 6E). The role of TIGAR, also known as fructose 2,6-bisphosphatase, has been recently investigated in cancer, in which it promotes a glycolytic bottleneck resulting in the accumulation of early Embden-Meyerhof intermediates, in turn fueling the pentose phosphate pathway.30 In so doing, TIGAR mediates the generation of reducing equivalents such as NADPH for antioxidant purposes and keeps in check redox homeostasis and cell survival under stress conditions. Here, increased steady-state levels of fructose bisphosphate, decreased glucose 6-phosphate, and increased ribose phosphate in HTS and HTS+CMX SAECs depict increased fluxes through the pentose phosphate pathway (Figure 6F). This observation was confirmed with tracing experiments with U-13C-glucose (Figure 7A), indicating increased incorporation of the heavy M+5 isotopologue of ribose phosphate in HTS and HTS+CMX SAECs despite decreased uptake of glucose and generation of glucose 6-phosphate (both M+6 isotopologues, Figure 7B). This phenotype is consistent with the observed decreases in reduced glutathione levels (GSH) after HTS treatment, either consistent with increased oxidative stress (observed only in response to HTS+CMX; Figure 4K) or decreased glutathione synthesis (explained at the protein level by decreased levels of glutathione biosynthesis enzymes in response to HTS treatment; Figure 5C). Lowest steady-state (Figure 6F) and de novo fluxed (heavy isotopologues, M+2, e.g., malate; Figure 7B) levels of Krebs cycle intermediates were detected in HTS-treated cells, consistent with a less-active mitochondrial metabolism (Figure 4J). Merging metabolomics data with results from proteomics analyses (Figure 6G) indicated that CMX and HTS effect on glycolysis (lactate accumulation (Figure 6F), especially upon HTS treatment, as determined through tracing experiments; Figure 7B) may be mediated by the up-regulation of glycolytic enzymes (Figure 5C), as mentioned above. Increased amino acid uptake (e.g., glutamine; Figure 6F) in response to HTS treatment may be mediated by the increased protein expression of amino acid transporters (SLC1, SLC6, SLC38 and LAT1; Figure 6G). Despite apparent decreases in mitochondrial metabolism, HTS cells did not appear to be energetically challenged, displaying higher levels of ATP than control or CMX-treated SAECs (Figure S4).
Figure 6.

HTS promotion of significant metabolic reprogramming UHPLC-MS metabolomics analyses were performed on SAECs cells and supernatants (n = 5, panel A). Partial least-squares-discriminant analysis shows significant alterations of the metabolic phenotype induced by the HTS and CMX treatments (B), as detailed by the heat maps for supernatants (C) and cells (D) showing impaired nucleotide metabolism and increased amino acid uptake and decreased amino acid catabolism in HTS-treated cells. In the heat map, Z-score normalized relative metabolite levels across samples are graphed in a blue-to-red (low-to-high) color coding. Western blot analysis showed that protein expression of TIGAR, a downstream target of the p53-p21 transcriptional axis, was induced by HTS alone (E). Specifically (F), this translated into increased steady-state levels of fructose bisphosphate, decreased glucose 6-phosphate, and increased ribose phosphate in HTS and HTS+CMX cells, suggestive of increased fluxes through the pentose phosphate pathway. The lowest levels of Krebs cycle intermediates were detected in HTS-treated cells, suggestive of depressed mitochondrial metabolism. Merging metabolomics data with results from proteomics analyses (G) indicated that HTS’s effect on glycolysis may be mediated by the up-regulation of glucose and lactate transporters (GLUT1 and MCT4), hexokinase, and biphosphoglycerate mutase. The effect on amino acid uptake in response to HTS treatment may be mediated by the increased protein expression of amino acid transporters (solute carriers SLC1, SLC6, SLC38, and large amino acid transporter 1 (LAT1, panel G). However, down-regulation of Krebs cycle (acetyl-coA lyase, citrate synthase, oxoglutarate dehydrogenase, and fumarate hydratase) and glutathione biosynthetic enzymes (γ glutamyl cysteine lyase and glutathione synthase) may contribute to explaining the metabolic observations on depressed Krebs cycle and decreased glutathione levels in response to HTS treatment (G).
