

Abstract
Extracellular nanovesicles (ENV) released by many cells contain lipids, proteins and nucleic acids that contribute to intercellular communication. ENV have emerged as biomarkers and therapeutic targets but they have also been explored as drug delivery vehicles. However, for the latter application clinical translation has been limited by low yield and inadequate targeting effects. ENV vectors with desired targeting properties can be produced from parental cells engineered to express membrane-bound targeting ligands, or they can be generated by fusion with targeting liposomes, however, neither approach has met clinical requirements. In this study, we demonstrate that mechanical extrusion of ~107 cells grafted with lipidated ligands can generate cancer cell-targeting ENV and can be prepared in ~1 hour. This rapid and economic approach could pave the way for clinical implementation in the future.
Keywords: Extracellular vesicles, mechanical extrusion, aptamer, targeted drug delivery, chemotherapeutics
Introduction
Extracellular nanovesicles (ENVs) are cell-derived lipid bilayer-enclosed entities with size ranging from 30 to 300 nm (1). ENVs are secreted by many cell types and have been identified in diverse body fluids (2). They are specialized in intercellular communications facilitating transfer of cargo proteins and nucleic acids (3). In tumor, growing evidence indicates that ENVs can regulate tumor immune response, initiate formation of pre-metastatic niche, determine organotropic metastasis, contribute to chemotherapeutic resistance, and enable liquid biopsy for cancer diagnostics (4). Moreover, ENVs have been exploited as drug vehicles for drug delivery (5). Compared with micelles, liposomes, and polymeric nanoparticles, ENVs as a natural delivery system can evade phagocytosis, have extended blood half-life, and exhibit optimal biocompatibility without potential long-term safety issues (6). ENVs can fuse with the cell membrane and deliver drugs directly into cytoplasm. By evading the engulfment by lysosomes, ENVs remarkably enhance delivery efficiency of vulnerable molecules (5,7,8). Additionally, the small size of ENVs facilitates their extravasation, translocation through physical barriers, and passage through extracellular matrix (9). Although ENVs-based drug delivery is promising, two major challenges exist. First, the yield of ENVs is low (10). In general, ENVs can be collected from either cultured cell supernatant or autoplasma of patients. Given the secretion rate of ENVs ranges from 50 to 150 per cell per hour, the mass-production of ENVs for potential clinical translation would be costly (11,12). On the other hand, the prevalent ENV isolation techniques, such as ultracentrifugation, fail to generate highly pure ENVs (13), let alone the low isolation efficiency. Second, the preparation procedure for current ENVs-based targeted drug delivery systems are tedious and labor-intensive. The typical approach is to secrete targeting peptide or protein grafted ENVs by transfecting the donor cells with corresponding recombinant virus or plasmid (14,15). To note, these transfection approaches have been criticized for inefficiency, potential toxicity, and insertional mutagenesis (16). The ‘click chemistry’ has been used to conjugate targeting moieties onto EV surface in one-step (17). However, the conjugation procedure is time consuming, and the reaction conditions must be well controlled to avoid EV disruption and aggregation (18,19). Hence, direct surface modification of EV is nontrivial either. In brief, for cancer treatment, nevertheless, simple approaches for large-scale production of tumor-targeting ENVs are desirable.
Here, we report a method for preparing aptamer grafted ENVs for paclitaxel (PTX) loading (Fig. 1). A nucleolin-targeting aptamer AS1411 was covalently conjugated to cholesterol-poly(ethylene glycol) (cholesterol-PEG) followed by anchoring the compounds onto living mouse dendritic cell (DC) membrane. Afterward, cells were extruded by passing through micro-constrictions to obtain exosome-mimetic ENVs, and PTX was subsequently loaded into ENVs with sonication. Using this aptamer-grafted ENVs, we investigated the ENVs biodistribution and whether PTX can be more effectively delivered both in vitro and in vivo.
Figure 1. Schematics showing steps of experiments (not drawn to scale).
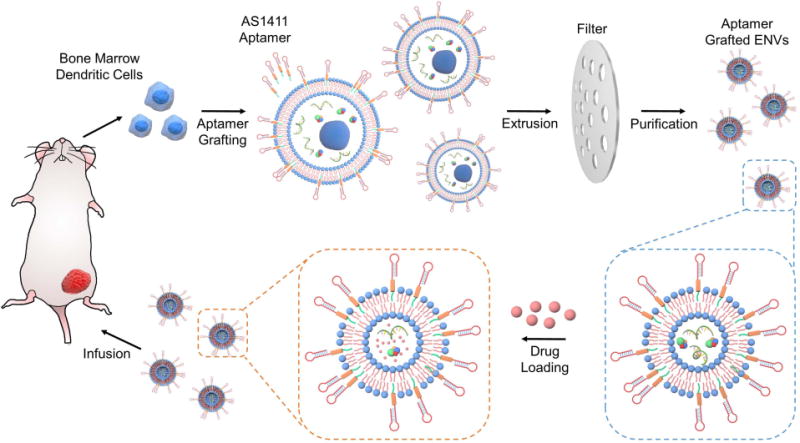
The DCs were isolated from mouse BM and multiplied in vitro. Cells were homogeneously labeled with aptamer conjugated PEGylated cholesterol and then repeatedly filtrated through two filters with 10- and 5-µm pore size for generating ENVs. After loading paclitaxel, tumor-targeting ENVs were intravenously administered for tumor treatment in vivo.
Materials and Methods
Cell culture
Cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C. The MDA-MB-231 cells (ATCC, received in May 2016, passed fluorescence testing for mycoplasma contamination on August 1st 2017, no more than 50 passages) were maintained in DMEM supplied with 10% FBS. Immature mouse DCs were isolated from bone marrow (BM) (20). BALB/c mouse was euthanized. After removal of the femurs, both ends of femurs were trimmed. The contents of marrow was flushed with 2 ml of HBSS with a needle, and BM cells were washed with HBSS thrice. ~2×106 cells were cultured in RPMI1640 containing 10% FBS and 0.2 µg rmGM-CSF. Surface expression of CD11c (Santa Cruz, sc-23951) was analyzed.
Selection of lipid
FITC-tagged stearoyl-PEG (C18-PEG2000), distearoyl phosphethanolamine-PEG (DSPE-PEG2000), and cholesterol-PEG (chol-PEG2000) were dissolved in ethanol at 1 mM. 3 nmol of each lipid probe was added to 107 DCs in 250 µl of PBS, respectively. The samples were incubated at 4 °C for 5 min followed by washing thrice. Cells were fixed, stained with DAPI, rinsed with PBS, and finally resuspended in 500 µl of PBS. 20 µl of cell suspension was added onto slips for imaging.
Synthesis of AS1411-PEG2000-Chol and DCs labeling
The 100 nmol of disulphide-tagged AS1411 aptamers (Sangon) were cleaved by TCEP solution on ice for 30 min (21), followed by incubation with 500 nmol of Chol-PEG2000-maleimide (NanoCS) in HEPES buffer (Sigma-Aldrich) overnight at 4 °C. The excess Chol-PEG2000-maleimide was removed by filtration (MWCO 5000) at 10,000 g for 15 min. The purified FITC-AS1411-PEG2000-Chol was examined by MALDI-TOF-MS. 1-5 nmol of FITC-AS1411-PEG2000-Chol was added to 107 DCs in 250 µl of PBS, respectively. Cells were incubated at 4 °C for 5 min followed by washing with PBS thrice. The fluorescence intensity was measured using an Infinite M200.
