

Abstract
DNA-protein crosslinks (DPCs) are complex DNA lesions that induce mutagenesis and cell death. DPCs are created by common antitumor drugs, reactive oxygen species, and endogenous aldehydes. Since these agents create other types of DNA damage in addition to DPCs, identification of the mechanisms of DPC repair is challenging. In this study, we created plasmid substrates containing site-specific DPC lesions, as well as plasmids harboring lesions that are selectively repaired by the base excision or nucleotide excision repair (NER) pathways. These substrates were transfected into mammalian cells and a quantitative real-time PCR assay employed to study their repair. This assay revealed that DPC lesions were rapidly repaired in wild-type human and Chinese hamster derived cells, as were plasmids harboring an oxoguanine residue (base excision repair substrate) or cholesterol lesion (NER substrate). Interestingly, the DPC substrate was repaired in human cells nearly three times as efficiently as in Chinese hamster cells (>75% vs ∼25% repair at 8 hours post-transfection), while there was no significant species-specific difference in the efficiency with which the cholesterol lesion was repaired (∼60% repair). Experiments revealed that both human and hamster cells deficient in NER due to mutations in the xeroderma pigmentosum A or D genes were five to ten-fold less able to repair the cholesterol and DPC lesions than were wild-type control clones, and that both the global genome and transcription-coupled sub-pathways of NER were capable of repairing DPCs. In addition, analyses using this PCR-based assay revealed that a 4 kDa peptide DNA crosslink was repaired nearly twice as efficiently as was a ∼38 kDa DPC, suggesting that proteolytic degradation of crosslinked proteins occurs during DPC repair. These results highlight the utility of this PCR-based assay to study DNA repair and indicate that the NER machinery rapidly and efficiently repairs plasmid DPC lesions in mammalian cells.
Introduction
DNA-protein crosslinks (DPCs) are unusually bulky lesions formed upon covalent trapping of proteins on DNA strands (1). These helix-distorting complexes are mutagenic, toxic, and are able to block essential cell processes such as transcription and replication (2,3). Proteins of various sizes and functions are capable of becoming crosslinked to DNA via multiple mechanisms (4,5). For example, endogenous DPCs can be formed by the trapping of repair proteins recruited to sites of DNA damage or as a byproduct of lipid peroxidation of reactive oxygen species in the blood (6-9). Exogenous agents such as ionizing radiation, UV light, cigarette smoke, and chemotherapeutics such as cisplatin also create DPCs (5,10-15). Despite their common occurrence and cytotoxic effects, the exact mechanism(s) by which DPCs are repaired is still not well understood.
In order to gain insight into the repair mechanism of DNA-protein crosslinks, DPC-forming agents have been used to assess hypersensitivity in repair mutants (11). Results from these experiments have provided evidence for the roles of nucleotide excision repair (NER) and homologous recombination (HR) in DPC repair (16). However, there are contradictory reports in the literature regarding the relative contributions of the two repair pathways. Specifically, genetic studies performed in Escherichia coli revealed that uvrA and recA mutants deficient in NER or HR were hypersensitive to the DPC-inducing agent formaldehyde (17,18). However, only recA and not uvrA mutants were hypersensitive to DPCs induced by azacytidine (19,20). In Saccharomyces cerevisiae, mutants deficient in NER, but not HR, were sensitive to formaldehyde (21,22). Similarly, human cells from xeroderma pigmentosum patients possessing mutations in the NER pathway were sensitive to DPC-inducing agents (23,24).
Since all known DPC-forming agents induce other types of DNA damage, such as DNA monoadducts and DNA-DNA cross-links, it is difficult to conclude if sensitivity to these drugs is influenced by lesions other than DPCs. In an effort to overcome this potential limitation, investigators have directly examined the kinetics of DPC formation and removal from wild-type and repair-deficient clones following exposure to DNA damaging agents (25). However, these studies have yielded contradictory results. For example, Quievryn et al. failed to detect differences in the kinetics of formaldehyde-induced DPC removal between NER-deficient human fibroblasts and control (26). Conversely, DPCs induced by nornitrogen mustard accumulated at higher rates in human cells deficient in the NER gene XPA compared to HR-deficient or wild-type clones (4).
To more directly assess the involvement of NER in DPC repair, Minko et al. incubated DPC-containing oligonucleotides with UvrABC nucleases from Escherichia coli and saw slower incision rates of DNA containing a 16 kDa protein compared to smaller DNA-peptide crosslinks (27,28). In vitro studies performed with human nucleases saw similar results where 4 and 12 amino acid peptide-crosslinks were recognized by the NER machinery but were unable to remove a 16 kDa protein-crosslink (29). Nakano et al. later reported a size limit of 8-10 kDa for the excision of crosslinked proteins by NER in mammalian cells while Baker et al. saw NER-directed repair of a 38 kDa attached to plasmid DNA (30,31). Currently, the role of NER in the repair of DPCs (specifically those consisting of proteins larger than 10 kDa) remains unclear. However, it is hypothesized that the decreased efficiency of NER to repair larger protein-crosslinks is caused by steric hindrance of damage recognition proteins and suggests that proteolytic degradation is necessary prior to repair. While it was originally proposed that the proteasome is responsible for proteolysis of full size DPCs, more recent studies have suggested that a different protease (Spartan in humans) is involved (32-37).
To clarify the role of NER in the repair of DPCs in mammalian cells, as well as to address more specific questions regarding how size and location influence DPC repair, we employed a PCR-based assay we term Strand-Specific Primer Extension-Quantitative Polymerase Chain Reaction (SSPE-qPCR). This assay is capable of quantifying the repair kinetics of a broad range of lesions present on plasmid DNA transfected into repair deficient and corrected mammalian cells. This assay provides significant advantages over previously utilized approaches in that it is rapid, highly quantitative, and extremely flexible. Importantly, this method directly measures repair activity, in contrast to other plasmid-based strategies that rely on indirect measures such as host-cell reactivation of gene function. Results from our initial analyses provide new insight into the ability of the global genome and transcription-coupled NER pathways to repair DPCs in hamster and human cells.
Material and Methods
Materials
Chemicals and enzymes
Oligodeoxynucleotides (ODNs) containing 8-oxo-2′-deoxyguanosine (8-oxo-dG) or cholesterol modifications were obtained from Midland Certified Reagent (Midland, TX). All other ODNs were purchased from the University of Minnesota Genomic Center. Human oxoguanine glycosylase 1 (OGG1) was expressed and purified from BL21(DE3) bacteria (Thermo Fisher) using a pET-28a expression vector (38). Single-stranded M13 vector and all enzymes were obtained from New England Biolabs (Beverly, MA) unless specified otherwise. Chemicals were purchased from Sigma Chemical (St. Louis, MO) unless indicated.
Cell lines
Chinese hamster lung fibroblast cell lines V79 (GM16136) and V-H1 (GM16141) were obtained from the Coriell Institute for Medical Research (Camden, NJ). V79 are wild-type cells from which the V-H1 clones were derived following an ethylnitrosourea-induced mutagenesis screen (39-41). V-H1 cells belong to nucleotide excision repair complementation group 2 and lack a functional XPD gene (42). These cells are deficient in the ERCC2 gene, which codes for the XPD protein involved in the helicase activity that unwinds duplex DNA during nucleotide excision repair (42). Chinese hamster lung fibroblast cells were cultured in Ham's F-12 modified essential Eagle's media (Life Technologies, Grand Island, NY) supplemented with 9% fetal bovine serum (Atlanta Biologics, Atlanta, GA). Chinese hamster ovary CHO-K1 cells were a kind gift from Professor Harry Orr (University of Minnesota). Immortalized human dermal fibroblasts from xeroderma pigmentosum patients with inactivating mutations in the NER XPD gene (GM08207) or XPA gene (GM04312), as well as gene-corrected clones (GM15877 and GM15876, respectively) derived from these lines were obtained from the Coriell institute. Human and CHO-K1 cells were cultured in Dulbecco's modified Eagle's media (Life Technologies, Grand Island, NY) supplemented with 9% fetal bovine serum. All cells were maintained in a humidified atmosphere of 5% carbon dioxide, 95% air, at 37 °C.
Methods
Taq polymerase extension assay
To confirm that Taq polymerase permanently stalls at abasic sites (and by extension, lesions attached to abasic sites) 200 pmol of the oligodeoxynucleotide M13-RSV-Zeo-8oxo (template) was annealed to 200 pmol of primer C (Table 1) in restriction enzyme buffer 2 (New England Biolabs) and water in a volume of 10 μL at 95°C for 5 minutes and slowly cooled to room temperature. The creation of a duplex is necessary because OGG1 protein will not react with single-stranded DNA. The duplex DNA was then incubated in the presence or absence of OGG1 (200 pmol) in buffer containing 100 mM NaCl, 1mM MgCl2, and 20 mM Tris-HCl pH 7.0 at 37°C (final volume 15 μL) for 30 minutes. Samples incubated with OGG1 excise the 8-oxo-dG residue leaving an abasic site at its position. Each sample was divided in half and incubated in the presence or absence of primer Z (100 pmol, Table 1) with 10 mM dNTPs (Thermo Scientific), Taq reaction buffer (New England Biolabs), and Taq polymerase (20 units) in a final volume of 10 μL at 95°C for 5 minutes. Samples were then slow cooled to 75°C for 15 minutes, resolved on a NuPAGE 12% Bis-Tris Gel (Invitrogen) at 18V/cm at 55°C, and stained with ethidium bromide.
Table 1.