Figure 7.

Tracing experiments with U-13C-glucose and confirmation that HTS decreases glucose uptake and glucose catabolism through the TCA cycle, which is in turn promoted by CMX. Hypertonic saline treatment in combination with CMX promotes activation of the pentose phosphate pathway, as indicated by the accumulation of isotopologue M+5 of ribose phosphate. Experiments were performed by incubating SAECs with HTS, alone or in combination with CMX, in the presence of uniformly labeled glucose for up to 4 h (240 min). Lines indicate median of time course tracing experiments, color coded according to the legend in panel A (top right corner).
DISCUSSION
With post-injury incidences as high as 50%, postshock lung dysfunction remains a challenging clinical complication in the trauma patient population, where it significantly contributes to the increased morbidity and extended hospitalization and thus represents an obvious economic burden for the healthcare system.3 Post-injury lung dysfunction seems to be associated with unregulated inf;lammatory responses to the initial injury or secondary insults or both.3 Currently, anti-inflammatory therapies rest on steroids or nonsteroidal anti-inflammatory drugs. Because HTS is inexpensive and exposures can be timed, it could offer pulmonary critical care a novel means of regulating inflammatory gene expression. Improved understanding of the molecular mechanisms mediating the beneficial effects promoted by HTS treatment in this patient population could lead to agents more effective than HTS or hyperosmolarity. Inhaled therapy with HTS has been shown to be beneficial in other conditions in which inflammatory responses in the lung and chronic airway inflammation play a major role, such as cystic fibrosis13 and asthma44 but not chronic obstructive pulmonary disease.45 These inconsistencies in the effectiveness of the treatment despite the shared inflammatory mechanisms underlying the etiology of the aforementioned pathologies are attributable to the incomplete understanding of the mechanisms through which HTS deregulates pro-inflammatory cascades.
Although HTS has been shown to impair cytokine signaling in A549 lung cancer cells by disrupting the I-γBα-NF-γB signaling cascade,18 molecular mechanisms driving HTS-mediated prevention of phenotypic changes triggered by pro-inflammatory stimuli in primary lung cells are currently missing. Recently, we noted that HTS retarded the activation of NF-kB and IRF1 transcription factors against inflammatory stimuli TNFα and IL-1β in A549 cells.18 Because of the aneuploidy of A549, we selected human SAEC and the robust proinflammatory stimulus CMX (TNFα, IL-1β, and IFNγ) to better model in vivo inflammatory conditions observed, for example, in post-trauma and hemorrhagic shock ALI or ARDS. The goal of the present study was to confirm the effect of HTS on the NF-kB or IRF1 signaling axis17,18 or, rather, identify novel signaling cascades affected by HTS that did not emerge from our previous investigations on lung cancer cell lines. Therefore, we performed RNaseq analyses on HTS-stimulated SAECs to further our understanding of the global effects of HTS on the cellular transcriptome and confirmed a potential anti-inflammatory role of HTS by negatively regulating several cytokines at the transcriptional level. For the first time, unsupervised analysis of RNaseq data suggested a role for HTS treatment in the modulation of the p53-p21 signaling axis, as we further confirmed through RT-PCR, WB, and fluorescence microscopy. Of note, hyperosmolarity has been very recently associated with increased p53 protein expression in mouse fibroblasts,28 where p53 protein was stabilized through a post-translational mechanism (phosphorylation at serine 15), decreasing its degradation via MDM2-mediated ubiquitinylation. Here, similar results were observed because RNaseq and proteomics data showed increased transcription and protein expression of genes downstream to p53, above all p21, while only protein levels of p53 (but not its direct mRNA transcription) were significantly up-regulated by HTS alone.
