

SUMMARY
The interleukin-17 (IL-17) family cytokines have emerged as critical players in inflammatory diseases. Among them, IL-25 has been shown to be important in allergic inflammation and protection against parasitic infection. Here we have demonstrated that IL-17B, a poorly understood cytokine, functions to inhibit IL-25-driven inflammation. IL-17B and IL-25, both binding to the interleukin-17 receptor B (IL-17RB), were upregulated in their expression following acute colonic inflammation. Individual inhibition of these cytokines revealed opposing functions in colon inflammation: IL-25 was pathogenic but IL-17B was protective. Similarly opposing phenotypes were observed in Citrobacter rodentium infection and allergic asthma. Moreover, IL-25 was found to promote IL-6 production from colon epithelial cells, which was inhibited by IL-17B. Therefore, our data demonstrate that IL-17B is an anti-inflammatory cytokine in the IL-17 family.
INTRODUCTION
Members of the interleukin-17 (IL-17) family of cytokines have recently emerged as critical players in inflammation. IL-17A and IL-17F are predominately expressed by CD4+ T helper 17 (Th17) cells, but can be produced by other lymphocytes as well (Dong, 2008; Cupedo et al., 2009; Takatori et al., 2009; Reynolds et al., 2010; Sawa et al., 2010). Recently, IL-17C and its receptor IL-17 receptor E (IL-17RE) have been described to regulate Th17 cell responses and epithelial cell-dependent colon immunity (Chang et al., 2011; Ramirez-Carrozzi et al., 2011; Song et al., 2011; Reynolds et al., 2012). IL-25 (IL-17E) is unique in that it induces T helper 2 (Th2) cell-mediated mediated inflammation (Fort et al., 2001; Angkasekwinai et al., 2007; Zaph et al., 2008). IL-25 thus is protective against parasitic infection (Fallon et al., 2006; Owyang et al., 2006; Moro et al., 2010; Neill et al., 2010; Price et al., 2010; Saenz et al., 2010). IL-25 signals through a heterodimeric receptor comprised of IL-17 receptor A (IL-17RA) and IL-17 receptor B (IL-17RB), both of which are necessary for IL-25 to promote cytokine production from target cells, including Th2 cells and group 2 innate lymphoid cells (Rickel et al., 2008; Angkasekwinai et al., 2010). Outside of parasitic infection, much less is known about the roles of IL-25 in the colon. Injection of IL-25 throughout the course of DSS-induced colitis results in a protective response as mice exhibit less inflammation and increased survival (McHenga et al., 2008). Conversely, in a model of oxazolone-induced colitis, neutralizing IL-25 or IL-17RB can decrease colonic inflammation (Camelo et al., 2012). Finally, commensal flora drives the expression of IL-25 in the colon, which may serve to limit the numbers of Th17 cells and IL-22-expressing RORγt+ group 3 innate lymphoid cells (Zaph et al., 2008; Sawa et al., 2011).
Little is known about IL-17B. The initial cloning and characterization reveals that human IL-17B can induce tumor necrosis-α (TNF-α) and IL-1β production by THP-1 cells (Li et al., 2000). Moreover, IL-17B is substantially expressed in the paws of arthritic mice and polyclonal anti-IL-17B antibody treatment ameliorates collagen-induced arthritis (Yamaguchi et al., 2007). Indeed, mice injected with IL-17B exhibit a neutrophilia phenotype similar to those injected IL-17A (Schwarzenberger et al., 1998; Shi et al., 2000). However, IL-17B and IL-25 share a common receptor, IL-17RB (Shi et al., 2000; Huang et al., 2013), suggesting that there may be unexplored and unique functions of IL-17B, although the affinity of IL-17B for IL-17RB is weaker compared to IL-25 (Li et al., 2000; Lee et al., 2001). Non-immune functions have been attributed to IL-17B as well, including roles in development (You et al., 2005), fracture (Kokubu et al., 2008), and cancer (Sanders et al., 2010; Huang et al., 2013).
Given that IL-17RB is expressed by mucosal epithelial cells (Shi et al., 2000; Lee et al., 2001; Zhao et al., 2010), we have investigated the potential role of IL-17B and IL-25 in the regulation of mucosal inflammation. Opposing disease phenotypes by IL-17B- and IL-25-deficient animals were observed in models of acute colitis, airway inflammation, and Citrobacter rodentium infection. Overall, our studies have identified a critical inhibitory function for IL-17B in IL-25-mediated mucosal inflammation.
RESULTS
IL-17B and IL-25 are expressed by colon epithelial cells
To determine the function of IL-17B and IL-25 in colon inflammation, we first examined mRNA expression of Il17b and Il25 in total colon tissues isolated from healthy C57BL/6 mice (WT) and as well as WT mice administered DSS for 8 d (Figure 1A). We found that there was basal expression of both cytokines in the colon under steady-state conditions. However, induction of acute colitis by DSS led to a substantial increase in the expression of Il17b (~10 fold) and Il25 (~35 fold) mRNA (Figure 1A) as well as protein (Figure S1). Given that multiple tissues have been shown to express both cytokines (Li et al., 2000; Shi et al., 2000; Lee et al., 2001; You et al., 2005; Sanders et al., 2010), we next examined the contribution of hematopoietic and non-hematopoietic cells in the production of each. We first isolated colons from DSS-induced WT animals and then separated cells on the basis of CD45 and CD16/32 expression. We found that the majority of IL-17B and IL-25 mRNA expression was clearly in the colon epithelial cell (CEC) (CD45−CD16/32−) fraction (Figure 1A), which is consistent with our previous report that epithelial cells are a major source of IL-25 (Angkasekwinai et al., 2007).
Figure 1. Colon epithelial cells produce IL-17B and IL-25 in acute colitis.
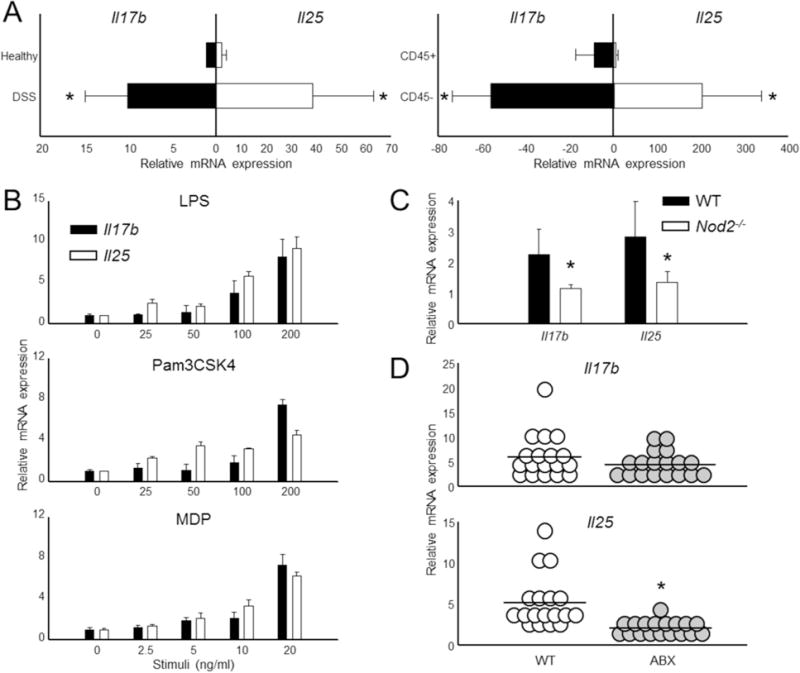
(A) Colon samples were derived from healthy controls or from animals following 8 d DSS-induced colitis (left). DSS samples were further processed and separated into leukocyte (“CD45+”) and epithelial cell (“CD45−”) fractions (right). The relative expression of Il17b and Il25 were then measured by qPCR. n = 5 mice per group. (B) Primary colon epithelial cells derived from healthy mice were activated through TLR (LPS, Pam3CSK4) and NOD2 (MDP) pathways for 6 h. Il17b and Il25 mRNA expression was assessed by real-time RT-PCR. (C) Total colon tissue was isolated from healthy WT and NOD2-deficient animals and then assessed for Il17b and Il25 expression by qPCR. n = 5 mice per group. (D) C57BL/6 animals were left untreated (normal) or administered an antibiotic regiment (ABX) for 4 wk to deplete microbiota. Total colon tissue was then obtained and the expression of Il17b and Il25 was measured by qPCR. n = 18 mice per group. All gene values were normalized to the expression of the house keeping gene Actb. Data are presented as mean + SD and are representative of at least 3 independent experiments. * Student’s t test; p < 0.05. See also Figure S1.
