

Abstract
We produced 8 lines of transgenic (Tg) rats expressing one of two different rhodopsin mutations in albino Sprague-Dawley (SD) rats. Three lines were generated with a proline to histidine substitution at codon 23 (P23H), the most common autosomal dominant form of retinitis pigmentosa in the United States. Five lines were generated with a termination codon at position 334 (S334ter), resulting in a C-terminal truncated opsin protein lacking the last 15 amino acid residues and containing all of the phosphorylation sites involved in rhodopsin deactivation, as well as the terminal QVAPA residues important for rhodopsin deactivation and trafficking. The rates of photoreceptor (PR) degeneration in these models vary in proportion to the ratio of mutant to wild-type rhodopsin. The models have been widely studied, but many aspects of their phenotypes have not been described. Here we present a comprehensive study of the 8 Tg lines, including the time course of PR degeneration from the onset to one year of age, retinal structure by light and electron microscopy (EM), hemispheric asymmetry and gradients of rod and cone degeneration, rhodopsin content, gene dosage effect, rapid activation and invasion of the outer retina by presumptive microglia, rod outer segment disc shedding and phagocytosis by the retinal pigmented epithelium (RPE), and retinal function by the electroretinogram (ERG). The biphasic nature of PR cell death was noted, as was the lack of an injury-induced protective response in the rat models. EM analysis revealed the accumulation of submicron vesicular structures in the interphotoreceptor space during the peak period of PR outer segment degeneration in the S334ter lines. This is likely due to the elimination of the trafficking consensus domain as seen before as with other rhodopsin mutants lacking the C-terminal QVAPA. The 8 rhodopsin Tg lines have been, and will continue to be, extremely useful models for the experimental study of inherited retinal degenerations.
Keywords: P23H, S334ter, rhodopsin, animal model, rat, retinitis pigmentosa
The clinical problem of inherited and age-related retinal degeneration (RD) is enormous. It is estimated that different forms of hereditary retinitis pigmentosa affect as many as 1:3,500 worldwide, and the incidence of the much more prevalent age-related macular degeneration rises with age, affecting as many as 1:4 by the age of 75. The loss of vision in both classes of diseases is due to the death of photoreceptor (PR) cells, but the retinal pigment epithelium (RPE) is involved or is the site of mutant gene expression in some forms of each type of disease. Retinitis pigmentosa has currently been associated with 67 different genetic loci, and 60 genes have been identified (Daiger, 2017). More than 150 different mutations have been identified in the rhodopsin gene alone (Farrar et al., 2012). Although major progress has been made in elucidating the genetic and molecular nature of many human forms of retinitis pigmentosa (see below), the key issue of how these gene defects lead to PR cell death is largely unanswered.
Many vertebrates and invertebrates have gene defects that lead to RD. Vision scientists have taken advantage of RD mutations in laboratory animals, which have played a prominent role as experimental models in vision research in the past several decades and from which much has been learned about the cellular mechanisms of PR degeneration (Baehr and Frederick, 2009; Chader, 2002; Colley et al., 1995; Stuck et al., 2016; Sung and Tai, 2000; Tanna et al., 2017; Veleri et al., 2015). Among the various species with RDs, mice and rats have been used most extensively, primarily because of the experimental advantages of small animal size with relatively low costs, short gestation time, powerful genetic control in the form of several readily available RD mutants, multiple inbred and congenic strains with genetic controls and different eye pigmentation types, and the potential to carry out certain embryological genetic procedures such as the production of experimental chimeras, transgenic (Tg) and gene knock-in animals (Baehr and Frederick, 2009; Flannery, 1999; Sakami et al., 2011; Veleri et al., 2015).
Among rodents, the number of mouse mutants that may serve as relevant animal models for human diseases is remarkable (Baehr and Frederick, 2009; Chang et al., 2002; Chen et al., 1999; Hafezi et al., 2000; Veleri et al., 2015). Some naturally occurring mouse mutants have human counterparts with orthologous gene defects, such as the rd1, rd2 and Mertk mutants. In addition, many Tg mouse mutants carry constructs that lead to overexpression or disruption of candidate genes for RDs (Chader, 2002; Fauser et al., 2002; Hafezi et al., 2000), as well as knock-in rhodopsin models (Price et al., 2011; Sakami et al., 2011).
The goal of RD research is ultimately to develop therapeutic means to prevent or slow the rate of RD. At present, no generally accepted treatment exists for most of the RDs. However, in the past 2–3 decades, many areas of experimental therapy have arisen and continue to expand significantly to prevent PR degeneration or restore visual function. These include: 1) neuroprotective therapy with direct application of various survival-promoting factors (Abed et al., 2015; Faktorovich et al., 1990; LaVail et al., 1992; Wen et al., 2012), 2) gene-based therapy of recessively and dominantly inherited RDs, as well as viral vector delivery vehicles (Acland et al., 2001; Bennett et al., 1996; Dalkara et al., 2016; Dalkara and Sahel, 2014; Farrar et al., 2012; Laemmli, 1970; Lau et al., 2000; Lewin et al., 1998; Thompson et al., 2015; Trapani et al., 2015; Yang et al., 2015), 3) nanoparticles that act as antioxidants and biodegradable microspheres as non-viral delivery vectors for drug, gene and trophic factor delivery (Adijanto and Naash, 2015; Fernandez-Sanchez et al., 2017; Trapani et al., 2014; Wong et al., 2015; Zarbin et al., 2013; Zulliger et al., 2015), 4) transplantation and cell-based therapy with the use of retinal, RPE and stem cells (Aramant and Seiler, 2002; Li and Turner, 1988; Seiler et al., 2017; Thompson et al., 2015; Yang et al., 2015; Zarbin, 2016), 5) the development of visual prostheses using silicon chip technology (da Cruz et al., 2016; Duncan et al., 2017; Marc et al., 2014; Stingl and Zrenner, 2013), and 6) the field of optogenetics (Dalkara and Sahel, 2014; Duebel et al., 2015; Marc et al., 2014; Zarbin et al., 2013). The need for animal models has increased concomitantly with this research.
Although some therapeutic studies can take advantage of the mouse as an animal model, the small size of the eye is severely limiting for some approaches, particularly when surgical procedures are required. The problem is exacerbated by the early onset of many rodent RDs, thus requiring the use of a particularly small, young mouse eye. Indeed, even the relatively simple delivery of neurotrophic factors by intravitreal injections can be inconsistent or ineffective with very small mouse eyes (LaVail et al., 1998). By contrast, the rat eye is 6–12 times the volume of the mouse eye, depending upon age (LaVail et al., 1998), so the larger eye size of a rat is highly desirable or necessary for many types of therapeutic RD research. The RCS rat is a widely studied model of RD (LaVail, 2001; Strauss et al., 1998), but for decades it had been the only rat model with an inherited RD. Although the RCS rat has an orthologous human gene defect (Gal et al., 2000), and it is particularly interesting because of its mutant gene expression in the RPE (Mullen and LaVail, 1976; Vollrath et al., 2001), there had been no rat model with an RD gene defect intrinsic to the PR cell.
With the goal of creating rat models with gene defects expressed in rod PR cells, like some human forms of retinitis pigmentosa, and to expand the number of rat models available for RD therapeutic research with different rates of RD, we developed a number of Tg rat lines with mutant opsin genes identified in (or similar to) autosomal dominant retinitis pigmentosa (adRP) in human patients (Steinberg et al., 1996). We chose to produce transgenic rats that carry one of two different rhodopsin mutations. The first, P23H, had a proline to histidine substitution at codon 23. This was the first and most common rhodopsin mutation identified in patients with adRP (Dryja et al., 1990), accounting for approximately 12% of adRP cases in the United States (Berson et al., 1991; Sung et al., 1991). We produced 3 lines of P23H rats with different levels of transgene expression and different rates of RD. Second, we produced 5 lines of rats with a mouse opsin transgene bearing a termination codon at residue S334 (S334ter), resulting in a C-terminal truncated opsin protein lacking the last 15 residues. The S334ter opsin lacks all of the phosphorylation sites (Chen et al., 1995) and the QVAPA residues needed for rhodopsin trafficking (Concepcion and Chen, 2010; Sung et al., 1994). We chose these different genetic constructs because they represent two of the different classes of rhodopsin mutations with respect to molecular conformation, cellular processing and localization, and probable pathogenic function in PRs (reviewed by Sung and Tai, 2000), and they generally have different degrees of clinical expression in human adRP patients (Sandberg et al., 1995). In general, the P23H mutation, as a member of several other mutations located in or near the N-terminus of rhodopsin, is considered to result in RD due to defective rhodopsin folding in the endoplasmic reticulum (ER), aggregation and retention within the ER, with resulting ER stress and activation of the unfolded protein response (UPR) (Chiang et al., 2012; Illing et al., 2002; Lin et al., 2007). The RD resulting from truncation of the rhodopsin C-terminus, such as in the S334ter lines, is considered to result in RD due to defective rhodopsin trafficking to the ROSs and mis-localization of the protein (Green et al., 2000; Sung et al., 1994). This truncated protein with an absence of phosphorylation sites also fails to deactivate the rhodopsin molecule and has prolonged responses to light absorption (Chen et al., 1995). Having both type of mutations on the same Sprague-Dawley (SD) background allows for controlled comparison of the different pathological processes.
These Tg rat models were produced in 1996 (Steinberg et al., 1996) and since have been made available to be used by various studies by numerous vision scientists and have used them for various studies (see Discussion). Although some aspects of the RDs in these animals have been described in these studies, only 4 of the 8 lines have been described to any extent; most of these have only been described for a short period of the degeneration; and only a few phenotypic features have been presented or assayed for each line. In the current study, we present relatively comprehensive data on many structural and functional phenotypic characteristics of each of these lines up to at least one year of age. These data should allow vision scientists to use the Tg rats effectively to gain insight into mechanisms of RD caused by rhodopsin mutations and to have rat models with different mechanisms and rates of degeneration with which to carry out experimental therapeutic studies that may be applicable to many different forms of RD. In addition, we also present ultrastructural data that confirms the presumed cytopathological process in the S334ter lines that results from defective rhodopsin transport does not occur in the P23H lines. Moreover, we demonstrate that injury-induced endogenous neuroprotection that occurs in some RDs, and which can interfere with results from utilizing intraocular injections, do not occur in the rhodopsin Tg lines, at least not at ages in which PR degeneration occurs.
2. Materials and methods
2.1 Production of transgenic rats
All work with the animals followed the guidelines of the National Institutes of Health for the care and use of laboratory animals and the UCSF Committee on Animal Research. The Tg rats were generated by Chrysalis DNX Transgenic Sciences (Princeton, NJ; later Xenogen Biosciences) using constructs of two different mouse rhodopsin mutations, P23H (proline to histidine amino acid substitution at codon 23) produced by M.I.N. and S334ter (bearing a termination codon at residue 334, resulting in a C-terminal truncated opsin protein lacking the last 15 amino acid residues and, thus, all of the phosphorylation sites of the molecule) produced by J.C. These constructs have previously been described, (S334ter Chen et al., 1995; P23H Orhan et al., 2015). Three founder rats were generated with the P23H construct and 10 with the S334ter construct, as determined by PCR analysis showing they carried the transgene. We crossed each of these founder rats to wild-type (WT) SD rats and examined their retinas histologically and with PCR (see below). All 3 of the P23H founders produced rats with RD phenotypes in about 50% of the progeny consistent with a Mendelian inheritance pattern of the inserted transgene; the rate of degeneration was different in each of the lines, but all within a given line were virtually identical. The lines were named based on their original cages, with formal nomenclature of SD-Tg(P23H)1Lav, 2Lav or 3Lav (each abbreviated P23H-1, or -2 or -3, or referred to as P23H Line 1, etc.; Table 1). Of the 10 S334ter founders that were crossed by SD rats, 1 did not breed and 9 produced progeny with RD phenotypes in about 50% of each litter, as expected. The RD in 5 of the founders was identical among littermates, whereas it differed widely among littermates from the other 4 founders. We assumed these multiple phenotypes within individual litters indicated multiple transgene insertion sites, so these founders and the infertile one were removed from the study. The remaining 5 S334ter lines were named for their original cages, SD-Tg(S334ter)3Lav, 4Lav, 5Lav, 7Lav and 9Lav (abbreviated S334ter-3, etc., or referred to as S334ter Line 3, etc.; Table 1). The laboratory code “Lav” has been assigned to us by the Institute of Laboratory Animal Research (ILAR). While we and others have not used this in the past, its use as a part of the strain name is now recommended (http://www.informatics.jax.org/mgihome/nomen/strains.shtml).
Table 1.
Lines of rhodopsin transgenic (Tg) rats, terminology, and ratio of the amount of mutant opsin to wild-type, endogenous opsin in the retinas of Tg rats
| Line | Abbreviation | RRRC*# | Rate of Degeneration | RT-PCR Ratio of Tg to Endogenous Opsin mRNA | Westerns Ratio of Tg to Endogenous Opsin |
|---|---|---|---|---|---|
| SD-Tg(P23H)1Lav | P23H-1** | 639 | Moderate | 0.1:1 | NA |
| SD-Tg(P23H)3Lav | P23H-3 | 641 | Slow | <0.05:1 | NA |
| SD-Tg(P23H)2Lav | P23H-2 | 640 | Very Slow | Trace | NA |
|
| |||||
| SD-Tg(S334ter)7Lav | S334ter-7** | 642 | Extremely Fast | 5:1 | 1.4:1 |
| SD-Tg(S334ter)3Lav | S334ter-3 | 643 | Very Fast | 1.5:1 | — |
| SD-Tg(S334ter)5Lav | S334ter-5 | 644 | Fast | 0.4:1 | 1:1 |
| SD-Tg(S334ter)4Lav | S334ter-4 | 645 | Moderate | 0.1:1 | 0.7:1 |
| SD-Tg(S334ter)9Lav | S334ter-9 | 646 | Very Slow | <0.05:1 | 0.05:1 |
Rat Resource & Research Center Strain #, Columbia, Missouri
or P23H Line 1, S334ter Line 7, etc.
To eliminate the need to identify every Tg animal from its WT littermate by PCR, we attempted to breed the Tg rat lines to homozygosity by intercrossing two hemizygotes, discarding those not carrying a rhodopsin transgene (approximately 25% of the litter) and identifying hemizygotes (assumed to be approximately 50% of the original progeny) from homozygotes (approximately 25% of the progeny) by progeny-testing Tg animals by backcrossing with SD rats. Homozygous rats (carrying transgene insertions at both alleles) then were bred by one another to maintain the lines with no need for PCR, and they were bred to WT SD rats for experiments to produce all affected hemizygous Tg rats, thereby avoiding the need for PCR for every litter and giving twice the number of usable animals for experiments. All lines were bred to homozygosity except S334ter-5 and S334ter-7, which could not be accomplished despite 20+ attempts for each line over a 5-year span. The explanation for this is unknown, but presumably it relates to fertility issues or embryonic lethality as a consequence of the transgene insertion sites.
The rats were maintained in a 12:12 hr light:dark environment with cyclic white illumination provided from overhead fluorescent lamps in the ceiling. The illuminance levels within the cages varied considerably, from < 1 ft-c up to 15 ft-c, depending upon the position of the cage in the rack and the position of the rack in the room. The housing rooms were temperature controlled at 23 ± 1°C and the animals were fed various rat chows ad libitum over the years.
The production of these animals was announced previously (Steinberg et al., 1996), as was their availability to vision scientists. In 2015, all of the Tg rat lines were moved to the Rat Resource and Research Center (RRRC) at the University of Missouri, where they are maintained either live or cryopreserved and are available to scientists. The RRRC# for each line is given in Table 1. The website of the RRRC is http://www.rrrc.us/ and information from the RRRC can be obtained at RRRC@missouri.edu.
2.2 RNA analysis
Six-week-old F1 transgenic offspring from founders (and nontransgenic littermate controls) were sacrificed, and total cellular RNA was prepared from retina using RNA stat-60 (Tel-Test, Inc) or RNeasy kit (Qiagen). Total RNA was reverse transcribed using RT-PCR kit (Stratagene) or Advantage RT-for PCR kit (Clontech). An aliquot of this RT product was then used as template for quantitative PCR analysis. Alternatively, a small amount of tail tissue was used.
For the P23H lines, the qRT-PCR amplification procedure described by Orhan et al. (2015) was followed. Each amplification was performed with two sets of primers designed to amplify a 300 base pair segment from the mouse P23H opsin transcript (5′-AACCTTATCTTCTGGGAAAGGC-3′ and 5′-GAAGACCAGCACTAGCGGTG-3′) and two primers to amplify a 450 base pair segment from the native rat opsin transcript (5′-GGGGCCTATGACAATCCACCTG-3′ and 5′-CCCCCTGAATCCTGCTTTGTAA-3′).
For the S334ter lines, the qRT-PCR procedure described by Chen et al. (1995) was followed. Each amplification was performed with two primers, designed to amplify a 350 base pair segment from the mouse opsin transcript (5′-TGGGAGATGACGACGCCTAA-3′ and 5′-TGAGGGAGGGGTACAGATCC-3′) and two primers to amplify a 450 base pair segment from the native rat opsin message (5′-GGGGCCTATGACAATCCACCTG-3′ and 5′-CCCCCTGAATCCTGCTTTGTAA-3′).
The PCR products were resolved on a 1.5% agarose gel, stained with ethidium bromide, and photographed. The pictures were scanned and the band intensity was determined with IP gel pro 3.0 software.
2.3 Protein analysis
For Western Blot analysis, rat retinas were immediately frozen following separation from the RPE. The procedure used for the S334ter lines was as described by (Green et al., 2000). The difference in protein expression in the P23H lines cannot be seen by Western Blot analysis because of the similarity in the size of mutant and WT opsin.
2.4 Electroretinographic (ERG) analysis
Rats were dark-adapted overnight and then anesthetized with intramuscular injections of xylazine (13 mg/kg) and ketamine (87 mg/kg) in dim red light. The corneas of the rats were anesthetized with a drop of 0.5% proparacaine hydrochloride, and the pupils were dilated with 1% atropine and 2.5% phenylephrine hydrochloride. Full-field scotopic and photopic ERGs were elicited and recorded as described previously (Lewin et al., 1998; Roddy et al., 2012), To measure scotopic ERG responses, stimuli were presented at intensities from -4.6 to 2.4 log cd sec m−2 at inter-stimulus intervals ranging from 5 sec at lowest intensities to 60 seconds at the highest intensity, although the S334ter-9 rats with the lowest amount of mutant rhodopsin (Table 1) showed prolonged recovery and required much longer inter-stimulus intervals (see Results).
2.5 Retinal histology
As described previously (LaVail and Battelle, 1975), rats were euthanized at various ages by overdose of carbon dioxide inhalation and immediately enucleated and immersed into a mixture of combined aldehydes (2% paraformaldehyde and 2.5% glutaraldehyde). Alternatively, some rats were intravascularly perfused with the fixative immediately after being euthanized, as described above, after which their eyes were dissected out of the orbit. Eyes were bisected, post-fixed in osmium tetroxide, and embedded in epoxy resin, and single 1-μm thick histological sections were made along the vertical meridian. The thickness of the outer nuclear layer (ONL) was taken as a measure of PR number (Michon et al., 1991). For each retina, the mean ONL thickness was obtained from 54 measurements taken at equidistant points around the eye; these values were averaged to obtain a single ONL value for each retina, which was used for used for statistical analysis as described (Faktorovich et al., 1992). In addition, groups of 3 measurements in each 440-μm microscopic field were averaged and the data were plotted to show specific regional differences across the section (Faktorovich et al., 1992), as first done by Rapp and Williams (1980) and later known as a retinal spidergram. After light microscopic examination, selected areas were thin-sectioned and examined by transmission electron microscopy as described (Carter-Dawson and LaVail, 1979a). For the identification and quantification of cone PR nuclei, we used the criteria developed for analysis of light-damaged (LaVail, 1976b), RCS inbred (LaVail, 1981) and RCS congenic strains (LaVail et al., 1975) of rats.
2.6 Rhodopsin measurements
Rhodopsin content in each Tg line was measured and in control SD rats at different ages. The method was that described by Smith (1992). Two eyecups from each dark-adapted rat were pooled, homogenized in buffer followed by centrifugation, and the supernatant was collected and its absorbance spectrum measured from 450–550 nm using a Spectronics Genesys 5 spectrophotometer. The retina sample was bleached by light (60 W at a distance of 10 cm for 1 min), and then the absorbance spectrum was measured; the decrease in absorbance at 498 nm was used to assess the amount of rhodopsin in the tissue.
3. Results
3.1 General phenotypic features of the Tg lines
3.1.1 Mutant opsin in the retina
The rates of degeneration of the 8 lines of mutant rhodopsin are shown in Figure 1 and described in Table 1. The rate of PR degeneration in the Tg rats is approximately proportional to the amount of mutant opsin in the retinas expressed as a ratio of mutant:WT opsin when both the mRNA and protein levels are measured (Table 1). The Tg opsin mRNA in the slowest RDs (P23H-2 and S334ter-9) is only a trace or very low amount, whereas the fastest RDs contain 1.5 (S334ter-3) or 5 (S334ter-7) times the WT endogenous opsin mRNA (Table 1). Thus, much greater Tg opsin protein is found in the retinas of the fastest degenerating lines than in the slower ones (Table 1).
Figure 1.