Collection and characterization of nanovesicles
107 AS1411-PEG2000-Chol labeled DCs in 1 ml PBS were continuously filtered with 10-µm and 5-µm track-etched membrane (Whatman) at 2000 rpm for 5 times, and each filtration took 5 minutes. The supernatant was centrifuged at 300 g for 5 min followed by 16,500 g for 20 min. The supernatant was filtered using 0.22 µm filter. The collection of ENVs spontaneously secreted from DCs follows a general protocol. In brief, DCs were cultured in serum free medium (SFM) for 48 h. The medium was centrifuged at 16,500 g for 20 min. Afterwards, the medium was filtered using a 0.22 µm filter followed by ultracentrifugation at 100,000 g and 4 °C for 2 h. The ENV pellets were suspended in 500 μl of PBS.
5 µl of AS1411-ENV samples were seeded onto substrate and fixed. The morphology of AS1411-ENVs was confirmed under Zeiss FESEM. 5 µl of AS1411-ENV sample was placed on 400 mesh grids and incubated for 3 min at RT. Excess samples were blotted with filter paper and stained with 1% uranyl acetate for 1 min. Samples were then examined in a FEI Tecnai TEM. For cryo-TEM, 5 µl of AS1411-ENV sample was applied to a 200 mesh grids blotted for 1 sec with FEI Vitrobot before plunging into liquid ethane. Samples were visualized in FEI Tecnai F20 TEM. The number of AS1411-ENVs was measured using Nanosight LM10 (Malvern). The characterization of ENVs follows the same protocol.
After lysis with RIPA buffer, protein amount in AS1411-ENVs and ENVs were determined using Micro BCA Protein Assay (Pierce). Protein samples were analyzed using acrylamide gels, and then transferred onto PVDF membranes. The protein blot was blocked for 1 h at RT with 5% non-fat dry milk in PBS/0.05% Tween and incubated overnight at 4 °C with Santa Cruz antibodies against Annexin II (sc-28385), TSG101 (sc-7964), HSC70 (sc-7298), CD9 (sc-13118), CD59 (sc-133170), and CD55 (sc-51733). Afterward, secondary antibodies were incubated for 1 h at RT. Samples were washed with PBS/0.05% Tween 20 for 10 min thrice. Blots were developed with chemiluminescent.
10 µl of AS1411-ENV or ENV samples were seeded onto slips and incubated at 4 °C for 1 h. The slip was blocked with BSA (10mg/ml) for 20 min followed by PBS rinsing. A complementary DNA probe of AS1411 aptamer (Cy5-5′-CCA CCA CCA CCA CAA CCA CCA CCA CC-3′) and a control random DNA probe (Cy5-5′-TGC TGT GAG TGA ACC TGC TGT GTT GA-3′) in 50 µl of solution (pH 8.4, 50 mM KCl and 1.5 mM MgCl2) was incubated with AS1411-ENVs or ENVs in a humidified chamber at 37 °C for 2 h (22-24). After hybridization, the slip was washed with PBS for fluorescence analysis.
Drug loading and release
PTX and nanovesicles were mixed in number ratio of 106. The mixture was sonicated using a Model 505 Sonic Dismembrator with .25” tip with the following settings: 20% amplitude, 6 cycles of 30 s on/off for 3 min with a 2 min cooling period between each cycle. After sonication, the mixture was incubated at 37 °C for 1 h. Alternatively, PTX and nanovesicles were mixed in the same ratio, stirred and incubated for 1 h at RT. Excess free drug was removed from PTX-loaded AS1411-ENVs or ENVs with a Sephadex G25 column. The amount of loaded PTX in respective group was measured by HPLC. To measure PTX release, freshly prepared PTX-loaded AS1411-ENVs or ENVs was placed in a 300K MWCO float-A-lyzer G2 device (Spectrum Laboratories). The device was then placed in PBS at RT with stirring. Samples were taken at timepoints and analyzed by HPLC.
Therapeutic efficacy in vitro and in vivo
The cytotoxicity of PTX, PTX-loaded ENVs, and PTX-loaded AS1411-ENVs in MDA-MB-231 cells was evaluated using MTT assay (ThermoFisher). Approximately 4,000 MDA-MB-231 cells were treated with PTX in various concentrations, ranging from 0 to 3300 nM. After 24 h, 20 µl of MTT solution (5 mg/ml) was added to each well and incubated for 4 h. Medium was discarded, and formazan precipitate was dissolved in 10% sodium dodecyl sulfate in DMSO containing 0.6% acetic acid. The microplates were shaken in the dark for 30 min, and absorbance at 570-nm was measured. The IC50 was determined from the dose-response curves.
MDA-MB-231 cells in 6 groups were respectively treated with PBS, ENVs without PTX, AS1411-ENVs without PTX, 200 nM bare PTX, PTX-loaded ENVs, and PTX-loaded AS1411-ENVs for 24 h. 5×105 harvested MDA-MB-231 cells in 100 µl 1× binding buffer (BD Biosciences) were incubated with 10 µl PI and 5 µl of Annexin V-FITC for 30 min at RT. Each sample was analyzed by flow cytometry. With the same experimental setup, cells in 6 groups were stained with PI only and photographed with high-content imaging system (Molecular Device).
Approximately 2×106 MDA-MB-231 cells in 50 µl PBS mixed with 50 µl of matrigel were inoculated subcutaneously to the flanks of BALB/c mice (~18-22 g, 6 weeks), and allowed to grow to a tumor size ~100 mm3. The mice were then randomly divided into 6 groups. Drug was intravenously administrated every 2 days (7.5 mg of PTX-equiv per kg of body weight per dose) for 3 weeks. Mice were euthanized to harvest tumors. To study pharmacokinetics and biodistribution of PTX, 0.1-0.3 g tissue samples were collected at 6 timepoints after treatment. The analysis of PTX by HPLC follows the published protocol (25). All animal experiments were approved by and performed in accordance with guidelines from the Institutional Animal Care and Use Committee (IACUC) of the Model Animal Research Center of the Second Affiliated Hospital of Southeast University.
Statistical analyses
Results are presented as mean±SD. Statistical comparisons were performed by paired Student’s t-test or ANOVA.
Results
Preparation and characterization of AS1411-ENVs
To efficiently immobilize lipid conjugated aptamers onto cell membrane for subsequent generation of aptamer grafted ENVs, we first determined the lipid molecule type and optimal dose. PEGylated monoacyl lipid, diacyl lipid, and cholesterol are frequently used (26,27). Hence, we chose FITC-tagged C18-PEG2000, DSPE-PEG2000, and cholesterol-PEG2000 for optimization. The three lipids can spontaneously insert into cellular lipid bilayer, but show differential labelling effects (Fig. 2a). The average fluorescence intensity of cells labeled with chol-PEG2000 was higher than that of the others (p<0.01, Fig. 2b). There was no difference in fluorescence intensity between C18-PEG2000 and DSPE-PEG2000. Since C18-PEG2000 and DSPE-PEG2000 can readily assemble forming micelles or liposomes due to the long hydrophobic tail(s) and hydrophilic head, the self-assembly impairs cell labeling effect. In contrast, Chol-PEG2000 can alleviate that to a certain extent as cholesterol itself is amphiphilic and relatively rigid. In addition, cholesterol also can increase the rigidity and stability of liposomes or EVs by enhancing the hydrophobic-hydrophobic interactions in lipid bilayers (28).