In the present study, fluorescence microscopy experiments suggest that p53 protein stabilization is achieved in HTS-treated SAECs only in response to a second hit by CMX, resulting in decreased phosphorylation at T155, which is known to promote cytosolic localization and proteasomal degradation of p53 via MDM2-dependent ubiquitinylation.29 In the model we propose, stabilization of the p53 signaling promotes p21 transcription and expression. Additionally, phosphorylation of p21 at T14546 here corresponds to its cytosolic localization and inhibition of proliferation in HTS treated SAECs, although HTS treatment alone results in depression of mitochondrial activity without induction of apoptosis, which in turn is moderately triggered(<5% over HTS alone) by the CMX treatment in HTS-preconditioned SAECs.
Of note is the fact that an anti-inflammatory role has been previously proposed for p21 in stratified epithelia and IL-1β activated macrophages.47 Intertwinement of tumor suppressor signaling (especially p53), metabolic reprogramming, and inflammatory cascades has been increasingly appreciated in the past few years in the field of cancer research.30,31 Notably, the concept of metabolic reprogramming as a key “hallmark of cancer” has been borrowed by immunologists to explain the highly proliferative phenotype of activated neutrophils, lymphocytes, and macrophages.32 In particular, reprogrammed mitochondrial metabolism results in the accumulation of specific pro-inflammatory metabolites, such as succinate.33 Of note is the fact that here we show that treating SAECs with a cocktail of pro-inflammatory cytokines promotes a hyper-metabolic state in mitochondria. Preconditioning with HTS prevents mitochondrial hyperpolarization induced by CMX, depresses mitochondrial metabolism in the absence of significant impairments of energy metabolism, suggesting metabolic reprogramming as a potential therapeutic avenue by which to correct the pro-inflammatory phenotype driving ALI and ARDS in trauma patients and improve the effectiveness of the HTS treatment.
Mitochondrial uncoupling mediated by HTS results from the selective downregulation of ETC components of complexes I, III, IV, and V, while components of complex II (succinate dehydrogenase) were in fact up-regulated by HTS. In this model, selective increase of mitochondrial enzymes and decrease in specific electron transport chain complexes components in the absence of significant alterations of mitochondrial counts are suggestive of other mechanisms than mitophagy (mitochondrial autophagy) at play, in contrast to what was previously proposed in other models.48 However, further studies on the LKB1 signaling cascades49 in response to HTS may be necessary to rule out the activation of this mechanism in the present model.
RNaseq, proteomics, and metabolomics analyses all suggested an effect of HTS on membrane transporters, resulting in the increased activation of the pentose phosphate pathway to cope with CMX-induced oxidative stress, depressed nucleotide metabolism, increased amino acid uptake, and decreased catabolism. These observations suggest a predictable yet unexplored regulatory mechanism that cells may exploit to counteract hyperosmotic stress induced by HTS. Overall, these data suggest that anti-inflammatory mechanisms mediated by HTS in SAECs are not just limited to a negative transcriptional control of pro-inflammatory cytokines. For example, decreased amino acid catabolism, especially in the case of glutamine, would result in decreased levels of pro-inflammatory Krebs cycle intermediates such as succinate, of which glutamine represents an elective substrate in activated immune cells.33
Although preliminary, the results we presented in this paper suggest an unanticipated role for the p53-p21 signaling axis in mediating the beneficial anti-inflammatory effects of HTS. Further mechanistic evidence will be provided by follow-up mechanistic studies on in vitro and in vivo models of inflammation and lung injury following trauma and hemorrhagic shock, in which p53 and p21 will be silenced through molecular biology approaches or targeted with specific drugs.50
CONCLUSIONS
Treatment with systemic or nebulized HTS has emerged as an effective clinical strategy with which to mitigate the inflammatory injury in the lungs of patients suffering from cystic fibrosis or traumatic brain injury. HTS treatments may represent a viable inexpensive and safe therapeutic opportunity to treat ALI and ARDS in the trauma patient populations. Although previous studies have shown significant anti-inflammatory properties of HTS in vitro and in vivo,11,18,23 the molecular mechanisms mediating this anti-inflammatory activity are still unclear.