Outer nuclear layer (ONL) thickness of WT and Tg rats. ONL thickness is proportional to the number of PR nuclei, in Sprague-Dawley albino WT (WT) rats and the 3 different lines of Tg rats carrying the P23H (A) and 5 different S334ter (B) mutant rhodopsin transgenes. The values are the means of 54 measurements taken along the vertical meridian of one retina from 3–5 rats at each age. The variance from these means in most cases falls within the symbol, so error bars have been omitted for clarity. In most cases, the SEM was <1.0 mμ, and in all cases after P10, it was <2.0 mμ except for S334ter-3 at P15, where the SEM was 3.3 mμ. For conversion to rows of PR nuclei, in the 1-μm thick plastic sections, each rod nucleus is approximately 4.5 μm in diameter.
3.1.2 Progressive loss of photoreceptor cells
The loss of PRs is first indicated by the presence of pyknotic nuclei (Table 2) followed by progressive thinning of the ONL (Figs. 1–3). The onset and pace of degeneration among the 8 lines of rhodopsin Tg lines range from extremely fast (S334ter-7), with earlier onset and rate of RD faster than even that in the rd1 mouse, to those with very slow rates of RD in which the retinas are indistinguishable from normal WT for up to 2 (P23H-2) or 4 months of age (S334ter-9), followed by a very slow degeneration thereafter (Table 2, Figs 1 and 2).
Table 2.
Degenerative retinal changes in P23H and S334ter rhodopsin Tg rats
| Line | P4 | P6 | P8 | P10 | P12 | P15 | P21 | P30 | P60 | P90 | P120 | P180 | P240 | P365 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P23H-1 | = | = | = | PNx | PN, ONL* | PN, ONL76, IS, OS, MG, MGo | PN, ONL, IS, OS, MGi, MGo | PN, ONL51, IS, OS, MGi | PNx, ONL27, ISx, OSx, MG | PNx, ONL†, ISx, OS†, RPEa | ONL†, ISx, OS†, MCh | ONL†, IS†, OS†, MCh, RPEa | ONL†, IS†, OS†, MCh, MCs, INL, IPL, MN, RPEa, RPEn | ONL†, IS†, OS†, MCh, MCs, INL, IPL, MN, RPEa, RPEn |
| P23H-3 | – | = | = | = | PNx, ONL* | PNx, ONL, OS | PN, ONL, IS, OS MGo | PN, ONL77, IS, OS MGo | PN, ONL, IS, OS, MCh. MG, MGi, MGo | PN, ONL60, IS, OS, MCh, MGi, MGo | ONL40, IS, OS, MCh, MGi, MGo | ONL, ISx, OSx, MCh | ONL28x, ONL†, ISx, OS†, MCh | ONL†, IS†, OS†, MCh, MCs, RPEn, RPEa, IPL |
| P23H-2 | – | – | – | – | = | = | PNx | PNx, MGi**, MGo** | PNx, ONL*, MGi**, | PNx, ONL | PNx, ONL, IS, OS | PNx, ONL73, IS, OS, MCh | ONL, PNx, IS, OS, MCh | PNx, ONL61, ISx, OSx, MCh |
| S334ter-7 | PNx, fewMG | PN, dPR, IS, MG, MGi | PN, ONL80*, dPR, ISx, OS†, MG, MGi | PN, ONL61, dPR, ISx, OS†, MG, MGi | PN, ONL24x ONL†, dPR, ISx, OS†, MG, MGi | PN, dPR, ISx, OS†, ONL†, fewMG | PNx, ONL†, dPR, IS†, OS†, fewMG | PNx, ONL†, IS†, OS† MG** | ONL†, IS†, OS†, MCh, RPEa | ONL†, IS†, OS†, MCh, RPEa | ONL†, IS†, OS†, MCh, RPEa | ONL†, IS†, OS†, MCh, RPEa | – | ONL†, IS†, OS†, MCh, RPEa |
| S334ter-3 | PNx | PNx | PN, dPR, OS† | PN, ONL*, dPR, IS, OS†, MG MGo | PN, ONL55*, dPR, ISx, OS†, MG, MGo | PN, ONL34, dPR, ISx, OS†, MG | PNx, ONL†, ISx, OS†, MG** | PNx, ONL†, IS†, OS†, MCh | PNx, ONL†, IS†, OS†, MCh, IPL, MN | ONL†, IS†, OS†, MCh, IPL, MN, RPEa | ONL†, IS†, OS†, MCh, IPL, MN, RPEa, RPEn | ONL†, IS†, OS†, MCh, IPL, MN, RPEa, RPEn | – | ONL†, IS†, OS†, MCh, IPL, MN, RPEa, RPEn |
| S334ter-5 | = | = | dPR, OS | PNx, dPR, OS, fewMG | PN, ONL*, dPR, IS, OS, OSd, MG, MGo | PN, ONL75, dPR, IS, OS, OSd, MG, MGo | PN, ONL50, dPR, ISx, OS, OSd, fewMG | PNx, ONL40, dPR, ISx, OSx, fewMG, MGi** | ONL†, ISx, OS† | ONL†, IS†, OS†, MCh, MN, MG** | ONL†, IS†, OS†, MCh, RPEa, RPEn | ONL†, IS†, OS†, MCh, RPEa, RPEn, IPL | ONL†, IS†, OS†, MCh, RPEa, RPEn, IPL | ONL†, IS†, OS†, MCh, RPEa, RPEn, IPL |
| S334ter-4 | – | = | = | = | PNx | PNx, OSv | PN, ONL*, OSv, fewMG, fewMGo | PNx, ONL76, OS, OSv, fewMG | PNx, ONL50, IS, OSx, MG**, fewMGo | PNx | PNx, ONL34x, ONL†, IS†, OS†, MCh, MG** | ONL†, IS†, OS†, MCh, RPEa | ONL†, IS†, OS†, MCh, RPEa, RPEn, IPL | ONL†, IS†, OS†, MCh, RPEa, RPEn, IPL |
| S334ter-9 | – | – | – | – | = | = MG** | = | = | = | = | ONL* | ONL, PNx, MG** | ONL, PNx, IS, OS | ONL75, PNx, ISx, OSx, MG** |
=, no difference from normal SD retina; –, not examined.
For each transgenic line, the observations are based on examination of the eyes from 2–5 rats at each age, and compared to those of 3 SD control rats at each age.
Note: Where superior/inferior asymmetry exists, the earliest changes are typically seen in the superior posterior retina, which are indicated here as the first degenerative changes; these are not seen in the peripheral or inferior retina until slightly later ages.
PN, pyknotic nuclei number in ONL greater than normal (2+ per 180-μm field). PNx, more PN than normal, but < 2 per 180-μm field.
ONL*, overall outer nuclear layer first thinner than normal (P < 0.5). ONL, ONL thinner than normal. ONLxx, % of control of ONL as close to 75%, 50% and 25% of control as was measured. ONL†, ONL reduced to less than one complete row in most degenerated region. (Note: the quantitative measure of % of control ONL thickness is an overall measurement {see Methods}, whereas the ONL† is descriptive of the most degenerated region. Thus, P23H-3 at P240, for example, has an ONL that is 28% of control overall, but is less than one complete row in the superior posterior retina.)
IS, inner segments shorter than normal. ISx, IS reduced to very short, thin nubs. IS†, IS missing.
OS, outer segments shorter than normal. OSx, OS reduced to very short nubs. OS†, OS missing. OSd, OS more disorganized than normal. OSv, many OS vacuolated or pale-staining.
dPR, displaced photoreceptors in outer segment zone greater number than normal.
INL, inner nuclear layer thinner than normal.
MCh, Müller cell hypertrophy in the IPL.
MCs, Müller cell seal in outer retina.
MN, apparent microneuromas in INL.
RPEn, RPE neovascularization from retinal capillaries.
RPEa, RPE attenuation at focal points.
IPL, strands of INL nuclei cross the IPL.
Figure 3.

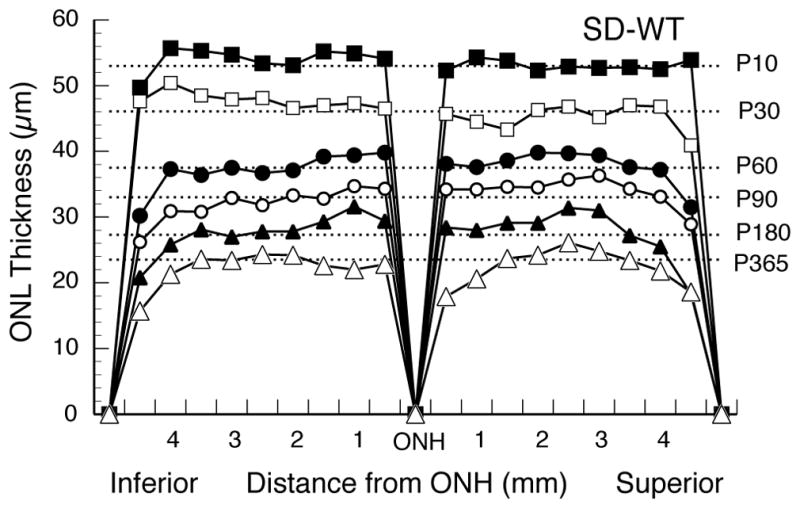
Retinal Spidergrams of the ONL thickness of Sprague-Dawley WT rats at different ages. Each ONL data curve is the mean of 3–5 rats, and each data point is the mean of 3 adjacent measurements; thus, the 54 ONL measurements are reduced to 18 points across the retina, 9 in each of the superior and inferior hemispheres along the vertical meridian. Error bars are omitted for clarity. Dashed lines are given for the mean ONL thickness at ages ranging from P10 to P365. The curves illustrate the normal retinal thinning with age, the symmetrical ONL thickness in the two hemispheres along the vertical meridian, and the thinning of the ONL near the periphery of the retina compared to that of the central retina.
Figure 2.

Different phenotypic characteristics of WT and Tg rats at ages up to P365. The values are expressed as a percent of control (WT), shown as a dashed line. The P23H lines (AC) and S334ter lines (D–H) are each arranged in descending order of PR degeneration rate, with the most rapidly degenerating given first. ONL, solid line; X, ONL of homozygote; ROS length, shaded bars; whole-eye rhodopsin, open circles. ERG amplitudes in each of the Tg rat lines up to P365 or until the waves are extinguished. Scotopic a-wave (blue line); scotopic b-wave (green line); photopic b-wave (red line). The amplitudes are in response to saturating stimuli. The most rapidly degenerating lines, S334ter-7 and S334ter-3, showed no ERG responses. The values are the means of 3–5 rats at each age. The scotopic b-waves shown are from stimuli presented at an intensity of 0.4 log cd-s/m2, followed by a stimulus at 2.4 log cd-s/m2 to elicit the scotopic a-wave. The rats were then exposed to a background light of 29 cd/m2 for 10 minutes before they were exposed to stimuli of 0.4 log cd-s/m2 to elicit the photopic b-wave. The error bars have been omitted for clarity. As noted in the legend to Fig. 1, the variance in the raw data upon which these percent of control values were calculated was small for the ONL, most being <1.0 mμ and all but one being <2.0 mμ. The SEMs of all of the rhodopsin and rod outer segment lengths after P10 were mostly <1.0 mμ, with a few <2.0 mμ. The ERG measurements were intrinsically more variable, but the SEMs were still mostly <20.0 μV, with almost all <40μV.
A note should be made about the interpretation and limitations of retinal spidergrams (Figs. 3 and 4) and the overall average of 52 ONL measurements in a given retinal section. Retinal spidergrams give an excellent visual image of the distribution of surviving PR nuclei in one plane of a retina (vertical meridian in this case). However, each plotted data point is the mean of 3–5 values, each from a different animal. In the case of identification of the first age at which the ONL is totally obliterated at one region, that region may be slightly different in different retinas, thus masking a zero value. For example, in the rapidly degenerating S334ter-3 line at P19, the ONL does not reach zero in the spidergram (Fig. 4E), yet in individual rats at P19-P20, it is possible to see total loss of the ONL in small focal regions (Fig. 11F). Likewise, the overall average of all 52 ONL measurements in each retina (Figs. 1 and 2) obscures the minimal ONL thickness, but microscopic observation of a series of ages shows the first observed total loss of ONL in any region of the retina (ONL† in Table 2).
Figure 4.

Retinal spidergrams of each of the Tg lines illustrate hemispheric asymmetry in rate of degeneration. In each of the P23H (A–C) and P334ter (D–H) lines, the superior hemisphere degeneration is greater than that in the inferior hemisphere at some point in the degenerative period, to a greater or lesser extent in each line. Different ages are plotted, depending upon the rate of degeneration, but dashed lines are given for the mean ONL thickness at ages ranging from P12 to P365.
Figure 11.
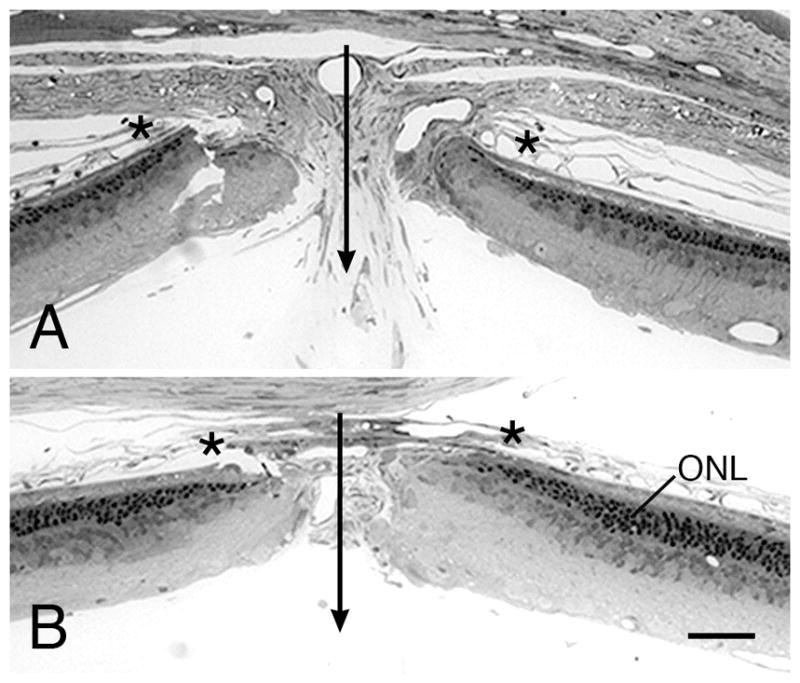
S334ter-3 rats - Light micrographs of the outer retina at ages ranging from P6 (A) to P30 (G). Ages given on the micrographs. All micrographs are taken from the superior posterior to equatorial regions of the eyes, except H, which is from the inferior posterior hemisphere in the same retinal section as F. (A) Developing ONL at P6, which is indistinguishable from that of normal controls, except for a greater number of pyknotic nuclei (arrows). Displaced PR cells (d) are present in the IPS from P8-P15 (B–E). PR nuclei are lost from the ONL during this time, and at the period of most rapid PR loss, P10-P12, the greatest number of pyknotic nuclei in the ONL is in the innermost region of the layer (C, D, arrows). At this time, presumptive microglia are abundant in the ONL (C, D arrowheads). Occasionally, a very large, pale nucleus is found in the ONL (*), which may be a cell in some phase of mitotic division, as the invading microglia are known to proliferate in the ONL. An incomplete single row of nuclei is first seen in the superior posterior retina at P20 (F); a complete row is still present in the same region of the inferior hemisphere (H). At P30 (G) and thereafter, only scattered PR nuclei are present in the retinas. r, rod cell nucleus; c, cone cell nucleus. Magnification bar = 20 μm.
3.1.3 Hemispheric asymmetry and gradients of ONL degeneration
As is widely appreciated, the ONL of WT rats becomes thinner with age, particularly during the first 2 months (Figs. 1 and 3), due to rapidly expanding eye size without the generation of new cells. Retinal spidergrams of ONL thickness along the vertical meridian and at various ages of WT rats demonstrate typical uniformity of ONL thickness in both hemispheres of the retina and thinning of the ONL near the periphery in comparison to that of the central retina (Fig. 3). This is shown by the 1–2 most peripheral data points being somewhat smaller than those in the posterior retina.
The onset and pace of PR degeneration in the Tg lines proceeds in a central-peripheral gradient. This is most readily seen at early stages of the most rapidly degenerating Tg lines, S334ter-7, S334ter-3 and S334ter-5, where the central retina has already lost most PRs, while fewer have been lost in the peripheral retina (Figs. 4D–F). Unlike the WT retina and at all ages, the Tg lines with a more moderate rate of PR loss, P23H-1, P23H-3 and S334ter-4 (Figs. 4A, B and G) at most ages have a peripheral ONL thickness the same or similar to that in the central retina, unlike that in WT rats (Fig. 3), thus indicating that the central retina has degenerated more rapidly than the peripheral retina. The P23H-2 line with very slow PR degeneration shows a more subtle central-peripheral gradient based on this criterion (Fig. 4C), and the S334ter-9 line (Fig. 4H) shows a nearly normal WT ONL thickness, as expected from the low level of transgene expression (Table 1).
A hemispheric difference exists in each of the Tg lines. Thinning of the ONL due to degeneration and loss of PRs is greater in the superior hemisphere than in the inferior hemisphere in each of the Tg lines, at least at some age(s) in the degenerative period (Fig. 4). This asymmetry in rate of degeneration is subtle in the most rapidly degenerating lines, such as S334ter-7 (Fig. 4D) and S334ter-3 (Fig. 4E), and in the very slowly degenerating line S334ter-9 (Fig. 4H). However, it is much more obvious in the other lines (Fig. 4). The S334ter-4 line (Fig. 4G) shows the greatest asymmetry at all ages beginning at P21. This line was explored further at P60, when the superior-inferior asymmetry was more obvious (Figs. 4G and 5A). Diagonal sections taken approximately 45° from the vertical meridian either along the inferior-temporal to the superior-nasal axis (Fig. 5B), or from the inferior-nasal to the superior-temporal axis (Fig. 5C) reveals that most of the inferior hemisphere has the slowest rate of degeneration throughout the hemisphere. A horizontal section taken immediately above the horizontal meridian (Fig. 5D) shows that the faster rate of degeneration in the superior hemisphere (Figs. 5A–C) begins immediately above the horizontal meridian, and that combined with Figs. 5A–C, it can be seen that the degeneration is faster throughout the superior than in the inferior hemisphere.
Figure 5.

Positional asymmetry in ONL of S334ter-4 retinas at P60. Section along the vertical meridian as in Fig. 4. Diagonal sections approximately 45° from the vertical meridian are shown in B and C. Thus, most of the inferior hemisphere has the slowest rate of degeneration throughout the hemisphere. Horizontal section just above the optic nerve head (D) reveals that the faster rate of degeneration in the superior hemisphere (A–C) begins immediately above the horizontal meridian and is faster throughout that hemisphere than in the inferior hemisphere.
3.1.4 Rod outer segment length and rhodopsin content in the degenerating retinas
In the two lines with earliest onset and most rapid loss of PR cells, S334ter-7 and S334ter-3, no rod outer segments (ROSs) could be detected (Figs. 2D and 2E), and this was further confirmed in S334ter-3 rats by immunocytochemistry (Martinez-Navarrete et al., 2011) and electron microscopy (data not shown). All of the other lines showed reduction in overall length of ROSs that roughly paralleled the ONL thickness (Fig. 2), even though there was variation within the eye, as with the ONL thickness.
Our measurements of whole-eye rhodopsin content were generally 10–15% lower in percent of WT control than (OS) length, but the levels generally fell as PRs were lost, as did OS length. In another study of the P23H-3 line at P60 (Bicknell et al., 2002), the rhodopsin content was 50% of that in WT retinas, as was ours (by interpolation) (Fig. 2B).
In that study, as well, the percent of control number of PRs surviving was estimated to be greater than that of rhodopsin, as seen in the present study. In the degenerations with earliest onset (S334ter-7 and S334ter-5) where rhodopsin was measured at very early age (P8), even in the absence (Fig. 2D) or extremely short (Fig. 2F) PR OSs, the rhodopsin levels were higher than normal (P=0.01 and P=0.0175 in the S334ter-7 and S334ter-5 lines, respectively), and this was found in 3 separate experiments with each of these lines. Each of these sets of measurements compared 4–6 retinas from Tg and WT littermates. The rhodopsin content of S334ter-3 retinas was also greater at P8 than WT, whereas those of all slower lines were not greater than WT at this age; since littermates (exactly the same age) were not compared and there was some variation in rhodopsin content of SD retinas measured on different days, we could not do a statistical analysis on these groups. Nevertheless, the lines with the fastest RDs consistently showed a greater amount of rhodopsin at P8 than those with slower RDs. Presumably, the higher than normal levels of rhodopsin in the rapidly degenerating retinas results in the PR degeneration, because overexpression of either mutant or WT rhodopsin is toxic to PRs (Chen et al., 1995; Li et al., 1996; Olsson et al., 1992; Tan et al., 2001).
3.1.5 Gene dosage effect on degeneration rates in the Tg lines
As noted in the Methods, we produced homozygous Tg rats of all lines except S334ter-5 and S334ter-7. When we examined the dose effect of the transgene on the ONL thickness by assessing hemizygotes versus homozygotes for each line (Fig 2), it was clear that the retinas from the homozygotes (two copies of the transgene) from all lines were more degenerated than the hemizygotes (single copy of the transgene) of the same age. Two copies of the transgene affected the P23H lines less than they did the S334ter lines (where homozygous rats could be produced). For example, in the P23H-1 line, the ONL was hardly changed from that of the hemizygotes, the means of all measurements in the P23H-1 line being only 5.6% less than those of the hemizygotes (Fig. 2A). In the slower degenerating P23H-3 and P23H-2 lines, the means of the 3–4 homozygote ONL measurements were 20.6% and 22.4%, respectively, thinner than the values for the hemizygotes (Figs. 2B, C). By contrast, the means of the ONL measurements in the S334ter-3, S334ter-4 and S334ter-9 lines were 52.8%, 41.6% and 59.8%, respectively, thinner than the values of the hemizygotes (Figs. 2E, G, H).
The S334ter-9 line, in which the hemizygotes have the lowest amount of truncated opsin expression (Table 1), have an almost normal structure throughout the first year of life (Figs. 2H and 14D). However, their homozygote counterparts at P180 and P300 showed a marked effect on the number of surviving PRs (Fig. 2H). This suggests that in the S334ter-9 line, there is a threshold of mutant opsin that affects PR viability. Another unsuspected difference in hemizygotes and homozygotes occurs in the rapidly degenerating S334ter-3 line. As noted by Li et al. (2010), the homozygous S334ter-3 line had little or no difference in structural appearance to the hemizygotes. However, in neuroprotective studies, various factors had a greater protective effect in hemizygous animals than in homozygotes, which sometimes had no effect (LaVail et al., unpublished observations; confirmed by Wen, personal communication). Thus, even in the appearance of minimal negative effect, higher dose of the mutant opsin protein can have a more deleterious effect on the PRs.
Figure 14.