Figure 2. Optimization of lipid conjugated aptamer and characterization of nanovesicles.
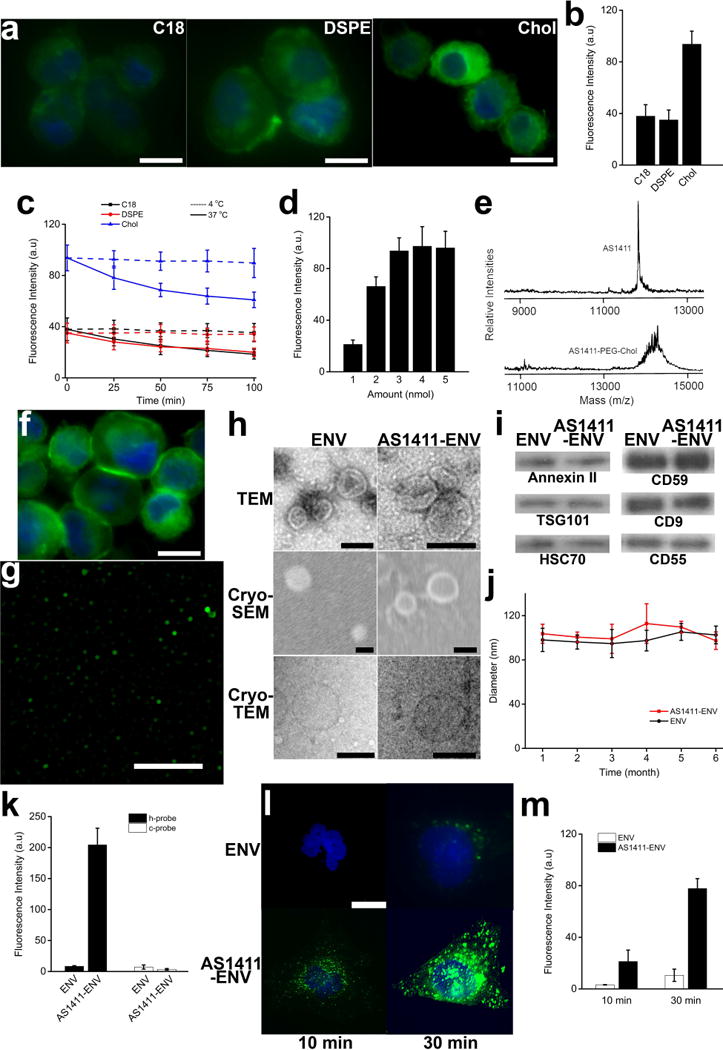
a–b, fluorescent images of DCs labelled with three types of PEGylated lipids (C18, DSPE, and cholesterol) in PBS (scale bar corresponds to 10 µm), and quantified data of fluorescence intensity of each group (n = 150). c, changes of fluorescence intensity of cells in each group at 4 °C and 37 °C, respectively, reflecting retention of each type of lipid on cell membrane (n = 150). d, fluorescence intensity of cells labeled with various amount of cholesterol (n = 150). e, confirmation of conjugation of AS1411 aptamer with PEGylated cholesterol with MALDI-TOF mass spectrometry. f, fluorescence image of AS1411-PEG-cholestrol labeled DCs (scale bar corresponds to 10 µm). g, fluorescence image of AS1411 conjugated ENVs (scale bar corresponds to 5 µm). h, morphological characterization by electron microscopy (scale bar corresponds to 100 nm). i, Annexin II, TSG101, HSC70, CD59, CD9, and CD55 were extracted from spontaneous released ENVs and extruded AS1411-ENVs, respectively, and identified by western blotting. j, changes in size of ENVs and AS1411-ENVs stored at −80 °C in PBS for up to 6 months (n = 5). k, fluorescence intensity of dots of ENVs and AS1411-ENVs hybridized with h-probes and c-probes, respectively (n = 20). l–m. fluorescence images showing ENVs and AS1411-ENVs taken up by MDA-MB-231 cells in 30 min, respectively (scale bar corresponds to 10 µm), and quantified data of fluorescence intensity of each group (n = 25).
Since the stable retention of lipids conjugated aptamer on cell surface favors generation of AS1411-ENVs, the retention of three lipids over time was evaluated with the corresponding decay of fluorescence at 4 °C and 37 °C, respectively. At 4 °C the fluorescence intensity of cells labeled with C18-PEG2000, DSPE-PEG2000, and chol-PEG2000, respectively, decreased to 93.2%, 97.71%, and 95.74% indicating excellent retention over 100 minutes at low temperature (Fig. 2c). In contrast, at 37 °C the fluorescence intensity of cells labeled with C18-PEG2000, DSPE-PEG2000, and chol-PEG2000, respectively, gradually decreased to 48.63%, 56.98%, and 65.04% over the same period (Fig. 2c). We did not further measure the fluorescence intensities beyond 100 minutes as the following extrusion of labeled cells for generating adequate ENVs only takes less than 30 minutes. Considering chol-PEG2000 demonstrated a superior labeling effect and an excellent retention at low temperature, we chose chol-PEG2000 for the following studies.
Next, we optimized the dose of chol-PEG2000 for cell membrane labeling. The insufficient label with cholesterol-conjugated aptamers causes unsaturated aptamer immobilization on cell surface. On the contrary, over labeling might trigger plasma membrane bubbling and destroy the integrity of the lipid bilayer. The fluorescence intensity significantly increased with the dose of chol-PEG2000 ranging from 1 nmol to 4 nmol (p<0.05) (Fig. 2d). To identify the labeling efficiency of chol-PEG2000, we compared the fluorescence intensity of FITC-tagged chol-PEG2000 at various dose before and after labeling (Supplementary Fig. 1a). The labeling efficiency (fraction of fluorescence intensity difference over initial FITC-tagged chol-PEG2000) of chol-PEG2000 ranging from 1 nmol to 5 nmol (1–5 µl of 1 mM chol-PEG2000 added into 250 µl of cell suspension, Supplementary Fig. 1b) was 15.33%, 20.03%, 20.05%, 22.09%, and 18.73%, respectively (Supplementary Fig. 1c). Therefore, we chose 4 nmol of chol-PEG2000 for labeling of ~107 cells, and accordingly we determined ~5.3×107 chol-PEG2000 molecules were incorporated into an individual DC.