Oligodeoxynucleotides (ODNs) used is this study (5′➔3′).
| ODN | Sequence | Use |
|---|---|---|
| M13-8oxo | AGGGTTTTCCA(8-oxo-dG)TCACGACGTT | Primer Extension |
| M13-cholesterol | CCGGGTACCGAGCTCGAATTC(cholesterol)GTAATC TTGGTCATAGCTG | Primer Extension |
| M13-RSV-Zeo-8oxo (template) | ACTGGTCAACTTGGCCAT(8-oxo-dG)TTGGCC TTGGAGGTCGACACC | Primer Extension |
| M13-RSV-Zeo-8oxo (coding) | CACCTCCAAGGCCAACAT(8-oxo-dG)GCCAAG TTGACCAGTGCCGTT | Primer Extension |
| M13 Primer R | CGGCTCGTATGTTGTGTG | qPCR of M13 plasmid |
| M13 Primer L | GCTGCAAGGCGATTAAGT | qPCR of M13 plasmid |
| RSV-Zeo 1 | TATCCGAGATCCGAGGAA | Topo 2.1 PCR Cloning Kit |
| RSV-Zeo 2 | TATGGATCGTCGAGACTC | Topo 2.1 PCR Cloning Kit |
| M13-Zeo R Primer | ACGCCATTTGACCATTCAAA | qPCR of M13-Zeo plasmid |
| M13-Zeo L Primer | CCGGTCGGTCCAGAACTC | qPCR of M13-Zeo plasmid |
| Primer C | GGCCAACATGGCCAA | Taq extension assay |
| Primer Z | GGTGTGCACCTCCAA | Taq extension assay |
| Zeo F1 | CAAGTTGACCAGTGCCGTTC | RT PCR |
| Zeo R1 | TGATGAACAGGGTCACGTCG | RT PCR |
| Abasic complement | GTCGACCTCCAA | Taq extension assay |
| Abasic | ACTGGTCAACTTGGCCAT(abasic)TTGGCCTTGGA GGTCGAC | Taq extension assay |
| Abasic correct | ACTGGTCAACTTGGCCATGTTGGCCTTGGAGGTC GAC | Taq extension assay |
Construction of plasmid DNA repair substrates
Synthetic oligodeoxynucleotides (80 pmol) containing either cholesterol (NER positive control) or 8-oxo-dG modifications (M13-cholesterol or M13-8oxo, Table 1) were phosphorylated with T4 PNK (10 units) in ligase buffer (New England Biolabs) for 30 minutes at 37°C in a total volume of 50 μL. Phosphorylated oligonucleotides were then annealed to single-stranded M13 (8 pmol), extended with Taq polymerase (100 units), Taq reaction buffer (New England Biolabs), 100 mM ATP (Teknova), 10 mM dNTPs (Thermo Scientific), restriction enzyme buffer 2 (New England Biolabs), and 8 μg bovine serum albumin for 15 minutes at 75°C in 260 μL. The temperature was then lowered to 37°C at which point T4 polymerase (60 units) and T4 ligase (8000 units) were added and the reaction incubated at 37°C overnight in 375 μL (Figure 2). Extension products were resolved on a 0.8% low melting temperature agarose gel. Supercoiled DNA was excised with a razor blade, digested with β-agarase (10 units) according to manufacturer's protocol, purified by phenol: chloroform extraction and ethanol precipitation, and resuspended in water. DNA containing an 8-oxo-dG residue (0.36 pmol) was crosslinked to OGG1 (36 pmol) in buffer containing 100 mM NaCl, 1 mM MgCl2, 20 mM Tris-HCl pH 7.0, and 10 mM sodium cyanoborohydride at 37°C for 30 minutes (43,44). During this reaction, the 249K active residue attacks the C1 position of the 8-oxo-dG lesion, expelling the base to create an abasic site. Addition of sodium cyanoborohydride reduces the intermediate Schiff base formed, trapping the protein to the ribose (Figure 4). To study of the effect of protein size on DPC repair efficiency, DPC substrates created as described above were exposed to 0.5 μg of trypsin (Promega) at 37°C in 50 mM Tris-HCl pH 8 overnight prior to transfection. Molecular weight of the digestion product was confirmed by crosslinking 500 pmol (1 μL) of OGG1 protein to 500 pmol of duplex oligo containing an 8-oxo-dG modification using the same conditions stated above. This material was subsequently treated with 5 μg trypsin (10 μL) in 50 mM Tris-HCl pH 8 buffer (20 μL) overnight at 37°C, followed by addition of 10 U DNASE I (5 μL) in DNASE I buffer (15μL) at 37°C for 30 minutes. This material was subsequently resolved on a 16% Tris-Tricine Gel (Bio-Rad) and stained with simply blue (Invitrogen).
Figure 2. An AP site blocks Taq-mediated primer extension.
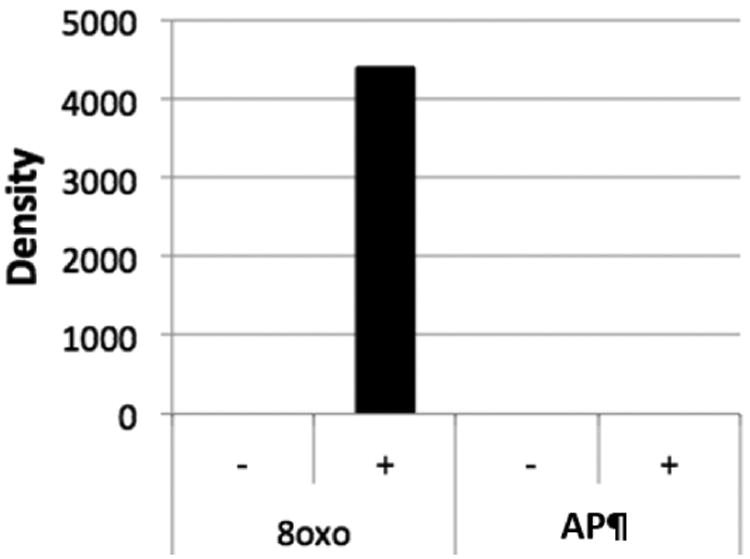
Taq polymerase extension reactions were performed as described in the text on 40-mer oligonucleotides containing either an 8-oxo dG residue (8oxo) or an AP site (AP), see methods section for details, in the absence (-) or presence (+) of complementary primer Z (15-mer, see methods). The products were resolved by electrophoresis, stained with ethidium bromide and subject to scanning densitometry. The relative amount of full-length product (40 nt) was quantitated using image J and plotted on the y axis (arbitrary units).
Figure 4. Cholesterol-DNA adducts are repaired with reduced efficiency in NER-deficient cells.
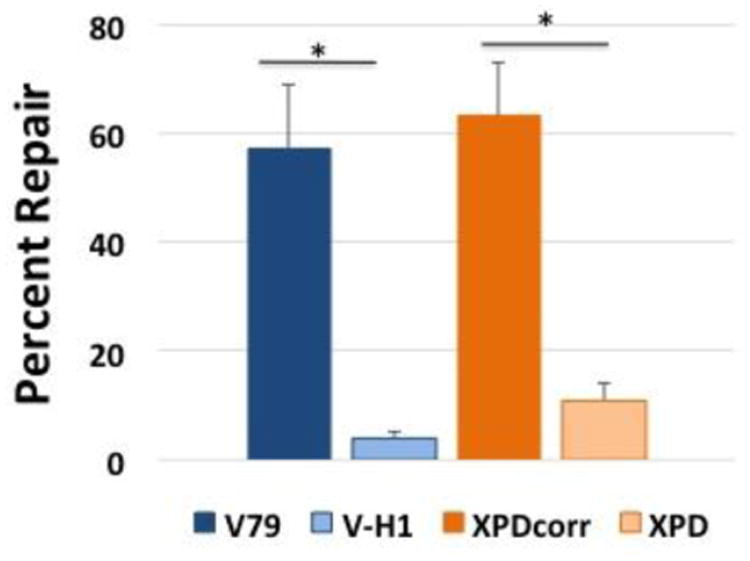
Plasmid DNA containing a cholesterol adduct was transfected into Chinese hamster (blue) and human (orange) cells lines proficient (dark bars) or deficient (light bars) in NER via lipofection and low molecular weight DNA recovered after 8 hours. DNA repair assays were performed as described above. Values depict mean percent repair, ± SEM, N=4, *P < 0.05.
KCl/SDS precipitation of DNA-protein crosslinks
To confirm the presence of the OGG1 protein crosslink, lesion-containing M13 plasmids (prepared as described above) were digested with BspDI for 1 hour at 37°C to generate two restriction fragments, one containing the DPC (4.4 kb) and a second, protein-free fragment (2.8 kb). The digest was treated with SDS at a final concentration of 0.5% and incubated at 65°C for 10 minutes to facilitate protein binding. The samples were then divided in two, and KCl (final concentration 100 mM) was added to one sample and omitted from the other. The reactions were incubated on ice for 5 minutes followed by centrifugation at 12K × G for 5 minutes at 4°C (45). The supernatant from both samples was resolved by agarose gel electrophoresis and stained with ethidium bromide. Crosslinking efficiency was determined by scanning densitometry using ImageJ to determine the amount of plasmid DNA recovered from solution following KCl/SDS precipitation, as depicted in Figure 5.
Figure 5. Creation of an OGG1-plasmid DNA crosslink substrate.