S334ter-9 rats - Light micrographs of the outer retina at ages ranging from P90 (A) to P365 (D). Ages given on the micrographs. All micrographs are taken from the superior posterior to equatorial regions of the eyes. At P90 (A), the retina appears normal. Thereafter, (B–D) the ONL progressively thins, pyknotic nuclei (arrow) are occasionally seen, and the ROSs shorten slightly. Phagosomes (p) are seen in the RPE cells. c, cone cell nucleus; r, rod cell nucleus. Magnification bar = 20 μm.
3.1.6 Invasion of phagocytic cells to the outer retina
In many forms of RD and other neuronal degenerations, cell death attracts immunocompetent cells that phagocytize cellular debris, and may play important roles in the degenerations. The bulk of evidence concludes that these are resident microglia that are activated, after which they migrate to the site of injury/degeneration, proliferate, phagocytize remains of degenerating cells and often enlarge (see Discussion). This invasion of presumptive microglia has been examined at a few ages of 3 lines of the rhodopsin Tg rats, S334ter-3 (Zhu et al., 2013a), S334ter-4 (Glybina et al., 2010; McGill et al., 2012b) and the homozygous P23H-3 (Noailles et al., 2014) lines. In this study, we examined all of the lines for the presence of presumptive microglia at several ages listed in Table 2.
The invading cells are characterized by a pale-staining nucleus with a sharp peripheral rim of heterochromatin, sometimes with a prominent nucleolus (e.g., Figs. 7N–P and Figs. 10B–F). The cells can be found in the outer plexiform layer (MGo in Table 2; e.g., Fig. 7N, arrowhead), ONL (MG; e.g., Figs. 11C, D) and interphotoreceptor space (IPS) (MGi; e.g., Figs. 7P and 13H), with a few apparently migrating through the inner nuclear layer (Fig. 7O).
Figure 7.

P23H-1 rats - Light micrographs of the outer retina at ages ranging from P10 (A) to P280 (G, H). Ages given on the micrographs. Regions of retina: (A–F) superior posterior; (G, I, K) inferior posterior; (H) superior ora serrata; (L) inferior ora serrata. An increase in pyknotic nuclei is present at P10 and P12 (A, B, arrows). (C, D, F) PR nuclei are progressively lost, along with the shortening of rod inner and OSs, with the first obliteration of the ONL in foci at P120 (F). The greater degree of PR degeneration in the posterior superior hemisphere than that in the same regions of the inferior hemisphere is shown at P30 (D, I), P50 (E, J) and P120 (F, K). A central-to-peripheral gradient of degeneration is shown at P120 between the more degenerated inferior posterior region (K) and the region near the ora serrata of the same eye section (L). Phagosomes (p) in the RPE and the RPE processes are indicative of OS disc shedding; the small number is typical of the daytime, when most of the eye specimens were taken (D, J), but many are present when the eye was taken during the burst of shedding within the first 90 minutes of light onset in the morning (M). When most PR nuclei have degenerated and disappeared, a few rods (r) and cones (c) persist. At late stages (G), the RPE is frequently vascularized (v) and strands of inner nuclear layer nuclei are displaced through the inner plexiform layer (IPL). (H) A small cluster of PR nuclei typically persists at the ora serrata for up to at least a year of age. Both rod (r) and cone (c) PR nuclei are present at all stages of degeneration, even in the oldest ages (G, H). Invading presumptive microglia (arrowheads) are present in the outer plexiform layer (N), ONL (O), INL (O) and IPS (I, O, P). The cells are phagocytic, and the pale microglial cell process shown among the PR ISs in (O) is filled with ingested debris; it cannot be determined whether this process communicates with the nearby microglial cell nucleus in the ONL, or with another cell out of the plane of section. The cell in the IPS of (I) contains internal phagosomes and appears to be ingesting a displaced rod cell. INL, inner nuclear layer; IPL, inner plexiform layer; pep, RPE cell processes. Magnification bar = 20 μm.
Figure 10.

S334ter-7 rats - Light micrographs of the outer retina at ages ranging from P4 (A) to P30 (L). Ages given on the micrographs. Regions of eye: (A–D and F–H), superior posterior; (E) superior peripheral; (I, K) inferior posterior; (J) inferior peripheral; (L) inferior ora serrata. (A) Pyknotic nuclei (arrows) are seen in the developing outer nuclear layer (ONL) at P4, and then many of the innermost immature PR nuclei in the ONL rapidly become pyknotic at P6 (B), P7 (C), P8 (D) and P10 (E). At these same ages, many displaced PR nuclei (d) are present in the IPS (B–E). Beginning at about P8, pyknotic nuclei rapidly disappear (D–F), so that by P12 in the superior posterior retina, there is a loss of PRs (F), which becomes more extensive at P15 (G) and P21 (K). A cluster of PRs persists at the ora serrata at P30 (L). Invading cells, presumptive microglia, (arrowheads) that are presumably phagocytic and remove pyknotic cells are widely present in the inner part of the ONL at most ages, with some in the subretinal space (E). Pyknotic displaced PR nuclei are present in the IPS (1), and the RPE apparently participates in the removal of dead cells by phagocytosis (2). Once PR inner and OSs disappear (G, H), the IPS is filled with RPE cell processes (pep). c, cone nucleus; r, rod nucleus. Magnification bar = 20 μm.
Figure 13.
S334ter-4 rats - Light micrographs of the outer retina at ages ranging from P14 (A) to P120 (E, G). Ages given on the micrographs. All micrographs are taken from the superior posterior to equatorial regions of the eyes, except G and H, which are from the inferior posterior hemisphere in the same retinal sections as D and E, respectively. At P14, the ONL is the same thickness as SD controls, although pyknotic nuclei (arrows) are more common than in SD retinas. As PR nuclei are lost, the ONL thins by P21 (B) and P30 (C), and thereafter. The hemispheric asymmetry in rate of PR degeneration is most marked in this S334ter-4 line than in all the rhodopsin Tg lines, with the superior hemisphere degenerating much more rapidly than the inferior hemisphere, as evident in (D) and (G) at P60. The ROSs (os) are as long as those in SD retinas at P15, P21, P30 and P60 (A–C, G), but they show some irregular and vesicular profiles and appear slightly frothy in some cases, with alternating ROS and pale spaces parallel to them. Phagosomes are present within the RPE cell bodies and processes (F, G). (H) Presumptive migrating and phagocytic microglia are shown at arrowheads in the ONL and the interphotoreceptor space (IPS). c, cone cell nucleus; pep, RPE cell processes; p, phagosomes; r, rod cell nucleus. Magnification bar = 20 μm.
These presumptive microglia are present in the outer retinas where the degeneration and disappearance of PR nuclei is most actively occurring (Table 2). Thus, in the very slowly degenerating P23H-2 and S334ter-9 lines, only 2–3 such cells were seen in the entire collection of sections at all ages (Table 2), and this was the same as the 2 such cells found in all of the WT SD retinas (data not shown). In the rapid S334ter-7 and S334ter-3 degenerations, where the highest incidence of pyknotic nuclei is in the innermost ONL, that is the same location of the majority of presumptive microglia (Figs. 10C–E and Figs. 11C, D, respectively). The overall highest incidence coincided with the ages where PR degeneration was greatest. For P23H-1, these were P15-P60, with the highest incidence at P21-P30. For P23H-3, these were P21-P120, with the highest incidence at P30-P60, but the density of presumptive microglia in any one section of the P23H-3 rats was lower than in the faster degenerating P23H-1 line. This same pattern occurred in the S334ter lines, where the highest number was present at P6-P10 in the S334ter-7 line and at P10-P12 in the S334ter-3 line, with only a few present thereafter. The somewhat slower degenerating S334ter-5 line had the highest density at a slightly later age, from P10-P15, with almost none present by P21. The even slower RDs showed a similar, but later pattern, with the S334ter-4 line having the most presumptive microglia between P21-P30, with a few present up to P120. Thus, it appears that the greater stimulus of a higher density of degenerating cells attracts a greater number of presumptive microglia.
The invasion of presumptive microglia occurred rapidly, soon after the onset of PR cell death, particularly with the early-onset S334ter-7 and S334ter-3 lines, where there is massive early degeneration. In fact, with line S334ter-7, the earliest we examined (P4) already had some PR cell death (Fig. 10A), and a few presumptive microglia could be found at that age (not shown), although they were more difficult to distinguish from the relatively undifferentiated cells just as the outer neuroblastic layer was separating into an inner and. By P6, they were readily evident (Fig. 10B, arrowhead). A similar rapid invasion following the onset of PR degeneration was seen in the other lines (Table 2).
Likewise, once the majority of pyknotic PR nuclei had disappeared, the number of presumptive microglia was quickly reduced, e.g., line S334ter-7 at P15 (Table 2 and Fig. 10G). A similar fairly rapid reduction in incidence of the cells following the loss of most pyknotic PR nuclei occurred in the other lines, as well (Table 2).
3.1.7 Rod outer segment disc shedding and phagocytosis by the RPE
The role of the RPE in ROS renewal is widely known, and the phagocytic ingestion of shed packets of OS discs is under circadian control in rats, with a major burst of disc shedding soon after the onset of light in the morning (LaVail, 1976a). From about 2 hrs after light onset, the phagosomes in the RPE cell processes and cell bodies are lower in incidence and typically smaller than during the burst of shedding. The earliest age at which disc shedding is seen is about P12 (Tamai and Chader, 1979).
Since almost all of the rats studied in the present study had their eyes taken after the burst of shedding, most microscopic fields in normal SD (Figs. 6E–I) and Tg rats showed the expected low incidence of phagosomes, and some showed none. However, phagosomes could be found in the RPE in all of the Tg lines, such as P23H-1 (Figs. 7D, J, N, P), P23H-3 (Figs. 8A, C, E, I), P23H-2 (Figs. 9A, C, E, G), S334ter-3 (Fig. 11E), S334ter-5 (Fig. 12F), and S334ter-9 (Figs. 14A, C). An example of a retina taken during the early burst of shedding is shown in a P23H-1 rat in Figure 7M. Thus, as expected from a RD due to a rhodopsin defect, ROS disc phagocytosis in the Tg rats appears to function relatively normally.
Figure 6.

WT SD rats - Light micrographs of the outer retina at different ages ranging from neonatal P6 (A) to one year of age (I) for comparison with the rhodopsin mutants. Ages in postnatal days (P), given on the micrographs. All micrographs are taken from the superior posterior or superior equatorial regions of the eyes. PR OSs (OS) begin to be elaborated at P8 (B), and they elongate and become more organized (D, E) until they reach the adult length by about P30 (F). The outer nuclear layer (ONL) comprises approximately 98.5% rod PR nuclei and 1.5% cone (c) nuclei that are slightly larger than those of rods. During the postnatal development, the PR nuclei have a spotty chromatin appearance until about P21 (E) to P30 (F). Some naturally occurring cell death is seen as pyknotic nuclei (arrow). The RPE (RPE) contains ingested ROS tips, known as phagosomes (p). As the retina ages, the retina thins slowly, particularly after P30 (F–I), as shown in the ONL thickness in Fig. 1. INL, inner nuclear layer; IPL, inner plexiform layer. Note: The layer designations apply to the same labels in Figs. 7–14, below. Magnification bar = 20 μm.
Figure 8.

P23H-3 rats - Light micrographs of the outer retina at ages ranging from P14 (A) to one year of age (K–L). Ages given on the micrographs. Regions of retina: (A–G) superior posterior; (H) near superior ora serrata; (I–K) inferior posterior; (L) inferior ora serrata. At early ages, pyknotic nuclei (arrows) are present in the ONL (A–C) at a greater incidence than in SD WT retinas (Fig. 6). As PR nuclei are lost from the ONL, the surviving PR inner and OSs get progressively shorter in the superior posterior hemisphere at P30 (C), P60 (D), P90 (E) and P120 (F), until they are essentially missing by P240 (G). The greater degree of PR degeneration in the posterior superior hemisphere than that in the same regions of the inferior hemisphere is shown at P90 (E, I) and P120 (F, J). Cone cell nuclei (c) are found at all ages in the ONL (e.g., E). Most PRs are missing at one year of age (K), when the retina is disrupted and the RPE is vascularized (v). However, clusters of persisting rod (r) and cone (c) nuclei are present in the ONL at the ora serrata at older ages, up to one year (L). p, phagosomes; pep, RPE cell processes. Magnification bar = 20 μm.
Figure 9.

P23H-2 rats - Light micrographs of the outer retina at ages ranging from P60 (A) to P380, greater than one year of age (D, E, H). Ages given on the micrographs. Regions of retina: (A–D) superior posterior; (E) superior equatorial to peripheral; (F, G, H) inferior posterior. At P60 (A), the retina appears almost normal, although a slightly higher incidence of pyknotic nuclei (arrows) and the ONL first measures slightly thinner than that in SD control retinas (Table 2). Thereafter, PRs are relatively slowly lost up to P180 (B–E), where rod inner and OSs are somewhat shortened. Phagosomes (p) are present in the RPE and RPE cell processes in many of the retinal sections (C, E). At P380, 1–2 rows of PR nuclei comprise the ONL in the most degenerated region of the superior posterior region of the eye (D), but significantly more PR nuclei survive just more peripheral to the most degenerated region (E). Both rod (r) and cone (c) nuclei are present at all ages. Magnification bar = 20 μm.
Figure 12.