The increased expression of IL-17B and IL-25 during colitis led us to investigate which signal(s) regulated expression. In vitro stimulation of CEC with toll-like receptor-4 (TLR4) agonist (LPS) and TLR1 and 2 agonist (Pam3CSK4) led to a dose-dependent mRNA increase of both cytokine genes (Figure 1B). We also stimulated CEC with muramyl dipeptide (MDP) and saw a similar enhancement of IL-17B and IL-25 (Figure 1B), suggesting that the nucleotide oligomerization domain-2 (NOD2) pathway is important for the expression of both by CEC as well. Indeed, deficiency in NOD2 led to reduced basal Il17b and Il25 expression in healthy colons compared to WT controls (Figure 1C).
Since IL-17B and IL-25 were both expressed in colons from healthy animals we thought that expression may be regulated by the microbiota. To investigate this hypothesis, we depleted the gut microbiota using a broad antibiotic regiment (Ivanov et al., 2008). Similar to a previous report (Sawa et al., 2011), we found that antibiotic depletion of WT animals led to a substantial decrease of Il25 expression in the colon (Figure 1D). Surprisingly, antibiotic depletion only moderately reduced Il17b, suggesting that the signals governing IL-17B production are more restricted.
Deficiency in IL-25 confers protection against DSS-induced colitis
We next tested WT and IL-25-deficient mice (Il25−/−) (Angkasekwinai et al., 2010) for the development of colon inflammation 8 d post DSS administration. Both groups started to lose weight ~5 days following treatment, however, Il25−/−mice exhibited considerable protection from the wasting disease manifesting in WT controls (Figure 2A). Further analysis following euthanasia revealed that the moderate weight loss observed in Il25−/− animals was directly attributable to decreased colonic inflammation; characteristic inflammatory parameters such as colon length and H&E histology revealed that Il25−/− mice were protected from inflammatory destruction, tissue remodeling, and leukocyte infiltration typical of this mouse model (Figures 2B-D).
Figure 2. IL-25-deficient mice are protected from DSS-induced colitis.
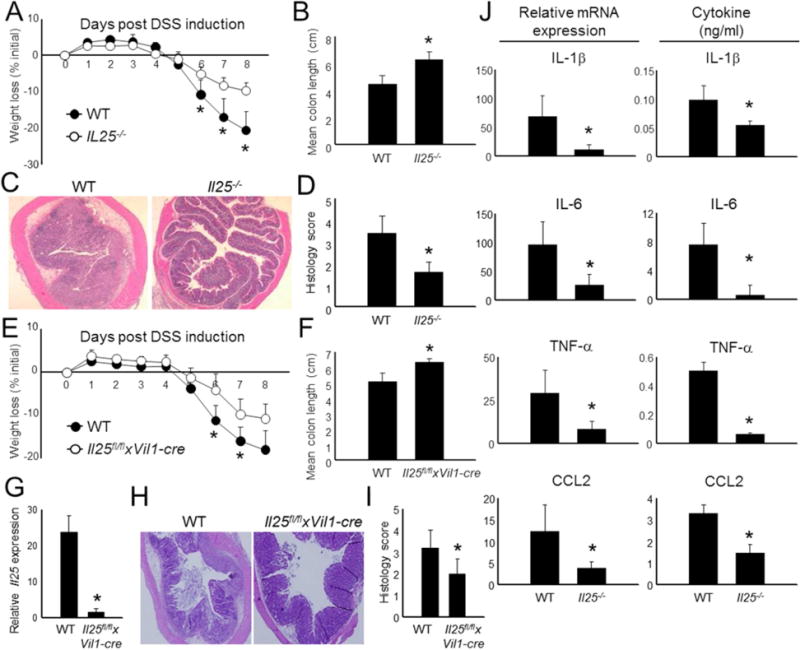
(A) Acute colitis was induced in WT and Il25−/− mice by the addition of DSS in drinking water (n = 5 – 6 per group). Weight loss during colitis progression is shown. (B) Mice with colitis were euthanized on day 8 and colon length was measured from individual mice and data was combined. (C) Representative H & E histology from intermediate colon sections derived from the mice in (A). (D) Combined histological scores of all mice and all sections (proximal, intermediate, and distal) in (A). (E) Weight loss of WT and Il25fl/flxVi1l-cre mice throughout the course of DSS-induced colitis. n = 5-6 mice per group. (F) Combined individual colon lengths from the mice in (E). (G) Combined expression of total colon IL-25 mRNA from each mouse in (E). (H) Representative H & E histology from middle colon sections from mice in (E). (I) Combined histological scores from all colon sections of DSS mice presented in (E). (J) Total colon tissue was isolated from mice 8 d following the induction of DSS colitis and equivalent biopsy sections were taken for inflammatory mRNA analysis (qPCR) or were cultured in media overnight for cytokine analysis in supernatants (ELISA). The data from individual mice were combined for each group. All gene values were normalized to the expression of Actb. Data from individual mice are presented as mean + SD and are representative of at least 3 independent experiments. *Student’s t test; p < 0.05 for comparisons between WT control and Il25−/− groups.
After establishing the pathogenic function of IL-25 in colonic inflammation, we next hypothesized that this phenotype is driven by the action of CEC (Figure 1A). Therefore, we generated a mouse strain with epithelial-specific deletion of IL-25 by crossing mice with a floxed Il25 gene to mice with a cre recombinase under control of the villin 1 promoter (Vil1-cre). The resulting epithelial cell-specific conditional Il25−/−mice (Il25fl/flxVi1l-cre) were then subjected to DSS-induced colitis. As expected, Il25fl/flxVi1l-cre animals exhibited resistance to DSS-induced weight loss and colon shortening compared to WT controls (Figures 2E-F). Importantly, the expression of Il25in the colons derived from Il25fl/flxVi1l-cre animals following DSS treatment was ~4% of the expression found in co-housed WT littermates, further demonstrating the importance of IL-25 expression by CEC (Figure 2G). Similar to Il25−/− animals, colon tissue was less inflamed in Il25fl/flxVi1l-cre mice compared to WT (Figures 2H-I).
We next asked if IL-25 regulates inflammation through the production of pro-inflammatory mediators following the induction of colitis. Indeed, colons isolated from Il25−/− mice following DSS administration were markedly reduced in mRNA and protein expression of the IL-1β, IL-6, TNF-α cytokines, and the CCL2 chemokine (Figure 2J). Thus, our results indicate that IL-25, whose expression in epithelial cells is enhanced in DSS colitis, plays a pathogenic role by regulating pro-inflammatory mediators.