In the present study, we applied an integrated transcriptomics, proteomics, and metabolomics approach to investigate the molecular mechanisms underlying the anti-inflammatory properties of treatments based upon the administration of a hyperosmolar and hypertonic saline solution on primary small airways epithelial cells. Treatment with HTS significantly decreased cytokine expression at the transcriptional level, consistent with previous findings in A549 cells and rat lungs.11,18,23 Omics analyses suggested a role for HTS treatment in priming the activation of the p53-p21 signaling axis, resulting in the stabilization and cytosolic translocation of p21, which in turn inhibited cell proliferation in vitro. Here, we show that preconditioning SAECs with HTS protects them from TNFα, INFγ, and IL-1β cytokine-induced mitochondrial hyperactivation, a hallmark of inflammatory responses. Such an effect is mediated by the promotion of mitochondrial membrane depolarization, with a minor (<5% over untreated controls) induction of caspase 3/7-dependent intrinsic mitochondrial pathway of apoptosis in cytokine-treated cells preconditioned with HTS. Integration of transcriptomics, proteomics, and metabolomics data suggests that the mechanisms underlying these observations may involve key metabolic enzymes that are known to respond to p53-p21 signaling, such as TIGAR.42 HTS-dependent decreased expression of mitochondrial electron transport chain complexes I, III, and IV may account for the deregulation of mitochondrial activity. A parallel increase in membrane transporter and alterations of mitochondrial metabolism here resulted in increased amino acid uptake and decreased amino acid and nucleotides metabolism, suggesting a compensatory mechanism that cells may use to counteract increased osmolarity in response to HTS treatment. Future flux balance analysis metabolomics studies using heavy-labeled amino acid in culture will test this hypothesis. Additionally, further evidence will continue to elucidate the centrality of the p53-p21 signaling axis in mediating the anti-inflammatory beneficial effects of HTS in vitro and in vivo; genetic or pharmacological therapies targeting this regulatory pathway could be used to replace or prolong the clinical effectiveness of HTS treatment of trauma patients with ALI and ARDS symptoms.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award nos. P50GM049222 and T32GM008315 and grant no. P50 GM049222 from NIGMS, NIH (C.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions
F.G. and C.A. performed immunofluorescence and WB analyses, cell cultures, and treatments. S.M. performed RT-PCR experiments. T.N. and A.D. performed metabolomics experiments. J.A.R. and A.D. performed tracing experiments. K.L.J. performed RNaseq experiments. M.D. and K.C.H. performed proteomics experiments. A.D., A.B., and F.G. designed the experiments. A.D. interpreted the results, prepared figures and tables, and wrote and revised the paper. All of the authors critically commented on and reviewed the final version of the paper.
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00602.
Figures showing fluorescent microscopy analysis of apoptosis, TP53 family members up-regulated at the protein level in SAECS following HTS and HTS+CMX treatment, proteasome and protein degradation machinery components increasing in SAECs in response to HTS treatment, hierarchical clustering analysis of metabolite levels in SAECs treated with HTS (alone or in combination with CMX), and hierarchical clustering analysis of metabolite levels in culture media of SAECs treated with HTS (alone or in combination with CMX). Tables showing functionally enriched pathways as determined from gene ontology elaboration of RNA sequencing data; relative quantitation of proteins in SAECs cells in the presence and the absence of a cytokine cocktail (cytomix), with or without preconditioning with hypertonic saline; metabolomics report of supernatants of SAECs cells, cultured in the presence and the absence of a cytokine cocktail (cytomix), with or without preconditioning with hypertonic saline; and a metabolomics report of cell extracts of SAECs cells, cultured in the presence and the absence of a cytokine cocktail (cytomix), with or without preconditioning with hypertonic saline. (PDF)