S334ter-5 rats - Light micrographs of the outer retina at ages ranging from P6 (A) to P90 (G). Ages given on the micrographs. All micrographs are taken from the superior posterior to equatorial regions of the eyes, except H, which is from the inferior posterior hemisphere in the same retinal section as E. (A) Developing ONL at P6, which is indistinguishable from that of normal controls. (B–E) The IPS contains an increasing number of displaced immature PRs (d), some in clusters (D, E), but the ONL does not show many pyknotic nuclei (arrows) until P12 (D). A pyknotic nucleus is shown internalized within the RPE (D). Coincident with the appearance of pyknotic nuclei at P12 and P15 is the appearance of invading presumptive microglia into the ONL (Figs. 12D, E; arrowheads). Very short, disorganized PR OSs (os) are shown at ages from P10-P30 (C–F, H). The inferior posterior hemisphere (H) at P15 is thicker than superior posterior in the same retina (E). Magnification bar = 20 μm.
In lines S334ter-7 and S334ter-3, where no ROSs are generated, a few phagosomes could be found in the RPE, which presumably represented ingestion of extracellular debris (S334ter-3, Fig. 11E). In addition, the RPE was capable of ingesting the degenerating nuclei of PR cells displaced into the IPS (S334ter-7, Figs. 10D, J, K; S334ter-5, Fig. 12D), even at P8 and P10 (Figs. 10D and J, respectively) before disc shedding of OSs begins in normal retinas (Tamai and Chader, 1979).
3.1.8 Rod cell death precedes cone cell death
Since both the P23H and S334ter mutations are expressed in rhodopsin, rod PRs die before cone PRs (see Discussion). To gain quantitative evidence of this in the Tg lines at different ages, we assessed the percentage of surviving rod and cone nuclei in the ONL in the plastic sections (Table 3). In retinas with rod PR defects, the percentage of cones in the ONL rises, such as in the extreme example of the rd1 mouse, where cones rapidly become 100% of the PR population with the loss of all rods, and the cones are much more slowly lost (Carter-Dawson et al., 1978).
Table 3.
Percentage and number of rod and cone cell nuclei in Tg rat retinas of selected ages
| Line | Age | N | % Cone Sup | % Cone Inf | P | # rods/# cones Sup (% of WT remaining) | # rods/# cones Inf (% of WT remaining) |
|---|---|---|---|---|---|---|---|
| SD | P130 | 3 | 2.2 ± 0.2 | 2.3 ± 0.2 | 0.225 | 1021/23 | 1058/25 |
| P240 | 4 | 2.5 ± 0.4 | 2.5 ± 0.3 | 0.824 | 949/24 | 940/24 | |
| P23H-3 | P130 | 3 | 6.9 ± 2.2 | 4.3 ± 1.2 | 0.020 | 315/22 (31%/92%) | 471/18 (50%/92%) |
| P240 | 3 | 19.7 ± 4.0 | 10.7 ± 1.6 | 0.014 | 67/16 (7%/67%) | 185/22 (20%/92%) | |
| S334ter-4 | P240 | 3 | 32.5 ± 9.9 | 21.1 ± 0.7 | 0.084 | 31/13 (8%/55%) | 65/17 (7%/73%) |
| P23H-1 | P170 | 1 | 86.0 | 73.8 | NA | 15/92 (<1%/90%) | 56/158 (<1%/+144%) |
| P263 | 1 | 93.2 | 90.2 | NA | 5/69 (<1%/65%) | 15/138 (<1%/+130%) | |
| P23H-3 | P370 | 1 | 77.8 | 25.3 | NA | 22/77 (<1%/73%) | 227/77 (6%/73%) |
| S334ter-7 | P161 | 2 | 90.7 | 97.8* | NA | 2/20 (<1%/20%) | 1/18* (<1%/16%) |
| S334ter-3 | P63 | 3 | 97.3 ± 1.1 | 93.2 ± 2.5 | 0.067 | 2/81 (<1%/79%) | 6/87 (<1%/79%) |
| S334ter-5 | P120 | 1 | 100 | 77.2 | NA | 0/95 (0%/93%) | 38/129 (<1%/+117%) |
| P250 | 1 | 97.4 | 92.0 | NA | 1/37 (<1%)/35%) | 2/23 (<1%/22%) |
WT = wild-type SD
Above thick line, values are the total of 5 172-μm contiguous fields (100x oil immersion objective lens), or a total of approximately 860 μm in each hemisphere, starting 440 μm from the optic nerve head and moving peripherally. The rod and cone nuclei of the superior (Sup) and inferior (Inf) hemispheres were counted in the same eyes. One retinal section from each animal was counted, and the number of animals (sections) is given as N; the % cones is the mean of each 860-μm, and S.D of the mean is given where 3 or 4 sections were counted. Below the thick line, in retinas where many fewer PR cells were present, we counted all of the cells in the retinal section, tabulating the Sup and Inf hemispheres separately. Any cells that had non-typical cone nuclear structure were omitted from the counts, as they may have been invading cells.
The rod and cone counts in the two methods of measurements can be directly approximated as follows: for those shown above the thick line, the length of retina in each of the Sup and Inf counts is 860 μm; thus, multiplying that sum by 4.42 will result in the number of cells in a 3,800-μm retinal hemisphere (the approximate length of the entire retinal hemisphere), which is used in the measurements below the thick line. The number of cones in the Inf hemisphere In some instances (below the thick line) is greater than the number in the WT retinas, due in part to the cluster of surviving cells near the ora serrata that are not present in the counts above the thick line.
One of the 2 retinal sections had no cones in this hemisphere.
In the WT SD retinas, we found about 2.2–2.5% of the ONL to be cones (Table 3). This is somewhat higher than the 1.5% we have previously found (LaVail, 1976b), but it may be that our earlier lighting conditions led to a slightly stressful environment. Nevertheless, in all of the rhodopsin Tg lines measured, all of the retinas showed preferential rod degeneration with a consequential elevation in percentage of surviving cone cells (Table 3) Although there was a much greater loss of rod PRs, some survived for most of the life of the Tg animals.
As seen in Table 3, the correlation of a greater survival of cones and rods at earlier stages of degeneration can also be seen in the hemispheric asymmetry within individual sections, particularly in the slower degenerating lines where greater asymmetry exists (Fig. 4). In almost every case, the more rapidly degenerating superior hemisphere shows a greater loss of both rods and cones than the more slowly degenerating inferior hemisphere, but far greater loss of rods than cones (Table 3). Thus, the rod loss can be very rapid, with relatively slower cone loss.
3.1.9 Müller cell hypertrophy and other late changes in the retina
In most RDs, the loss of PRs leads to many responses and remodeling of the synaptic connectivity and organization of the remaining retinal cells (Cuenca et al., 2014; Jones and Marc, 2005; Jones et al., 2003). The most widely recognized secondary change is the hypertrophy of retinal Müller cells, the main retinal glial cell type that spans the retina. As noted in Table 2, Müller cell hypertrophy is found in all Tg lines following extensive ONL thinning or loss, except for the very slowly degenerating S334ter-9. Activated Müller cells can overgrow into the OS layer by P30 in the S334ter-4 line, earlier than expected (McGill et al., 2012b). As described by Jones et al. (2003), microneuromas are also found in some of the retinas, particularly in the latest stages of the most rapidly degenerating of the P23H lines (P23H-1) and two of the most rapid S334ter lines (S334ter-3 and S334ter-5) (Table 2).
3.2 Specific phenotypic features of the different Tg lines
The focus of this study was primarily on the changes in PRs and their degenerations. We have previously reported on the relationship of PR development and degeneration to retinal vascularization and the later events of RPE neovascularization and attenuation (Pennesi et al., 2008). In lines where neovascularization of the RPE occurred (all except S334ter-7), it happened with a latency of 60–180 days after the loss of most PRs (Pennesi et al., 2008).
As with Tables 1 and 2, the lines are described in descending rate of degeneration, the fastest first, within each Tg type. Many of the phenotypic features given in Table 2 are illustrated.
3.2.1 SD WT
The background strain in which the Tg rats were produced was the SD albino strain. Light micrographs of the outer retina at various ages are given in Fig. 6 for comparison with retinas from the Tg lines. Retinal cell genesis in the rat is complete at the posterior retina by the first week of life (Rapaport et al., 2004), and most of the young PRs have reached their destination in the ONL by P6 (Fig. 6A). At this age and up through about P16 (Fig. 6D), most of the PR nuclei have a spotty heterochromatin pattern; as the PRs mature, at P21 and P30, most rods have developed the typical single clump of heterochromatin (Figs. 6E and F, respectively). PR OSs begin to be elaborated at P8, and they elongate and become more organized (Figs. 6D and E) until they reach the adult length by about P30 (Fig. 6F). Cone nuclei can be seen at or near the outer border of the ONL, identified by their slightly larger diameter than that of rods and by their multiple lobes of heterochromatin and greater amount of euchromatin than in rod nuclei (Fig. 6G). With optimal staining, the euchromatin of rod nuclei can be seen to be more intensely stained than that of cone nuclei. As the retina ages, the retina thins slowly as the eye enlarges, particularly after P30 (Figs. 6F–I), as shown in the ONL thickness in Fig. 1. The RPE contains ingested ROS tips (phagosomes), so OS turnover appears to occur normally, as expected.
3.2.2 P23H-1
The P23H-1 line has the earliest onset and fastest rate of degeneration of the 3 P23H lines (Fig. 1A). The early histogenetic sorting of PR nuclei into the definitive ONL in the P23H-1 line (not shown) is indistinguishable from that seen in littermate WT rats from P4-P8. Likewise, the early elongation of the rod inner segments (RIS) and elaboration of ROS membranes at P8 and P10 are indistinguishable in the P23H-1 (Fig. 7A) and WT SD (Fig. 6C) retinas. The first clear signs of degeneration are the slight increase in the incidence of pyknotic nuclei in the P23H-1 retinas at P10 (Table 2 and Fig. 7A, arrow). There is a higher incidence of pyknotic nuclei in the ONL of P23H-1 rats at P12 (Fig. 7B, arrows), and the overall ONL thickness is first seen as thinner than that of the SD retina at that age (P<0.05; Table 2).
By P15, the P23H-1 retina has lost significantly more PRs (Fig. 7C), with the ONL thickness only about 76% of that of the SD retina, overall (Fig. 6D and Table 2). The rod ISs and OSs are also decidedly shorter than WT at this age. At progressively later ages, the P23H-1 retinas lose more PR cells (Figs. 7D–G), and the ONL is obliterated at focal points by P120 (Fig. 7F). As the ONL is lost, the rod ISs and OSs progressively shorten (when a given region of the eye is compared), e.g., Figs. 7D–F. As long as ROSs are still present, even if very short, the OSs maintain their characteristic shape (Figs. 7D, E, J, L). Moreover, whenever ROSs are present, some phagosomes (p) are present in the RPE cell bodies (Fig. 7D, N, P). Soon after the onset of light, there is an abundance of phagosomes within the RPE cell bodies, and RPE cell processes become more noticeable (Fig. 7M).
The greater degree of PR degeneration in the posterior superior hemisphere than that in the same regions of the inferior hemisphere is shown at P30 (Figs. 7D and E), P50 (Figs. 7E and J) and P120 (Figs. 7F and K). A central-to-peripheral gradient of degeneration is shown at P120 between the more degenerated inferior posterior region (Fig. 7K) and the region near the ora serrata of the same eye hemisphere (Fig. 7L).
Invading presumptive microglia are present in the outer plexiform layer (Fig. 7N), ONL (Fig. 7O), inner nuclear layer (INL, Fig. 7O) and IPS (Figs. 7I, O, P). The cells are phagocytic, and the pale microglial cell process shown among the PR ISs in Figure 7O is filled with ingested debris; it cannot be determined whether this process communicates with the nearby microglial cell nucleus in the ONL, or with another cell out of the plane of section. The cell in the IPS of Figure 7I contains internal phagosomes and appears to be ingesting a displaced rod cell.
When PR inner and OSs are mostly (Fig. 7F) or completely missing (Fig. 7G), the IPS is filled with a pale zone that that is seen by electron microscopy to be RPE cell processes (Figs. 17A–B). When almost all PRs have been lost, a few rods and cones still persist (Fig. 7K). At such late stages, strands of inner nuclear layer nuclei can be seen crossing the inner plexiform layer, and RPE vascularization is evident (Fig. 7G). In these same retinas, a small cluster of PRs typically survives at the ora serrata (Fig. 7H); most are rods, with a few cones surviving.
Figure 17.

Electron micrographs illustrating compacted pigment epithelial cell processes (pep) at late stages of degeneration in P23H-1 (A, P120) and S334ter-4 (B, P120). (A) This configuration can be seen when disorganized OS tips (os) are present. (B) When PRs have degenerated and disappeared, a Müller (glial) cell seal (MC seal) is present in the outer retina, with Müller cell processes interdigitating with the pep (arrows). RPE, retinal pigment epithelium. Magnification bar for both = 2.0 μm.
A fairly high percentage of the P23H-1 rats have been shown to have auditory deficits, with auditory loss in about 26% of the rats at P50, and about 45% having total hearing loss by P200 (Sotoca et al., 2014). As far as we are aware, none of the other rhodopsin Tg lines (P23H or S334ter) have been examined for this sensory impairment.
3.2.3 P23H-3
Phenotypic characteristics of the P23H-3 Tg line are very similar to those of the P23H-1 line, with the major difference being the slower rate of degeneration (Fig. 1A). As seen in Fig. 1A and in the descriptions in Table 2, the P23H-3 line has a steep and early slope of degeneration similar to that of the P23H-1 line, but after about P21, the rate slows from that of the P23H-1 line. For example, the P23H-1 line reaches the overall loss of about 25% of PR nuclei in the ONL at P15, whereas the P23H-3 line reaches approximately the same degree of loss at P30, a difference of 15 days (Table 2). By contrast, the difference to reach a 75% loss in the 2 lines is about 180 days (Table 2).
In the P23H-3 line, the ONL is already slightly thinner than the WT SD at P14 (Figs. 8A and 6D), and the ROSs are slightly less well organized. By P21 (Fig. 8B), P30 (Fig. 8C), P60 (Fig. 8D) and P90 (Fig. 8E), there is a progressive loss of PRs in the ONL and a shortening of PR ISs and OSs throughout the retina, but particularly in the superior posterior region of the retina. The ONL in the P23H-3 line is first significantly thinner than WT due to loss of PRs at P21 (P<0.05; Table 2). The less severe degeneration in the inferior posterior hemisphere seen in retinal spidergrams (Fig. 4B) is more evident at P90 (Figs. 8E and I) and P120 (Figs. 8F and J). The reduction of the ONL to less than one complete row of PR nuclei is reached by P240, when both rod and cone nuclei are present, and RPE cell processes fill the former PR inner and OS layer (Fig. 8G). In retinas of this same age, a cluster of mostly rod cells persists at the ora serrata (Fig. 8H), similar to that seen in the P23H-1 retina, but with a somewhat greater number (cf. Figs. 8H and 7H); at one year of age, fewer rods and cones persist at the ora serrata (Fig. 8L). At this age, many disruptions in the INL are present, as are profiles indicating neovascularization of the RPE (Fig. 8K).
As in the P23H-1 line, when ROSs are present, even if lying down due to the loss of most of them (e.g., Fig. 8F), they still maintain their characteristic cylindrical shape, albeit slightly distorted and sometimes with a greater diameter than normal (Fig. 8F). Likewise, when ROSs are present, packets of shed OS disks (phagosomes) are frequently found in the RPE processes and cell bodies at most stages of degeneration (Figs. 8A–C, E, F, I).
3.2.4 P23H-2
The P23H-2 line has the latest onset and slowest rate of degeneration of the 3 P23H rhodopsin Tg lines (Figs. 1A, 9). Its pattern of PR degeneration is similar to that of the P23H-1 and P23H-3 lines, albeit slower. It has a few more pyknotic nuclei in the ONL than normal WT retinas at all ages from about P21 (Table 2). The P23H-2 retina appears almost normal at P60 (Fig. 9A), but the overall thickness of its ONL is significantly thinner than WT at that age (P<0.00005; Table 2). The overall loss of about 25% of PR nuclei does not occur until P180 (Table 2). As the animal ages, the loss of PR nuclei and the concomitant shortening of the PR inner and OSs occurs, as shown in the superior posterior region of the retina in Figs. 9B–D at P120, P180 and P380. Like the other P23 lines, 1) the inferior posterior retina is less severely affected at each age (cf. Figs. 9B with F at P120, 9C with G at P180, 9D with H at P380), 2) the retinas display a central-peripheral gradient, as shown at P380, where the superior equatorial retina (Fig. 9E) is less severely affected than in the same section in the superior posterior retina (Fig. 9D), and 3) at all ages when ROSs are present, they can be clearly distinguished by their cylindrical shape (Figs. 9D, E H), and the frequently presence of phagosomes (Figs. 9A, C, E, G).
3.2.5 S334ter-7
The S334ter-7 line has the earliest onset and most rapid degeneration of the S334ter lines and, in fact, of any known RD experimental mutant, as far as we are aware. At P4, the earliest we examined, more pyknotic nuclei are present in the developing ONL (Fig. 10A). The first signs of cell death occur at approximately the age at which the opsin promoter begins to be expressed (Treisman et al., 1988). (Note, the studies in Barnstable’s lab consider the day of birth as P1; thus, our P4 is equivalent to P5 in the Barnstable studies.) Presumably, the extremely high content of mutant rhodopsin in the S334ter-7 line (Table 1) is particularly toxic to the developing PR cells. In fact, in this line, the total whole-eye rhodopsin at the earliest age consistently measures significantly greater than in WT control retinas (Fig. 2D).
Soon after P4, and through P10, three features are prominent that are not present in the more slowly degenerating P23H lines. First, the abundance and rapid formation of pyknotic nuclei is seen in the innermost ONL, usually in the innermost 20–30% (Figs. 10B–E, I, J). Second, many immature PRs, identified by their spotty heterochromatin as in normally developing PRs (Figs. 5A–D), are displaced into the IPS, between the RPE and nascent rod ISs (Figs. 10A–D) or, when ISs are missing, between the RPE and outer limiting membrane (Figs. 10E–G, J, K). Profiles of individual cells and clusters of immature PRs are frequently seen protruding through the outer limiting membrane. (Figs. 10E, I) After entering the subretinal space, many of the immature PRs appear to become pyknotic (Figs. 10B, C, F). Third, PR ISs do not develop to longer than short nubs (Figs. 10A–E) and are virtually missing by P10 (Fig. 10E), and no observable PR OSs are evident (Figs. 10A–E). The ONL of the S334ter-7 retinas is first significantly thinner than WT at P8 (P<0.05; Table 2).
The developing PRs and pyknotic nuclei are lost from both the ONL and subretinal space at a remarkably fast rate, beginning at about P6 (Fig. 10B) and reaching the loss of a continuous ONL in the most degenerated superior posterior retina by P12 (Fig. 10F) and thereafter (Figs. 10G–H). The rapid elimination of the dying PR nuclei appears to occur in two ways. First, there is a massive invasion of the ONL by presumptive microglial cells that have a pale nuclear euchromatin with a peripheral rim of heterochromatin, often with a prominent nucleolus (Figs. 10B–F). Invasion of such phagocytic cells is typical of many RD models (see Discussion). These cells are also seen in the IPS (Fig. 10E), and some can be seen apparently penetrating the outer limiting membrane from the ONL (Figs. 10B, F). Second, the RPE cells appear to phagocytize many pyknotic nuclei that are located in the IPS (Figs. 10D, J, K).
The S334ter-7 line shows the same hemispheric differences as seen in the P23H lines. For example, the RD has a faster rate of degeneration in the superior hemisphere than in the inferior hemisphere as seen at P8 (Figs. 10D, I), P10 (Figs. 10E, J) and P12 (Figs. 10F, K). Likewise, a cluster of PRs, both rods and cones, persists for a time at the ora serrata (Fig. 10L) after most PRs have degenerated and disappeared in most of the retina. A very few scattered PR nuclei persist for up to one year of age. When PR ISs and OSs have disappeared, this layer is filled with RPE cell processes (Fig. 10G, K).
As noted by Pennesi et al. (2008), the rapidly degenerating S334ter-7 line shows aborted vascular development and no neovascularization of the RPE.
3.2.6 S334ter-3
The S334ter-3 line is the second fastest Tg rhodopsin mutant, PR with degeneration beginning at about P8 (Fig. 11B), and with the ONL reduced to less than a single row of PR nuclei by P20 in the superior posterior hemisphere (Fig. 11F); at this age a complete row of PR nuclei persists in the inferior posterior retina (Fig. 11H). Thereafter, only a few scattered PRs are found in the S334ter-3 retinas (Fig. 11G). This rate is similar to the widely studied rd1 mouse (LaVail and Sidman, 1974).
The features of the RD in the S334ter-3 line are almost identical with those of the fastest S334ter-7 line, albeit about 2–8 days later (Table 2). For example, displaced PRs in the subretinal space are first found at P8 in the S334ter-3 retinas (Fig. 11B), compared to P6 in the faster S334ter-7 line (Fig. 10B). This was first described by Liu et al., as was the preponderance of pyknotic nuclei in the ONL being located at the innermost part of the layer (Fig. 11C) (Liu et al., 1999). The loss of PR nuclei in the ONL is first significantly see at P10 (P<0.05; Table 2). The development of PR ISs between P6-P10 (Figs. 11A–C) is slightly greater than in S334ter-7 retinas (Figs. 10A–E), but is still less than those in the SD or P23H-1 (Fig. 7A) lines. The ISs in S334ter-3 retinas are shorter at P10 than at earlier ages and are almost missing by P12 (Figs. 11A–D). Like the S334ter-7 line, no PR OSs are evident in the S334ter-3 retinas (Figs. 11A–E). Similarly, the earliest age where an absence of PRs is seen in the ONL is at P20 in the S334ter-3 line (Fig. 11F), compared to P12 in the faster S334ter-7 line (Fig. 10F). Otherwise, most features are similar, including there being a cluster of PR nuclei at the ora serrata at P20 (not shown).
A consequence of a somewhat slower degeneration than that in line S334ter-7 is that there are fewer dying cells at any given time. The result is that somewhat fewer displaced PRs (Figs. 11B–E), pyknotic PR nuclei in the innermost ONL (Figs. 11B–D), and invading phagocytic cells (Figs. 11C, D) are seen at any given age than in the faster S334ter-7 line (Fig. 10).
The developing rod ISs are approximately the same length as normal at P6, but not thereafter in the S334ter-3 line; they are about the same length only through P4 in the faster S334ter-7 line. In neither of these early onset RDs are any ROS membranes generated (confirmed by electron microscopy at P8-P10; Table 4).
Table 4.
Ages of retinas examined by electron microscopy and incidence of extracellular vesicles in the interphotoreceptor space
| Line | P8 | P9 | P10 | P12 | P14-15 | P20-22 | P30-32 | P60 | P120 | P240-365 |
|---|---|---|---|---|---|---|---|---|---|---|
| SD | 0 | – | 0 | 0 | 0 | 0 | – | – | – | – |
| P23H-1 | – | – | 0 | 0 | – | – | 0 | – | 0 | – |
| P23H-3 | – | – | – | 0 | 0 | 0 | – | – | – | 0 |
| P23H-2 | – | – | – | – | – | – | 0 | – | – | 0 |
| S334ter-7 | 0 | 0 | – | – | 0 | – | – | – | – | – |
| S334ter-3 | 0 | – | 0 | 0 | – | – | 0 | – | – | – |
| S334ter-5 | – | – | – | 0 | ++ | – | + | – | – | – |
| S334ter-4 | – | – | – | 0 | + | +++ | ++ | +, 0* | 0 | – |
| S334ter-9 | – | – | – | – | 0 | 0 | 0 | – | – | – |
| S334ter-9 (Homoz.) | – | – | – | – | – | – | +++ | – | – | – |
–, not examined by electron microscopy.
For each age studied, the observations are based on examination of 6–15 electron micrographs.
Ages examined and incidence of vesicles in the interphotoreceptor space: 0, none; +, few vesicles (4–7/mμ2), approximately 1/3 the maximal density; ++, significant number of vesicles (8–11/mμ2), approximately 2/3 the maximal density; +++, many vesicles (12–20/mμ2), maximal density (e.g., S334ter-4 at P21, Figs. V1B and E; S334ter-9 homozygote at P32, Figs. V2B and D).
Where 3–4 rows of photoreceptor nuclei existed, a few vesicles were present between the inner segments; none were found where only 1–2 rows of photoreceptor nuclei were present.
3.2.7 S334ter-5
The S334ter-5 line is another early onset RD, but with a somewhat different character than the two fastest lines. The overall RD time course is approximately P8-P90 (Fig. 1B). At P6 (Fig. 12A), the retina is indistinguishable from WT (Fig. 6A). At P8 (Fig. 12B), P10 (Fig. 12C) and P12 (12D), there is an increasing abundance of displaced immature PRs in the IPS, but a significant number of pyknotic nuclei in the ONL are not present until P12 (Fig. 12D), when the ONL is significantly reduced in thickness from that of WT controls (P<0.005; Table 2).
The ONL is thinner at P15 (Fig. 12E) than at P12 (Fig. 12D), and it becomes even thinner at P30 (Fig. 12F), reaching less than a full single row of PR nuclei by P90 (Fig. 4G). As indicated in the retinal spidergrams (Fig. 4F), the hemispheric differences seen in the other lines are found in the S334ter-5 line, with the ONL being thinner in the superior (Fig. 12E) than the inferior (Fig. 12H) hemisphere at any given age. A cluster of rod and cone nuclei persists at the ora serrata at P90 (not shown). A few scattered rod and cone nuclei are commonly present at 8 months of age, with only a few remaining at a year of age.
A significant difference in the S334ter-5 line from the two more rapidly degenerating lines is that some PR OS membranes are generated in line S334ter-5 (Figs. 12C–E, H), but not as many as in the SD (Figs. 6B–D), P23H-1 (Figs. 7A–D) or P23H-3 (Figs. 8A–D) retinas. When viewed by electron microscopy at P12, P15 and P31, OSs were decidedly present, but in small fragments and much shorter than in SD retinas (data not shown).
3.2.8 S334ter-4
The development of the S334ter-4 retina is indistinguishable from that in WT rats through P12, as judged by light and electron microscopy, although a few more pyknotic nuclei are present in the ONL at P12 and later (Table 2). In the S334ter-4 retinas at P14-P16, when the PR OSs are being abundantly elaborated (Fig. 13A), they are almost of the same length as those in WT SD rats (Fig. 6D), and at P21 the OSs are even more regularly arranged, parallel to one another, and almost the same length at P21 in both the S334ter-4 and SD animals (Figs. 13A, B and 6E, respectively). However, from about P14 onward, the OSs are somewhat more irregular in diameter and uniformity, with some appearing slightly frothy (Figs. 13A–C), particularly near the junction of the inner and OSs and in the inner half of the OSs (Fig. 13B). In some cases, where the OSs were fairly regular in shape, the IPS between them was pale-staining and prominent (Figs. 13C, G).
With the loss of PR nuclei, the ONL is thinner than in normal SD rats beginning at P21 (P<0.05; Table 2), being reduced to a single continuous row of PR nuclei by P120 (Table 2; Fig. 13E). Thereafter, a few scattered PR nuclei are seen (Fig. 13F), and hypertrophied Müller cell processes are seen crossing the inner plexiform layer in these and other late-stage Tg retinas (Table 2).
As noted above, the S334ter-4 line displays the greatest superior-inferior asymmetry in ONL thickness (Figs. 4G and 5). This is shown at P60 (Figs. 13D, H) and P120 (Figs. 13E, G). The difference is large enough that differences in rhodopsin content in the superior and inferior hemispheres can be measured in vivo by fundus reflectometry (Jiang and Laties, personal communication).
3.2.9 S334ter-9
Line S334ter-9 displays the slowest RD of all the Tg lines (Fig. 1B). It is indistinguishable from SD WT retinas at all ages (e.g., Fig. 14A) up to P120 (Fig. 14B), when the ONL is first thinner than normal (P<0.05; Table 2), although the PR inner and OSs appear no different from normal. Thereafter, as the animals age, more pyknotic nuclei are seen (Fig. 14C) and the ONL slowly becomes thinner and OSs concomitantly become shorter (Fig. 14D). By one year of age, the ONL is still about 75% of normal. The ROSs in S334ter Line 9 appear normal, and the inner and OS zone does not show vacuolization or appear frothy, as in the other S334ter lines where ROSs are seen (Lines 5 and 4).
3.3 Electron microscopic examination: similarities and differences in P23H and S334ter Tg lines
Several structural similarities and differences between P23H and S334ter Tg retinas as seen by light microscopy and in the probable differences in mechanisms of mutant gene action led us to examine the Tg lines at a number of different ages by transmission electron microscopy (Table 4).
3.3.1 Rod outer segment integrity
As seen by light microscopy, the S334ter-4 line appeared to have pale-staining, vertical spaces alongside the ROSs at most ages where the OSs were present (Figs. 13B, C, G). By electron microscopy, the pale-staining spaces were abundant, and many were filled with small vesicles (Fig. 15B). In some fields, these extended most of the length of the OSs (data not shown).
Figure 15.