Defective IL-17B expression exacerbates the development of acute colitis
To examine the role of IL-17B in colon inflammation, we generated a mouse strain containing a genetrap in the upstream promoter of the Il17b gene (IL-17B-deficient). This strain develops normally and exhibits substantially reduced IL-17B mRNA and protein expression in mucosal tissues (Figure S2). Next we performed DSS-induced colitis in these animals. Surprisingly, defective IL-17B led to an opposing phenotype compared to Il25 deletion (Figure 2). IL-17B-deficient mice began to lose weight earlier than their WT counterparts (Figure 3A) and colons were consistently shorter (Figure 3B). Moreover, H&E analysis revealed greater leukocyte infiltration, thickening, and goblet cell destruction as a result IL-17B deficiency (Figures 3C-D). To determine the basis for these observations, we next isolated mRNA and protein from colons of WT and IL-17B-deficient mice (Figure 3E). In contrast to our observations in Il25−/− animals (Figure 2), we observed that inflammatory mRNA and protein (IL-1β, IL-6, TNF-α, and CCL2) was markedly increased in animals lacking IL-17B Overall, these results suggest that while both IL-25 and IL-17B bind to IL-17RB, they differentially regulate the development of colonic inflammation.
Figure 3. IL-17B-deficient mice exhibit exacerbated colitis.
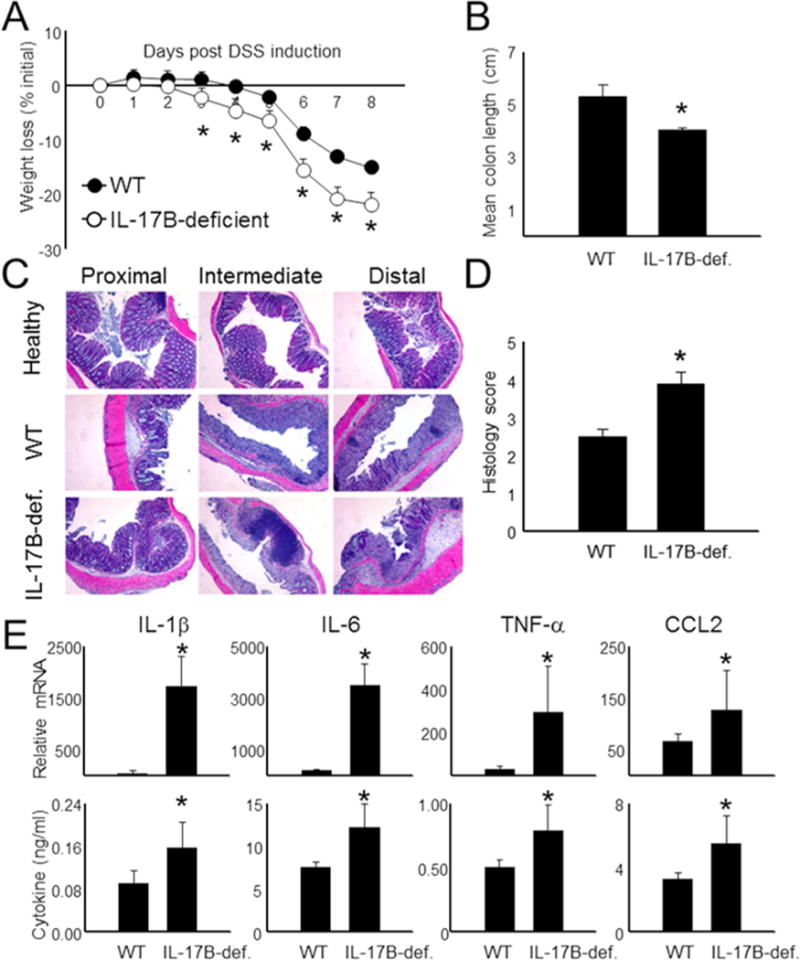
(A) Weight loss (% of starting weight) was assessed daily in WT and IL-17B-deficient mice following DSS administration. n = 5-6 mice per group. Colons were isolated from individual mice following sacrifice at d 8 and then measured and combined for each group (B). (C + D) Colon tissue derived from the mice in (A) were sectioned and then stained with H&E to measure infiltration and inflammation in individual mice following DSS onset at d 8. (E) Total colon tissue was divided into similar sections and then combined for each mouse. mRNA was then isolated and gene values were measured by qPCR using the expression of Actb as a reference. Supernatants from the colon were also assayed for cytokine expression by ELISA. Data are presented as mean + SD and are representative of at least 3 independent experiments. *Student’s t test; p < 0.05 in comparisons of IL-17B-deficient to WT controls. See also Figure S2.
IL-17B inhibits IL-25-induced IL-6 production
To further investigate the mechanisms underlying the opposing phenotypes in these animals, we first examined the contribution of IL-17RB signaling. Bone marrow chimeras were generated with IL-17RB present and/or absent (Watarai et al., 2012) in the hematopoietic and non-hematopoietic compartments. Mice deficient in IL-17RB in both compartments exhibited resistance to DSS, similar to Il25−/ mice (Figure 4A). Conversely, mice with IL-17RB deficiency only in leukocytes exhibited weight loss consistent with WT. Finally, animals with deletion of IL-17RB in the non-immune cells exhibited protection from DSS colitis (Figure 4A). Thus, CEC are not just the primary source of IL-25 (Figure 2), but are likely the primary cells responding to IL-25 in this model.
Figure 4. IL-17B inhibits IL-25-mediated IL-6 production in colon epithelial cells.
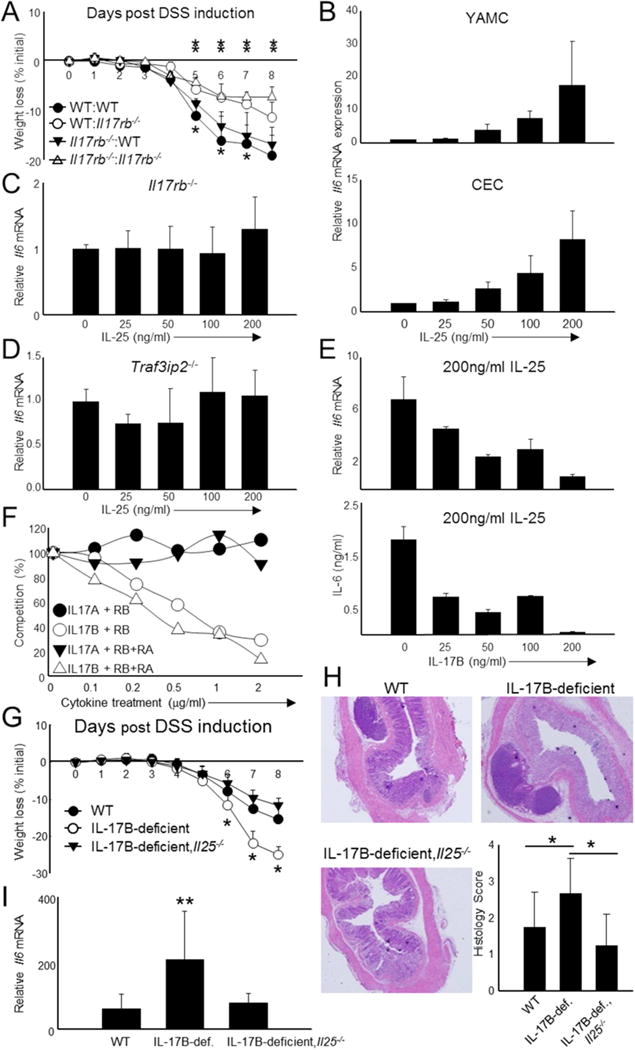
(A) Bone marrow chimeric mice were generated using WT and Il17rb−/ animals as donors and recipients. The first portion of the legend represents hematopoietic cell origin (WT or Il17rb−/−) while the second represents non-hematopoietic cell origin. (B) IL-6 expression was measured by qPCR following 6 h stimulation with recombinant IL-25 in a colon epithelial cell line (YAMC) and primary colon epithelial cells (CEC). (C+D) IL-6 mRNA expression was similarly measured from CEC derived from IL-17RB- (C) and Act1 (Traf3ip2−/−) - (D) deficient mice. (E) IL-6 was examined in CEC following fixed IL-25 stimulation (200ng/ml) followed by simultaneous treatment of increasing IL-17B concentrations. IL-6 was measured by qPCR (6 h stimulation, top) and ELISA (24 h stimulation, bottom). (F) Summary of competition experiments measuring the ability of increasing concentrations of IL-17B or IL-17A to displace IL-25-hIg bound to murine IL-17RB (RB) or IL-17RB and IL-17RA (RB+RA). Competition (%) is the percentage of IL-25 displacement at each IL-17B or IL-17 concentration compared those treated with only IL-25. (G) Combined weight loss data from WT, IL-17B-deficient, and IL-17B and IL-17E double deficient (IL-17B-deficient,Il25−/−) mice following DSS administration. n = 7-8 mice per group. (H) Representative H & E histology and combined histological scores of all colon sections from the mice shown in (G). (I) Combined IL-6 mRNA expression from total colon samples derived from the mice in (G). Data are presented as mean + SD and are representative of 2 independent experiments (A) and at least 3 independent experiments (B-I). mRNA data was obtained by examining relative gene expression to Actb. *Student’s t test; *p < 0.05 for daily comparisons of WT:WT mice to Il17rb−/−: Il17rb−/− mice and **p < 0.05 for daily comparisons of WT: Il17rb−/− mice to Il17rb−/−: Il17rb−/− mice (A); *p < 0.05 for daily comparisons of IL-17B-deficient,Il25−/− mice to WT (G); **p < 0.05 for comparison of IL-17B-deficient,Il25−/− mice to IL-17B-deficient and to WT (G-I). See also Figure S3.