References
- 1.Rhee P, Joseph B, Pandit V, Aziz H, Vercruysse G, Kulvatunyou N, Friese RS. Increasing trauma deaths in the United States. Ann Surg. 2014;260(1):13–21. doi: 10.1097/SLA.0000000000000600. [DOI] [PubMed] [Google Scholar]
- 2.Gunst M, Ghaemmaghami V, Gruszecki A, Urban J, Frankel H, Shafi S. Changing epidemiology of trauma deaths leads to a bimodal distribution. Proc Bayl Univ Med Cent. 2010;23(4):349–354. doi: 10.1080/08998280.2010.11928649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sauaia A, Moore EE, Johnson JL, Chin TL, Banerjee A, Sperry JL, Maier RV, Burlew CC. Temporal trends of postinjury multiple-organ failure: still resource intensive, morbid, and lethal. J Trauma Acute Care Surg. 2014;76(3):582–592. doi: 10.1097/TA.0000000000000147. discussion 592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calfee CS, Eisner MD, Ware LB, Thompson BT, Parsons PE, Wheeler AP, Korpak A, Matthay MA. Trauma-associated lung injury differs clinically and biologically from acute lung injury owing to other clinical disorders*. Crit Care Med. 2007;35(10):2243–50. doi: 10.1097/01.ccm.0000280434.33451.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizoli SB, Rhind SG, Shek PN, Inaba K, Filips D, Tien H, Brenneman F, Rotstein O. The immunomodulatory effects of hypertonic saline resuscitation in patients sustaining traumatic hemorrhagic shock: a randomized, controlled, double-blinded trial. Ann Surg. 2006;243(1):47–57. doi: 10.1097/01.sla.0000193608.93127.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulger EM, Jurkovich GJ, Nathens AB, Copass MK, Hanson S, Cooper C, Liu P-Y, Neff M, Awan AB, Warner K, et al. Hypertonic resuscitation of hypovolemic shock after blunt trauma: a randomized controlled trial. Arch Surg. 2008;143(2):139–148. doi: 10.1001/archsurg.2007.41. discussion 149. [DOI] [PubMed] [Google Scholar]
- 7.Bahrami S, Zimmermann K, Szelényi Z, Hamar J, Scheiflinger F, Redl H, Junger WG. Small-volume fluid resuscitation with hypertonic saline prevents inflammation but not mortality in a rat model of hemorrhagic shock. Shock. 2006;25(3):283–289. doi: 10.1097/01.shk.0000208808.03148.ea. [DOI] [PubMed] [Google Scholar]
- 8.Powers KA, Woo J, Khadaroo RG, Papia G, Kapus A, Rotstein OD. Hypertonic resuscitation of hemorrhagic shock upregulates the anti-inflammatory response by alveolar macrophages. Surgery. 2003;134(2):312–318. doi: 10.1067/msy.2003.246. [DOI] [PubMed] [Google Scholar]
- 9.Murao Y, Loomis W, Wolf P, Hoyt DB, Junger WG. Effect of dose of hypertonic saline on its potential to prevent lung tissue damage in a mouse model of hemorrhagic shock. Shock. 2003;20(1):29–34. doi: 10.1097/01.shk.0000071060.78689.f1. [DOI] [PubMed] [Google Scholar]
- 10.Shukla A, Hashiguchi N, Chen Y, Coimbra R, Hoyt DB, Junger WG. Osmotic regulation of cell function and possible clinical applications. Shock. 2004;21(5):391–400. doi: 10.1097/00024382-200405000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Wohlauer M, Moore EE, Silliman CC, Fragoso M, Gamboni F, Harr J, Accurso F, Wright F, Haenel J, Fullerton D, et al. Nebulized hypertonic saline attenuates acute lung injury following trauma and hemorrhagic shock via inhibition of matrix metalloproteinase-13. Crit Care Med. 2012;40(9):2647–2653. doi: 10.1097/CCM.0b013e3182592006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santry HP, Alam HB. Fluid resuscitation: past, present, and the future. Shock. 2010;33(3):229–241. doi: 10.1097/SHK.0b013e3181c30f0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev. 2009;(2):CD001506. doi: 10.1002/14651858.CD001506.pub3. [DOI] [PubMed] [Google Scholar]