Transmission electron micrographs of the outer retina of SD (A and D), S334ter-4 (B and E) and P23H-3 (C and F) at the age of P21. The upper row is at low magnification and the lower row is at higher magnification. In the S334ter-4 retina, many small extracellular vesicles (arrows) are present in the interphotoreceptor space (IPS) in the basal region of the OSs (OS) and apical region of the ISs (IS). These are absent in the SD (A and D) and P23H-3 (C and F) retinas. cc, connecting cilium; RPE, retinal pigment epithelium. Magnification bars = 5 μm in AC; 2 μm in D–F, with E at a slightly higher magnification than A and F.
The retinas of S334ter-9 rats appear normal by light microscopy, at least up to P120, and near normal thereafter. Electron microscopy of the S334ter-9 retinas at P30 confirmed that most ROSs have normal structure (Figs. 16A, C). However, a few OSs showed some irregularities in diameter and slightly disorganized regions where the disks were not fully compacted (Fig. 16A).
Figure 16.

Transmission electron micrographs of the outer retina of a S334ter-9 hemizygote (A and C) and a S334ter-9 homozygote (Hz; B and D) at P30 and P32, respectively. The upper row is at low magnification and the lower row is at higher magnification. The hemizygote appears almost normal, except for a few irregularities in diameter and slightly disorganized regions (d) of the OSs where the disks are not fully compacted (A, compare to SD WT, Fig. 15A). The hemizygote shows no extracellular vesicles (A and C), but the homozygote (B and D) has many vesicles (arrows) in the interphotoreceptor space, and the outer retina is significantly more disorganized and degenerated. cc, connecting cilium; RPE, retinal pigment epithelium. Magnification bars = 5 μm in A and B; 2 μm in C and D.
3.3.2 Accumulation of submicron vesicles in the IPS and its relation to different mechanisms of PR cell death in the two mutations
In P347S transgenic mice, Li et al. (Li et al., 1996) observed the accumulation of submicron-sized rhodopsin-containing vesicles in the extracellular (interphotoreceptor) space in the vicinity of basal OSs and apical ISs. They suggested these vesicles resulted from a defect in the vectorial transport of post-Golgi vesicles to the nascent OS discs. Others have found a similar observation in mutant animals with defects in the C-terminus of rhodopsin (see Discussion), so we examined many ages of WT SD and the Tg lines of rats for the presence of these vesicles.
As noted in Table 4, none of the vesicles were found in normal SD or P23H mutant retinas. Examples of these are illustrated in Fig. 15A and D and 15C and F, respectively. In the S334ter retinas, none were found in the very rapid and early onset degenerating S334ter-7 and S334ter-5 lines, but some were present in the S334ter-4 (Fig. 15B and E) and S334ter-5 lines (Table 4). The accumulation of these vesicles was highest during the period of greatest PR degeneration (cf. Table 4 and Fig. 1) and was not present in the earliest period of OS development (P10-P12, see Fig. 6) or after most PR OSs had disappeared (Table 4). In the S334ter-4 line, the vesicles can be seen in electron micrographs in the IPS in two previous studies (Green et al., 2000; Lau et al., 2000).
The S334ter-9 hemizygote line with lowest mutant rhodopsin expression did not show any extracellular vesicles in the ages examined (Table 1; Figs. 16A and C). Since the homozygous S334ter-9 rats displayed a much faster rate of degeneration than the hemizygous animals (Fig. 2H), we examined homozygous animals at one age. The retinas showed maximal accumulation of vesicles in the degenerating retina (Table 1; Figs. 16B and D), similar to those seen in the S334ter-4 line (Figs. 15B and E).
3.3.3 Structural relationship between RPE cell and retinal Müller cell processes
By light microscopy, the apical processes of RPE cells of normal rats that interact with the tips of the rod OSs and are involved in phagocytosis of shed packets of OS disks, are seen as a pale-staining area at the apex of the RPE cells (LaVail, 1976a), as shown in SD retinas between P30-P90 (Figs. 6G–I). In all of the Tg lines where there is loss of most or all of the PRs (i.e., other than P23H-2 and S334ter-9), the region of the RPE cell processes is much more prominent than in WT retinas. This can be found when the ONL is reduced to 2–3 rows of PR nuclei in the ONL, typically with few or no ISs (Figs. 7F, 10G, 12F), but more often when PR nuclei were totally missing, or nearly so (Figs. 7G, 8G & K, 10H, 11F & G, 12G, 13E).
By electron microscopy, it can be seen that when several rows of PR nuclei and short inner and OSs are present, the RPE cell process region consists only of RPE cell microvillous processes, as well as some OS membranes in the process of being ingested (Fig. 17A; P23H-1 P120). Where there are no PR nuclei and no inner or OSs, the RPE cell processes are apposed to the degenerated retina. In this case, the many electron-lucent RPE cell processes are interdigitated with more electron-dense Müller cell villous processes (Fig. 17B; S334ter-4 P120), which extend from the glial seal (Jones et al., 2003) at the outer border of the neural retina. In both cases, the determinant appeared to be the state of degeneration of the retina. For example, in the case where some PRs are present, virtually the same image seen in the P23H-1 retina at P120 (Fig. 17A) can be observed in the faster degenerating S334ter-5 retina at P30 (not shown). Likewise, virtually the same image with Müller cell processes interdigitating with RPE cell processes as seen in the S334ter-4 retina at P120 (Fig. 17B) can be observed in the faster degenerating S334ter-3 retina at P30 and the more slowly degenerating P23H-3 retina at P365 (not shown).
The presence of abundant RPE cell processes in all of the Tg rat lines that lose most PR cells is sometimes present across much of the retinal hemisphere. This is in contrast to the late stages of most other RD models, in which there are generally only short intervals with the abundant RPE cell processes, such as in rd1 (Karli et al., 1965), pcd (Blanks et al., 1982) and nr (White et al., 1993) mice and the RCS rat (LaVail et al., 1974).
3.4 Electrophysiological assessment of photoreceptor degeneration in the Tg lines
The ERG has been the most widely used measure of RD in the Tg rats, as shown in Table 5. The reason for this is that not only does the ERG give a measure of retinal function, measured as the rod-dominated (a-wave), cone (photopic b-wave) and post-receptoral activity (b-wave), but it also is a non-invasive measure that can be done repeatedly in a given animal to follow the course of a RD. For this reason, it is widely used in experimental therapeutic studies (Table 5).
Table 5.
Previous studies with P23H and S334ter rhodopsin Tg rats
| Characterization of the genotype | P23H-1: {Orhan, 2015 #2579} |
| Characterization of the RD phenotype |
P23H-1: {Caminos, 2015 #2725;Farinelli, 2013 #2726;Lee, 2003 #1987;Orhan, 2015 #2579;Traverso, 2002 #2703}{Stiles, 2016 #2672}{Anderson, 2002 #1990} P23H-3: {Lee, 2003 #1987;Martin, 2005 #2295;Traverso, 2002 #2703}; Hz: {Fernandez-Sanchez, 2015 #2620}{Anderson, 2002 #1990}{Acosta, 2010 #2747;Bicknell, 2002 #2377} P23H-2: {Anderson, 2002 #1990} S334ter-3: {Chen, 2014 #2727;Farinelli, 2013 #2726;Martinez-Navarrete, 2011 #2679}{Anderson, 2002 #1990} S334ter-5: {Lee, 2003 #1987}{Martinez-Navarrete, 2011 #2679}{Anderson, 2002 #1990} S334ter-4: {Martin, 2005 #2295;Traverso, 2002 #2703}{Anderson, 2002 #1990}{Green, 2000 #1894} S334ter-9: {Anderson, 2002 #1990} |
| ERG |
P23H-1: {Bush, 2000 #2270;Jayaram, 2014 #2668;Machida, 2000 #2271;Machida, 2008 #2652;Murray, 2015 #2653;Orhan, 2015 #2579;Pinilla, 2005 #2655;Vasireddy, 2011 #2650}{Stiles, 2016 #2672;Wong, 2015 #2580;Yang, 2009 #2565;Yang, 2010 #2674}; Pig: {Cuenca, 2014 #2619;Rahmani, 2013 #2659;Segura, 2015 #2660;Viringipurampeer, 2016 #2643}; Hz: {Cuenca, 2004 #2581;Parfitt, 2014 #2654;Pinilla, 2005 #2655} P23H-3: {Aleman, 2001 #1989;Chrysostomou, 2008 #2521;Chrysostomou, 2009 #2520;Chrysostomou, 2009 #2519;Gorbatyuk, 2007 #2481;Gorbatyuk, 2010 #2534;Jozwick, 2006 #2658;Kaldi, 2003 #2269;LaVail, 2000 #1936;Leonard, 2007 #2423;Machida, 2000 #2271;Ranchon, 2003 #2121;Valter, 2009 #2616}{Lewin, 1998 #1827;Valter, 2009 #2616}; Pig: {Leonard, 2007 #2423}; Hz: {Fernandez-Sanchez, 2012 #2649;Fernandez- Sanchez, 2012 #2646;Fernandez-Sanchez, 2015 #2647;Fernandez- Sanchez, 2011 #2648;Lax, 2014 #2675;Noailles, 2016 #2651;Parfitt, 2014 #2654} P23H-2: Pig: {McGill, 2012 #2555}; Hz: {Ozaki, 2013 #2657} S334ter-7: Pig: {McGill, 2012 #2555} S334ter-3: {Roddy, 2012 #2558} S334ter-5: Pig: {McGill, 2012 #2555} S334ter-4: {Glybina, 2010 #2630;Green, 2000 #1894;Guerin, 2008 #2665;Kaldi, 2003 #2269;Lau, 2000 #1979;Leonard, 2007 #2423;Ranchon, 2003 #2121}{Nagai, 2016 #2676}; Hz: {Ozaki, 2013 #2657}; Pig: {McGill, 2012 #2555;McGill, 2012 #2629} |
| PR cell death: rates & mechanisms | See section on PR cell death in Discussion |
| Cone cell loss & mosaic | See section on cone cell loss in Discussion |
| Microglia | See section on invading microglia in RDs of Tg rats in Discussion |
| ER and oxidative stress; UPR; mitochondrial dysfunction- characterization |
P23H-1: {Lin, 2007 #2399;Maleki, 2013 #2748} P23H-3: {Lin, 2007 #2399;Sizova, 2014 #2614}{Shinde, 2016 #2749} S334ter-3: {Kroeger, 2012 #2615} S334ter-5: {Kroeger, 2012 #2615} S334ter-4: {Kroeger, 2012 #2615}{Shinde, 2016 #2749;Shinde, 2012 #2556} |
| ER and oxidative stress; UPR; mitochondrial dysfunction-protection |
P23H-1: {Vasireddy, 2011 #2650}; Hz: {Parfitt, 2014 #2654} P23H-3: {Gorbatyuk, 2010 #2534}; Hz: {Parfitt, 2014 #2654} S334ter-3: {Dykens, 2004 #2413;Roddy, 2012 #2558} |
| Vascular development and late changes |
P23H-1: (Pennesi et al., 2008){Zhang, 2003 #2709}; Hz: {Garcia-Ayuso, 2010 #2728} P23H-3: (Pennesi et al., 2008) P23H-2: (Pennesi et al., 2008) S334ter-7: (Pennesi et al., 2008) S334ter-3: (Pennesi et al., 2008) S334ter-5: (Pennesi et al., 2008) S334ter-4: (Pennesi et al., 2008){Zhang, 2003 #2709} S334ter-9: (Pennesi et al., 2008) |
| Inner retinal changes and remodeling |
P23H-1: {Cuenca, 2004 #2581}{Jones, 2005 #2293;Jones, 2003 #1988}{Machida, 2008 #2652}; Pig: {Cuenca, 2014 #2619;Sekirnjak, 2009 #2681}{Lu, 2013 #2678}; Hz: {Cuenca, 2004 #2581}{Pinilla, 2016 #2793} P23H-2: {Jones, 2005 #2293;Jones, 2003 #1988} P23H-3: {Aleman, 2001 #1989}{Jones, 2005 #2293;Jones, 2003 #1988}{Vessey, 2014 #2705}; Hz: {Zhu, 2013 #2710} S334ter-7: {Jones, 2005 #2293;Jones, 2003 #1988} S334ter-3: {Martinez-Navarrete, 2011 #2679}{Jones, 2005 #2293;Jones, 2003 #1988}; Pig: {Ray, 2010 #2732} S334ter-4: {Jones, 2005 #2293;Jones, 2003 #1988}{McGill, 2012 #2629} S334ter-5: {Martinez-Navarrete, 2011 #2679}{Jones, 2005 #2293;Jones, 2003 #1988} |
| Retinal ganglion cells, central projections and cortex |
P23H-1: {Fransen, 2014 #2719;Kolomiets, 2010 #2736}; Pig: {Sekirnjak, 2009 #2681;Sekirnjak, 2011 #2682}; Hz: {Garcia-Ayuso, 2010 #2728}; Hz: {Jensen, 2016 #2794} S334ter-3: {Chan, 2011 #2737;Froger, 2012 #2730}{An, 2002 #2733}; Pig: {Sekirnjak, 2011 #2682}{Chen, 2016 #2738}{Yu, 2017 #2799} |
| ipRGCs |
P23H-1: {Garcia-Ayuso, 2015 #2734}; Hz: {Lax, 2016 #2797} P23H-3: {Garcia-Ayuso, 2015 #2734}; Hz: {Esquiva, 2013 #2735} |
| Pigmented vs albino eyes |
P23H-1: {Lowe, 2005 #2422}{Leonard, 2007 #2423} P23H-3: {Lowe, 2005 #2422} P23H-2: {Lowe, 2005 #2422} S334ter-7: {Lowe, 2005 #2422} S334ter-3: {Lowe, 2005 #2422} S334ter-5: {Lowe, 2005 #2422} S334ter-4: {Lowe, 2005 #2422}{Leonard, 2007 #2423} S334ter-9: {Lowe, 2005 #2422} |
| ER and oxidative stress; UPR; mitochondrial dysfunction-characterization |
P23H-1: {Lin, 2007 #2399;Maleki, 2013 #2748} P23H-3: {Lin, 2007 #2399;Sizova, 2014 #2614}{Shinde, 2016 #2749} S334ter-3: {Kroeger, 2012 #2615} S334ter-5: {Kroeger, 2012 #2615} S334ter-4: {Kroeger, 2012 #2615}{Shinde, 2016 #2749;Shinde, 2012 #2556} |
| ER and oxidative stress; UPR; mitochondrial dysfunction-protection |
P23H-1: {Vasireddy, 2011 #2650}; Hz: {Parfitt, 2014 #2654} P23H-3: {Gorbatyuk, 2010 #2534}{Yu, 2004 #2335}; Hz: {Parfitt, 2014 #2654} S334ter-3: {Dykens, 2004 #2413;Roddy, 2012 #2558} |
| Neuroprotective agents |
P23H-1: {Froger, 2012 #2730;Vasireddy, 2011 #2650;Yang, 2009 #2565}{Ohguro, 2008 #2731}; Hz: {Parfitt, 2014 #2654} P23H-3: Hz: {Parfitt, 2014 #2654}{Fernandez-Sanchez, 2012 #2649;Fernandez-Sanchez, 2012 #2646;Fernandez-Sanchez, 2015 #2647;Fernandez-Sanchez, 2011 #2648} S334ter-3: {Dykens, 2004 #2413;Ohguro, 2008 #2731;Roddy, 2012 #2558;Song, 2003 #2697;Tao, 2002 #1999;Wen, 2012 #2564;Xia, 2011 #2633}; Pig: {Yang, 2010 #2674}{Seiler, 2008 #2695}{Vargas, 2017 #2798} |
| Protection by inhibitors |
P23H-1: {Aguila, 2014 #2745;Stiles, 2016 #2672} S334ter-3: {Shin, 2016 #2746} |
| Neuroprotective agents – AAV delivery |
P23H-1: {Liang, 2001 #2006;Raz-Prag, 2009 #2729}{Peterson, 1998 #1771} P23H-3: {Green, 2001 #2031} S334ter-4: {Green, 2001 #2031;Lau, 2000 #1979;Liang, 2001 #2006;McGee Sanftner, 2001 #2029} |
| Topical application and electroporation of protective agents |
P23H-1: {Picard, 2015 #2656} P23H-2: Hz: {Ozaki, 2013 #2657} S334ter-4: {Nagai, 2016 #2676}; Hz: {Ozaki, 2013 #2657} |
| Neuroprotective agents – from biodegradable microspheres | P23H-3: Hz: {Fernandez-Sanchez, 2017 #2795} |
| Systemic application of protective agents |
P23H-1: {Athanasiou, 2017 #2712;Froger, 2012 #2730;Vasireddy, 2011 #2650}; Pig: {Viringipurampeer, 2016 #2643} P23H-3: {Martin, 2004 #2744} S334ter-4: {Yanai, 2012 #2706}{Martin, 2004 #2744} |
| Protection by nanoceria | P23H-1: {Wong, 2015 #2580} |
| Targeted knock-down of P23H rhodopsin expression |
P23H-1: {Murray, 2015 #2653} P23H-3: {Drenser, 1998 #1899;LaVail, 2000 #1936;Lewin, 1998 #1827;Tessitore, 2006 #2677}{Gorbatyuk, 2007 #2481}; Hz: {Gorbatyuk, 2012 #2560} |
| Gene therapy-other |
P23H-1: {AAV-XIAP \Leonard, 2007 #2423}; Pig: {Leonard, 2007 #2423} S334ter-3: {CRISPR/Cas9 gene editing \Bakondi, 2016 #2612} S334ter-4: {AAV-XIAP \Leonard, 2007 #2423}; Pig: {Leonard, 2007 #2423} |
| Transplantation |
P23H-1: {Yang, 2010 #2674} S334ter-3: Pig: {Qiu, 2005 #2740;Seiler, 2010 #2688;Seiler, 2012 #2685;Seiler, 2010 #2690;Seiler, 2005 #2694;Seiler, 2008 #2695;Seiler, 2008 #2696}{Thomas, 2006 #2300}{Yang, 2010 #2707}{Sagdullaev, 2003 #2069}{Seiler, 2017 #2800} S334ter-5: {Qiu, 2005 #2740}; Pig: {Seiler, 2005 #2694;Thomas, 2004 #2702} |
| Electrical current therapy | P23H-1: Pig: {Hanif, 2016 #2713;Rahmani, 2013 #2659} |
| Retinal prosthetics |
P23H-1: {Salzmann, 2006 #2724}; Pig.: {Adekunle, 2015 #2723} P23H-3: {Linderholm, 2013 #2714} S334ter-3: {Light, 2014 #2720}; Pig: {Fransen, 2014 #2719} S334ter-4: {Finlayson, 2010 #2718} |
| OCT imaging |
P23H-1: {Orhan, 2015 #2579;Picard, 2015 #2656}; Hz: {Pinilla, 2016 #2793} S334ter-3: Pig: {Seiler, 2010 #2690;Thomas, 2006 #2300}{Seiler, 2017 #2800} |
| Behavior and OKR |
P23H-1: Pig: {McGill, 2012 #2555}{Segura, 2015 #2660} P23H-3: Pig: {McGill, 2012 #2555} P23H-2: Pig: {McGill, 2012 #2555} S334ter-7: Pig: {McGill, 2012 #2555} S334ter-3: Pig: {McGill, 2012 #2555}{Thomas, 2007 #2698;Thomas, 2007 #2701;Thomas, 2004 #2344;Thomas, 2010 #2547} S334ter-5: Pig: {McGill, 2012 #2555} S334ter-4: Pig: {McGill, 2012 #2555;McGill, 2012 #2629} S334ter-9: Pig: {McGill, 2012 #2555} |
| Light-induced stress and degeneration, including susceptibility differences |
P23H-1: {Machida, 2001 #1947} P23H-3: {Organisciak, 2006 #2741;Organisciak, 2003 #2251;Vaughan, 2003 #2360}{Ablonczy, 2000 #2742;Bicknell, 2002 #2377;Duncan, 2006 #2743;Ranchon, 2003 #2121;Yu, 2007 #2708} P23H-2: {Organisciak, 2006 #2741;Organisciak, 2003 #2251;Vaughan, 2003 #2360} S334ter-3: {Thomas, 2007 #2701} S334ter-4: {Organisciak, 2006 #2741;Organisciak, 2003 #2251;Vaughan, 2003 #2360}{Ranchon, 2003 #2121} S334ter-9: {Organisciak, 2006 #2741;Organisciak, 2003 #2251;Vaughan, 2003 #2360} |
| Light management of RD | P23H-3: {Valter, 2008 #2704;Valter, 2009 #2616;Walsh, 2004 #2367}{Nir, 2001 #2371} |
| Gene dosage | P23H-1: {Pinilla, 2005 #2655} |
| Broad reviews of cell death and therapeutic approaches | {Cuenca, 2014 #2619;Stone, 1999 #2370} |
| Reviews of specific areas | {Leveillard, 2014 #2631;Lin, 2010 #2536}{Yu, 2005 #2304}{Wen, 2012 #2564} |
| Genetic modification of line | S334ter-3: Pig: {RD nude; \Seiler, 2014 #2689}{Seiler, 2017 #2800} |
| Hearing deficit | P23H-1: {Sotoca, 2014 #2645} |
Hz, Homozygous
Pig, Pigmented
We and others have published extensively on the retinal function of most of the Tg rat lines (Table 5), so we limit the presentation in this work to the amplitudes of the light responses to saturating (or near saturating) stimulus intensities at a number of ages (Fig. 2) and the recovery kinetics following a flash of light in the S334ter-9 line (Fig. 18), which have not yet been described. The P23H lines have been examined more extensively than the S334ter lines, and the most detailed single study has been by Machida et al. (2000). Other studies with comprehensive ERG analysis of P23H lines (Chrysostomou et al., 2009a; Murray et al., 2015; Orhan et al., 2015; Wong et al., 2015), including homozygous P23H rats (Lax et al., 2014; Pinilla et al., 2005), and S334ter line 4 (Kaldi et al., 2003; Leonard et al., 2007; Ranchon et al., 2003), including homozygous S334ter-4 rats (Ozaki et al., 2013), are among those in Table 5. The general pattern of most of the lines is for the rod-dominated scotopic a-wave to decline earliest, followed by the scotopic b-wave, and then the photopic b-wave (Figs. 2A–C and 2F–H). The photopic b-wave is the same as that of control SD rats at least to P30 in the P23H-3 and P23H-2 lines (Figs. 2B–C), and it stays the same to at least P120 in the S334ter-9 line (Fig. 2H), the oldest we examined.
Figure 18.