To investigate how IL-17RB promotes colonic inflammation, we next examined the effect of IL-25 on the production of IL-6 by CEC. We focused on IL-6 production due to the correlation of expression with disease severity (Figure 2J and Figure 3E) and its importance in colon inflammation and cancer (Atreya et al., 2000; Gay et al., 2006; Yen et al., 2006; Tajima et al., 2008; Grivennikov et al., 2009). When treating either the young adult mouse colon (YAMC) cell line or primary CEC with IL-25, we observed a dose-dependent effect of inducing IL-6 production (Figure 4B). In contrast, IL-17B had no effect in promoting IL-6 or other mRNAs from YAMC or CEC (Figure S3A). Since IL-25 induced IL-6, we next asked whether this effect was dependent on the IL-17RA and IL-17RB heterodimer as well as the Act1 adaptor, all of which are known to be required for IL-17RB signaling in other cells (Chang et al., 2006; Qian et al., 2007; Rickel et al., 2008; Claudio et al., 2009; Angkasekwinai et al., 2010). Stimulation of IL-17RA- (data not shown), IL-17RB- (Figure 4C) or Act1 (Traf3ip2)-deficient (Figure 4D) CEC with IL-25 resulted in little to no IL-6, indicating that IL-25 signaling in CEC is comparable to other cell types.
Although we could not observe a discernible effect of IL-17B, we investigated if IL-17B influences IL-25-mediated IL-6 expression, considering the opposing colitis phenotypes (Figures 2–3). We isolated primary CEC and stimulated with a fixed concentration of IL-25 (200ng/ml) accompanied by simultaneous treatment of increasing IL-17B. We found that IL-17B treatment resulted in a reduction of IL-6 (Figure 4E). High-dose IL-17B almost completely abolished IL-6 mRNA and protein, suggesting that IL-17B may restrict IL-25 signaling. Conversely, IL-17B could not inhibit IL-6 expression induced by IL-17 (Figure S3B). These results suggest that IL-17B inhibits IL-25 binding to IL-17RB as a means to dampen IL-25 signaling. Therefore, we analyzed the binding of IL-25-hIg to IL-17RB or IL-17RB and IL-17RA in the presence of increasing IL-17B (Figure 4F and Figure S3C). Low-dose IL-17B was unable to inhibit IL-25 binding to HEK 293 cells expressing IL-17RB alone. However, high doses of IL-17B substantially inhibited binding. Moreover, when cells expressed both IL-17RB and IL-17RA, IL-17B could inhibit IL-25 binding at lower concentrations. These data suggest that IL-25 binds to IL-17RB regardless of IL-17RA, though dimerization of IL-17RB to IL-17RA is critical for the transmission of signal (Figure 4C). IL-17B preferentially inhibited IL-25 binding to the heterodimeric form of the receptor (Figure 4F). However, IL-17A had no effect on IL-25 binding to IL-17RB or the IL-17RB and IL-17RA complex.
Since IL-17B deficiency led to greater colitis severity, we asked whether treatment of WT or IL-17B-deficient mice with IL-17B could improve disease severity. We found that treatment of mice by i.p. IL-17B injection at the start of DSS was sufficient to improve weight loss (Figure S3D) in WT animals. Furthermore, IL-17B treatment in IL-17B-deficient animals led to a partial rescue from wasting disease.
To test if the severe DSS phenotype in IL-17B-deficient mice exaggerated IL-25 signaling, we crossed IL-25- and IL-17B-deficient to create double-deficient mice (IL-17B-deficient, Il25−/−). We treated these mice with DSS and found that additional IL-25 deletion greatly improved wasting in IL-17B-deficient animals (Figure 4G). Moreover, colonic inflammation was identical in the WT and IL-17B-deficient, Il25−/− animals (Figure 4H) as was IL-6 mRNA expression in the colon (Figure 4I). Thus, the deficiency of both IL-17B and IL-25 reverts a single-deficient animal back to the WT phenotype.
Opposing roles of IL-17B and IL-25 in response against C. rodentium
To determine if IL-17B and IL-25 functions are specific to acute colitis or further manifest in general inflammatory responses, we orally infected WT, IL-17B-deficient, and Il25−/− mice with Citrobacter rodentium, a murine-specific, Gram-negative model for human E. coli infection (Luperchio and Schauer, 2001). All experiments were performed on mice co-housed or on a rotating cage system to minimize differential microbiota effects (Elinav et al., 2011). IL-17B-deficient mice consistently exhibited a mild, but significant, weight loss compared to WT animals (Figure 5A). This weight loss correlated with an extensive bacterial burden in the feces, indicating a failure to properly control the pathogen (Figure 5B). The bacterial load in the liver and spleen, an indicator of dissemination, was also higher with IL-17B deficiency; however, only a modest count was obtained from these organs, possibly due to the relatively low CFU for infection (Figure S4A). Thus, IL-17B deficiency is pathogenic for Citrobacter rodentium infection, similar to the DSS-induced colitis, although the mechanism may be different due to the importance of IL-6 in combating C. rodentium (Dann et al., 2008).
Figure 5. IL-17B is protective against Citrobacter rodentium infection.