AS1411-PEG2000-chol was synthesized and confirmed by MALDI-TOF-MS (Fig. 2e). Excess free lipids were removed, and then the amount of AS1411-PEG2000-chol was determined by UV-Visible spectroscopy. Meanwhile, DC cells were isolated and expanded in vitro in 5 days (Supplementary Fig. 2a). Following the optimized protocol of cell labeling, we identified FITC-tagged AS1411-PEG2000-chol can be grafted onto DC membrane (Fig. 2f). After labeling, we loaded ~107 DC cells (purity: approximately 90%, characterized with CD11c, Supplementary Fig. 2b) into an extruder for generating AS1411-ENVs (29). The morphology of partial AS1411-ENV was observed with a super-resolution microscopy (Nikon) in SIM mode (Fig. 2g). The average diameter of AS1411-ENV measured by microscopy ranged from 110 nm to 310 nm. Nanosight revealed the size distribution of AS1411-ENVs ranged from 50 nm to 270 nm with peak concentration at 103 nm. In contrast, in the control group the diameter of ENVs spontaneously secreted from unlabeled DCs was 30-240 nm with peak concentration at 98 nm. In addition, ~8.7×1010 AS1411-ENVs were prepared by extrusion of ~107 cells in 25 min. Under the assumption that the whole cell membrane would participate in forming ENVs during extrusion, the production efficiency of AS1411-ENVs using our protocol was nearly 30%, and these is still room for improvement. In comparison, only 3.4×1010 ENVs were harvested from ~6.75×107 cells cultured in SFM for 48 h. The generation efficiency of nanovesicles by extrusion was approximately 17-fold higher and 115-fold faster than that of spontaneous secretion from cells. Under electron microscopy, there was no significant difference in morphology between AS1411-ENVs and ENVs (Fig. 2h). Both displayed a typical saucer-shaped morphology. The cryo-TEM and cryo-SEM images also demonstrate both were spherical and enclosed by a lipid bilayer.
We also directly incubated bare ENVs with FITC-tagged AS1411-PEG2000-chol and determined the labeling efficiency. Given we used 4 nmol to label ~107 cells, following the same ratio of lipid amount to total membrane surface area we labeled ~7×1010 bare ENVs with 2 nmol of AS1411-PEG2000-chol in 250 µl suspension at 4 °C for 5 min. After removal of free aptamer conjugated lipids with ultracentrifugation, we recovered ~3×1010 AS1411-ENVs. The size at peak concentration was 105 nm (Supplementary Fig. 3a). Moreover, we found the fluorescence intensity of AS1411-ENVs prepared by extrusion (~5×108 in 10 µl) was higher than that of equivalent AS1411-ENVs prepared by direct labeling (p<0.05, Supplementary Fig. 3b). We speculated that under the same experimental setting labeling ENVs with nanoscale lipids might be inefficient than microscale cells labeling. The operation of ENV labeling is not comparable to cell labeling either. The additional purification with ultracentrifugation not only requires at least 2-hour processing time but also further decreases the final output. Therefore, direct ENV labeling with targeting ligands works despite low efficiency.
Cargo proteins extracted from ~2×108 AS1411-ENVs and ENVs were analyzed by western blot. Three cytosolic proteins including Annexin II, TSG101, and HSC70 and three surface proteins including CD59, CD9 and CD55 were identified in both AS1411-ENVs and ENVs (Fig. 2i), indicating these proteins can be enclosed into AS1411-ENVs during extrusion of cells. The CD55 and CD59 protect extracellular vesicle (EV) from attack of the complement system, making them stable in the blood (30,31). Tetraspanin CD9 facilitates direct membrane fusion with the target cell and contents-delivery directly into cytosol (32). In addition, we chose immature DCs as they are devoid of lymphocyte stimulatory molecules such as MHC II, CD80 or CD86 (33). The stability of AS1411-ENVs and ENVs were examined by monitoring changes in size at respective peak concentration. The cryopreserved AS1411-ENVs and ENVs with approximately 100 nm in diameter did not show significant aggregation or degradation in PBS at −80 °C over 6 months (Fig. 2j), indicating AS1411-ENVs could be prepared in large-scale, cryopreserved in batches for at least 6 months, and then thawed for use.
We further used Cy5 conjugated hybridization probe (h-probe) of AS1411 and random probe as a control (c-probe) to reconfirm the existence of AS1411 on conjugated ENVs. The fluorescence intensities demonstrated only the dot blots of AS1411-ENVs incubated with h-probes showed strong red fluorescence signals (Fig. 2k). In comparison, due to minute amount of residual probes weak fluorescence signals were detected in the rest three groups. Next, ~8×105 AS1411-ENVs and ENVs were incubated with MDA-MB-231 cells expressing nucleolin on cell membrane, respectively (34). We identified that AS1411-ENVs efficiently bound MDA-MB-231 cells in only 10 min (Fig. 2l and 2m). In contrast, equal amount of PKH67 labeled ENVs barely fused with MDA-MB-231 cells at the same timepoint. After 30 min a few ENVs attached and fused with cell membrane, while significant amount of AS1411-ENVs were observed in the experimental group (p<0.01). The findings indicate that AS1411-ENVs can rapidly recognize and bind onto cancer cell surface nucleolin, and thus would facilitate targeted drug delivery.
Characterization of PTX-loaded nanovesicles
PTX was loaded into AS1411-ENVs and ENVs (in absolute number ratio of PTX/vesicle: 106), respectively, using either sonication or simple incubation at RT. The average PTX loading efficiency with sonication was 21.33±1.53% in AS1411-ENVs and 26.4±2.67% in ENVs. In comparison, PTX loading efficiency with simple mixing, stirring and incubation was 5.06±1.12% in AS1411-ENVs and 7.73±2.38% in ENVs, respectively. During sonication, the membrane microviscosity significantly decreases, allowing drug molecules to enter vesicle, and the membrane integrity can be restored immediately after sonication. It does not significantly affect the membrane-bound proteins or the lipid contents of the ENVs (35). To note, the negatively charged aptamers on the outer surface might affect encapsulation of negatively charged PTX, causing slightly lower loading efficiency in comparison with that of ENVs. As the loading efficiency of sonication was much higher than that of incubation, we used sonication for PTX loading in the following studies. After PTX loading, the whole size distribution showed a slight rightward shift, and the size of AS1411-ENVs and ENVs at peak concentration increased to 111 nm and 109 nm (black curves), respectively (Fig. 3a). In addition, after loading of PTX the mean zeta potential of AS1411-ENVs decreased from −23.7 mv to −25.6 mv, and that of ENVs decreased from −11.8 mv to −16.1 mv (Fig. 3b). The three cytosolic proteins including Annexin II, TSG101, and HSC70 were barely detectable in ~2×108 AS1411-ENVs and ENVs after PTX loading, respectively. These syntenins might be released from vesicles during sonication (Fig. 3c).
Figure 3. Characterization of PTX-loaded nanovesicles and cancer cell treatment in vitro.