(A) Representative recordings of scotopic ERG responses from P90 SD, P23H-2 and S334ter-9 rats. The responses are from flashes of 1.9 log cd-s/m2 presented in darkness with different intervals between stimulus presentations (the interstimulus interval, ISI). At ISI intervals of 1 and 2 min, the downward going scotopic a-wave was similar in all three lines. However, when the ISI was shortened, the a-wave became progressively reduced (“a” at 30 and 10 sec) in the S334ter-9 line, but not in the SD or P23H-2 lines. (B) Amplitudes of the a-wave as a function of ISI. The values represent the mean ± SEM of 3–4 rats of each line.
The extremely rapid RDs with no ROSs generated, lines S334ter-7 and S334ter-3, showed no responses to light (not shown in Figs. 2D–E), as previously reported for the S334ter-3 line (Roddy et al., 2012) and the pigmented S334ter-7 line (McGill et al., 2012a). The next fastest RD with very short ROSs, line S334ter-5, showed no scotopic a-wave or b-wave responses, but did give measurable cone dominated photopic b-wave responses (Fig. 2F).
In all of the lines with ERG responses, the decline was relatively close to the decline in number of PRs (ONL thickness) (Figs. 2A–C and 2F–G), except for line S334ter-9 (Fig. 2H). S334ter-9 had a nearly normal ONL for most of the first year, yet the a- and b-waves declined as if the retina was degenerating. The cone-mediated photopic b-wave remained nearly normal in this line (Fig. 2H). It should be noted that Chen et al. (1995) produced 5 Tg mouse lines using the same S334ter construct as in the present study, and 4 of the 5 lines expressed high amounts of S334ter rhodopsin, in which PRs degenerated rapidly, failing to make OSs. The one line with the least mutant rhodopsin expressed had a much slower PR degeneration, relatively similar to our Tg rat line S334ter-9. In their mouse line, they found that the rod-mediated scotopic a-wave had a significantly prolonged recovery period following the light stimulus (Chen et al., 1995). In these mice, the intervals between the flash presentations (interstimulus interval, ISI) needed to be extended to up to several minutes to result in response amplitudes to reach their maximal size. Similar ISI effects were found in the S334ter-9 rats, but not in normal SD rats or in P23H-2 rats with slow degeneration comparable to that of S334ter-9, as shown in Figs. 18A–B.
The most significant consequence of the prolonged recovery phase in the S334ter-9 line was that with a decreasing ISI, the rod-mediated a-wave amplitude progressively decreased (see “a” in Fig. 18A). This became so severe that it ultimately resulted in lower b-wave amplitudes, as well (see Fig. 18A, S334ter-9 at 30 and 10 sec).
3.5 Lack of injury-induced protective response in the transgenic rat retinas
A prominent feature of RCS rat retinas and those of light-damaged SD rats is that retinal injury from injections of neuroprotective agents, PBS buffer, or even a dry needle, results in a transient local rescue of degenerating PRs (Faktorovich et al., 1990, 1992). This is seen as a thickening of the ONL as the injection site is approached. This increased thickness of the ONL at or near the injection site can sometimes confound results based on ONL thickness. Thus, we have asked whether the same phenomenon occurs in the rhodopsin Tg rat retinas.
In more than 1,000 experiments where we injected intravitreally either PBS buffer, CNTF, bFGF, BDNF, LIF, or NT-3 into each of the Tg lines, except the two slowest-degenerating lines, P23H-2 and S334ter-9, we found 26 clear injection sites (e.g., Figs. 19A, B). The rats were mostly P8-P16 at the time of injection into the eye through the far peripheral superior retina, with a few as old as P45. The eyes were taken mostly at the ages of P28-P59, with a few as old as P130. Thus, postinjection intervals were mostly 15–40 d, but some were as long as 115 d. Each of the 26 injection sites showed a thinning of the ONL as the injection site was approached (asterisks in Figs. 19A, B), the opposite of that seen in RCS rats and in light-damaged retinas (Faktorovich et al., 1990, 1992).
Figure 19.