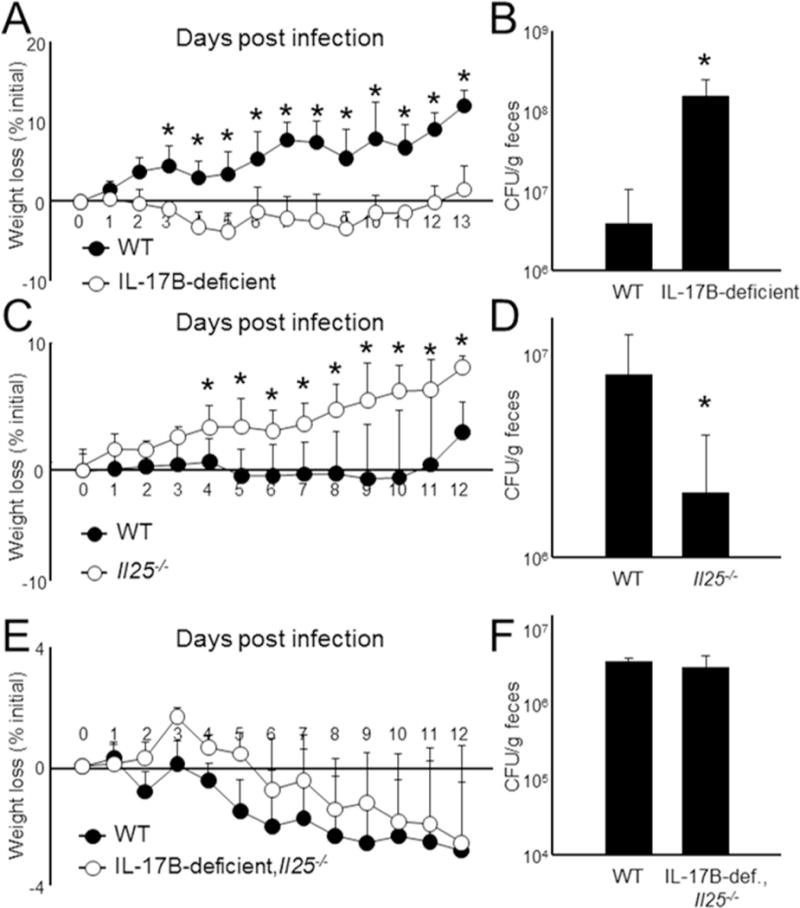
(A) WT and IL-17B-deficient, WT and IL-25KO (C), or WT and IL-17B-deficient,Il25−/− mice (E) mice were orally infected with 2 × 109 CFU of Citrobacter rodentium and weighed daily for 12-13 d. n = 7-18 mice per group. (B-F) After harvest, feces were collected, homogenized, and then examined for Citrobacter rodentium growth on MacConkey agar after overnight incubation. Data are presented as mean + SD and are representative of at least 3 independent experiments. *Student’s t test; p< 0.05. See also Figure S4.
We next performed similar experiments using Il25−/− mice. For reasons unknown, in these experiments WT animals had a moderate weight loss (Figure 5C). Nonetheless, Il25−/− mice did exhibit a slight, but significant, improvement in weight compared to WT mice (Figure 5C), indicating that IL-25 is important for C. rodentium defense. Further analysis revealed a likewise trend where fecal bacteria were significantly reduced in Il25−/− mice (Figure 5D). The protective effect observed in Il25−/− mice, however, did not reach the threshold for statistical significance for spleen and liver comparisons (Figure S4A).
IL-22 is critical cytokine for combating Citrobacter infection (Zheng et al., 2008). Therefore, we analyzed IL-22 mRNA and protein in infected colons (Figure S4B). We found that IL-22 expression was decreased in Il25−/ mice and increased in IL-17B-deficient mice, which was somewhat surprising considering that IL-17B-deficient mice were susceptible and Il25−/ mice were protected against Citrobacter infection. Thus, the functions of IL-25 and IL-17B in the innate response to pathogens are complex and likely to involve other factors outside of IL-6 and IL-22. When analyzing IL-17B-deficient, Il25−/− animals, we found that deletion of both cytokines during infection resulted in a phenotype equivalent to WT animals, including weight loss, bacteria load, and IL-22 production (Figure 5E-F and Figure S4A-B), similar to that observed in DSS experiments (Figure 4G-I).
Opposing functions of IL-17B and IL-25 in airway inflammation
Previous work has demonstrated that IL-25 mediates allergic inflammation by promoting Th2 cell responses and facilitating eosinophil recruitment (Fort et al., 2001; Angkasekwinai et al., 2007; Zaph et al., 2008; Angkasekwinai et al., 2010). Consequently, animals lacking IL-25 are protected against the development of experimental asthma. In allergic asthma induced by OVA and alum, IL-17B is also induced in the lung tissue (Figure S5A). While lack of IL-25 led to a reduction of eosinophils, the bronchoalveolar lavage fluid (BALF) of IL-17B-deficient mice was marked by substantial eosinophil influx (Figure 6A). When we examined various Th2 cell mediators, we expectedly found that the IL-4, IL-5, and IL-13 cytokines were reduced in Il25−/− and exacerbated in IL-17B-deficient mice (Figure 6B). A similar profile was obtained when analyzing the total lung mRNA (Figure 6C). IL-9, regulated by IL-25 (Angkasekwinai et al., 2010), was increased in the lungs of IL-17B-deficient animals as well (Figure 6C). Similarly, the BALF of Il25−/− mice exhibited a loss of the Th2 cell compartment upon OVA restimulation while IL-17B-deficient BALF contained increased Th2 cells (Figure 6D). To further examine Th2 cell-mediated responses, we restimulated total splenic and mediastinal lymph node cells with OVA for 3 d and analyzed cytokine production. We found a similar exacerbation of Th2 cell responses in the cells isolated from IL-17B-deficient mice (Figure 6E). Thus, IL-25 strongly promotes allergic airway inflammation through enhanced Th2 cell responses while IL-17B conversely restrains Th2 cell responses in the airway.
Figure 6. IL-17B and IL-25 have opposing roles during allergic asthma.
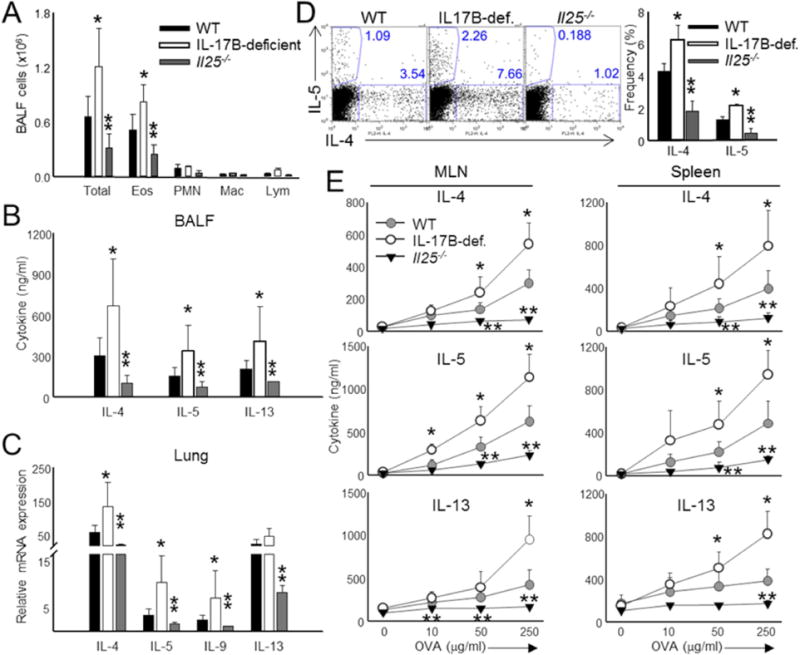
(A) Total cell counts in the BAL fluid of WT, IL-17B- and IL-25-deficient animals sensitized with OVA in alum at day 0 and 14, followed by intranasal challenge with OVA at day 14, 25, and 26. (B) BALF derived from the animals in (A) was assayed for the presence of IL-4, IL-5, and IL-13 by ELISA. (C) Combined mRNA analysis of Th2 and Th9 cell-associated genes from total lung samples derived from the mice in (A). (D) Representative (left) and combined analysis (right) of Th2 cell frequencies in the BALF of OVA and alum sensitized animals. T cells isolated from the BALF were restimulated with 50μg/ml OVA prior to intracellular cytokine staining for IL-4 and IL-5. (E) Analysis of splenic and mediastinal lymph node Th2 compartments following 3 d restimulation with increasing doses of OVA. IL-4, IL-5, and IL-13 protein was measured from the supernatants by ELISA. Data are presented as mean of data obtained from individual mice + SD and are representative of at least 3 independent experiments. *Student’s t test; *p < 0.05 for comparisons of WT to IL-17B-deficient and **p < 0.05 for comparisons of WT to Il25−/−. See also Figure S5.