- 14.Williams HD, Behrends V, Bundy JG, Ryall B, Zlosnik JEA. Hypertonic Saline Therapy in Cystic Fibrosis: Do Population Shifts Caused by the Osmotic Sensitivity of Infecting Bacteria Explain the Effectiveness of this Treatment? Front Microbiol. 2010;1:120. doi: 10.3389/fmicb.2010.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bulger EM, May S, Kerby JD, Emerson S, Stiell IG, Schreiber MA, Brasel KJ, Tisherman SA, Coimbra R, Rizoli S, et al. Out-of-hospital hypertonic resuscitation after traumatic hypovolemic shock: a randomized, placebo controlled trial. Ann Surg. 2011;253(3):431–441. doi: 10.1097/SLA.0b013e3181fcdb22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Junco DJ, Bulger EM, Fox EE, Holcomb JB, Brasel KJ, Hoyt DB, Grady JJ, Duran S, Klotz P, Dubick MA, et al. Collider bias in trauma comparative effectiveness research: The stratification blues for systematic reviews. Injury. 2015;46(5):775–780. doi: 10.1016/j.injury.2015.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee A, Moore EE, McLaughlin NJ, Lee L, Jones WL, Johnson JL, Nydam TL, Silliman CC. Hyperosmolarity attenuates TNF-α-mediated proinflammatory activation of human pulmonary microvascular endothelial cells. Shock. 2013;39(4):366–372. doi: 10.1097/SHK.0b013e3182894016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright FL, Gamboni F, Moore EE, Nydam TL, Mitra S, Silliman CC, Banerjee A. Hyperosmolarity invokes distinct anti-inflammatory mechanisms in pulmonary epithelial cells: evidence from signaling and transcription layers. PLoS One. 2014;9(12):e114129. doi: 10.1371/journal.pone.0114129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciesla DJ, Moore EE, Biffl WL, Gonzalez RJ, Silliman CC. Hypertonic saline attenuation of the neutrophil cytotoxic response is reversed upon restoration of normotonicity and reestablished by repeated hypertonic challenge. Surgery. 2001;129(5):567–575. doi: 10.1067/msy.2001.113286. [DOI] [PubMed] [Google Scholar]
- 20.Junger WG, Coimbra R, Liu FC, Herdon-Remelius C, Junger W, Junger H, Loomis W, Hoyt DB, Altman A. Hypertonic saline resuscitation: a tool to modulate immune function in trauma patients? Shock. 1997;8(4):235–241. [PubMed] [Google Scholar]
- 21.Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14(6):523–535. doi: 10.1016/s1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 22.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202(2):145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 23.Nydam TL, Moore EE, McIntyre RC, Wright FL, Gamboni-Robertson F, Eckels PC, Banerjee A. Hypertonic saline attenuates TNF-alpha-induced NF-kappaB activation in pulmonary epithelial cells. Shock. 2009;31(5):466–472. doi: 10.1097/SHK.0b013e31818ec47d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Held HD, Boettcher S, Hamann L, Uhlig S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med. 2001;163:711–716. doi: 10.1164/ajrccm.163.3.2003001. [DOI] [PubMed] [Google Scholar]
- 25.Ochi H, Masuda J, Gimbrone MA. Hyperosmotic stimuli inhibit VCAM-1 expression in cultured endothelial cells via effects on interferon regulatory factor-1 expression and activity. Eur J Immunol. 2002;32(7):1821–1831. doi: 10.1002/1521-4141(200207)32:7<1821::AID-IMMU1821>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 26.McLaughlin NJD, Banerjee A, Kelher MR, Gamboni-Robertson F, Hamiel C, Sheppard FR, Moore EE, Silliman CC. Platelet-activating factor-induced clathrin-mediated endocytosis requires beta-arrestin-1 recruitment and activation of the p38 MAPK signalosome at the plasma membrane for actin bundle formation. J Immunol. 2006;176(11):7039–7050. doi: 10.4049/jimmunol.176.11.7039. [DOI] [PubMed] [Google Scholar]