Light micrographs of injection sites (arrows) in the superior far peripheral retinas of LIF-injected eyes. Both are from line S334ter-5. (A) Injected at P13, eye taken at P31. (B) Injected twice, at P10 and P19, eye taken at P30. Immediately adjacent to the damaged area due to the injection, the ONL tapers near the injection site (asterisks). Magnification bar = 50 μm.
The reason for the difference between the injury response in RCS rats and light-damaged retinas and the lack of one in the rhodopsin Tg retinas is not fully understood. On the one hand, injury-induced gene expression of many protective molecules in SD rats is minimal at early postnatal ages, but increases with age and peaks at P60-P90 (Cao et al., 2001). Most experimental injections in the Tg rats are made earlier than this age, so they might not be expected to produce an injury-induced response. On the other hand, many injections in the RCS rat have been made at about P20-P25 (Faktorovich et al., 1990), when an injury-induced protective response would not be expected in a developing rat retina (Cao et al., 2001).
4. Discussion
4.1 PR cell death: rates and mechanisms
The P23H and S334ter Tg lines described here show a wide range of PR cell death and disappearance (Fig. 1). The overall rate of cell death is proportional to the relative amount of the mutant to WT rhodopsin (Table 1), the greater the Tg rhodopsin, the faster the pace of PR degeneration. In all of the lines that show a frank PR degeneration (all except P23H-2 and particularly S334ter-9), the rate of degeneration appears to be biphasic, with a rapid early phase and a much slower late phase (Figs. 1 and 2). Remarkably, the early rapid phase appears to be similar in all of these lines, at least up to about P20-P30, whereas the slower late phase varies widely among the lines. A similar biphasic rate of degeneration is seen in P23H knock-in mice (Chiang et al., 2015; Sakami et al., 2014).
The initial rapid phase of PR cell death and loss coincides with the abundance of DNA fragmentation detected TUNEL staining in all examined lines, suggesting apoptosis as the major early mechanism of degeneration. For example, the rapidly degenerating S334ter-3 line shows a peak in TUNEL staining at about P12 (Kaur et al., 2011; Liu et al., 1999), when PRs are rapidly degenerating (Fig. 1B). Likewise, the slightly slower degenerating in the S334ter-5 line (Fig. 1B) has a peak of TUNEL-labeled PR nuclei at about P17. The more slowly degenerating P23H lines, despite their overall slower loss of PR cells, show a similar very early rate of degeneration up to about P30 (Fig. 1A), and the peak of TUNEL-labeled PRs in the P23H-1 line is at P15-P18 (Kaur et al., 2011; Lee et al., 2003), and the peak in the slower P23H-3 line is at about P20 (Lee et al., 2003; Yu et al., 2004). There is evidence, however, for non-apoptotic cell death in some of these lines (Arango-Gonzalez et al., 2014; Farinelli et al., 2014; Kaur et al., 2011). Although we and other investigators assume the early phase of rod PR cell death in the Tg rats is a cell autonomous event (Bakondi et al., 2016), it should be noted that other potentially related events occur at or near the early peak of cell death; for example, in the S334ter-3 line caspase-3-like activity and activated caspase-3 are present at the peak of degeneration (Liu et al., 1999). In addition, invading presumptive microglia are seen in the ONL at or preceding the peak of cell death in the rapid degenerations (Figs. 10–12) that may influence PR cell death (Discussed below). This is complicated by the normally developing hypoxic state in the rat between P15-P22 (Maslim et al., 1997).
Importantly, in most of the TUNEL studies, the incidence of labeled dying PR cells after the peak drops almost as rapidly as the rise to the peak (Kaur et al., 2011; Lee et al., 2003; Liu et al., 1999; Yu et al., 2004). This suggests that the slower, later phase of cell death is due to, at least in part, on cellular mechanisms other than apoptosis.
The rapidly degenerating lines, S334ter-7, S334ter-3 and S334ter-5, probably succumb to the overexpression of rhodopsin (Fig. 2) (Roddy et al., 2012), despite the absence of RR OSs in the S334ter-7 and S334ter-3 lines. This would be consistent with the much greater proportion of mutant rhodopsin than in the other lines (Table 1), as well as the total eye rhodopsin being extremely high soon after the onset of rhodopsin expression (Treisman et al., 1988) in the S334ter-7 and S334ter-5 lines (not measured in S334ter-3) (Figs. 2D and F). The overexpression of rhodopsin also correlates with the displacement of PR nuclei into the IPS seen in these 3 lines (Figs. 10–12). The mechanism of this mislocalization of PR nuclei remains to be determined.
The early onset and rapid pace of degeneration in the S334ter-7 and S334ter-3 lines shows an apparent developmental regulation of rod PR cell death. The abundance of pyknotic nuclei is in the innermost ONL (Figs. 10 and 11) (Farinelli et al., 2014), and the highest incidence of TUNEL labeling (Liu et al., 1999) and DNA hypermethylation (Farinelli et al., 2014) co-localize there. Likewise, the highest incidence of TUNEL labeling in the ONL of S334ter-5 rats is in the innermost part of the layer, albeit at a slightly lower incidence and later time than in the somewhat faster S334ter-3 line (Martinez-Navarrete et al., 2011). The earliest generated PRs are ultimately localized in the innermost ONL (Carter-Dawson and LaVail, 1979b; Ilia and Jeffery, 2000; Sidman, 1961; Young, 1985). This first appearance of pyknotic PR nuclei in the innermost ONL is also seen in heterozygous and homozygous P23H knock-in mice (Chiang et al., 2015) and in I-255/256 rhodopsin mice with a 3 bp deletion (Penn et al., 2000). Thus, the earliest-generated rod PRs are the first to degenerate.
The slow phase of PR degeneration is almost certainly complex, with numerous extrinsic or epigenetic factors involved. For example, Pennesi et al. (Pennesi et al., 2008) determined in the 8 Tg rat lines that early loss of PRs had a profound effect on the vascularization of the outer retina that was proportional to the rate of PR cell loss. This, in turn, has further consequences and creates a much thinner outer retinal structure, which results in a hyperoxic state that has been implicated in further PR cell loss and metabolic changes of the retina that follow (Yu et al., 2004; Yu and Cringle, 2005). Numerous studies have demonstrated other changes in degenerating PRs and in the retina that may play a role in the slow phase of RD, including changes in the expression of autophagy proteins, Bcl2 family of proteins and the mTOR/AKT pathways (Sizova et al., 2014); oxidative stress (Cuenca et al., 2014); hyperoxic environment (Yu et al., 2004); activation of the unfolded protein response (Kroeger et al., 2012; Lin et al., 2007); DNA hypermethylation (Farinelli et al., 2014); environmental light regimen and intensity (Organisciak et al., 2003; Valter et al., 2009; Walsh et al., 2004), seen also in VPP (P23H) mice (Naash et al., 1996a; Wang et al., 1997); endogenous upregulation of protective factors, such as CNTF, FGF-2 and FGF-2/FGFR1 (Yu et al., 2004); Müller cell and astrocyte reactive responses (Fernandez-Sanchez et al., 2015a); microglial activation and invasion (see below); microglial interactions with Müller cells (Bringmann et al., 2006; Harada et al., 2002; Zhu et al., 2013a); genetic modifier genes (Naash et al., 1996b); and other factors (Reviewed by Cuenca et al., 2014). Thus, there are many changes in the retinal environment that may influence PR degeneration after the peak of early cell death.
Numerous other studies of PR cell death mechanisms have been carried out with the Tg rats. For example, it has been found that misregulation of rhodopsin phosphorylation may be involved as an important step in RD of P23H-2 rats, and the Ca2+ channel blocker, nilvadipine, had a protective effect in this line (Saito et al., 2008); however, D-cis-Diltiazem did not have a protective effect in P23H-1 rats (Bush et al., 2000). A relationship between mitochondrial DNA damage and PR cell death has been found in P23H-3 rats (Bravo-Nuevo et al., 2007), and that mitochondrial μ-calpain and the AIF pathway may be involved in PR degeneration in homozygous P23H-2 and S334ter-4 rats (Ozaki et al., 2013). Most recently, it has been reported that in P23H-1 rats, rod PRs die by necroptosis via an RIP1/RIP3/DRP1 mechanism, whereas genetically normal cone PRs die via NLRP3 inflammasome activation (Viringipurampeer et al., 2016).
4.2 Cone PR loss in the rhodopsin Tg rats
In retinitis pigmentosa, rod PR degeneration precedes cone PR degeneration in most cases (Milam et al., 1998), and more so in models with rhodopsin mutations. This is clearly seen in each of the Tg lines, as shown in Table 3. The question of why cone PRs die, even though mutant rhodopsin is expressed only in rod PRs, has been the subject of important study and debate. These include the reduction of rod-derived cone viability factors, oxidative stress, elevated oxygen status with the loss of rods, release of cytotoxic agents by invading microglia or degenerating rods, activation of the NLRP3 inflammasome, and nutrient deprivation and metabolic dysregulation of the cones (Campochiaro, 2007; Cuenca et al., 2014; Leveillard et al., 2014; Punzo et al., 2009; Punzo et al., 2012; Sahel, 2005; Stone et al., 1999; Viringipurampeer et al., 2016; Yu et al., 2004; Yu and Cringle, 2005). These proposed mechanisms of cone cell death are not mutually exclusive. In fact, each of them can be supported by experimental studies in animal models. Most of them support an indirect or direct relationship to rod cell loss, as do our data on survival of substantial numbers of cones following the death of most rods (Table 3). Indeed, the slower degenerating lines, e.g., P23H-1, P23H-3 and S334ter-4 that have a larger complement of surviving rods, particularly in the inferior hemisphere, have a higher percentage of surviving cones. Chrysostomou et al. (2009a) have reported that in dim light 100% of cones survive for the life of the animal in P23H-3 rats. Similarly, in pigmented P23H-1 rats, cone function was normal and did not change until the thickness of the ONL was reduced by 75% (Lu et al., 2013). Clearly, there is a relationship of rod degeneration to cone degeneration (Chrysostomou et al., 2008; Chrysostomou et al., 2009a; Chrysostomou et al., 2009b). Nevertheless, it must be recognized that in longstanding cases of retinitis pigmentosa, some cones persist in the apparent absence of rods.
The use of various histochemical and immunohistochemical methods, particularly using retinal wholemounts, has uncovered a remarkable spatiotemporal phenotype in the rapidly degenerating S334ter-3 Tg line. That is the formation of “cone rings” as a result of outer retina remodeling following massive rod cell death. It is thought by most that hot spots of rod cell death combined with a later cone cell migration lead to the orderly mosaic of “cone rings” (Ji et al., 2014; Ji et al., 2012; Lee et al., 2011; Li et al., 2010; Zhu et al., 2013a). Müller cell interactions likely maintain the cones in ring orientation as shown experimentally by transiently blocking Müller cell metabolism with alpha-aminoadipic-acid (AAA) (Lee et al., 2011). Similarly, suppression of the adherens-junction protein, Zonula occludens-1 (ZO-1) with AAA and by using siRNA to inhibit ZO-1 expression (Yu et al., 2016) both transiently disrupt the cone rings.
In these studies, several questions are raised that can be explained by our current work, or with which our findings are consistent. The degeneration in Tg rats has been considered to follow the same rate in superior and inferior hemispheres (Machida et al., 2000). In wholemounts of S334ter-3 retinas, the number and size of cone rings increase more quickly in the superior hemisphere than in the inferior (Ji et al., 2014). We demonstrate that in all of the Tg lines, the rate of degeneration in the superior hemisphere is greater than in the inferior hemisphere (Fig. 4 and Table 3). This is also consistent with the presumption that the increase in size and number of cone rings is a result of rod cell death and rearrangement of cones.
The second feature of the S334ter-3 studies is that despite the rapid, massive rod cell death, cones were found to persist with little (Hombrebueno et al., 2010; Li et al., 2010) or no (Ji et al., 2012) cell death for 80–180 days. It is significant that although the cone PRs lose their OSs at very early stages (Hombrebueno et al., 2010), their OSs can be stimulated to regenerate with the application of two members of the IL-6 family of cytokines, CNTF (Li et al., 2010) or oncostatin M (Xia et al., 2011). In our present study, we confirm that cones survive for an extended period in the S334ter-3 line (and others). For example, at P63, when less than 1% of rods survive, almost 80% of cones are still present (Table 3).
In the lone study where cone rings were sought in slower degenerating P23H-1 rats, it was found that the formation of cone rings also occurs. They were first seen at a somewhat later time, after P30 and by P90 (Garcia-Ayuso et al., 2013), than in the much faster S334ter-3 degeneration, where they are detectible by P10-P20 (Ji et al., 2012; Li et al., 2010; Zhu et al., 2013a). In the P23H-1 retina, the cone rings are much larger in the superior hemisphere than in the inferior hemisphere, just as in the S334ter-3 line. It remains to be determined whether the formation of cone rings is a common pathologic process in other RD models, which requires immunohistochemistry on retinal wholemounts for detection.
4.3 Invading microglia in the outer retina of Tg rats
A potentially important modifier of both the rapid and slow phases of the RDs is the invasion of microglia into the ONL and IPS. Microglia are the major immunocompetent cells of the CNS, including the retina, which represent an acutely responsive, tissue-based system for scavenging viruses, bacteria, dead neurons and any cell debris (Glybina et al., 2010). In addition to their phagocytic role, activated microglia often proliferate (Zeiss and Johnson, 2004) and release many toxic agents (Cuenca et al., 2014; Langmann, 2007; Roque et al., 1999; Schwartz et al., 2006), leading to death of neighboring cells in a bystander effect (Gupta et al., 2003). However, some microglia may secrete protective molecules (Butovsky et al., 2005; Harada et al., 2002; Noailles et al., 2014; Polazzi and Monti, 2010; Schwartz et al., 2006), so there has been an ongoing debate on the precise role of microglia (Reviewed by Cuenca et al., 2014). What is known is that the activated microglia are found in virtually all RDs where they have been sought (Cuenca et al., 2014; Harada et al., 2002; Langmann, 2007; Zabel et al., 2016; Zeiss and Johnson, 2004; Zhang et al., 2005), including human retinas from patients with retinitis pigmentosa, late-onset RD and AMD (Gupta et al., 2003). After the loss of PRs, these cells may persist in the retina and eye for the life of the animal with immune consequences (Noailles et al., 2016).
The source of the microglia in the outer retina is also known, first from the pioneering work of Thanos (1992), who axotomized retinal ganglion cells and injected a fluorescent dye into the region of the optic nerve stump in RCS rats with inherited retinal dystrophy. The dye was transported in retrograde direction to the ganglion cell bodies, and when the ganglion cells degenerated due to the axotomy, the resting microglia became activated and ingested the fluorescent dye along with retinal ganglion cell debris. These cells were then attracted to the ONL, where they phagocytized massive amounts of degenerating PR debris. The same result was obtained with light-induced RD using axotomy, fluorescent dye and a monoclonal antibody, 5D4, to identify microglia (Ng and Streilein, 2001), providing further evidence that activated microglia in the inner retina migrate into the outer retina in response to damaged PRs.
Later, more specific antibodies to microglia were developed and they were used (primarily Iba-1) to demonstrate microglia immunohistochemcally in the Tg rats described in the present study. These have been the S334ter-3 (Zhu et al., 2013a), S334ter-4 (Glybina et al., 2010; McGill et al., 2012b), P23H-1 (Di Pierdomenico et al., 2017) and P23H-3 (Noailles et al., 2014) lines. These studies illustrate two points about microglia that are consistent with our observations. First, microglia respond to degenerating cells rapidly, typically within 6–24 hrs.(Glybina et al., 2010; Zhang et al., 2005), and second, when the activating stimulus is eliminated, they quickly return to the resting state (Glybina et al., 2010; Zhang et al., 2005). While we have not used immunohistochemistry, we have found invading cells in the ONL at times that precisely match these studies. In the rapidly degenerating S334ter-3 line, Zhu et al. (2013a) found that in wholemounts at P14 and P17, the holes in the surviving rod PR mosaic contained clusters of Iba-1-positive microglia, and these cells were gone by P21. We found invading cells with the nuclear characteristics of microglia to be abundant early, up through P15 in the ONL of S334ter-3 retinas, but only one such cell at P21 and none later (Fig. 11). It should be noted that the very high contrast of immunohistochemistry, particularly in sections 5–10 μm in thickness, allows a much greater chance to see immunopositive cells than in 1-μm thick plastic sections. Thus, the frequent finding of such invading cells at P10 and P12 in our study indicates the probable very high density of such cells at these early stages. It should be noted that microglial cells also proliferate near the end of PR cell death, presumably to generate more phagocytic cells to clear the debris of dead and dying cells (Di Pierdomenico et al., 2017).
In the rapidly degenerating line S334ter-7, we observed such invading cells in the ONL at P6, P7, P8, P10 and P12, but very few thereafter (Fig. 10). In the somewhat slower degenerating S334ter-5 line, we found invading microglia also early in the degeneration at P12 and P15, but none thereafter (Table 2). Presumptive microglia have been shown with similar kinetics in P23H knock-in mice (Chiang et al., 2015).
4.4 Accumulation of vesicular structures in the IPS of S334ter model
The S334ter lines carry a Class I mutation, which is thought to result in defective vectorial transport of rhodopsin from the PR IS Golgi apparatus to the PR OSs, and thus is considered a sorting defect. The P23H lines carry a Class II rhodopsin mutation, which results in abnormal folding of the newly formed rhodopsin molecule and its retention in the endoplasmic reticulum (See Introduction). The S334ter mutation results in the production of a truncated rhodopsin molecule missing the last 15 residues of the C-terminus (S334 to A348) (Chen et al., 1995). The P347S transgenic mouse, with its defect in the C-terminus of rhodopsin, shows a remarkable accumulation of small (100–370 μm), membrane-bound, rhodopsin-containing vesicles in the IPS, primarily among the apical ISs and basal OSs (Li et al., 1996). It was thought by Li et al. that this accumulation represented aberrant transport of mutant rhodopsin from the ISs to the nascent disc membranes of the OSs (1996). Much research supports this notion. We have found numerous extracellular vesicles in several lines of S334ter rats (Table 4), and evidence that PR cell death in the S334ter-4 line is due to rhodopsin mis-sorting has been found with immunogold labeling (Green et al., 2000).
The phenotype of extracellular vesicle accumulation has been found in other RDs. These include the Q344ter mouse (Concepcion and Chen, 2010), P347L transgenic rabbit (Kondo et al., 2009); rds mouse (Chakraborty et al., 2014; Cohen, 1983; Jansen and Sanyal, 1984); pcd mouse (Blanks et al., 1982); erd dog (Acland and Aguirre, 1987); tubby (rd5) mouse (Heckenlively et al., 1995); and the tulp1−/− mouse (Hagstrom et al., 1999). While the mechanism of PR cell death, or even the gene product in a few cases, is not known for each of these, the aberrant transport of rhodopsin has been suggested for each, or is plausible given the cytopathology. What is clear is that vesicle accumulation is not a general phenomenon for all RDs. Many mutants with gene defects other than those above (i.e., in the case of rhodopsin, gene defects located away from the C-terminal and near the N-terminal), do not show vesicle accumulation. These include the following rhodopsin mutants: P23H mouse (Olsson et al., 1992); VPP (P23H, V20G, P27L) mouse (Liu et al., 1997); rhodopsin knock-out mouse (Humphries et al., 1997); P23H knock-in mouse (Sakami et al., 2014; Sakami et al., 2011); K296E mouse (Li et al., 1996); T17M mouse (Hagstrom et al., 1999); G90D mouse (Naash et al., 2004); and P23H rats (present study, Table 4). Moreover, no mis-sorting was seen in P23H transgenic rats (Green et al., 2000).
Several features of the vesicle accumulation are common to these mutants. In each case that has been studied, the vesicles have been shown to contain rhodopsin using immunogold labeling. These include the P347S mouse (Li et al., 1996); the rds mouse (Jansen et al., 1987; Nir and Papermaster, 1986; Usukura and Bok, 1987); and the pcd mouse (Blanks and Spee, 1992). It is highly probable that the vesicles seen in the same region of the retina of other mutants also contain rhodopsin.
In all cases, the origin of the vesicles appears to be from budding or blebbing from the ISs (Acland and Aguirre, 1987; Hagstrom et al., 1999; Kondo et al., 2009), or in some cases, protrusions from the ISs (Blanks et al., 1982; Blanks and Spee, 1992). Most of the vesicles are ultimately extracellular, as confirmed by scanning electron microscopy (Blanks and Spee, 1992; Li et al., 1996).
The various mutants also share an increased incidence of extracellular vesicles at or near the period of maximal PR cell death, where a time course was studied (Hagstrom et al., 1999; Li et al., 1996). In the present study, vesicle accumulation was seen at the peak of cell death in the S334ter-4 and S334ter-5 lines (Table 4). Likewise, mis-sorting in the S334ter-4 line was greatest at this age (Green et al., 2000). Note that no vesicles were seen in the very rapid degenerations with early onset, lines S334ter-7 and S334ter-3; no OSs were present in these retinas, and the ISs showed early abortive changes. Presumably, the massive overexpression of mutant rhodopsin led to very early cell death before vectorial machinery and target OSs could be developed, or the energy to carry out this task was missing in these early-degenerating PR cells. Also, the extremely slowly degenerating S334ter-9 line that appeared almost normal showed no extracellular vesicles. Increasing the rate of degeneration by doubling the transgene complement, as in the S334ter-9 homozygote, did show an abundance of vesicles. A similar dose-dependency was found in homozygous P347S Tg mouse retinas (Li et al., 1996).
In the two very slow RD mutants, rds and pcd, the incidence of vesicles peaked at early ages, then declined at later ages, with the peak density seen at P10-P20 in rds (Jansen and Sanyal, 1984; Usukura and Bok, 1987) and at approximately P25-P40 in pcd (Blanks et al., 1982; Blanks and Spee, 1992). However, the rds and pcd retinas degenerate over the course of about a year. This apparent discrepancy can be understood, at least in part, by these RDs having rapid early rates of degeneration, with much slower rates thereafter (LaVail, 1981). In pcd, for example, by P53, very few vesicles remained (Blanks et al., 1982), and at that age, 50% of rod PRs had degenerated (LaVail et al., 1982). The precise mechanisms of the two phases of PR cell death in the faster-degenerating mutants are poorly understood. Presumably, the defective sorting of IS proteins to the OS is involved in PR cell death, but the exact molecular and cellular mechanisms, and how they relate to the phenotype, need to be resolved. It is likely they are complex (Fliesler et al., 1984; Grossman et al., 2011; Sammons and Gross, 2013).
4.5 Practical considerations in selection of rat models for experimental studies
Before the construction of the Tg rats, two rat models were widely available and have contributed important insights and findings for vision science. These are the RCS rat and the light-induced model of PR cell death. The RCS rat has been and continues to be extremely valuable in many ways, but its PR degeneration is a non-cell-autonomous process, as the Mertk gene defect is expressed in the RPE cells (D’Cruz et al., 2000) resulting in a failure of OS membrane phagocytosis and subsequent PR cell death. There are, also, some human retinitis pigmentosa patients with defects in the Mertk gene (Gal et al., 2000; Thompson et al., 2002). Likewise, light-induced degeneration has been extensively studied and has the real advantage of being able to control the intensity and exposure time components to produce a given degree of RD in a given period of time (Organisciak and Winkler, 1994; Wenzel et al., 2005). Indeed, the model has been extremely useful to test the neuroprotective effects of various agents (LaVail et al., 1992). However, it is now known that a number of neuroprotective agents are effective in the light-induced model, but not in some other naturally occurring or genetically modified rats (Kaldi et al., 2003; Machida et al., 2001b; Ranchon et al., 2003) and mice (Grimm et al., 2004; LaVail et al., 2008; LaVail et al., 1998).
The Tg rats described here offer two different rhodopsin defects that are expressed in rod PRs resulting in an autonomous cell degeneration in the same SD background. Both of the P23H and S334ter models also offer a range of RD times of onset and overall rates of PR degeneration that can effectively be used to carry out many types of experiments. Obviously, if a given approach is specific to one type of mutation, that directs the use to one of the sets of lines. For instance, the P23H lines would be used if an approach attempts to modify mutant P23H alleles or knock down the P23H rhodopsin, such as the use of a P23H-directed ribozyme in either an allele-specific (LaVail et al., 2000; Lewin et al., 1998) or allele-independent (Gorbatyuk et al., 2007) approach, siRNA (Tessitore et al., 2006) or antisense oligonucleotide (Murray et al., 2015). Moreover, these different rat models with mutations located at different ends of the rhodopsin molecule allow the comparison of the two.
While the Tg rat lines have been studied for many aspects of retinal biology (Table 5), they were developed initially as therapeutic disease models. For this purpose, the key factors for choice of line(s) are the time (age) of initial rod cell degeneration and the pace of degeneration, as well as the expected effective life of the agent or procedure being tested. These are essential to establish the age of application and endpoints for the experiments. For example, the most widely used line for initially testing various neuroprotective agents has been the S334ter-3 line (Dykens et al., 2004; Roddy et al., 2012; Song et al., 2003; Xia et al., 2011), with its early age of onset and rapid degeneration, and relative ease of intravitreal injection. For P23H therapeutic studies with neuroprotective agents, the most rapid P23H-1 line has been the most heavily used. However, for most neuroprotective agents with relatively short half-lives, the P23H (and slower S334ter) lines require some sort of sustained delivery, such as the use of encapsulated cell-based delivery of CNTF (Tao et al., 2002) or the use of AAV vectors to deliver various protective agents (Ali, 2012; Conlon et al., 2013; Liang et al., 2001; McGee Sanftner et al., 2001). The use of AAV vectors requires careful consideration of age of onset and pace of PR degeneration, since the most widely used AAV-2 requires a 2- to several-week time for full gene expression of the neuroprotective agent (Bennett et al., 1997; Bennett et al., 1999). There are, however, AAV of different serotypes and isoforms (and other vectors) that have a shorter time to achieve full gene expression (Byrne et al., 2015; Deng et al., 2012; Gorbatyuk et al., 2010; Kolstad et al., 2010; Leonard et al., 2007), as well as different modes of delivery (Byrne et al., 2015).
One of the principal findings of the present study is that the initiation of rod cell death (Table 2) begins earlier than one would expect from most published studies. For example, most studies using the S334ter-3 model have injected a therapeutic agent at about P8-P10 and have examined the retina at about P18-P20 (Dykens et al., 2004; Roddy et al., 2012). Significant cell death has already begun by P10. So, it is reasonable to assume that if the protective agent had been (or could have been) applied earlier, a greater degree of protection might have been achieved.