To determine the outcome of deleting both cytokines in this model, we performed a similar set of experiments in IL-17B-deficient, Il25−/− mice (Figure S5B-C). We found that respiratory infiltration as well as Th2 cell cytokine production was equivalent between WT and IL-17B-deficient, Il25−/− animals. These results indicated that removal of both cytokines counteracts the phenotypes observed in single deficient mice, similar to what was observed for DSS (Figure 4) and Citrobacter rodentium infection (Figure 5).
DISCUSSION
The pro-inflammatory functions of IL-17 have been well-documented (Dong, 2008). Here, we have demonstrated further functional complexity amongst the IL-17 family of cytokines. Adding to a well-established role in type 2 immunity, we found that IL-25 was pathogenic in acute colitis, while IL-17B was conversely protective. These opposing effects were similar in Citrobacter rodentium infection and allergic asthma. Notably, we have found a unique role for IL-17B in inhibiting IL-25-mediated IL-6 production in CEC, making IL-17B an antagonistic and anti-inflammatory cytokine in this family.
IL-25 is generally thought of as a Th2 cell-derived factor, promoting allergic asthma responses but also protecting against parasitic infection (Reynolds et al., 2010). Here, we have shown that IL-25 in acute colitis promoted IL-6 production from CEC, which is consistent with a report of IL-25 promoting the release of IL-6 from eosinophils (Wong et al., 2005). Throughout the course of this study, IL-25-dependent IL-6 production was the most consistently inhibited by IL-17B treatment. However, at this time we cannot rule out the influence of other mediators in promoting the protective and pathogenic phenotypes observed in IL-25- and IL-17B-deficient mice, respectively. Most likely IL-17B antagonism of IL-25 is influencing multiple facets of the mucosal immune response, which is the subject of our current investigations.
Our finding that IL-25 deletion protects against DSS differs from another showing that injecting IL-25 protects against the development of DSS colitis (McHenga et al., 2008). Though we cannot fully explain the discrepancy between reports, IL-25 stimulation of primary CEC in vitro promoted the production of IL-6, consistent with decreased inflammation observed in Il25−/− mice. Indeed, when we injected WT mice directly with IL-25 (not shown) or IL-17B, we observed phenotypes opposite to those found in cytokine-deficient mice. Thus, different housing conditions or microbiota may influence results obtained from recombinant protein injection.
IL-17B has long been a mysterious member of the IL-17 cytokine family. Previous work has demonstrated that IL-17RB may be oncogenic while IL-17B may contribute to tumor cell growth (Tian et al., 2000; Furuta et al., 2011; Huang et al., 2013). The original report characterizing IL-17B demonstrates that this cytokine stimulates the release of TNF-α and IL-1β from THP-1 cells. However, we did not observe production of pro-inflammatory mediators following IL-17B stimulation, consistent with IL-17B being unable to induce IL-6 from fibroblasts (Li et al., 2000). Instead, the primary effect we observed for IL-17B treatment in CEC was direct inhibition of IL-25-mediated IL-6 production. This of course begs the question of how exactly IL-17B can inhibit IL-25-mediated IL-6 production in CEC. One possibility is that IL-17B competes with IL-25 for binding IL-17RB. Indeed, we found that IL-17B inhibited IL-25 binding to IL-17RB or IL-17RA and IL-17RB complexes. Since previous work has demonstrated that IL-17A and IL-17F form heterodimers, we investigated this possibility as well. However, exhaustive attempts to identify IL-17B and IL-25 heterodimers were unsuccessful (not shown); suggesting that competition for IL-17RB binding causes functional antagonism. IL-17B had higher activity in blocking IL-25 binding to the IL-17RA and IL-17RB heterodimer compared to IL-17RB alone. Conversely, IL-25 was capable of binding IL-17RB in the absence of IL-17RA, suggesting that IL-17RA was more important for signaling. These binding assays have limitations considering that we cannot rule out the contribution of endogenous IL-17 receptors that may be expressed in the human HEK cells as we do not know if the murine IL-17RB could form heterodimers with endogenous human IL-17RA. Another caveat is that only mouse receptors were co-expressed in these cells and more direct studies need to be performed in the future on both mouse and human IL-17RA and IL-17RB similar to studies on IL-17 and IL-17F (Toy et al., 2006; Kuestner et al., 2007). Finally, we need to determine if IL-17RB is internalized following binding of IL-17B, IL-25, or both. Differential IL-17RB internalization could represent another mechanism governing IL-17B regulation of IL-25.
Our current model is that colon inflammation enhances IL-25 pro-inflammatory signaling while IL-17B is concurrently induced to restrain IL-25. IL-25 has a higher affinity for IL-17RB alone compared to IL-17B. However, treatment with IL-17B throughout the course of DSS colitis led to protection in WT animals, indicating that a shift in the IL-25 to IL-17B ratio can influence the course of inflammation. Previous work has demonstrated that IL-17F in some situations antagonizes IL-17 signaling, including forming heterodimeric complexes with IL-17 that have weaker activity compared to IL-17 alone (Wright et al., 2008). We believe that the antagonism exerted on IL-25 signaling by IL-17B, however, is unique to this family as IL-17F by itself can activate signaling pathways important for inflammation (Yang et al., 2008) while we have yet to find a standalone function for IL-17B. Moreover, IL-17B could not antagonize IL-6 production induced by IL-17 in CEC, demonstrating the specificity of IL-17B action. Therefore, further study is needed to determine if IL-17B alone can transmit a functional signal to any cell type and to determine the precise mechanism for how IL-17B is inhibiting IL-25 binding to IL-17RB.
The prospect of modulating IL-17RB signaling for the treatment of colitis at this time seems promising. Our findings that recombinant IL-17B injections throughout the course of DSS administration can improve disease outcome in WT animals further supports this theory Whether IL-17B is as important in human IBD is currently under investigation. The production of IL-25 has been linked to human IBD even though polymorphism is not believed to be a risk factor (Buning et al., 2003). Two recent reports have shown that inflammatory bowel disease (IBD) patients have decreases in IL-25 (Caruso et al., 2009; Su et al., 2013). These results may suggest that IL-25 is protective factor in human IBD; however, IL-17B expression in these patients has yet to be determined. Decreased IL-25 expression in human IBD may also be representative of a failure of IL-17B to control inflammation. Thus, further investigation is necessary to determine the role of IL-17B in human IBD as well as determining if IL-17RB represents is a viable therapeutic target.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 mice were purchased from NCI, and Nod2−/−, and Vil1-cre mice were purchased from The Jackson Laboratory. Il25fl/fl and IL-25-deficient mice have been previously described (Angkasekwinai et al., 2010). Il25fl/fl mice were crossed with Vil1-cre mice on the B6 background to generate Il25fl/flxVil1-cre mice. IL-17B-deficient mice were generated by gene trapping in the R1 ES cell line (clone 371C12; Toronto Centre for Phenogenomics, Centre for Modeling Human Disease). Both IL-25- and IL-17B-deficient mice have been backcrossed to B6 background for >8 generations. Il17rb−/− and Traf3ip2−/− (Act1) mice have previously been described (Chang et al., 2011; Watarai et al., 2012). All experiments were conducted on 6-12 wk-old animals using protocols approved by the Rosalind Franklin University of Medicine and Science and MD Anderson Cancer Center Institutional Animal Care and Use Committees. Additional information on experimental mice can be found in the Supplemental Procedures.