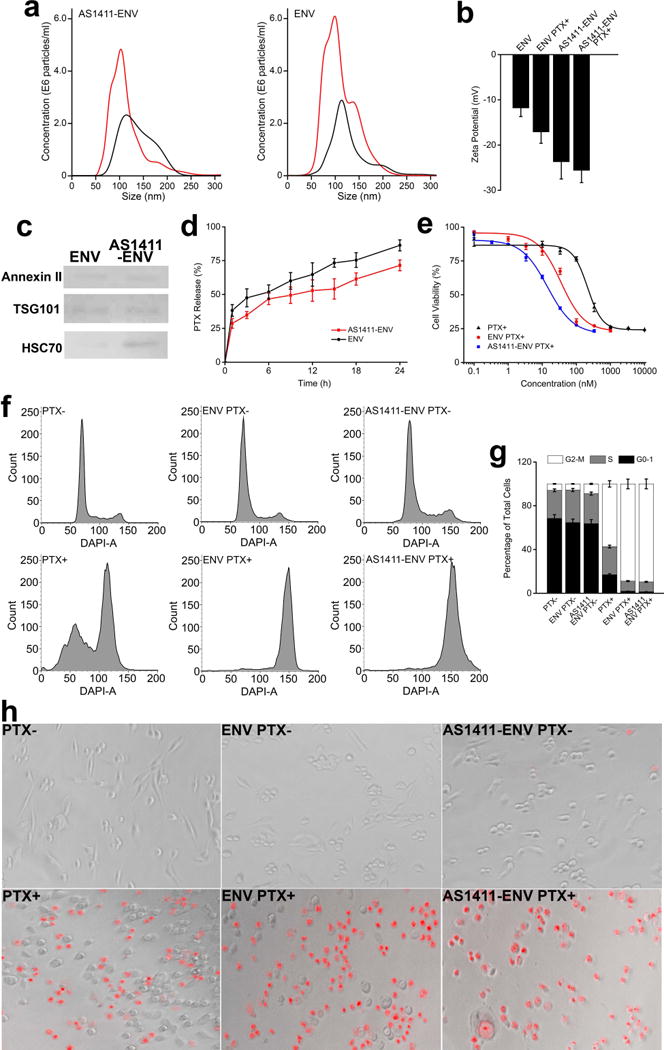
a, size distribution of AS1411-ENVs and ENVs before PTX loading (red curve) and after PTX loading (black curve). b, changes of zeta potential of ENVs and AS1411-ENVs before and after PTX loading. c, Annexin II, TSG101, and HSC70 extracted from PTX-loaded ENVs and AS1411-ENVs, respectively and confirmed with western blotting. d, in vitro release of PTX from ENVs and AS1411-ENVs at 37 °C, respectively. e, respective IC50 of bare PTX, PTX-loaded ENVs, and PTX-loaded AS1411-ENVs after 24 h cell incubation. f-g, cell cycle of MDA-MB-231 cells treated with 200 nM bare PTX or PTX-equiv in nanovesicles for 24 h, respectively, and quantified data of cell cycles of each group. h, fluorescence images of MDA-MB-231 cells treated with 200 nM bare PTX, or PTX-equiv loaded ENVs, and PTX-loaded AS1411-ENVs, stained with PI, and scanned with high-content screening platform.
Moreover, we investigated the release kinetics of PTX from AS1411-ENVs and ENVs at 37 °C, respectively (Fig. 3d). Both PTX-loaded nanovesicles showed burst release within the first one hour, and then displayed a sustained release profile thereafter. At 24 h timepoint, PTX-loaded AS1411-ENVs and ENVs released approximately 71.47% and 86.29% PTX, respectively. The slow release of PTX from nanovesicles is favorable for drug administration. Especially in targeted drug delivery, more drugs in vectors thus can be effectively delivered to the lesion. The ability to deliver PTX into target cells in vitro was studied by identifying IC50 of PTX-loaded AS1411-ENVs, PTX-loaded ENVs, and bare PTX, respectively after cell treatment for 24 h, and we determined the respective IC50 of PTX in the above three groups as 14.24 nM, 35.08 nM, and 212.69 nM (Fig. 3e). The treatment efficacy of PTX-loaded AS1411-ENVs and PTX-loaded ENVs exhibited a 15-fold and 6-fold enhancement compared with bare PTX in vitro. To note, AS1411 as therapeutic effects, and AS1411 for acute myeloid leukemia is being evaluated in phase-II clinical trial (36). On the basis of generated AS1411-ENVs amount and AS1411 grafting amount, we deduced that each individual AS1411-ENV bears ~2000 AS1411 molecules. The amount is adequate for tumor targeting in vivo (37), but has very limited therapeutic effects. The dose of AS1411 in tumor therapy in vivo is 10 mg/kg/day or even higher, while the amount of AS1411 for targeting is less than 4% of the therapeutic dose. We speculated the significant decrease of IC50 in group of AS1411-ENVs was mainly attributed to high delivery efficiency of PTX to cells through the targeted ENVs.
Cancer treatment efficacy in vitro and in vivo
We used 200 nM PTX-loaded AS1411-ENVs, PTX-loaded ENVs, bare PTX and three negative controls to treat MDA-MB-231 cells, respectively. In groups of PTX-loaded AS1411-ENVs and PTX-loaded ENVs approximately 90% of cells were effectively blocked in G2-M phase, while in bare PTX group approximately 60% of cells had been retained in G2-M phase (Fig. 3f and 3g). The PTX-loaded AS1411-ENVs and PTX-loaded ENVs demonstrated higher therapeutic efficacy in comparison with bare drugs as PTX-loaded nanovesicles could efficiently deliver drugs to target cells by membrane fusion and AS1411 mediated internalization. In contrast, bare negatively charged PTX molecules rely on passive diffusion for crossing negatively charged cell membrane. The entry mode bypasses the endosomal-lysosomal pathway (38), and thus both AS1411-ENVs and ENVs as delivery vehicles can circumvent the need for endosomal-escape strategies. The high-content screening also confirmed significantly more dead cells (stained in red) in groups of PTX-loaded AS1411-ENVs and PTX-loaded ENVs in comparison to that of bare PTX group and three negative control groups (Fig. 3h). To note, after PTX treatment dead cells detached from flask surface during staining and rinsing, and thus less cells left in the three experimental groups.