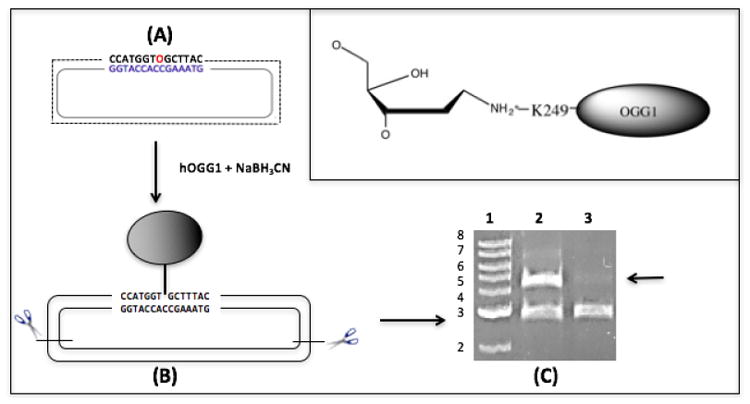
(A) Plasmid DNA containing an 8-oxo-dG residue (O, in red) was reacted with OGG1 in the presence of sodium cyanoborohydride to create a covalent bond between lysine residue 249 and a deoxyribosyl moiety on the plasmid (see Methods for details, chemical structure depicted in inset). (B) This product was cut with restriction enzymes to generate two fragments: a 2800 bp protein-free DNA fragment and a 4400bp fragment crosslinked to OGG1. (C) The restriction digested material was resolved by agarose gel electrophoresis prior to (lane 2) or following (lane 3) precipitation in the presence of K-SDS (see Methods), and stained with ethidium bromide. The arrow illustrates the selective loss of the OGG1-crosslinked DNA fragment following K-SDS precipitation. Lane 1; molecular weight marker in kilobase pairs.
Cloning of a transcriptional unit into M13mp18
Two pg of plasmid psF-CMV-RSV-Zeo Asc1 (Sigma) was amplified with RSV-ZEO 1 and 2 primers (1 μM, Table 1) using Pfu Master Mix (G-Biosciences). PCR conditions were as follows: pre-treatment at 94°C for 3 minutes followed by 25 cycles at 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute. The 925 bp PCR product containing the RSV promoter and zeocin gene (RSV-Zeo) was cloned using a Topo 2.1 PCR Kit (Thermo) and transformed into Top 10 F′ bacteria (Thermo). Clones harboring the PCR product were isolated and the insert recovered by digestion with EcoRI followed by gel purification and ligation into pLC118. (The plasmid pLC118 was created by cloning the neomycin phosphotransferase gene from pSV2neo (46) into a EcoRI-linearized M13mp18. One microliter of the ligation mixture was transformed into NEB Turbo electrocompetent bacteria (New England Biolabs), and individual clones harboring recombinant M13 molecules containing the RSV-Zeo insert were screened by restriction digestion analysis of double-stranded virion DNA to determine orientation. Single-stranded virion DNA was subsequently recovered from cultures harboring the insert in the two different orientations (referred to, respectively, as pLC119 and pLC120) and primer extension reactions performed as described above. M13-RSV-Zeo-8oxo template or M13-RSV-Zeo-8oxo coding primers (Table 1) were used to generate double-stranded plasmid substrates in which the 8-oxo-dG residue is present on, respectively, the template or coding strands of the RSV-Zeo transcriptional unit (Figure S1). Transcription of these plasmids following transfection into wild-type V79 cells was confirmed via quantitative reverse transcriptase PCR using SuperScript VILO (Invitrogen) according to manufacturer's instructions and primers Zeo F1 and Zeo R1 (Table 1) for amplification (47).
Purification of single-stranded DNA
Approximately 10 ng of double-stranded pLC119 or pLC120 was electroporated into NEB Turbo bacteria according to manufacturer instructions and shaken in 1 mL Luria-Bertani broth (LB) for 1 hour at 37°C. Either 50 or 100 μL of culture broth was added to 100 μL of an overnight culture of non-transfected bacteria and 3 mL of top agar containing 5 mM MgCl2. This solution was mixed, poured onto an LB plate, and incubated at 37°C overnight. Plaques formed were picked using a Pasteur pipette and incubated in 1 mL of LB for 3 hours at room temperature followed by 4°C overnight. The next day, 100 μL of an overnight culture of NEB Turbo bacteria was diluted into 5 mL of LB containing 5 mM MgCl2 and shaken for 2 hours at 37°C. The 5 mL culture was then diluted into 45 mL of LB containing 5 mM MgCl2 and divided into 1 mL aliquots. Next, 100 μL of diluted plaques were added to each aliquot and shaken at 37°C for 5 hours (48). Cultures were then spun at 12K × G in a table-top centrifuge for 5 minutes at room temperature (49). The supernatants were collected and single-stranded DNA purified using a QiaPrep Spin M13 kit (Qiagen).
Transfection
Mammalian cells were plated at 0.5 × 106 cells/well in a 6-well plate the night prior to transfection. For a one well transfection, 15 μL (1.2 pmol) of purified, DNA repair substrates prepared as described above was incubated with 6 μL of lipofectamine (Invitrogen) and 300 μL of serum-free culture media for 5 minutes at room temperature. (Note that 1 μL of the DNA substrate is reserved as non-transfection (i.e. 0 hour time point) controls. This material is diluted in 500 μL of water and subsequently included in the SSPE-qPCR assay described below.) 250 μL of DNA complexes were added to the well and allowed to recover for either 1.5, 3, 5, 7, or 8 hours. For multiple well transfections, the above conditions were doubled or tripled as needed. Following incubation, cell culture media was removed and 1 mL of 0.6% SDS/0.01 M EDTA was added to each well for 10-15 minutes at room temperature. Lysed cells were scraped with a rubber policeman and transferred to a 1.5 mL centrifuge tube. Sodium chloride was added to a final concentration of 1 M, inverted gently 5 times, and incubated at 4°C overnight (50). The next day, samples were spun at full speed in a table top centrifuge at 4°C for 30 minutes. The supernatant was ethanol precipitated, resuspended in 100 μL water and stored at -20°C until SSPE-qPCR.
Strand-specific primer extension-quantitative polymerase chain reaction (SSPE-qPCR) repair assay
Low molecular weight DNA recovered from transfected cells (1 μL) as well as non-transfected DNA samples (0 hour controls) were mixed with a PCR primer (1μM) complementary to the damaged strand of the plasmid (M13 Primer R, Table 1, Figure 1) in SYBR green master mix buffer (Invitrogen). Following a 10-minute melting step at 90°C, the DNA was melted at 90°C for 15 seconds and then reduced to 65°C for 1 minute to permit primer annealing/extension. This step was repeated an additional seven times, at which point a primer complementary to the undamaged strand of the plasmid (M13 Primer L, Table 1, Figure 1) was added (1μM). Cycle threshold (Ct) values were then measured via quantitative real-time PCR on an Applied Biosystems StepOnePlus Real Time PCR System using the reaction conditions described above. In parallel, a qPCR experiment analogous to that above was performed in which the eight rounds of primer extension were omitted. The Ct values from the latter experiment were subtracted from that of the former to calculate the delta Ct value ΔCt = [Ct] – [Ct primer extension]. Because the presence of an abasic site, cholesterol, or DPC lesion blocks primer extension, this delta Ct value can be used to calculate the percent of lesion repair using the formula: percent DNA repair = (2ΔCt/23) × 100 (47,51). The percent repair calculated at each time point using this method is then subtracted from the ‘apparent percent repair value’ calculated using the 0 hour time point (i.e. non-transfected DNA repair substrate sample) to correct for PCR background noise induced by the strand-specific primer extension.
Figure 1. Quantification of damaged plasmid via strand-specific primer extension/qPCR (SSPE-qPCR).
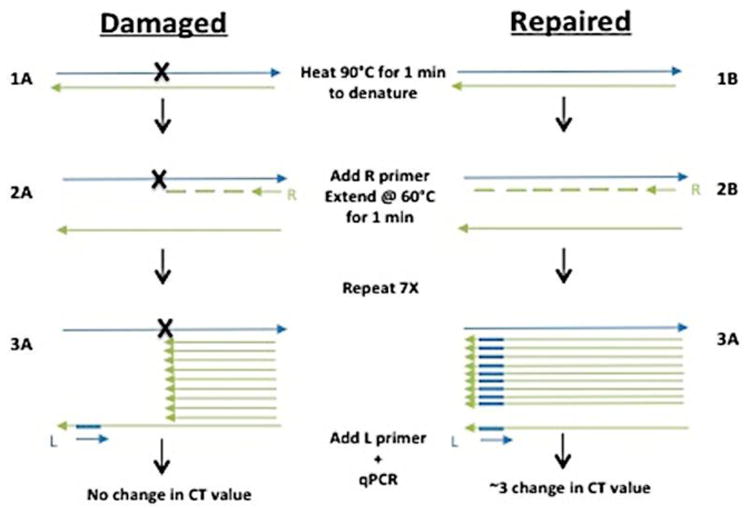
Repair of damaged plasmids is quantified using SSPE-qPCR: (1) Plasmid DNA is denatured, (2) a primer extension reaction is performed using primer R, (3) the denaturation/primer extension step is repeated and additional 7 cycles. After 8 cycles, primer L is added and Ct values determined using qPCR. The presence of an abasic site, cholesterol, or DPC (A) will cause Taq polymerase to stall, resulting in no full-length product strands. In contrast, when repair has occurred (B), Taq polymerase will produce full-length product strands containing the binding site for primer L.
Measurement of base excision repair (BER) activity in wild-type and NER-deficient cell lines
A plasmid containing an 8-oxo-dG modification was transfected into Chinese hamster V79 and V-H1 cell lines and low molecular weight DNA recovered at 1.5 and 3 hours post-transfection as described above. This material was then treated with 80 pmol of OGG1 enzyme for 30 minutes at 37°C. Under these conditions, the OGG1 enzyme quantitatively excises any unrepaired 8-oxo-dG residues from the plasmid, thereby creating an abasic site that blocks primer extension by Taq polymerase. It is thus possible to use the strand-specific primer extension-qPCR assay described above to calculate the respective levels of BER activity present in wild-type and NER-deficient cell lines.