Antibiotic depletion of commensal flora
Commensal flora was depleted using a previously described regiment (Ivanov et al., 2008). Briefly, mice were administered water containing a cocktail of ampicillin (Sigma-Aldrich), neomycin (Fisher Scientific), vancomycin (Fisher), and metronidazole (Fisher) for 4 weeks.
DSS-induced colitis
All experiments were performed using 6-12wk old male mice on the C57BL/6 background that were fed DSS in drinking water at a concentration of 2-3.5% (w/v) for a total period of 5 days. Starting weights were recorded and then mice were weighed and monitored daily until the experimental endpoint (d 8). For analysis, total colon tissue was harvested from animals and colon lengths were recorded. Colon sections were then divided into proximal, intermediate, and distal sections for H&E histological analysis. Sections from each region were also combined in plated in media overnight for supernatant analysis of cytokines by ELISA. Sections were also combined and lysed in TRIzol (Life Technologies) for analysis of mRNA expression by qPCR. Additional analysis methods, including mRNA, histology, and protein analysis, can be found in the Supplemental Procedures.
Cell lines and CEC
YAMC cells were a generous gift from Dr. Dingzhi Wang at the University of Texas MD Anderson Cancer Center. The isolation of CEC has been described previously (Reynolds et al., 2012). Primary CEC were plated and rested in 96-well dishes prior to stimulation with LPS (Sigma-Aldrich), Pam3CSK4 (Invivogen), MDP (Invivogen), or the indicated concentration of mouse IL-17B and/or IL-25 (R & D Systems) for 6h (mRNA) or 24h (ELISA). Additional experimental details can be found in the Supplemental Procedures.
Cytokine binding
To analyze IL-25 binding to IL-17RB, a recombinant IL-25 tagged with human Ig (IL-25-hIg) was raised in Drosophila S2 cells as described previously (Angkasekwinai et al,. 2007). Briefly, HEK293T cells were retrovirally transfected with constructs encoding the murine Il17ra or Il17rb genes. Following transfection, the indicated concentrations of IL-17A, IL-17B (R&D Systems), or IL-25-hIg were added simultaneously for 30min followed by the determination of IL-25-hIg surface binding by flow cytometery using anti-hIg conjugated to APC. Additional details may be found in the Supplemental Procedures.
Generation of bone marrow chimeras
Recipient WT or Il17rb−/− mice (6-8 weeks) were irradiated with 750 rad and injected intravenously with 5 × 106 donor cells as indicated. After 6 weeks, reconstituted mice were administered 3% DSS in drinking water as described above.
Citrobacter rodentium infection
C. rodentium (ATCC 51459) infection was performed using 2 × 109 CFU as previously described (Hu et al., 2013). Mice were weighed for 13-14d following infection and colon mRNA and cytokine production was analyzed as described above for DSS colitis. Bacterial load was assessed by plating fecal, liver, or splenic homogenates overnight on MacConkey agar. Additional details for this model can be found in the Supplemental Procedures.
OVA-induced asthma
Mice were immunized i.p. with 50μg of grade VII OVA (Sigma) emulsified in alum (Thermo) at d0 and 14 followed by i.n. administration of 50μg OVA at d 14, 25 and 26 as described previously (Angkasekwinai et al., 2010). Briefly, at d28 BALF was collected for IL-4, IL-5, and IL-13 determinations by ELISA (BD Biosciences). BALF was also analyzed for leukocyte infiltration using flow cytometry and Dif-Quick (Fisher) histological staining. Finally, lymphocytes isolated from the BALF were restimulated with OVA to determine Th2 cell generation by staining with antibodies specific for IL-4 and IL-5 (eBioscience). Lung tissue was collected and homogenized in TRIzol for the determination of cytokine gene expression by qPCR. Mediastinal lymph nodes and spleens were collected from each mouse, homogenized, and then stimulated with the indicated concentrations of OVA for 3d. IL-4, IL-5, and IL-13 expression was then measured in the supernatants by ELISA.
Statistical analysis
Statistical comparisons were performed using two-tail paired Students t test. P values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
This work was in part supported by a National Cancer Institute training grant T32CA009598 (J.M.R.), a SPORE grant from MD Anderson Cancer Center (5 P50 CA142509 04 to Y.H.L.), a Crohn’s and Colitis Foundation of America Career Development Award (#3633 to S.H.C.), and the Cancer Prevention and Research Institute of Texas (to S.H.C. and C.D.). C.D. is a Bayer Chair Professor at Tsinghua University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
C.D., J.M.R, and Y.H.L conceived the project, designed the experiments and wrote the manuscript. J.M.R. and Y.H.L. performed most of the experiments. Y.S., X.W., P.A., K.C.N, S.F., and S.H.C contributed to specific experiments. H.W. provided mice and intellectual input.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
References
- Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflammatory bowel diseases. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angkasekwinai P, Chang SH, Thapa M, Watarai H, Dong C. Regulation of IL-9 expression by IL-25 signaling. Nature immunology. 2010;11:250–256. doi: 10.1038/ni.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, Liu YJ, Zhu Z, Dong C. Interleukin 25 promotes the initiation of proallergic type 2 responses. The Journal of experimental medicine. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nature medicine. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- Buning C, Genschel J, Weltrich R, Lochs H, Schmidt H. The interleukin-25 gene located in the inflammatory bowel disease (IBD) 4 region: no association with inflammatory bowel disease. European journal of immunogenetics: official journal of the British Society for Histocompatibility and Immunogenetics. 2003;30:329–333. doi: 10.1046/j.1365-2370.2003.00411.x. [DOI] [PubMed] [Google Scholar]
- Camelo A, Barlow JL, Drynan LF, Neill DR, Ballantyne SJ, Wong SH, Pannell R, Gao W, Wrigley K, Sprenkle J, McKenzie AN. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J Gastroenterol. 2012;47:1198–1211. doi: 10.1007/s00535-012-0591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso R, Sarra M, Stolfi C, Rizzo A, Fina D, Fantini MC, Pallone F, MacDonald TT, Monteleone G. Interleukin-25 inhibits interleukin-12 production and Th1 cell-driven inflammation in the gut. Gastroenterology. 2009;136:2270–2279. doi: 10.1053/j.gastro.2009.02.049. [DOI] [PubMed] [Google Scholar]
- Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. The Journal of biological chemistry. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35:611–621. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. Journal of immunology. 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nature immunology. 2009;10:66–74. doi: 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ, Hanson E, Eckmann L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. Journal of immunology. 2008;180:6816–6826. doi: 10.4049/jimmunol.180.10.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. Regulation and pro-inflammatory function of interleukin-17 family cytokines. Immunol Rev. 2008;226:80–86. doi: 10.1111/j.1600-065X.2008.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, McIlgorm A, Jolin HE, McKenzie AN. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. The Journal of experimental medicine. 2006;203:1105–1116. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- Furuta S, Jeng YM, Zhou L, Huang L, Kuhn I, Bissell MJ, Lee WH. IL-25 causes apoptosis of IL-25R-expressing breast cancer cells without toxicity to nonmalignant cells. Science translational medicine. 2011;3:78ra31. doi: 10.1126/scitranslmed.3001374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay J, Kokkotou E, O’Brien M, Pothoulakis C, Karalis KP. Interleukin-6 genetic ablation protects from trinitrobenzene sulfonic acid-induced colitis in mice. Putative effect of antiinflammatory cytokines. Neuroimmunomodulation. 2006;13:114–121. doi: 10.1159/000096656. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, Xiao Y, Cheng X, Chang M, Fu YX, et al. OTUD7B controls non-canonical NF-kappaB activation through deubiquitination of TRAF3. Nature. 