Next, we investigate the potential of PTX-loaded AS1411-ENVs and PTX-loaded ENVs in drug delivery using the MDA-MB-231 xenograft BALB/c mouse model. Tissue samples (heart, liver, spleen, lung, kidney and tumor) were collected, and PTX concentration in each organ was analyzed with HPLC. At 30 min post-administration, PTX underwent rapid distribution to the main organs in each group (Fig. 4a). In bare PTX group PTX easily diffused into other organs but less PTX accumulated in tumors. In the next 12 hours, bare PTX was readily cleared from main organs and tumors. In groups of PTX-loaded AS1411-ENVs and PTX-loaded ENVs, PTX concentration in tumors was significantly higher than that in bare PTX group at all timepoints, indicating encapsulation of PTX into AS1411-ENVs and ENVs can achieve in vivo re-distribution of PTX. The clearance rate of PTX-loaded AS1411-ENVs and PTX-loaded ENVs were also slower than that of bare PTX. We noted that both PTX-loaded nanovesicles show relatively high concentration of PTX in liver and spleen at the 12 h timepoint. A previous study also reported that EVs displayed a predominant localization of intravenously administered EVs in the spleen and the liver (39). In general, the intravenous injection of nanoparticles with size over 50 nm results in intensive recognition by microphages which take nanoparticles to the liver, meanwhile 150-250 nm nanoparticles also frequently accumulate in the spleen (40). In addition, PTX-loaded AS1411-ENVs continued to accumulate in tumors and reached peak concentration at 12 h timepoint, showing significantly different (p<0.01) pharmacokinetics compared to the other two groups (Fig. 4a). The tumor volumes of mice treated with PBS (from 167.06±12.20 to 1041.59±238.3 mm3), ENVs without PTX (from 167.55±12.36 to 1059.67±339.68 mm3), and AS1411-ENVs without PTX (from 168.53±18.04 to 1110.32±284.27 mm3) rapidly increased, and there was no significant difference among the three negative controls (Fig. 4b and 4c). The final tumor weight of mice treated with PBS, ENVs without PTX, and AS1411-ENVs without PTX were 0.94±0.20 g, 0.96±0.32 g, and 1.00±0.26 g respectively. Tumors that received bare PTX showed a slight growth inhibition due to low bioavailability, and thus tumor volume and weight increased from 168.96±16.56 mm3 to 283.28±90.65 mm3 by the end of treatment. PTX-loaded ENVs inhibit the growth of tumor, and the volume decreased from 169.7±17.01 mm3 to 97.99±30.08 mm3. PTX-loaded AS1411-ENVs significantly restrained growth of grafted tumors, and the tumor volume decreased from 169.58±18.04 mm3 to 35.34±3.38 mm3 (p<0.05), which could be attributed to the active targeting effect in tumor treatment (Fig. 4b and 4c). At the end of treatment, the relative tumor volume of each group was 6.19±1.01 (without PTX), 6.24±1.49 (ENVs without PTX), 6.58±1.4 (AS1411-ENVs without PTX), 1.66±0.43 (bare PTX), 0.57±0.15 (PTX-loaded ENVs), and 0.21±0.04 (PTX-loaded AS1411-ENVs), respectively. Average tumor weight of PTX-treated three groups was 0.18±0.089 g (bare PTX), 0.05±0.016 g (PTX-loaded ENVs), and 0.02±0.002 g (PTX-loaded AS1411-ENVs), respectively (Fig. 4d). The in vivo therapeutic efficacy of PTX-loaded AS1411-ENVs was improved approximately 9- and 3-fold compared to that of bare PTX and PTX-loaded ENVs, respectively (p<0.05). There was no significant difference in mouse body weight during the 3-week administration (Fig. 4e). Furthermore, tumor proliferation and the histological structures of tumors and organs from all groups were analyzed (Fig. 4f and Supplementary Fig. 4). Compared with other formulations, PTX-loaded AS1411-ENVs caused remarkable tumor tissue damage and reduced the percentage of proliferating Ki67-positive tumor cells, indicating the enhanced treatment efficacy. We also measured the systemic toxicity. BM suppression is the major dose-limiting toxicity of PTX, and dose-/schedule-dependent neutropenia is the typical hematologic toxicity. Over 10 days after intravenous administration, a routine blood test was performed to exam leukocytes counting and function enzymes of liver and kidney. The mean counts of leukocyte, erythrocyte, and platelet were relatively lower in bare PTX-treated mice, meanwhile the average level of five major indexes of kidney and liver function was also relatively higher (Fig. 4g). However, no significant difference was observed among 6 groups, and their levels were well within the normal physiological range. The findings also indicated PTX-loaded AS1411-ENVs or PTX-loaded ENVs overcame the systemic toxicity. The tumor-bearing mice could tolerate higher dose of PTX-encapsulated in targeting ENVs without the occurrence of severe systemic toxicities. The encapsulation would increase the chemotherapeutic effects while minimizing exposure to normal and healthy cells.
Figure 4. Tumor treatment with PTX-loaded nanovesicles in vivo.
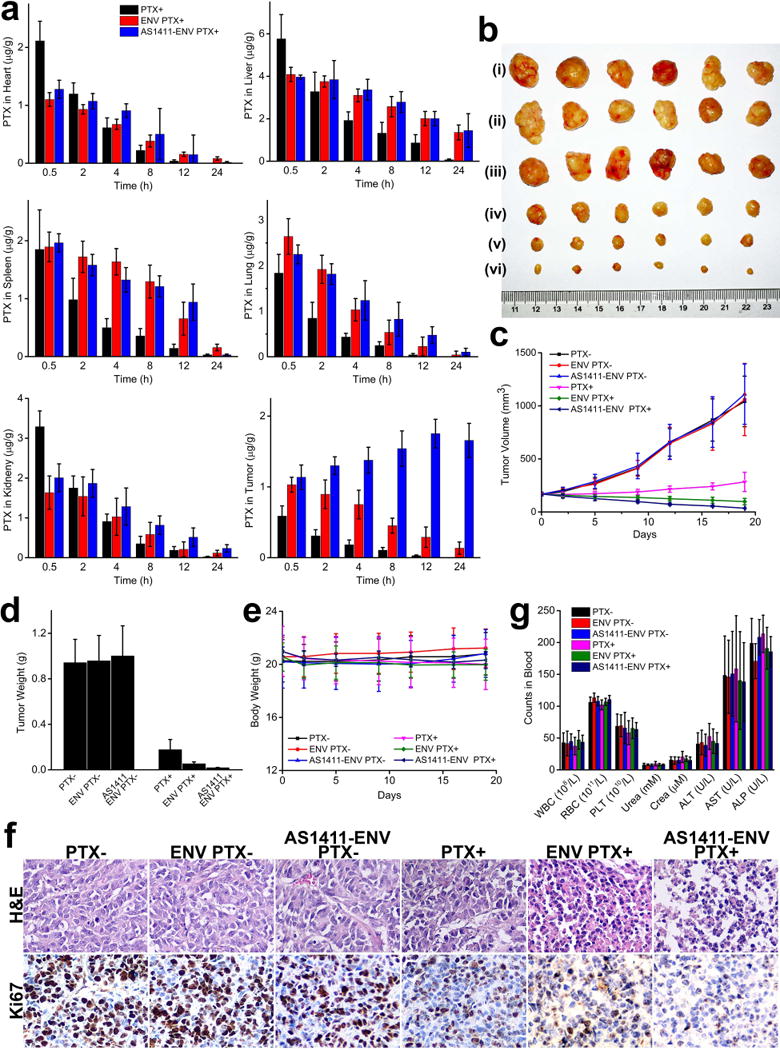
a, biodistribution of PTX in heart, liver, spleen, lung, kidney, and tumor in three groups, respectively, after an intravenous administration (n=6). b, MDA-MB-231 tumor tissues obtained from each group euthanized mice 20 days after administration of PTX- (i), ENV PTX- (ii), AS1411-ENV PTX- (iii), PTX+ (iv), ENV PTX+ (v), and AS1411-ENV PTX+ (vi), respectively, (n=6). c-d, Tumor volume and weight of MDA-MB-231 tumor xenograft in mice from each group after drug or placebo administration (n=6). e, average mass of mice from each group without significant difference (n=6). f, typical histological sections of tumors stained with H&E and Ki67, respectively. PTX-loaded AS1411-ENVs group demonstrated significant tumor tissue damage and the lowest Ki67 index, indicating high treatment efficacy (n=6). g, systemic toxicity profile of bare PTX, PTX-loaded ENVs, and PTX-loaded AS1411-ENVs. There was no significant difference in levels of leukocytes, erythrocytes, and liver and kidney function enzymes (n=6).