Measurement of cisplatin-induced DNA-protein crosslinks in Chinese hamster cell lines
Wild-type V79 and NER-deficient V-H1 Chinese hamster cells were grown to confluence on 10 cm dishes (2 per cell line). One dish each per cell line was maintained in serum-free media plus or minus 100 μM cisplatin for one hour at 37°C. Cells were then washed twice with PBS and re-suspended at a density of 1×107 cells/mL (45). Protein-linked DNA was isolated using 100 μL of cells lysed with 0.5 mL 2% SDS, 1mM PMSF, and 20 mM Tris-HCl pH 7.5 and frozen at -80°C. Cells were then thawed at room temperature, vortexed for 10 seconds, and incubated at 65°C for 10 minutes. Next, 0.5 mL of 200 mM KCl in 20 mM Tris-HCl pH 7.5 was added and the material passed through a 1 mL pipet tip five times. Samples were then put on ice for 5 minutes, spun at 3K × G for 5 minutes at 4°C, and the pellet resuspended in 1 mL of 100 mM KCl in 20 mM Tris-HCl pH 7.5 by pipetting 5 times to evenly disperse recovered material. Samples were heated to 65°C for 10 minutes, chilled on ice for 5 minutes, and spun at 4°C for 5 minutes at 3K × G. Resuspension of the pellet in 100 mM KCl in 20 mM Tris-HCl, heating, chilling, and spinning were repeated twice more before resuspending the final pellet in 1 mL of 100 mM KCl, 10 mM EDTA, 20 mM Tris-HCl pH 7.5 and proteinase K (final concentration 0.2 mg/mL). Samples were incubated at 50°C for 3 hours after which 100 μg of bovine serum albumin was added, chilled on ice, and spun at 10K × G for 10 minutes at 4°C. The supernatant was collected and mixed with 1 mL of freshly prepared 200 ng/mL Hoechst dye 33258 reagent (ChemCruz) in 20 mM Tris-HCl pH 7.5, vortexed, and incubated for 30 minutes in the dark at room temperature. Total DNA was isolated by lysing 100 μL of cells in 1 mL of 2M NaCl in 20 mM Tris-HCl pH 7.5 and mixed with the fluorescent agent as stated above. Fluorometer measurements of total DNA and protein-linked DNA were obtained using a Fluoromax 4 Spectrofluorometer (Horiba) at room temperature with 360 excitation and 450 emission filters. Protein-linked DNA values were divided by total DNA to obtain percent of protein-crosslinked DNA.
3. Results
3.1 Taq polymerase extension is blocked by abasic sites
The SSPE-qPCR assay utilized in this study is dependent on the ability of the lesions analyzed to permanently stall Taq polymerase extension. To confirm this, we performed two parallel primer extension reactions, one using a 40-mer oligonucleotide (M13-RSV-Zeo-8oxo (template), see Table 1) containing an 8-oxoguanine residue at position 19, and the other harboring an AP site created by OGG1 at this same position (see Methods for details). It has been reported that Taq polymerase is able to efficiently read-through an 8-oxoguanine residue but is blocked by abasic sites (52). We thus anticipated that a primer annealed to the 8-oxoguanine-containing strand would be extended by Taq polymerase, producing a 40-nucleotide product, while a primer annealed to the AP-containing oligonucleotide would not be extended to produce a full-length product. The densitometry results presented in Figure 2 (calculated using ImageJ) confirm this prediction, showing the presence of a full-length primer extension product when the 8-oxoguanine lesion-containing substrate (8oxo) was extended with Taq polymerase in the presence of a complementary primer. In contrast, no product was produced when the AP-containing substrate (AP, Figure 2) was extended with Taq polymerase and a complementary primer. We also observed a ∼ 21 nucleotide product (data not shown), as would be expected were the Taq polymerase stalled at the abasic site. It is noteworthy that creation of an AP site using OGG1 can also generate a single-strand break via beta-elimination by OGG1 (43). To confirm that Taq polymerase is blocked at abasic sites in the absence of single-strand breaks, the above experiment was repeated using an oligonucleotide ordered with an abasic modification (abasic, Table 1) or no lesion (abasic corrected, Table 1) in the presence or absence of a complementary primer (abasic complement, Table 1). These experiments confirmed an increase in full-length product during extension of an unmodified oligonucleotide but not with an abasic-containing substrate. Additionally, an 18 nucleotide product corresponding to the site of the abasic lesion was observed during extension of the damaged substrate, indicating halting of the Taq polymerase at the lesion, but not in the undamaged oligonucleotide extension (S2).
3.2 Repair of 8-oxo-dG in wild-type and NER-deficient cells
While our ultimate objective was to use the SSPE-qPCR assay to compare the relative efficiency with which NER proficient and NER-deficient clones repaired a DPC-containing plasmid substrate, we first performed two series of experiments to confirm: 1. That NER proficient and NER deficient clones would repair plasmids harboring a base excision repair substrate with equivalent efficiencies, and 2. That a plasmid harboring a lesion known to be selectively repaired by the NER pathway would be significantly more efficiently repaired in NER proficient cells than in isogenic clones deficient in NER.
As a first step, we transfected covalently closed circular plasmids harboring a site-specific 8-oxo-dG modification (see methods for details of construction and purification) into wild-type (V79) and NER-deficient (V-H1) Chinese hamster cell lines and, following incubation periods of 1, 1.5 or 3 hours, purified low molecular weight DNA from the cells. This material, as well as non-transfected DNA was treated with OGG1, was analyzed using SSPE-qPCR, as described in the Methods section. As illustrated in Figure 3, both wild-type and NER-deficient cells showed rapid and efficient repair of 8-oxo-dG lesions indicating that there is no appreciable difference in the relative BER activity in V79 and V-H1 clones. (It is critical to note that, as described in the Methods section, the percent repair calculated at 1, 1.5 and 3 hours post-transfection was corrected to subtract ‘apparent repair’ observed in the ‘0 hour’, i.e. no-transfection control.) This result is consistent with the interpretation that equivalent amounts of transfected DNA gain access to the cellular DNA repair machinery in the two cell lines. We next pursued a second series of experiments to test the hypothesis that NER-deficient clones would repair a plasmid substrate harboring a known NER substrate with reduced efficiently than would their isogenic wild-type counterparts.
Figure 3. 8-oxo-dG repair efficiency is similar in wild-type and NER-deficient Chinese hamster clones.
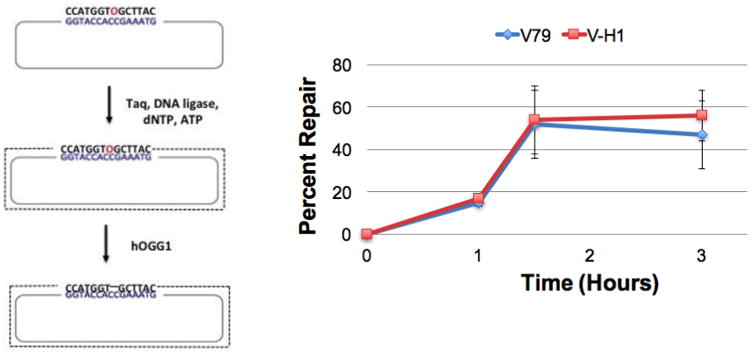
An oligonucleotide containing an 8-oxo-dG modification (O, in red) is annealed to single-stranded M13 and a primer extension reaction performed as described in the Methods. Covalently closed circular double-stranded DNA is gel-purified, and transfected into wild-type V79 (blue) and NER-deficient V-H1 (red) Chinese hamster cells via lipofection. Low molecular weight DNA was recovered 1, 1.5 and 3-hours post-transfection and treated with OGG1 to convert unrepaired 8-oxo-dG residues to abasic sites. DNA repair assays were then performed as described in the legend to Figure 1. Values depict mean percent repair, ± SEM, N=3.
3.3 Repair of cholesterol adducts in wild-type and NER-deficient cells
A plasmid containing a site-specific cholesterol-ribose lesion was prepared and purified as described in the methods. It has previously been shown that this lesion is subject to NER (53). This substrate was transfected into V79 (NER proficient) and V-H1 (NER deficient) cells. Low molecular weight DNA was recovered eight-hours post-transfection and subjected to the SSPE-qPCR assay to calculate DNA repair activity as described above. As depicted in Figure 4, the efficiency of repair of the cholesterol-containing plasmids in NER-deficient V-H1 cells was nearly 15-fold lower (4% vs 57%) than that detected in the NER proficient V79 cells. To confirm that this difference was indeed due to a defect in NER, an identical experiment was performed in human cell lines deficient (XPD) or proficient (XPDcorr) in NER. Results from this experiment indicated that repair of the cholesterol-containing plasmid in wild-type human cells was 6-fold more efficient than that detected in the isogenic NER-deficient clone (63% vs 11%, Figure 4). (As in the experiment above, repair efficiency values have been corrected by subtracting the background seen in the 0 hour, no transfection control sample.) These results confirmed the expectation that NER-deficient clones would show significantly decreased repair of a cholesterol moiety crosslinked to plasmid DNA, and that our SSPE-qPCR assay is capable of detecting differences in the repair of adducts processed by the NER pathway.
3.4 Crosslinking OGG1 to 8-oxo-dG-containing plasmid DNA substrates
As was outlined in the Introduction, proteins become crosslinked to DNA via multiple mechanisms. To address specific questions regarding DPC repair on plasmid DNA, an enzymatic reaction was chosen to crosslink the repair protein OGG1 to a deoxyribosyl residue. This biologically relevant lesion represents the main source of endogenous DPCs of apurinic/apyrimidinic (AP) site within nucleosome core particles (54). To quantify the crosslinking efficiency of these substrates, plasmids containing a site-specific OGG1-DNA crosslink were subjected to KCl/SDS precipitation as described in the Methods section. As the results in Figure 5 illustrate, the 4.4 kb fragment was quantitatively removed from the sample in which KCl was added while there is no evident reduction in the amount of the 2.8 kb fragment in this sample, compared to the untreated control. This result confirms that the crosslinking reaction is highly efficient, with nearly 100% of the plasmid molecules containing a site-specific DNA-protein crosslink.