2013;494:371–374. doi: 10.1038/nature11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CK, Yang CY, Jeng YM, Chen CL, Wu HH, Chang YC, Ma C, Kuo WH, Chang KJ, Shew JY, Lee WH. Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-kappaB-mediated antiapoptotic pathway. Oncogene. 2013 doi: 10.1038/onc.2013.268. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell host & microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubu T, Haudenschild DR, Moseley TA, Rose L, Reddi AH. Immunolocalization of IL-17A, IL-17B, and their receptors in chondrocytes during fracture healing. J Histochem Cytochem. 2008;56:89–95. doi: 10.1369/jhc.7A7223.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, Harder B, Okada S, Ostrander CD, Kreindler JL, et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. Journal of immunology. 2007;179:5462–5473. doi: 10.4049/jimmunol.179.8.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ho WH, Maruoka M, Corpuz RT, Baldwin DT, Foster JS, Goddard AD, Yansura DG, Vandlen RL, Wood WI, Gurney AL. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. The Journal of biological chemistry. 2001;276:1660–1664. doi: 10.1074/jbc.M008289200. [DOI] [PubMed] [Google Scholar]
- Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, Dowd P, Gurney AL, Wood WI. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc Natl Acad Sci U S A. 2000;97:773–778. doi: 10.1073/pnas.97.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luperchio SA, Schauer DB. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes and infection/Institut Pasteur. 2001;3:333–340. doi: 10.1016/s1286-4579(01)01387-9. [DOI] [PubMed] [Google Scholar]
- McHenga SS, Wang D, Li C, Shan F, Lu C. Inhibitory effect of recombinant IL-25 on the development of dextran sulfate sodium-induced experimental colitis in mice. Cell Mol Immunol. 2008;5:425–431. doi: 10.1038/cmi.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463:540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, Cua DJ, Goldschmidt M, Hunter CA, Kastelein RA, Artis D. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. The Journal of experimental medicine. 2006;203:843–849. doi: 10.1084/jem.20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A. 2010;107:11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nature immunology. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nature immunology. 2011;12:1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- Reynolds JM, Angkasekwinai P, Dong C. IL-17 family member cytokines: regulation and function in innate immunity. Cytokine Growth Factor Rev. 2010;21:413–423. doi: 10.1016/j.cytogfr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, Martinez GJ, Nallaparaju KC, Chang SH, Wang YH, Dong C. Cutting edge: regulation of intestinal inflammation and barrier function by IL-17C. Journal of immunology. 2012;189:4226–4230. doi: 10.4049/jimmunol.1103014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, Anders PM, Tocker JE, Comeau MR, Budelsky AL. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. Journal of immunology. 2008;181:4299–4310. doi: 10.4049/jimmunol.181.6.4299. [DOI] [PubMed] [Google Scholar]
- Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF, Jr, Tocker JE, Budelsky AL, Kleinschek MA, Kastelein RA, Kambayashi T, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464:1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AJ, Guo X, Mason MD, Jiang WG. IL-17B Can Impact on Endothelial Cellular Traits Linked to Tumour Angiogenesis. J Oncol. 2010;2010:817375. doi: 10.1155/2010/817375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, Eberl G. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665–669. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, Cua D, Di Santo JP, Eberl G. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nature immunology. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- Schwarzenberger P, La Russa V, Miller A, Ye P, Huang W, Zieske A, Nelson S, Bagby GJ, Stoltz D, Mynatt RL, et al. IL-17 stimulates granulopoiesis in mice: use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. Journal of immunology. 1998;161:6383–6389. [PubMed] [Google Scholar]
- Shi Y, Ullrich SJ, Zhang J, Connolly K, Grzegorzewski KJ, Barber MC, Wang W, Wathen K, Hodge V, Fisher CL, et al. A novel cytokine receptor-ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. The Journal of biological chemistry. 2000;275:19167–19176. doi: 10.1074/jbc.M910228199. [DOI] [PubMed] [Google Scholar]
- Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nature immunology. 2011;12:1151–1158. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- Su J, Chen T, Ji XY, Liu C, Yadav PK, Wu R, Yang P, Liu Z. IL-25 downregulates Th1/Th17 immune response in an IL-10-dependent manner in inflammatory bowel disease. Inflammatory bowel diseases. 2013;19:720–728. doi: 10.1097/MIB.0b013e3182802a76. [DOI] [PubMed] [Google Scholar]
- Tajima M, Wakita D, Noguchi D, Chamoto K, Yue Z, Fugo K, Ishigame H, Iwakura Y, Kitamura H, Nishimura T. IL-6-dependent spontaneous proliferation is required for the induction of colitogenic IL-17-producing CD8+ T cells. The Journal of experimental medicine. 2008;205:1019–1027. doi: 10.1084/jem.20071133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O’Shea JJ. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. The Journal of experimental medicine. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian E, Sawyer JR, Largaespada DA, Jenkins NA, Copeland NG, Shaughnessy JD., Jr Evi27 encodes a novel membrane protein with homology to the IL17 receptor. Oncogene. 2000;19:2098–2109. doi: 10.1038/sj.onc.1203577. [DOI] [PubMed] [Google Scholar]
- Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. Journal of immunology. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- Watarai H, Sekine-Kondo E, Shigeura T, Motomura Y, Yasuda T, Satoh R, Yoshida H, Kubo M, Kawamoto H, Koseki H, Taniguchi M. Development and function of invariant natural killer T cells producing T(h)2- and T(h)17-cytokines. PLoS biology. 2012;10:e1001255. doi: 10.1371/journal.pbio.1001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CK, Cheung PF, Ip WK, Lam CW. Interleukin-25-induced chemokines and interleukin-6 release from eosinophils is mediated by p38 mitogen-activated protein kinase, c-Jun N-terminal kinase, and nuclear factor-kappaB. American journal of respiratory cell and molecular biology. 2005;33:186–194. doi: 10.1165/rcmb.2005-0034OC. [DOI] [PubMed] [Google Scholar]
- Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, Tomkinson KN, Fitz LJ, Wolfman NM, Collins M, et al. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. Journal of immunology. 2008;181:2799–2805. doi: 10.4049/jimmunol.181.4.2799. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno NH, Takahashi K, Yamamoto K. IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. Journal of immunology. 2007;179:7128–7136. doi: 10.4049/jimmunol.179.10.7128. [DOI] [PubMed] [Google Scholar]
- Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PloS one. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. The Journal of experimental medicine. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. The Journal of clinical investigation. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, DuRaine G, Tien JY, Lee C, Moseley TA, Reddi AH. Expression of interleukin-17B in mouse embryonic limb buds and regulation by BMP-7 and bFGF. Biochem Biophys Res Commun. 2005;326:624–631. doi: 10.1016/j.bbrc.2004.11.087. [DOI] [PubMed] [Google Scholar]
- Zaph C, Du Y, Saenz SA, Nair MG, Perrigoue JG, Taylor BC, Troy AE, Kobuley DE, Kastelein RA, Cua DJ, et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. The Journal of experimental medicine. 2008;205:2191–2198. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A, Urban JF, Jr, Sun R, Stiltz J, Morimoto M, Notari L, Madden KB, Yang Z, Grinchuk V, Ramalingam TR, et al. Critical role of IL-25 in nematode infection-induced alterations in intestinal function. Journal of immunology. 2010;185:6921–6929. doi: 10.4049/jimmunol.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.