Disscussion
The field of EV based drug delivery has greatly expanded over the last few years, and many studies have eloquently demonstrated that EV can function as therapeutic nanocarriers for delivering a variety of cargos, including siRNAs, miRNAs, proteins, and chemotherapeutics to particular tissue (41-44). Compared with existing delivery systems, the outstanding advantage of EVs as delivery vehicles is their favorable lipid and surface protein composition. Currently, cells such as epithelial cells and model cancer cell lines have been used as donor cells for harvesting derived EVs. However, immunogenicity and biosafety concerns arise for in vivo applications of these donor cells derived EVs. Particularly, cancer cell lines derived EVs as drug delivery vehicles might possess tumorigenic potential due to incomplete depletion of cargos and/or inherent tumor-associated proteins imbedded on EV membrane. In comparison, DCs can be harvested from a patient’s BM or peripheral blood by standard protocols (45). They should thus be less immunogenic than any foreign delivery vehicle. The in vitro multiplication of DCs can constantly provide EVs, which is extremely important for multiple-episodes of treatment. On the other hand, at present only a small portion of published reports utilize a targeting strategy for direct delivery of the therapeutic cargos. The mainstream routine for harvesting derived EVs relies on expression of targeting protein or peptide on cell membrane after sophisticated gene transfection (8,14,33,46). Alternatively, targeting ligands can be covalently immobilized onto membrane proteins via click chemistry. However, both technique routines are quite challenging. Moreover, the workload required to upscale the production of cells and the resulting yield of EVs are currently not suitable for clinical use.
The method we developed can easily create adequate targeting ligands-grafted ENVs in 1 h for drug loading. Our approach has multiple advantages. First, both targeting ligands (lipid conjugated aptamers or palmitoylated antibodies) and donor cells (immune cells, stem cells, or others) can be arbitrarily geared to meet actual need. Theoretically, lipidated ligands targeting estrogen-related receptor beta 2 (ERRB2), prostate-specific antigen (PSA), carbonic anhydrase IX (CA IX), or other targets frequently used in chimeric antigen receptor (CAR) T-cell therapy could be produced in large-scale and grafted onto autologous immune cells for targeted therapy of different cancers. The whole procedure would be very flexible. On the contrary, the bioengineered cells through gene transfection only express designated protein or peptide. To construct cells expressing another specific membrane protein, the preparation of viral or plasmid vectors, transfection, and selection have to be performed once again. Second, donor cells potentially can be obtained from patient’s autologous cells to generate ENVs for drug loading. Genetic engineering is not necessary. Therefore, it is much safe than cell-based immunotherapy, such as CAR T-cell therapy. Although CAR T-cell therapy was recently approved, the efficacy, toxicity, potential mutagenesis and easy-to-operation are still big concerns (47,48). Third, we demonstrated generation efficiency and rate of ENVs via extrusion of cells is 17- and 115-fold higher than that of ENVs spontaneously released by donor cells. Cells harvested from a T75 flask can generate ~8×1010 ENVs via extrusion, satisfying a single dose, and the production can easily be scaled up. The relatively low cost and the ease of operation would benefit the clinical implementation. Forth, using our method donor cells can be safely, efficiently and homogenously labeled with lipid conjugated ligands in assigned ratio within 5 min, ensuring derived individual ENVs grafted with targeting ligands. Cell labeling with aptamer conjugated cholesterol only needs to determine the amount of cells and lipidated aptamers. In contrast, neither immobilization of targeting ligands onto EV membrane via click chemistry nor in fusion of EVs with ligands grafted liposomes can be easily and precisely performed at nanoscale (49). Altogether, the extrusion of targeting ligands grafted cells for generating ligands functionalized ENVs is a rapid, easy, and economic approach to prepare sufficient drug delivery vehicles. The enhanced in vivo therapeutic efficacy and low systemic toxicity make this approach conductive to potential clinical translation in the future.
Supplementary Material
Acknowledgments
S.Zheng thanks the Penn State Materials Research Institute, the Huck Institute of Life Sciences, the Penn State Microscopy and Cytometry Facility. This work was supported by Nanjing Science and Technology Development Foundation Number 201605031 to Q. Zheng, Jiangsu Provincial Medical Youth Talent Award QNRC2016054 to L. Wang, Nanjing Medical Science and Technology Development Foundation Number ZDX16008 to X. He, Natural Science Foundation of Jiangsu Province BK20170134 to C. Zhu, and National Cancer Institute of the National Institutes of Health under Award Number DP2CA174508 to S. Zheng.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest
References
- 1.Alderton GK. Diagnosis: Fishing for exosomes. Nat Rev Cancer. 2015;15:453. doi: 10.1038/nrc3990. [DOI] [PubMed] [Google Scholar]
- 2.Im H, Shao H, Park YI, Peterson VM, Castro CM, Weissleder R, et al. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat Biotech. 2014;32:490–5. doi: 10.1038/nbt.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Shen Z, Yu X-Y. Transport of microRNAs via exosomes. Nat Rev Cardiol. 2015;12:198. doi: 10.1038/nrcardio.2014.207-c1. [DOI] [PubMed] [Google Scholar]
- 4.Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–82. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kooijmans S, Vader P, van Dommelen SM, van Solinge WW, Schiffelers RM. Exosome mimetics: a novel class of drug delivery systems. Int J Nanomedicine. 2012;7:e41. doi: 10.2147/IJN.S29661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Che JI, Okeke C, Hu Z-B, Xu J. DSPE-PEG: A Distinctive Component in Drug Delivery System. Current Pharmaceutical Design. 2015;21:1598–605. doi: 10.2174/1381612821666150115144003. [DOI] [PubMed] [Google Scholar]
- 7.van Dommelen SM, Vader P, Lakhal S, Kooijmans S, van Solinge WW, Wood MJ, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. Journal of Controlled Release. 2012;161:635–44. doi: 10.1016/j.jconrel.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Ohno S-i, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Molecular Therapy. 2013;21:185–91. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Boorn JG, Schlee M, Coch C, Hartmann G. SiRNA delivery with exosome nanoparticles. Nat Biotech. 2011;29:325–6. doi: 10.1038/nbt.1830. [DOI] [PubMed] [Google Scholar]
- 10.van den Boorn JG, Schlee M, Coch C, Hartmann G. SiRNA delivery with exosome nanoparticles. Nature biotechnology. 2011;29:325. doi: 10.1038/nbt.1830. [DOI] [PubMed] [Google Scholar]
- 11.King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC cancer. 2012;12:421. doi: 10.1186/1471-2407-12-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi H, Liu C, Long L, Ren Y, Zhang S, Chang X, et al. Blood exosomes endowed with magnetic and targeting properties for cancer therapy. ACS nano. 2016;10:3323–33. doi: 10.1021/acsnano.5b06939. [DOI] [PubMed] [Google Scholar]
- 13.Liga A, Vliegenthart ADB, Oosthuyzen W, Dear JW, Kersaudy-Kerhoas M. Exosome isolation: a microfluidic road-map. Lab on a Chip. 2015;15:2388–94. doi: 10.1039/c5lc00240k. [DOI] [PubMed] [Google Scholar]