3.5 Repair of OGG1 crosslinked to plasmid DNA in Chinese hamster and human clones
To determine whether the M13 plasmid containing a covalent OGG1 crosslink is subject to repair, DPC-containing plasmids were transfected into wild-type V79 Chinese hamster cells. Low molecular weight DNA was recovered 3, 5, 7 and 8-hours post-transfection as described in Methods. SSPE-qPCR assays on this material as well as on a 0 hour, no transfection control, sample was used to calculate percent DNA repair as outlined in the Methods section. These results revealed essentially linear repair kinetics reaching 28% repair at the final time point (Figure 6, left panel), indicating that V79 cells are capable of repairing OGG1 crosslinked to a non-replicating plasmid DNA molecule. We performed similar experiments in a second Chinese hamster-derived cell line (CHO-K1), as well as in two human wild-type cell lines. In each instance, these clones were capable of repairing the plasmid DPC lesion (Figure 6, right panel). Interestingly, DPC repair efficiency in each of the hamster clones was in the range of ∼20-25%, whereas the corresponding repair efficiency in two human cell lines derived from unrelated donors was significantly greater (>70%). While there was no significant difference in the repair levels between species, the level of DPC repair detected in human cells was significantly greater than that detected in the Chinese hamster-derived clones. Interestingly, despite this species-specific difference in DPC repair activity there was no significant difference in the efficiency with which human and hamster cells repaired the cholesterol lesion (63% vs 57%, Figure 4).
Figure 6. OGG1-DNA crosslink repair in wild-type cells.
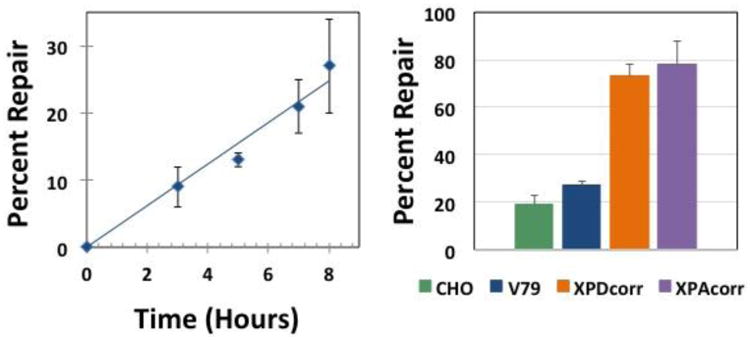
Left: Plasmid crosslinked to OGG1 was transfected into V79 cells via lipofection, low molecular weight DNA recovered 3, 5, 7, and 8 hours post-transfection, and repair assays performed as described above. The graph depicts mean percent repair, ± SEM, N=3. Right: Plasmid DNA containing an OGG1 crosslink was transfected into: Chinese hamster ovary (CHO-K1, green), Chinese hamster lung fibroblast (V79, blue), and human (XPDcorr, orange and XPAcorr, purple) cells, low molecular weight DNA recovered 8 hours post-transfection and repair assays performed as described above. Values depict mean percent repair, ± SEM, N=4.
3.6 Reduced DPC repair in Chinese hamster and human clones deficient in NER
To test the hypothesis that DPCs are repaired by the NER machinery, we transfected the DPC-containing plasmid into isogenic NER-deficient and NER-proficient Chinese hamster and human clones, and used the methodology outlined above to quantitatively measure cellular repair activity. Consistent with our prediction, DPC repair efficiency was dramatically lower in NER-deficient cells compared to their isogenic NER-proficient counterparts (Table 2). Chinese hamster V-H1 cells (XPD deficient) repaired the DPC 9-fold less efficiently than did their isogenic, wild-type counterpart V79 (3% vs 27%). Similarly, XPD deficient human cells repaired the DPC nearly four-fold less efficiently than did their gene-corrected counterpart (20% vs 73%). We extended this analysis to look at the effect of inactivation of another NER gene. As Table 2 illustrates, inactivation of the human XPA gene was associated with a five-fold reduction in DPC repair efficiency (16% vs 78%). In all instances the differences in DPC repair activity observed between NER-deficient and NER-proficient clones were statistically significant. These results indicate that in mammalian cells, the NER pathway is capable of rapidly and efficiently repairing plasmid DPC substrates.
Table 2.
OGG1-DNA crosslink repair in NER-deficient cells.
| Percent Repair* (mutated) | Percent Repair* (control) | Fold Reduction (vs control) |
|---|---|---|
| V-H1 (3 ± 1) | V79 (27 ± 2) | 9 |
| XPD (20 ± 7) | XPD Corr (73 ± 5) | 3.7 |
| XPA (16 ± 5) | XPA Corr (78 ± 10) | 4.9 |
Values depict mean percent repair, ± SEM, N=3.
3.7 Transcription-coupled repair of OGG1 crosslinked to a plasmid in Chinese hamster cells
The NER pathway is comprised of two distinct sub-pathways, respectively referred to as the transcription-coupled (TC) and global genome (GG) pathways (55). The DPC substrate studied in the experiments presented above was localized within a portion of the plasmid that was transcriptionally silent. Consequently, we conclude that the repair events we detected were catalyzed by the global genome NER pathway. To determine whether TC-NER is also capable of repairing DPCs, molecular cloning was employed to generate plasmids harboring an 8-oxo-dG-residue localized within a minigene encoding the bacterial zeocin-resistance gene driven by the RSV promoter. Because of the way these clones (pLC119/pLC120) were generated, the 8-oxo-dG residues were present, respectively, on the coding, or template strands of the DNA (Details of the creation of these clones, as well as how the activity of the RSV/zeocin transcription unit in Chinese hamster cells is described in the Methods section). We subsequently crosslinked recombinant OGG1 protein to the 8-oxo-dG residues in the pLC119 and pLC120 plasmids via the cyanoborohydride-trapping technique outlined above to generate plasmid substrates harboring a DPC with either the template or coding strand of the RSV/zeocin transcriptional unit. To confirm that the crosslinking efficiency of OGG1 was similar in both plasmids, KCl/SDS precipitation was performed on DPCs crosslinked to the template or coding strand. The soluble material remaining was resolved by agarose gel electrophoresis (see Methods for details), and scanning densitometry performed using ImageJ software. This analysis revealed that the crosslinking efficiency on the two clones was 94% and 95%, respectively (data not shown). The two plasmid DPC substrates were subsequently transfected into wild-type (V79) and NER-deficient (V-H1) Chinese hamster cell lines and their respective repair efficiencies determined using the strategy outlined above. Based on results from experiments investigating the repair of actively transcribing loci within mammalian cells exposed to UV radiation, we predicted that DPC lesions present on the template strand would be repaired more efficiently that would lesions present on the coding strand (56). The results depicted in Figure 7 are consistent with this expectation, showing that a DPC present on the template strand was repaired approximately 2.5-fold more efficiently than was an identical DPC lesion present on the coding strand. The data further illustrate that for both lesions, repair occurs significantly less efficiently in the NER-deficient V-H1 background than in its wild-type counterpart.
Figure 7. OGG1-DNA crosslinks are repaired more efficiently when present on a template strand than when present on a coding strand.
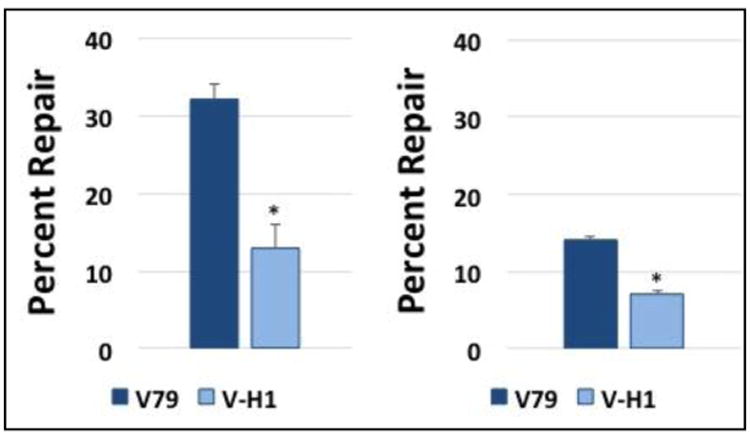
Plasmid pLC119/pLC120 DNA substrates containing an OGG1 crosslink present on the template strand (left), or coding strand (right) of a transcriptional unit (construction described in Methods) were transfected into wild-type (V79, dark bars) and NER-deficient (V-H1, light bars) cells, and repair assays performed as described above. The graph depicts mean percent repair, ± SEM, N=3, *P < 0.05.
3.8 A 4 kDa DNA-peptide crosslink is repaired more efficiency than a 38 kDa DPC
Results from our lab and others confirm that several agents are able to crosslink proteins of dramatically different sizes to DNA, both in vitro as well as in intact cells (57-59). It has been suggested that the size of the protein crosslink may influence the pathway through which a DPC lesion is repaired (16). We thus sought to examine whether a substantially smaller DPC substrate would also be subject to NER-mediated removal. To achieve this objective, OGG1-crosslinked plasmid DNA substrates were exhaustively digested with trypsin (see Methods) to generate a substrate in which a 39-amino acid residual (∼4.9 kDa) peptide fragment remained attached to the DNA (S3). This peptide-DNA crosslink substrate was transfected into Chinese hamster V79 and V-H1 cells and repair efficiency analyzed at three and eight hours post-transfection using the SSPE-qPCR assay, as outlined above. The results in Figure 8 indicate that repair of this DNA peptide substrate was significantly more efficient in the NER-proficient V79 cells than in their isogenic NER-deficient V-H1 counterparts. Interestingly, the DNA peptide (39 amino acid residue) crosslink was repaired approximately twice as efficiently as was the full length OGG1 DNA-protein (345 amino acid residue) crosslink (43% Figure 8 vs 27% Figure 6).