- 27.Junger WG, Rhind SG, Rizoli SB, Cuschieri J, Baker AJ, Shek PN, Hoyt DB, Bulger EM. Prehospital hypertonic saline resuscitation attenuates the activation and promotes apoptosis of neutrophils in patients with severe traumatic brain injury. Shock. 2013;40(5):366–374. doi: 10.1097/SHK.0000000000000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lambert IH, Enghoff MS, Brandi M-L, Hoffmann EK. Regulation of p53 in NIH3T3 mouse fibroblasts following hyperosmotic stress. Physiol Rep. 2015;3(6):e12412. doi: 10.14814/phy2.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berkers CR, Maddocks ODK, Cheung EC, Mor I, Vousden KH. Metabolic Regulation by p53 Family Members. Cell Metab. 2013;18(5):617–633. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.El Kasmi KC, Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol. 2015;27(4):267–275. doi: 10.1016/j.smim.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenmark KR, Tuder RM, El Kasmi KC. Metabolic Reprogramming and Inflammation Act in Concert to Control Vascular Remodeling in Hypoxic Pulmonary Hypertension. J Appl Physiol. 2015;119(10):1164–72. doi: 10.1152/japplphysiol.00283.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plecitá-Hlavatá L, Tauber J, Li M, Zhang H, Flockton AR, Pullamsetti SS, Chelladurai P, D’alessandro A, El Kasmi KC, Ježek P, et al. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2015;55(1):47–57. doi: 10.1165/rcmb.2015-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol Cancer Res. 2015;13(4):651–658. doi: 10.1158/1541-7786.MCR-14-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D’Alessandro A, Moore HB, Moore EE, Wither M, Nemkov T, Gonzalez E, Slaughter A, Fragoso M, Hansen KC, Silliman CC, et al. Early hemorrhage triggers metabolic responses that build up during prolonged shock. Am J Physiol Regul Integr Comp Physiol. 2015;308(12):R1034–R1044. doi: 10.1152/ajpregu.00030.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dzieciatkowska M, Hill R, Hansen KC. GeLC-MS/MS analysis of complex protein mixtures. Methods Mol Biol. 2014;1156:53–66. doi: 10.1007/978-1-4939-0685-7_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1(8):639–649. [PubMed] [Google Scholar]
- 40.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta, Bioenerg. 2009;1787(5):414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 43.Matoba S, Kang J-G, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 44.Mai X-M, Nilsson L, Kjellman N-IM, Björkstén B. Hypertonic saline challenge tests in the diagnosis of bronchial hyperresponsiveness and asthma in children. Pediatr Allergy Immunol Off Publ Eur Soc Pediatr Allergy Immunol. 2002;13(5):361–367. doi: 10.1034/j.1399-3038.2002.01011.x. [DOI] [PubMed] [Google Scholar]
- 45.Valderramas SR, Atallah AN. Effectiveness and safety of hypertonic saline inhalation combined with exercise training in patients with chronic obstructive pulmonary disease: a randomized trial. Respir Care. 2009;54(3):327–333. [PubMed] [Google Scholar]
- 46.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scatizzi JC, Mavers M, Hutcheson J, Young B, Shi B, Pope RM, Ruderman EM, Samways DSK, Corbett JA, Egan TM, et al. The CDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macrophages. Eur J Immunol. 2009;39(3):820–825. doi: 10.1002/eji.200838683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nunes P, Ernandez T, Roth I, Qiao X, Strebel D, Bouley R, Charollais A, Ramadori P, Foti M, Meda P, et al. Hypertonic stress promotes autophagy and microtubule-dependent autophagosomal clusters. Autophagy. 2013;9(4):550–567. doi: 10.4161/auto.23662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaw RJ. LKB1 and AMPK control of mTOR signalling and growth. Acta Physiol. 2009;196(1):65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy PJM, Galigniana MD, Morishima Y, Harrell JM, Kwok RPS, Ljungman M, Pratt WB. Pifithrin-α Inhibits p53 Signaling after Interaction of the Tumor Suppressor Protein with hsp90 and Its Nuclear Translocation. J Biol Chem. 2004;279(29):30195–30201. doi: 10.1074/jbc.M403539200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.