- 14.Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–90. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 15.Hartman ZC, Wei J, Glass OK, Guo H, Lei G, Yang X-Y, et al. Increasing vaccine potency through exosome antigen targeting. Vaccine. 2011;29:9361–7. doi: 10.1016/j.vaccine.2011.09.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim TK, Eberwine JH. Mammalian cell transfection: the present and the future. Analytical and bioanalytical chemistry. 2010;397:3173–8. doi: 10.1007/s00216-010-3821-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smyth T, Petrova K, Payton NM, Persaud I, Redzic JS, Graner MW, et al. Surface functionalization of exosomes using click chemistry. Bioconjugate chemistry. 2014;25:1777–84. doi: 10.1021/bc500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennan JL, Hatzakis NS, Tshikhudo TR, Dirvianskyte N, Razumas V, Patkar S, et al. Bionanoconjugation via click chemistry: the creation of functional hybrids of lipases and gold nanoparticles. Bioconjugate chemistry. 2006;17:1373–5. doi: 10.1021/bc0601018. [DOI] [PubMed] [Google Scholar]
- 19.Wang C-F, Mäkilä EM, Kaasalainen MH, Liu D, Sarparanta MP, Airaksinen AJ, et al. Copper-free azide–alkyne cycloaddition of targeting peptides to porous silicon nanoparticles for intracellular drug uptake. Biomaterials. 2014;35:1257–66. doi: 10.1016/j.biomaterials.2013.10.065. [DOI] [PubMed] [Google Scholar]
- 20.Lutz MB, Kukutsch N, Ogilvie AL, Rößner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. Journal of immunological methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 21.Ma H, Liu J, Ali MM, Mahmood MAI, Labanieh L, Lu M, et al. Nucleic acid aptamers in cancer research, diagnosis and therapy. Chemical Society Reviews. 2015;44:1240–56. doi: 10.1039/c4cs00357h. [DOI] [PubMed] [Google Scholar]
- 22.Wan Y, Kim Y-t, Li N, Cho SK, Bachoo R, Ellington AD, et al. Surface-Immobilized Aptamers for Cancer Cell Isolation and Microscopic Cytology. Cancer Research. 2010;70:9371–80. doi: 10.1158/0008-5472.CAN-10-0568. [DOI] [PubMed] [Google Scholar]
- 23.Wan Y, Mahmood MAI, Li N, Allen PB, Kim Y-t, Bachoo R, et al. Nanotextured substrates with immobilized aptamers for cancer cell isolation and cytology. Cancer. 2012;118:1145–54. doi: 10.1002/cncr.26349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan Y, Tan J, Asghar W, Kim Y-t, Liu Y, Iqbal SM. Velocity Effect on Aptamer-Based Circulating Tumor Cell Isolation in Microfluidic Devices. The Journal of Physical Chemistry B. 2011;115:13891–6. doi: 10.1021/jp205511m. [DOI] [PubMed] [Google Scholar]
- 25.Matuszewski B, Constanzer M, Chavez-Eng C. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC− MS/MS. Analytical chemistry. 2003;75:3019–30. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–22. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan Y, Cheng G, Liu X, Hao S-J, Nisic M, Zhu C-D, et al. Rapid magnetic isolation of extracellular vesicles via lipid-based nanoprobes. Nature Biomedical Engineering. 2017;1:0058. doi: 10.1038/s41551-017-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moghimi SM, Patel HM. Tissue specific opsonins for phagocytic cells and their different affinity for cholesterol-rich liposomes. FEBS letters. 1988;233:143–7. doi: 10.1016/0014-5793(88)81372-3. [DOI] [PubMed] [Google Scholar]
- 29.Jeong D, Jo W, Yoon J, Kim J, Gianchandani S, Gho YS, et al. Nanovesicles engineered from ES cells for enhanced cell proliferation. Biomaterials. 2014;35:9302–10. doi: 10.1016/j.biomaterials.2014.07.047. [DOI] [PubMed] [Google Scholar]
- 30.Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen‐presenting cell exosomes are protected from complement‐mediated lysis by expression of CD55 and CD59. European journal of immunology. 2003;33:522–31. doi: 10.1002/immu.200310028. [DOI] [PubMed] [Google Scholar]
- 31.Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017 doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hemler ME. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annual review of cell and developmental biology. 2003;19:397–422. doi: 10.1146/annurev.cellbio.19.111301.153609. [DOI] [PubMed] [Google Scholar]
- 33.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature biotechnology. 2011;29:341–5. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 34.Hovanessian AG, Soundaramourty C, El Khoury D, Nondier I, Svab J, Krust B. Surface expressed nucleolin is constantly induced in tumor cells to mediate calcium-dependent ligand internalization. PloS one. 2010;5:e15787. doi: 10.1371/journal.pone.0015787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine: Nanotechnology, Biology and Medicine. 2016;12:655–64. doi: 10.1016/j.nano.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mongelard F, Bouvet P. AS-1411, a guanosine-rich oligonucleotide aptamer targeting nucleolin for the potential treatment of cancer, including acute myeloid leukemia. Current opinion in molecular therapeutics. 2010;12:107–14. [PubMed] [Google Scholar]
- 37.Ko HY, Lee JH, Kang H, Ryu SH, Song IC, Lee DS, et al. A nucleolin-targeted multimodal nanoparticle imaging probe for tracking cancer cells using an aptamer. Journal of Nuclear Medicine. 2010;51:98–105. doi: 10.2967/jnumed.109.069880. [DOI] [PubMed] [Google Scholar]
- 38.Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nature reviews Immunology. 2002;2:569. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 39.Lai CP, Mardini O, Ericsson M, Prabhakar S, Maguire CA, Chen JW, et al. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS nano. 2014;8:483–94. doi: 10.1021/nn404945r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Storm G, Belliot SO, Daemen T, Lasic DD. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Advanced drug delivery reviews. 1995;17:31–48. [Google Scholar]
- 41.Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. Journal of translational medicine. 2005;3:9. doi: 10.1186/1479-5876-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Molecular therapy. 2008;16:782–90. doi: 10.1038/mt.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Escudier B, Dorval T, Chaput N, André F, Caby M-P, Novault S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. Journal of translational medicine. 2005;3:10. doi: 10.1186/1479-5876-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fais S, O’Driscoll L, Borras FE, Buzas E, Camussi G, Cappello F, et al. Evidence-based clinical use of nanoscale extracellular vesicles in nanomedicine. ACS nano. 2016;10:3886–99. doi: 10.1021/acsnano.5b08015. [DOI] [PubMed] [Google Scholar]
- 45.El-Sahrigy SA, Mohamed NA, Talkhan HA, Rahman AMA. Comparison between magnetic activated cell sorted monocytes and monocyte adherence techniques for in vitro generation of immature dendritic cells: an Egyptian trial. Central-European journal of immunology. 2015;40:18. doi: 10.5114/ceji.2015.50828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nedergaard MK, Hedegaard CJ, Poulsen HS. Targeting the epidermal growth factor receptor in solid tumor malignancies. BioDrugs. 2012;26:83–99. doi: 10.2165/11599760-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 47.Hey SP, Kesselheim AS. The FDA, Juno Therapeutics, and the ethical imperative of transparency. British Medical Journal Publishing Group. 2016 doi: 10.1136/bmj.i4435. [DOI] [PubMed] [Google Scholar]
- 48.Neelapu SS, Locke FL, Bartlett NL, Lekakis L, Miklos D, Jacobson CA, et al. Kte-C19 (anti-CD19 CAR T Cells) induces complete remissions in patients with refractory diffuse large B-cell lymphoma (DLBCL): results from the pivotal phase 2 Zuma-1. Am Soc Hematology. 2016 [Google Scholar]
- 49.Sato YT, Umezaki K, Sawada S, Mukai S-a, Sasaki Y, Harada N, et al. Engineering hybrid exosomes by membrane fusion with liposomes. Scientific reports. 2016;6:21933. doi: 10.1038/srep21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.