Figure 8. A DNA-peptide crosslink is repaired with reduced efficiency in NER-deficient Chinese hamster cells.
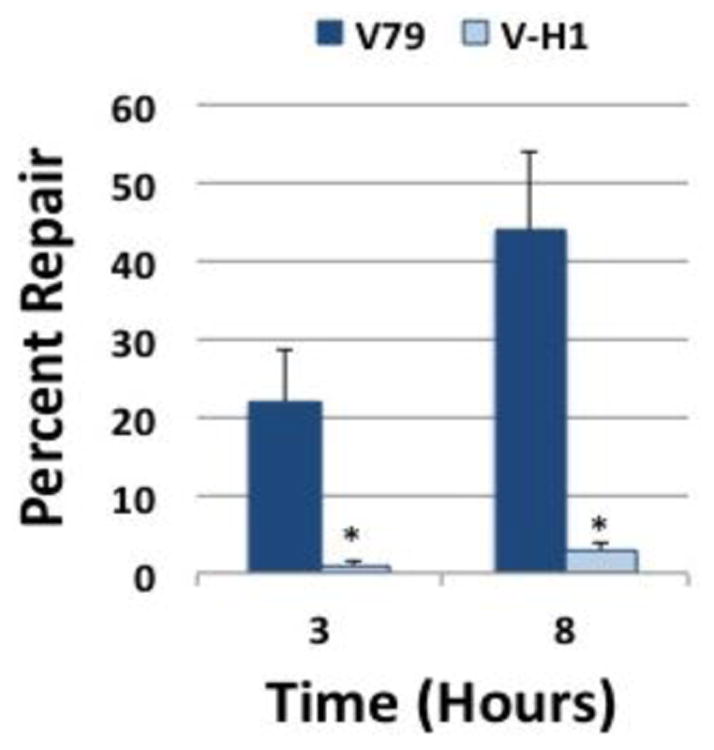
Plasmid DNA containing a DNA-peptide crosslink (prepared as described in the Methods) was transfected into wild-type (V79, dark bars) or NER deficient (V-H1, light bars) Chinese hamster cells via lipofection, low molecular weight DNA recovered at 3 and 8 hours post-transfection, and repair assays performed as described above. The graph depicts mean percent repair, ± SEM, N=4, *P < 0.05.
3.9 Repair of cisplatin-induced chromosomal DPCs in wild-type and NER-deficient Chinese hamster cells
The results presented above convincingly demonstrate that both the global genome and transcription-coupled NER pathways are able to efficiently repair a DPC comprised of the OGG1 protein (or a proteolytic fragment derived therefrom) covalently trapped onto a deoxyribosyl residue of a plasmid DNA molecule. However, it is conceivable that similar substrates crosslinked to genomic DNA may be subject to different DNA repair pathways, or that the nature of the chemical bond linking protein to DNA can influence the mechanism(s) through which DPCs are repaired. To confirm that results obtained using the SSPE-qPCR assay generally reflect the ability of the NER machinery to repair DPCs present on chromosomal DNA, we quantitated the level of accumulated DNA-protein crosslinks present in wild-type and NER-deficient cells following exposure to cisplatin. We hypothesized that if NER plays a significant role in repair of chromosomal cisplatin-induced lesions, levels of DPCs present following a one-hour exposure to 100 μM cisplatin would be elevated in V-H1 cells, compared to V79 cells. The results in Figure 9 illustrate an approximate 3.6-fold higher level of DPCs in cisplatin-treated V-H1 cells compared to their wild-type, isogenic counterpart, consistent with the interpretation that the NER pathway plays a significant role in the repair of drug-induced chromosomal DPCs.
Figure 9. Cisplatin-induced DNA-protein crosslinks accumulate at elevated levels in NER-deficient Chinese hamster cells.
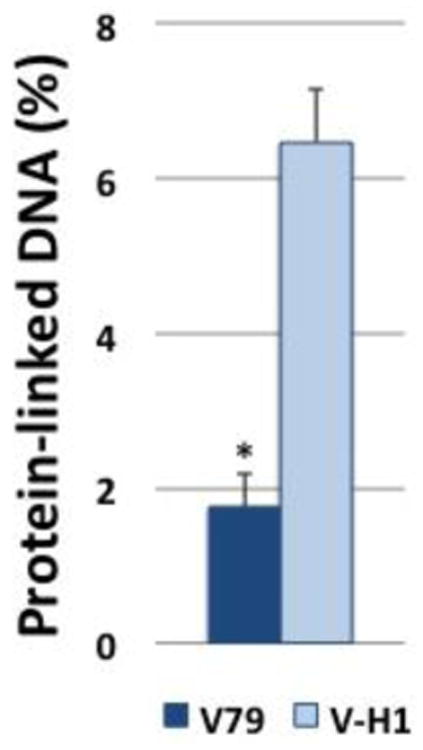
Wild type (V79, dark bar) and NER-deficient (V-H1, light bar) Chinese hamster cells were exposed to 100 μM cisplatin for one hour in serum-free media, and levels of chromosomal DPCs determined. The graph depicts mean percent of protein-crosslinked DNA (calculated as described in the Methods), ± SEM, N=3, *P < 0.05.
4. Discussion
In this report, we describe the development of a DNA repair assay we named strand-specific primer extension-quantitative PCR (SSPE-qPCR) and illustrate its utility in studying the repair of lesioned plasmids transfected into wild-type and NER-deficient human and Chinese hamster-derived cell lines. The versatility of this assay permits one to generate multiple distinct DNA repair substrates and subsequently measure the respective efficiencies with which they are repaired in different genetic backgrounds. In this way, we were able to show that both human and Chinese hamster-derived clones are able to efficiently repair episomal plasmids harboring a DPC, and that in both species this process was primarily dependent on the NER machinery. We presented evidence that both the GG and TC sub-pathways of NER can contribute to mammalian DPC repair. Our results also indicated that a DNA-crosslinked 39 amino acid polypeptide was repaired significantly more efficiently than was a 345 amino acid protein crosslink. While in this instance, our primary focus was on DNA protein crosslink repair, our results illustrate that with appropriate modifications this assay would be useful in the study of any number of DNA lesions.
This study was undertaken in an attempt to resolve a controversy in the literature regarding the role of nucleotide excision repair in the removal of DNA-protein crosslinks. Baker et al. (31) detected higher levels of host cell reactivation activity of a DPC containing plasmid transfected into NER-proficient cells compared to that detected in a NER-deficient clone. This group presented additional information leading them to conclude that larger protein crosslinks were proteolytically degraded to smaller peptide crosslink lesions that were subsequently repaired in an NER-dependent manner. In contrast, Nakano et al. (30) published results supporting a model in which the mammalian NER pathway was only capable of repairing DPCs smaller than approximately 10 kDa. In an effort to resolve this apparent paradox, we developed the SSPE-qPCR assay and used it to compare the relative efficiency with which NER-deficient and NER-proficient isogenic cell line pairs repaired a synthetic DPC substrate. Critically, as part of our experimental design, we also compared the relative efficiency with which these same cell line pairs repaired plasmid substrate harboring an 8-oxo-dG residue or a site-specific cholesterol moiety. While the former lesion is repaired by the base excision repair machinery, the latter lesion is known to be subject to NER repair. We were thus able to use these latter repair substrates as, in effect, negative and positive controls for the SSPE-qPCR assay. Were the assay faithfully monitoring cellular DNA repair function, there should be no difference in 8-oxo-dG repair activity between the NER-deficient and NER-proficient clones, however, these latter cells should display significantly greater repair efficiency directed towards the cholesterol substrate. Results presented above confirmed these predictions. Thus, our subsequent finding that repair of a DPC substrate was significantly reduced in the NER-deficient V-H1 cells, compared to their wild-type isogenic counterpart V79 is most consistent with the interpretation that the primary mechanism of repair of this substrate is NER. While our assay cannot definitively rule out the possibility that differential kinetics of plasmid nuclear uptake or transfection efficiency influence DPC repair efficiency, these potential explanations are inconsistent with our finding that there was no statistically significant difference in the cycle threshold values obtained from experiments in which plasmid DNA harboring 8oxoG or DPC lesions were transfected into WT and NER-deficient cell lines. Additional support for the interpretation that plasmid DPC repair activity in mammalian cells is primarily due to NER was obtained when we showed that human clones deficient in the XPA or XPD genes also showed greatly diminished efficiency of repair of the DPC substrate, compared to their isogenic wild-type counterparts.
Another advantage to using the SSPE-qPCR assay to study the repair of DNA substrates, is that parameters such as sequence context can be easily manipulated to assess their influence on repair. We demonstrated this by crosslinking a DPC to either the template or coding region of a transcriptional unit and measuring differences in repair efficiency using the SSPE-qPCR assay. Results from this experiment revealed that DPCs crosslinked to the template strand were repaired significantly higher than those attached to the coding strand and that in both cases, repair was significantly decreased in NER-deficient clones. Based on these observations we conclude that the TC-NER pathway is able to repair DPCs. It is notable that the repair efficiency of the template strand-containing DPC in this sequence context (32%) was not appreciably different from the ∼28% repair efficiency observed for a substrate present in a locus that does not support transcription (see Figures 6 and 7). Thus, it appears that additional factors besides transcription can influence DPC repair efficiency. For example, it is conceivable that additional factors, such as sequence context, %GC composition, etc., could potentially influence the efficiency of DPC repair. In addition to investigating these possibilities, it will be of interest to examine the effect on DPC repair of replication by generating a DPC on a plasmid containing an origin of replication.
Although our repair data refute the hypothesis that NER is incapable of repairing lesions larger than 10 kDa, we wanted to further investigate whether a DNA-peptide crosslink (∼4 kDa) would be more efficiently repaired than a DPC (38 kDa) using the SSPE-qPCR assay. Results presented above confirmed that the smaller, DNA-peptide crosslink was repaired twice as efficiently as the OGG1 crosslink in wild-type cells and that repair of either of these lesions was also dependent on the NER pathway. This data is consistent with the hypothesis that repair of DPC lesions requires one or more processing steps, and that a larger protein substrate requires more processing than does a smaller, peptide-crosslink substrate. Several publications have shown that the yeast WSS1 and its human homolog Spartan are DNA-dependent proteases that play critical roles in DPC repair (33,35,37). Currently Spartan's role in DNA damage tolerance and DPC repair is not fully understood but its thought to be replication-coupled (60-62). It has been shown that Spartan-deficient worms are hypersensitive to DPC-inducing agents, and that patients with Ruijs-Aalfs syndrome possessing germline mutations in the Spartan gene develop early onset hepatocellular carcinoma, as do mice hemizygous for the Spartan gene (34,62,63). It is conceivable that this cancer predisposition phenotype reflects a defect in repair of endogenously produced DPCs. Although our data are consistent with the hypothesis that non-replicating plasmids are also subject to proteolytic processing, additional experiments using the SSPE-qPCR assay will be required to rigorously test this. It will also be important to determine whether, as others have proposed (Baker et al.), the proteasome is capable of processing proteins crosslinked to DNA.
It is notable that results generated using the SSPE-qPCR assay uncovered a species-specific difference in DPC repair efficiency. While the overall repair efficiency of crosslinked OGG1 in two human cell lines obtained from unrelated donors exceeded 70%, repair efficiency of this substrate in two different Chinese hamster-derived clones was significantly lower (∼20%). As the data in Figure 4 illustrates, there is essentially no difference in the efficiency with which the cholesterol lesion is repaired in human versus Chinese hamster-derived cells. Thus, the species-specific difference in DPC repair efficiency is not due, per se, to reduced levels of NER activity in hamster cells. Based on data implicating the involvement of proteolysis in DPC repair, it is tempting to speculate that human cells possess a more active protease to generate smaller adducts that can be more easily excised by the NER machinery. Currently, experiments are underway to employ the SSPE-qPCR assay to determine the precise cause of this DPC repair deficit in Chinese hamster clones and determine whether it reflects a trend of reduced DPC repair activity in the cells of other short-lived mammals.
Finally, we tested the hypothesis that chromosomal DPC lesions formed following cisplatin treatment are subject to repair via the NER machinery. We determined that NER-deficient cells harbored significantly higher levels of cisplatin-induced DPCs than did isogenic wild-type cells immediately following a one hour incubation with 100μM cisplatin. We are currently using the SSPE-qPCR assay to assess the role of additional repair pathways, such as homologous recombination as a potential mechanism through which mammalian cells repair DPCs.
5. Conclusions
The SSPE-qPCR assay detailed above demonstrates a new way to quantitatively assess the repair mechanisms involved in DPC repair on plasmid substrates. This method is easy, sensitive, and capable of directly quantifying DPC repair at time points as early as 2 hours post-transfection. Using this assay, parameters such as DPC size, chemical crosslink, and DNA sequence context can be independently assessed to gain insight into the efficiency with which the plasmid DPC is repaired. Initial studies in our lab using the SSPE-qPCR assay have demonstrated that DPC lesions on transcribing and non-transcribing plasmids are repaired by a mechanism that is dependent on the nucleotide excision repair pathway and that DNA-peptide crosslinks are repaired at a higher efficiency than larger, DNA-protein crosslinks.
Highlights.
This SSPE-qPCR assay is a new method to study mechanisms of DPC repair in cells.
This plasmid-based assay is rapid, highly quantitative, and extremely flexible.
SSPE-qPCR provides significant advantages over previously utilized approaches.
This method can also evaluate repair of other types of DNA lesions in cells.
This approach definitively established a role for NER in repair of DPCs.
Acknowledgments
We acknowledge Professor Harry Orr (University of Minnesota) for providing CHO cells, and the Natalia Tretyakova (University of Minnesota) and Ashis Basu (University of Connecticut) labs for their support and technical advice during the early and intermediate stages of this work. We also thank Professor Natalia Tretyakova for her helpful editorial comments.
Funding: This work was funded by the National Institutes of Health (ES023350). Lisa N. Chesner is supported by Training Grant 5T32HL007741. Funding for open access charge: National Institutes of Health
Footnotes
Conflict of Interest: The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tretyakova NY, Groehler A, Ji S. DNA-Protein Cross-links: Formation, Structural Identities, and Biological Outcomes. Accounts of chemical research. 2015;48:1631–1644. doi: 10.1021/acs.accounts.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaz B, Popovic M, Ramadan K. DNA-Protein Crosslink Proteolysis Repair. Trends Biochem Sci. 2017;42:483–495. doi: 10.1016/j.tibs.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Tretyakova NY, Michaelson-Richie ED, Gherezghiher TB, Kurtz J, Ming X, Wickramaratne S, Campion M, Kanugula S, Pegg AE, Campbell C. DNA-reactive protein monoepoxides induce cell death and mutagenesis in mammalian cells. Biochemistry. 2013;52:3171–3181. doi: 10.1021/bi400273m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groehler At, Villalta PW, Campbell C, Tretyakova N. Covalent DNA-Protein Cross-Linking by Phosphoramide Mustard and Nornitrogen Mustard in Human Cells. Chemical research in toxicology. 2016;29:190–202. doi: 10.1021/acs.chemrestox.5b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gherezghiher TB, Ming X, Villalta PW, Campbell C, Tretyakova NY. 1,2,3,4-Diepoxybutane-induced DNA-protein cross-linking in human fibrosarcoma (HT1080) cells. Journal of proteome research. 2013;12:2151–2164. doi: 10.1021/pr3011974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connelly JC, Leach DR. Repair of DNA covalently linked to protein. Molecular cell. 2004;13:307–316. doi: 10.1016/s1097-2765(04)00056-5. [DOI] [PubMed] [Google Scholar]
- 7.Dizdaroglu M, Gajewski E. Structure and mechanism of hydroxyl radical-induced formation of a DNA-protein cross-link involving thymine and lysine in nucleohistone. Cancer research. 1989;49:3463–3467. [PubMed] [Google Scholar]
- 8.Quinones JL, Demple B. When DNA repair goes wrong: BER-generated DNA-protein crosslinks to oxidative lesions. DNA repair. 2016;44:103–109. doi: 10.1016/j.dnarep.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quinones JL, Thapar U, Yu K, Fang Q, Sobol RW, Demple B. Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase beta in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8602–8607. doi: 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barker S, Weinfeld M, Zheng J, Li L, Murray D. Identification of mammalian proteins cross-linked to DNA by ionizing radiation. The Journal of biological chemistry. 2005;280:33826–33838. doi: 10.1074/jbc.M502477200. [DOI] [PubMed] [Google Scholar]
- 11.Barker S, Weinfeld M, Murray D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutation research. 2005;589:111–135. doi: 10.1016/j.mrrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Johnson LA, Malayappan B, Tretyakova N, Campbell C, MacMillan ML, Wagner JE, Jacobson PA. Formation of cyclophosphamide specific DNA adducts in hematological diseases. Pediatric blood & cancer. 2012;58:708–714. doi: 10.1002/pbc.23254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groehler At, Degner A, Tretyakova NY. Mass Spectrometry-Based Tools to Characterize DNA-Protein Cross-Linking by Bis-Electrophiles. Basic & clinical pharmacology & toxicology. 2016;121:63–77. doi: 10.1111/bcpt.12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ming X, Groehler At, Michaelson-Richie ED, Villalta PW, Campbell C, Tretyakova NY. Mass Spectrometry Based Proteomics Study of Cisplatin-Induced DNA-Protein Cross-Linking in Human Fibrosarcoma (HT1080) Cells. Chemical research in toxicology. 2017;30:980–995. doi: 10.1021/acs.chemrestox.6b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chvalova K, Brabec V, Kasparkova J. Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic acids research. 2007;35:1812–1821. doi: 10.1093/nar/gkm032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakano T, Morishita S, Katafuchi A, Matsubara M, Horikawa Y, Terato H, Salem AM, Izumi S, Pack SP, Makino K, Ide H. Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-protein crosslinks. Molecular cell. 2007;28:147–158. doi: 10.1016/j.molcel.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 17.Nishioka H. Lethal and mutagenic action of formaldehyde in Hcr + and Hcr - strains of Escherichia coli. Mutation research. 1973;17:261–265. doi: 10.1016/0027-5107(73)90175-9. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, Morita T, Kawazoe Y. Mutagenic characteristics of formaldehyde on bacterial systems. Mutation research. 1985;156:153–161. doi: 10.1016/0165-1218(85)90058-8. [DOI] [PubMed] [Google Scholar]
- 19.Lal D, Som S, Friedman S. Survival and mutagenic effects of 5-azacytidine in Escherichia coli. Mutation research. 1988;193:229–236. doi: 10.1016/0167-8817(88)90033-8. [DOI] [PubMed] [Google Scholar]
- 20.Bhagwat AS, Roberts RJ. Genetic analysis of the 5-azacytidine sensitivity of Escherichia coli K-12. Journal of bacteriology. 1987;169:1537–1546. doi: 10.1128/jb.169.4.1537-1546.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chanet R, Izard C, Moustacchi E. Genetic effects of formaldehyde in yeast. II. Influence of ploidly and of mutations affecting radiosensitivity on its lethal effect. Mutation research. 1976;35:29–38. doi: 10.1016/0027-5107(76)90166-4. [DOI] [PubMed] [Google Scholar]
- 22.de Graaf B, Clore A, McCullough AK. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA repair. 2009;8:1207–1214. doi: 10.1016/j.dnarep.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumari A, Lim YX, Newell AH, Olson SB, McCullough AK. Formaldehyde-induced genome instability is suppressed by an XPF-dependent pathway. DNA repair. 2012;11:236–246. doi: 10.1016/j.dnarep.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Speit G, Schutz P, Merk O. Induction and repair of formaldehyde-induced DNA-protein crosslinks in repair-deficient human cell lines. Mutagenesis. 2000;15:85–90. doi: 10.1093/mutage/15.1.85. [DOI] [PubMed] [Google Scholar]
- 25.Ren X, Ji Z, McHale CM, Yuh J, Bersonda J, Tang M, Smith MT, Zhang L. The impact of FANCD2 deficiency on formaldehyde-induced toxicity in human lymphoblastoid cell lines. Archives of toxicology. 2013;87:189–196. doi: 10.1007/s00204-012-0911-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quievryn G, Zhitkovich A. Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis. 2000;21:1573–1580. [PubMed] [Google Scholar]
- 27.Minko IG, Kurtz AJ, Croteau DL, Van Houten B, Harris TM, Lloyd RS. Initiation of Repair of DNA-Polypeptide Cross-Links by the UvrABC Nuclease. Biochemistry. 2005;44:3000–3009. doi: 10.1021/bi0478805. [DOI] [PubMed] [Google Scholar]
- 28.Minko IG, Zou Y Fau - Lloyd RS, Lloyd RS. Incision of DNA-protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1905–1909. doi: 10.1073/pnas.042700399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reardon JT, Sancar A. Repair of DNA-polypeptide crosslinks by human excision nuclease. PNAS. 2006;103:4056–4061. doi: 10.1073/pnas.0600538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakano T, Katafuchi A, Matsubara M, Terato H, Tsuboi T, Masuda T, Tatsumoto T, Pack SP, Makino K, Croteau DL, Van Houten B, Iijima K, Tauchi H, Ide H. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. The Journal of biological chemistry. 2009;284:27065–27076. doi: 10.1074/jbc.M109.019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker DJ, Wuenschell G, Xia L, Termini J, Bates SE, Riggs AD, O'Connor TR. Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. The Journal of biological chemistry. 2007;282:22592–22604. doi: 10.1074/jbc.M702856200. [DOI] [PubMed] [Google Scholar]
- 32.Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, Drobnitzky N, Freire R, Amor DJ, Lockhart PJ, Kessler BM, McKenna GW, Gileadi O, Ramadan K. Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Molecular cell. 2016;64:704–719. doi: 10.1016/j.molcel.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SarahL, Tsutakawa SusanE, Borg A, Kjær S, Tainer JohnA, Skehel JM, Groll M, Boulton SimonJ. Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Molecular cell. 2016;64:688–703. doi: 10.1016/j.molcel.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez-Mosqueda J, Maddi K, Prgomet S, Kalayil S, Marinovic-Terzic I, Terzic J, Dikic I. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife. 2016;5:e21491. doi: 10.7554/eLife.21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 2014;158:327–338. doi: 10.1016/j.cell.2014.04.053. [DOI] [PubMed] [Google Scholar]
- 36.Duxin JP, Dewar JM, Yardimci H, Walter JC. Repair of a DNA-Protein Crosslink by Replication-Coupled Proteolysis. Cell. 2014;159:346–357. doi: 10.1016/j.cell.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stingele J, Habermann B, Jentsch S. DNA–protein crosslink repair: proteases as DNA repair enzymes. Trends in Biochemical Sciences. 2015;40:67–71. doi: 10.1016/j.tibs.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 38.Hill JW, Evans MK. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic acids research. 2006;34:1620–1632. doi: 10.1093/nar/gkl060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larminat F, Germanier M, Papouli E, Defais M. Impairment of homologous recombination control in a Fanconi anemia-like Chinese hamster cell mutant. Biology of the cell. 2004;96:545–552. doi: 10.1016/j.biolcel.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 40.Zdzienicka MZ, Arwert F, Neuteboom I, Rooimans M, Simons JW. The Chinese hamster V79 cell mutant V-H4 is phenotypically like Fanconi anemia cells. Somatic cell and molecular genetics. 1990;16:575–581. doi: 10.1007/BF01233098. [DOI] [PubMed] [Google Scholar]
- 41.Studzian K, Telleman P, van der Schans GP, Zdzienicka MZ. Mutagenic response and repair of cis-DDP-induced DNA cross-links in the Chinese hamster V79 cell mutant V-H4 which is homologous to Fanconi anemia (group A) Mutation research. 1994;314:115–120. doi: 10.1016/0921-8777(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 42.Kadkhodayan S, Salazar EP, Ramsey MJ, Takayama K, Zdzienicka MZ, Tucker JD, Weber CA. Molecular analysis of ERCC2 mutations in the repair deficient hamster mutants UVL-1 and V-H1. Mutation research. 1997;385:47–57. doi: 10.1016/s0921-8777(97)00030-x. [DOI] [PubMed] [Google Scholar]
- 43.Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Product-assisted catalysis in base-excision DNA repair. Nature structural biology. 2003;10:204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- 44.Huang R, Fang P, Kay BK. Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods. 2012;58:10–17. doi: 10.1016/j.ymeth.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhitkovich A, Costa M. A simple, sensitive assay to detect DNA-protein crosslinks in intact cells and in vivo. Carcinogenesis. 1992;13:1485–1489. doi: 10.1093/carcin/13.8.1485. [DOI] [PubMed] [Google Scholar]
- 46.Mulligan RC, Berg P. Expression of a bacterial gene in mammalian cells. Science. 1980;209:1422–1427. doi: 10.1126/science.6251549. [DOI] [PubMed] [Google Scholar]
- 47.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 48.Tang W, Liu JN, Gurewich V. Preparation of single-stranded phagemid DNA without chromosomal DNA contamination. BioTechniques. 1996;21:53–54. doi: 10.2144/96211bm10. [DOI] [PubMed] [Google Scholar]
- 49.Sambrook J, Russell DWDW, Laboratory CSH. Molecular cloning : a laboratory manual. 3rd. Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- 50.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. Journal of molecular biology. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 51.Lee HW, Lee HJ, Hong CM, Baker DJ, Bhatia R, O'Connor TR. Monitoring repair of DNA damage in cell lines and human peripheral blood mononuclear cells. Analytical biochemistry. 2007;365:246–259. doi: 10.1016/j.ab.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sikorsky JA, Primerano DA, Fenger TW, Denvir J. DNA damage reduces Taq DNA polymerase fidelity and PCR amplification efficiency. Biochemical and biophysical research communications. 2007;355:431–437. doi: 10.1016/j.bbrc.2007.01.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.George JW, Salazar EP, Vreeswijk MP, Lamerdin JE, Reardon JT, Zdzienicka MZ, Sancar A, Kadkhodayan S, Tebbs RS, Mullenders LH, Thompson LH. Restoration of nucleotide excision repair in a helicase-deficient XPD mutant from intragenic suppression by a trichothiodystrophy mutation. Mol Cell Biol. 2001;21:7355–7365. doi: 10.1128/MCB.21.21.7355-7365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sczepanski JT, Wong RS, McKnight JN, Bowman GD, Greenberg MM. Rapid DNA–protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proceedings of the National Academy of Sciences. 2010;107:22475–22480. doi: 10.1073/pnas.1012860108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Atanassov B, Velkova A Fau - Mladenov E, Mladenov E Fau - Anachkova B, Anachkova B Fau -Russev G, Russev G. Comparison of the global genomic and transcription-coupled repair rates of different lesions in human cells. Zeitschrift fur Naturforschung C, Journal of biosciences. 2004;59:445–453. doi: 10.1515/znc-2004-5-628. [DOI] [PubMed] [Google Scholar]
- 56.Ford JM, Lommel L, Hanawalt PC. Preferential repair of ultraviolet light-induced DNA damage in the transcribed strand of the human p53 gene. Molecular carcinogenesis. 1994;10:105–109. doi: 10.1002/mc.2940100208. [DOI] [PubMed] [Google Scholar]
- 57.Loeber RL, Michaelson-Richie ED, Codreanu SG, Liebler DC, Campbell CR, Tretyakova NY. Proteomic analysis of DNA-protein cross-linking by antitumor nitrogen mustards. Chemical research in toxicology. 2009;22:1151–1162. doi: 10.1021/tx900078y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michaelson-Richie ED, Ming X, Codreanu SG, Loeber RL, Liebler DC, Campbell C, Tretyakova NY. Mechlorethamine-induced DNA-protein cross-linking in human fibrosarcoma (HT1080) cells. Journal of proteome research. 2011;10:2785–2796. doi: 10.1021/pr200042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Michaelson-Richie ED, Loeber RL, Codreanu SG, Ming X, Liebler DC, Campbell C, Tretyakova NY. DNA-protein cross-linking by 1,2,3,4-diepoxybutane. Journal of proteome research. 2010;9:4356–4367. doi: 10.1021/pr1000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toth A, Hegedus L, Juhasz S, Haracska L, Burkovics P. The DNA-binding box of human SPARTAN contributes to the targeting of Poleta to DNA damage sites. DNA repair. 2017;49:33–42. doi: 10.1016/j.dnarep.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 61.Morocz M, Zsigmond E, Toth R, Enyedi MZ, Pinter L, Haracska L. DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic acids research. 2017;45:3172–3188. doi: 10.1093/nar/gkw1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maskey RS, Flatten KS, Sieben CJ, Peterson KL, Baker DJ, Nam HJ, Kim MS, Smyrk TC, Kojima Y, Machida Y, Santiago A, van Deursen JM, Kaufmann SH, Machida YJ. Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic acids research. 2017;45:4564–4576. doi: 10.1093/nar/gkx107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, Machida YJ. Spartan deficiency causes genomic instability and progeroid phenotypes. Nature communications. 2014;5:5744. doi: 10.1038/ncomms6744. [DOI] [PMC free article] [PubMed] [Google Scholar]