An important consideration for choice of Tg line is the potential of the rate of degeneration to be modified by retinal irradiance, either by environmental light modification or by eye pigmentation. As in mice, mutation in the N-terminus of rhodopsin, such as the P23H animals, are significantly more sensitive to ambient illumination than those with mutations in the C-terminus. For example, P23H-2 and P23H-3 rats are more susceptible to light-induced damage than are S334ter-4 and S334ter-9 rats (Organisciak et al., 2003; Vaughan et al., 2003). Conversely, dark-rearing slows the RD in P23H rats, but not in S334ter-334 rats (Organisciak et al., 2003). On occasion, and before learning this, we were surprised at the relatively slower degeneration than expected in experiments with P23H-3 rats where the ambient illumination was lower than usual (LaVail, unpublished observations). Within the same mutation type, the line with the greater mutant rhodopsin expression (Table 1) shows greater light-induced degeneration (Organisciak et al., 2003; Vaughan et al., 2003). Similarly, the protective effect of lowering light exposure in VPP mice (in which P23H is one mutation) has been demonstrated (Naash et al., 1996a; Wang et al., 1997).
The use of pigmented animals is beneficial for many experiments, particularly where visualization of the retina is needed (Lu et al., 2013), such as in most transplantation studies, and is essential with most behavioral studies, such as measuring the optokinetic tracking of eyes (McGill et al., 2012a; Segura et al., 2015; Thomas et al., 2010). Pigmented hemizygous Tg rats can easily be produced by crossing homozygous albino Tg rats on the Sprague-Dawley background to pigmented Long-Evans or Copenhagen rats, producing all pigmented hemizygous Tg animals. However, eye pigmentation slows the degeneration in P23H rats, but not in S334ter rats (Leonard et al., 2007; Lowe et al., 2005). In the case of VPP mice, pigmentation slows the degeneration compared to age-matched albino animals (Naash et al., 1996b).
Another consideration for experimental use of the Tg rats is gene dosage. We have encouraged the use of hemizygotes carrying one insertion of the transgene, as described mostly in this study. The rationale is that these animals carry two copies of the WT rhodopsin gene as well as one dose of the mutated rhodopsin gene. Having double the amount of the mutated rhodopsin in the homozygotes results in a genotype even further from the human condition than with the Tg hemizygotes, and an over-expression of rhodopsin is well-known to lead to PR degeneration. As shown in Table 1 and elsewhere, the greater the expression levels of the mutant rhodopsin, the faster and earlier onset of degeneration (Chen et al., 1995; Li et al., 1996; Olsson et al., 1992), and even overexpression to WT rhodopsin is toxic to PRs (Tan et al., 2001). Likewise, double the dose of mutant rhodopsin in homozygous Tg rats results in more severe degeneration than in hemizygotes to a greater or lesser degree among the different lines (Fig. 2). In some cases, the use of homozygous animals may be particularly useful, such as in the case of P23H-2 rats, where the hemizygous line is so slow that PR rescue may be difficult to detect in therapeutic studies, but would be much easier in the somewhat faster homozygous P23H-2 rats (Fig. 2) (Ozaki et al., 2013). In addition, the maintenance of homozygote lines requires less oversight than maintaining the homozygous lines and crossing them by WT rats to produce hemizygous animals. It should be noted that the recently produced P23H rhodopsin knock-in mouse represents the model that most closely matches the human condition (Sakami et al., 2011) and will be extremely useful, but it is still limited for some studies because of its small eye size (see Introduction).
4.6 Experimental uses of the Tg rats
In previous sections of the Discussion, we have considered in-depth the issues of PR cell death; cone cell death, specifically; the role of microglia in the Tg rhodopsin and other RDs; and phenotypic features (extracellular vesicles) that distinguish the S334ter from the P23H models. However, the rhodopsin Tg rats have been the subjects or experimental tools for the analysis of many more RD topics. These are listed in Table 5, which includes reference to all known experimental studies using the Tg rats, as far as we are aware. It should be noted that the assignment of a given study to a particular topic is somewhat artificial, as many or most studies consider multiple topics and use multiple approaches. Indeed, the rationale to test specific therapeutic approaches most often derives from cell death mechanisms. Clearly, the 8 rhodopsin Tg lines have been, and will continue to be, extremely useful models for the study of inherited RDs.
Highlights.
Rat models for retinitis pigmentosa due to rhodopsin mutations are described.
P23H and S334ter mutations are present in 8 transgenic lines.
Comprehensive study of photoreceptor degeneration from onset to 1 year of age.
Rhodopsin trafficking defect is seen in S334ter but not P23H models.
No injury-induced protective response occurs in the rat models.
Acknowledgments
The authors thank Nancy Lawson for assistance in managing the transgenic rat colony and to Jose Velarde and Dean Cruz for assistance in maintaining the colony. This work was supported by NIH grants EY01919, EY6842 and EY02162 (MML), EY00415 (JLD), EY010609, EY08656 and EY026499 (MIN), EY12155, EY027193, EY027387 (JC) and The Foundation Fighting Blindness (MML, JLD and JGF), That Man May See (MML) and The Macula Vision Research Foundation (MML). MML was an RPB Senior Scientist Investigator and JLD was an RPB Career Development Awardee during this study, and both were supported by an Unrestricted Grant from Research to Prevent Blindness to the Department of Ophthalmology, UCSF.
Footnotes
Conflict of Interest: None of the authors has any financial, personal or other relationship that could inappropriately influence, or appear to influence, this work.
Contributions of the authors:
Matthew M. LaVail, PhD, All aspects of the research; analysis of data; writing the manuscript; editing the manuscript.
Shimpei Nishikawa, MD, ERG data acquisition and analysis.
Roy H. Steinberg, MD, PhD†, Original idea for the research; planning the research; analysis of data.
Muna I. Naash, PhD, Constructed the P23H; established the screening protocol of the P23H rats, editing the final manuscript.
Jacque L. Duncan, MD, Planning and analysis of ERG; analysis of ERG data; statistical analysis; editing the final manuscript.
Nikolaus Trautmann, MD, Rhodopsin assays, including analysis and interpretation.
Michael T. Matthes, PhD, All histological and morphometric analyses; preparation of the manuscript.
Douglas Yasumura†, All histological, photomicrographic and morphometric analyses.
Cathy Lau-Villacorta, All PCR and planning of breeding in the development of the lines.
Jeannie Chen, PhD, Providing the S334ter construct; editing the final manuscript.
Ward M. Peterson, PhD, Intraocular injections to demonstrate lack of injury response.
Haidong Yang, OD, MS, FAAO ERG data acquisition and analysis; statistical analysis.
John G. Flannery, PhD, Planning of the studies; analysis of RNA and protein data; writing part of the original manuscript; editing the final manuscript.
All authors have approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abed E, Corbo G, Falsini B. Neurotrophin family members as neuroprotectants in retinal degenerations. BioDrugs. 2015;29:1–13. doi: 10.1007/s40259-014-0110-5. [DOI] [PubMed] [Google Scholar]
- Ablonczy Z, Knapp DR, Darrow R, Organisciak DT, Crouch RK. Mass spectrometric analysis of rhodopsin from light damaged rats. Mol Vis. 2000;6:109–115. [PubMed] [Google Scholar]
- Acland GM, Aguirre GD. Retinal degenerations in the dog: IV. Early retinal degeneration (erd) in Norwegian elkhounds. Exp Eye Res. 1987;44:491–521. doi: 10.1016/s0014-4835(87)80160-4. [DOI] [PubMed] [Google Scholar]
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J. Gene therapy restores vision in a canine model of childhood blindness. Nature Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- Acosta ML, Shin YS, Ready S, Fletcher EL, Christie DL, Kalloniatis M. Retinal metabolic state of the proline-23-histidine rat model of retinitis pigmentosa. Am J Physiol Cell Physiol. 2010;298:C764–774. doi: 10.1152/ajpcell.00253.2009. [DOI] [PubMed] [Google Scholar]
- Adekunle AN, Adkins A, Wang W, Kaplan HJ, de Castro JF, Lee SJ, Huie P, Palanker D, McCall M, Pardue MT. Integration of Perforated Subretinal Prostheses With Retinal Tissue. Transl Vis Sci Technol. 2015;4:5. doi: 10.1167/tvst.4.4.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adijanto J, Naash MI. Nanoparticle-based technologies for retinal gene therapy. Eur J Pharm Biopharm. 2015;95:353–367. doi: 10.1016/j.ejpb.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguila M, Bevilacqua D, McCulley C, Schwarz N, Athanasiou D, Kanuga N, Novoselov SS, Lange CA, Ali RR, Bainbridge JW, Gias C, Coffey PJ, Garriga P, Cheetham ME. Hsp90 inhibition protects against inherited retinal degeneration. Hum Mol Genet. 2014;23:2164–2175. doi: 10.1093/hmg/ddt613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman TS, LaVail MM, Montemayor R, Ying G, Maguire MM, Laties AM, Jacobson SG, Cideciyan AV. Augmented rod bipolar cell function in partial receptor loss: an ERG study in P23H rhodopsin transgenic and aging normal rats. Vision Res. 2001;41:2779–2797. doi: 10.1016/s0042-6989(01)00157-2. [DOI] [PubMed] [Google Scholar]
- Ali RR. Gene therapy for retinal dystrophies: twenty years in the making. Hum Gene Ther. 2012;23:337–339. doi: 10.1089/hum.2012.2506. [DOI] [PubMed] [Google Scholar]
- An GJ, Asayama N, Humayun MS, Weiland J, Cao J, Kim SY, Grebe R, de Juan E, Jr, Sadda S. Ganglion cell responses to retinal light stimulation in the absence of photoreceptor outer segments from retinal degenerate rodents. Curr Eye Res. 2002;24:26–32. doi: 10.1076/ceyr.24.1.26.5432. [DOI] [PubMed] [Google Scholar]
- Anderson RE, Maude MB, McClellan M, Matthes MT, Yasumura D, LaVail MM. Low docosahexaenoic acid levels in rod outer segments of rats with P23H and S334ter rhodopsin mutations. Mol Vis. 2002;8:351–358. [PubMed] [Google Scholar]
- Aramant RB, Seiler MJ. Transplanted sheets of human retina and retinal pigment epithelium develop normally in nude rats. Exp Eye Res. 2002;75:115–125. doi: 10.1006/exer.2002.2001. [DOI] [PubMed] [Google Scholar]
- Arango-Gonzalez B, Trifunovic D, Sahaboglu A, Kranz K, Michalakis S, Farinelli P, Koch S, Koch F, Cottet S, Janssen-Bienhold U, Dedek K, Biel M, Zrenner E, Euler T, Ekstrom P, Ueffing M, Paquet-Durand F. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS One. 2014;9:e112142. doi: 10.1371/journal.pone.0112142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiou D, Aguila M, Opefi CA, South K, Bellingham J, Bevilacqua D, Munro PM, Kanuga N, Mackenzie FE, Dubis AM, Georgiadis A, Graca AB, Pearson RA, Ali RR, Sakami S, Palczewski K, Sherman MY, Reeves PJ, Cheetham ME. Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum Mol Genet. 2017;26:305–319. doi: 10.1093/hmg/ddw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr W, Frederick JM. Naturally occurring animal models with outer retina phenotypes. Vision Res. 2009;49:2636–2652. doi: 10.1016/j.visres.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakondi B, Lv W, Lu B, Jones MK, Tsai Y, Kim KJ, Levy R, Akhtar AA, Breunig JJ, Svendsen CN, Wang S. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol Ther. 2016;24:556–563. doi: 10.1038/mt.2015.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Duan D, Engelhardt JF, Maguire AM. Real-time, noninvasive in vivo assessment of adeno-associated virus-mediated retinal transduction. Invest Ophthalmol Vis Sci. 1997;38:2857–2863. [PubMed] [Google Scholar]
- Bennett J, Maguire AM, Cideciyan AV, Schnell M, Glover E, Anand V, Aleman TS, Chirmule N, Gupta AR, Huang Y, Gao GP, Nyberg WC, Tazelaar J, Hughes J, Wilson JM, Jacobson SG. Stable transgene expression in rod photoreceptors after recombinant adeno-associated virus-mediated gene transfer to monkey retina. Proc Natl Acad Sci USA. 1999;96:9920–9925. doi: 10.1073/pnas.96.17.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Wilson J, Sun D, Forbes B, Maguire A. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nature Med. 1996;2:649–654. doi: 10.1038/nm0696-649. [DOI] [PubMed] [Google Scholar]
- Berson EL, Rosner B, Sandberg MA, Dryja TP. Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His) Arch Ophthalmol. 1991;109:92–101. doi: 10.1001/archopht.1991.01080010094039. [DOI] [PubMed] [Google Scholar]
- Bicknell IR, Darrow R, Barsalou L, Fliesler SJ, Organisciak DT. Alterations in retinal rod outer segment fatty acids and light-damage susceptibility in P23H rats. Mol Vis. 2002;8:333–340. [PubMed] [Google Scholar]
- Blanks JC, Mullen RJ, LaVail MM. Retinal degeneration in the pcd cerebellar mutant mouse. II. Electron microscopic analysis. J Comp Neur. 1982;212:231–246. doi: 10.1002/cne.902120303. [DOI] [PubMed] [Google Scholar]
- Blanks JC, Spee C. Retinal degeneration in the pcd/pcd mutant mouse: accumulation of spherules in the interphotoreceptor space. Exp Eye Res. 1992;54:637–644. doi: 10.1016/0014-4835(92)90019-o. [DOI] [PubMed] [Google Scholar]
- Bravo-Nuevo A, Williams NK, Valter K, Stone J. Relationship between mitochondrial DNA damage and photoreceptor death in developing and adult retina, assessed in normal and degenerative rat strains. Mitochondrion. 2007;7:340–346. doi: 10.1016/j.mito.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Bush RA, Kononen L, Machida S, Sieving PA. The effect of calcium channel blocker diltiazem on photoreceptor degeneration in the rhodopsin Pro213His rat. Invest Ophthalmol Vis Sci. 2000;41:2697–2701. [PubMed] [Google Scholar]
- Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci. 2005;29:381–393. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Byrne LC, Dalkara D, Luna G, Fisher SK, Clerin E, Sahel JA, Leveillard T, Flannery JG. Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration. J Clin Invest. 2015;125:105–116. doi: 10.1172/JCI65654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caminos E, Vaquero CF, Martinez-Galan JR. Relationship between rat retinal degeneration and potassium channel KCNQ5 expression. Exp Eye Res. 2015;131:1–11. doi: 10.1016/j.exer.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA. Seeing the light: new insights into the molecular pathogenesis of retinal diseases. J Cell Physiol. 2007;213:348–354. doi: 10.1002/jcp.21213. [DOI] [PubMed] [Google Scholar]
- Cao W, Li F, Steinberg RH, LaVail MM. Development of normal and injury-induced gene expression of aFGF, bFGF, CNTF, BDNF, GFAP and IGF-I in the rat retina. Exp Eye Res. 2001;72:591–604. doi: 10.1006/exer.2001.0990. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neur. 1979a;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. II. Autoradiographic analysis of cell generation using tritiated thymidine. J Comp Neur. 1979b;188:263–272. doi: 10.1002/cne.901880205. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978;17:489–498. [PubMed] [Google Scholar]
- Chader GJ. Animal models in research on retinal degenerations: past progress and future hope. Vision Res. 2002;42:393–399. doi: 10.1016/s0042-6989(01)00212-7. [DOI] [PubMed] [Google Scholar]
- Chakraborty D, Conley SM, Al-Ubaidi MR, Naash MI. Initiation of rod outer segment disc formation requires RDS. PLoS One. 2014;9:e98939. doi: 10.1371/journal.pone.0098939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LL, Lee EJ, Humayun MS, Weiland JD. Both electrical stimulation thresholds and SMI-32-immunoreactive retinal ganglion cell density correlate with age in S334ter line 3 rat retina. J Neurophysiol. 2011;105:2687–2697. doi: 10.1152/jn.00619.2010. [DOI] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–525. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- Chen CY, Lam BL, Bhattacharya SK. Mass spectrometric analyses of phospholipids in the S334ter-3 rat model of retinal degeneration. Mol Vis. 2014;20:1605–1611. [PMC free article] [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor D, Simon MI. Mechanisms of rhodopsin inactivation in vivo as revealed by COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- Chen J, Simon MI, Matthes MT, Yasumura D, LaVail MM. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness) Invest Ophthalmol Vis Sci. 1999;40:2978–2982. [PubMed] [Google Scholar]
- Chen K, Wang Y, Liang X, Zhang Y, Ng TK, Chan LL. Electrophysiology Alterations in Primary Visual Cortex Neurons of Retinal Degeneration (S334ter-line-3) Rats. Sci Rep. 2016;6:26793. doi: 10.1038/srep26793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Kroeger H, Sakami S, Messah C, Yasumura D, Matthes MT, Coppinger JA, Palczewski K, LaVail MM, Lin JH. Robust Endoplasmic Reticulum-Associated Degradation of Rhodopsin Precedes Retinal Degeneration. Mol Neurobiol. 2015;52:679–695. doi: 10.1007/s12035-014-8881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Messah C, Lin JH. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol Biol Cell. 2012;23:758–770. doi: 10.1091/mbc.E11-08-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrysostomou V, Stone J, Stowe S, Barnett NL, Valter K. The status of cones in the rhodopsin mutant P23H-3 retina: light-regulated damage and repair in parallel with rods. Invest Ophthalmol Vis Sci. 2008;49:1116–1125. doi: 10.1167/iovs.07-1158. [DOI] [PubMed] [Google Scholar]
- Chrysostomou V, Stone J, Valter K. Life history of cones in the rhodopsin-mutant P23H-3 rat: evidence of long-term survival. Invest Ophthalmol Vis Sci. 2009a;50:2407–2416. doi: 10.1167/iovs.08-3003. [DOI] [PubMed] [Google Scholar]
- Chrysostomou V, Valter K, Stone J. Cone-rod dependence in the rat retina: variation with the rate of rod damage. Invest Ophthalmol Vis Sci. 2009b;50:3017–3023. doi: 10.1167/iovs.08-3004. [DOI] [PubMed] [Google Scholar]
- Cohen AI. Some cytological and initial biochemical observations on photoreceptors in retinas of rds mice. Invest Ophthalmol Vis Sci. 1983;24:832–843. [PubMed] [Google Scholar]
- Colley NJ, Cassill JA, Baker EK, Zuker CS. Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc Natl Acad Sci USA. 1995;92:3070–3074. doi: 10.1073/pnas.92.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion F, Chen J. Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: a mouse model of Q344ter-induced retinal degeneration. PLoS One. 2010;5:e10904. doi: 10.1371/journal.pone.0010904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon TJ, Deng WT, Erger K, Cossette T, Pang JJ, Ryals R, Clement N, Cleaver B, McDoom I, Boye SE, Peden MC, Sherwood MB, Abernathy CR, Alkuraya FS, Boye SL, Hauswirth WW. Preclinical potency and safety studies of an AAV2-mediated gene therapy vector for the treatment of MERTK associated retinitis pigmentosa. Human gene therapy Clinical development. 2013;24:23–28. doi: 10.1089/humc.2013.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca N, Fernandez-Sanchez L, Campello L, Maneu V, De la Villa P, Lax P, Pinilla I. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog Retin Eye Res. 2014;43:17–75. doi: 10.1016/j.preteyeres.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Cuenca N, Pinilla I, Sauve Y, Lu B, Wang S, Lund RD. Regressive and reactive changes in the connectivity patterns of rod and cone pathways of P23H transgenic rat retina. Neuroscience. 2004;127:301–317. doi: 10.1016/j.neuroscience.2004.04.042. [DOI] [PubMed] [Google Scholar]
- D’Cruz PM, Yasumura D, Weir J, Matthes M, Abderrahim H, LaVail MM, Vollrath D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- da Cruz L, Dorn JD, Humayun MS, Dagnelie G, Handa J, Barale PO, Sahel JA, Stanga PE, Hafezi F, Safran AB, Salzmann J, Santos A, Birch D, Spencer R, Cideciyan AV, de Juan E, Duncan JL, Eliott D, Fawzi A, Olmos de Koo LC, Ho AC, Brown G, Haller J, Regillo C, Del Priore LV, Arditi A, Greenberg RJ, Argus IISG. Five-Year Safety and Performance Results from the Argus II Retinal Prosthesis System Clinical Trial. Ophthalmology. 2016;123:2248–2254. doi: 10.1016/j.ophtha.2016.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP. RetNet retinal information network. 2017 www.sph.uth.tmc.edu/RetNet/home.htm.
- Dalkara D, Goureau O, Marazova K, Sahel JA. Let There Be Light: Gene and Cell Therapy for Blindness. Hum Gene Ther. 2016;27:134–147. doi: 10.1089/hum.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkara D, Sahel JA. Gene therapy for inherited retinal degenerations. C R Biol. 2014;337:185–192. doi: 10.1016/j.crvi.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Deng WT, Dinculescu A, Li Q, Boye SL, Li J, Gorbatyuk MS, Pang J, Chiodo VA, Matthes MT, Yasumura D, Liu L, Alkuraya FS, Zhang K, Vollrath D, LaVail MM, Hauswirth WW. Tyrosine-mutant AAV8 delivery of human MERTK provides long-term retinal preservation in RCS rats. Invest Ophthalmol Vis Sci. 2012;53:1895–1904. doi: 10.1167/iovs.11-8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pierdomenico J, Garcia-Ayuso D, Pinilla I, Cuenca N, Vidal-Sanz M, Agudo-Barriuso M, Villegas-Perez MP. Early Events in Retinal Degeneration Caused by Rhodopsin Mutation or Pigment Epithelium Malfunction: Differences and Similarities. Front Neuroanat. 2017;11:14. doi: 10.3389/fnana.2017.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenser KA, Timmers AM, Hauswirth WW, Lewin AS. Ribozyme-targeted destruction of RNA associated with autosomal-dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1998;39:681–689. [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Duebel J, Marazova K, Sahel JA. Optogenetics. Curr Opin Ophthalmol. 2015;26:226–232. doi: 10.1097/ICU.0000000000000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JL, Richards TP, Arditi A, da Cruz L, Dagnelie G, Dorn JD, Ho AC, Olmos de Koo LC, Barale PO, Stanga PE, Thumann G, Wang Y, Greenberg RJ. Improvements in vision-related quality of life in blind patients implanted with the Argus II Epiretinal Prosthesis. Clin Exp Optom. 2017;100:144–150. doi: 10.1111/cxo.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan T, Wiggert B, Whittaker N, Darrow R, Organisciak DT. Effect of visible light on normal and P23H-3 transgenic rat retinas: characterization of a novel retinoic acid derivative present in the P23H-3 retina. Photochem Photobiol. 2006;82:741–745. doi: 10.1562/2005-10-05-RA-712. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Carroll AK, Wiley S, Covey DF, Cai ZY, Zhao L, Wen R. Photoreceptor preservation in the S334ter model of retinitis pigmentosa by a novel estradiol analog. Biochem Pharmacol. 2004;68:1971–1984. doi: 10.1016/j.bcp.2004.06.042. [DOI] [PubMed] [Google Scholar]
- Esquiva G, Lax P, Cuenca N. Impairment of intrinsically photosensitive retinal ganglion cells associated with late stages of retinal degeneration. Invest Ophthalmol Vis Sci. 2013;54:4605–4618. doi: 10.1167/iovs.13-12120. [DOI] [PubMed] [Google Scholar]
- Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM. Photoreceptor degeneration in inherited retinal dystrophy delayed by basic fibroblast growth factor. Nature. 1990;347:83–86. doi: 10.1038/347083a0. [DOI] [PubMed] [Google Scholar]
- Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM. Basic fibroblast growth factor and local injury protect photoreceptors from light damage in the rat. J Neurosci. 1992;12:3554–3567. doi: 10.1523/JNEUROSCI.12-09-03554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinelli P, Arango-Gonzalez B, Volkl J, Alesutan I, Lang F, Zrenner E, Paquet-Durand F, Ekstrom PA. Retinitis Pigmentosa: over-expression of anti-ageing protein Klotho in degenerating photoreceptors. J Neurochem. 2013;127:868–879. doi: 10.1111/jnc.12353. [DOI] [PubMed] [Google Scholar]
- Farinelli P, Perera A, Arango-Gonzalez B, Trifunovic D, Wagner M, Carell T, Biel M, Zrenner E, Michalakis S, Paquet-Durand F, Ekstrom PA. DNA methylation and differential gene regulation in photoreceptor cell death. Cell Death Dis. 2014;5:e1558. doi: 10.1038/cddis.2014.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar GJ, Millington-Ward S, Chadderton N, Humphries P, Kenna PF. Gene-based therapies for dominantly inherited retinopathies. Gene Ther. 2012;19:137–144. doi: 10.1038/gt.2011.172. [DOI] [PubMed] [Google Scholar]
- Fauser S, Luberichs J, Schuttauf F. Genetic animal models for retinal degeneration. Surv Ophthalmol. 2002;47:357–367. doi: 10.1016/s0039-6257(02)00314-4. [DOI] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Bravo-Osuna I, Lax P, Arranz-Romera A, Maneu V, Esteban-Perez S, Pinilla I, Puebla-Gonzalez MDM, Herrero-Vanrell R, Cuenca N. Controlled delivery of tauroursodeoxycholic acid from biodegradable microspheres slows retinal degeneration and vision loss in P23H rats. PLoS One. 2017;12:e0177998. doi: 10.1371/journal.pone.0177998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Campello L, Pinilla I, Cuenca N. Astrocytes and Muller Cell Alterations During Retinal Degeneration in a Transgenic Rat Model of Retinitis Pigmentosa. Front Cell Neurosci. 2015a;9:484. doi: 10.3389/fncel.2015.00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Esquiva G, Martin-Nieto J, Pinilla I, Cuenca N. Safranal, a saffron constituent, attenuates retinal degeneration in P23H rats. PLoS One. 2012a;7:e43074. doi: 10.1371/journal.pone.0043074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Isiegas C, Ayuso E, Ruiz JM, de la Villa P, Bosch F, de la Rosa EJ, Cuenca N. Proinsulin slows retinal degeneration and vision loss in the P23H rat model of retinitis pigmentosa. Hum Gene Ther. 2012b;23:1290–1300. doi: 10.1089/hum.2012.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Noailles A, Angulo A, Maneu V, Cuenca N. Natural Compounds from Saffron and Bear Bile Prevent Vision Loss and Retinal Degeneration. Molecules. 2015b;20:13875–13893. doi: 10.3390/molecules200813875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Pinilla I, Martin-Nieto J, Cuenca N. Tauroursodeoxycholic acid prevents retinal degeneration in transgenic P23H rats. Invest Ophthalmol Vis Sci. 2011;52:4998–5008. doi: 10.1167/iovs.11-7496. [DOI] [PubMed] [Google Scholar]
- Finlayson PG, Iezzi R. Glutamate stimulation of retinal ganglion cells in normal and s334ter-4 rat retinas: a candidate for a neurotransmitter-based retinal prosthesis. Invest Ophthalmol Vis Sci. 2010;51:3619–3628. doi: 10.1167/iovs.09-4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery JG. Transgenic Animal Models for the Study of Inherited Retinal Dystrophies. ILAR J. 1999;40:51–58. doi: 10.1093/ilar.40.2.51. [DOI] [PubMed] [Google Scholar]
- Fliesler SJ, Tabor GA, Hollyfield JG. Glycoprotein synthesis in the human retina: localization of the lipid intermediate pathway. Exp Eye Res. 1984;39:153–173. doi: 10.1016/0014-4835(84)90005-8. [DOI] [PubMed] [Google Scholar]
- Fransen JW, Pangeni G, Pardue MT, McCall MA. Local signaling from a retinal prosthetic in a rodent retinitis pigmentosa model in vivo. J Neural Eng. 2014;11:046012. doi: 10.1088/1741-2560/11/4/046012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froger N, Cadetti L, Lorach H, Martins J, Bemelmans AP, Dubus E, Degardin J, Pain D, Forster V, Chicaud L, Ivkovic I, Simonutti M, Fouquet S, Jammoul F, Leveillard T, Benosman R, Sahel JA, Picaud S. Taurine provides neuroprotection against retinal ganglion cell degeneration. PLoS One. 2012;7:e42017. doi: 10.1371/journal.pone.0042017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, Apfelstedt-Sylla E, Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nature Genet. 2000;26:270–271. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- Garcia-Ayuso D, Di Pierdomenico J, Esquiva G, Nadal-Nicolas FM, Pinilla I, Cuenca N, Vidal-Sanz M, Agudo-Barriuso M, Villegas-Perez MP. Inherited Photoreceptor Degeneration Causes the Death of Melanopsin-Positive Retinal Ganglion Cells and Increases Their Coexpression of Brn3a. Invest Ophthalmol Vis Sci. 2015;56:4592–4604. doi: 10.1167/iovs.15-16808. [DOI] [PubMed] [Google Scholar]
- Garcia-Ayuso D, Ortin-Martinez A, Jimenez-Lopez M, Galindo-Romero C, Cuenca N, Pinilla I, Vidal-Sanz M, Agudo-Barriuso M, Villegas-Perez MP. Changes in the photoreceptor mosaic of P23H-1 rats during retinal degeneration: implications for rod-cone dependent survival. Invest Ophthalmol Vis Sci. 2013;54:5888–5900. doi: 10.1167/iovs.13-12643. [DOI] [PubMed] [Google Scholar]
- Garcia-Ayuso D, Salinas-Navarro M, Agudo M, Cuenca N, Pinilla I, Vidal-Sanz M, Villegas-Perez MP. Retinal ganglion cell numbers and delayed retinal ganglion cell death in the P23H rat retina. Exp Eye Res. 2010;91:800–810. doi: 10.1016/j.exer.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Glybina IV, Kennedy A, Ashton P, Abrams GW, Iezzi R. Intravitreous delivery of the corticosteroid fluocinolone acetonide attenuates retinal degeneration in S334ter-4 rats. Invest Ophthalmol Vis Sci. 2010;51:4243–4252. doi: 10.1167/iovs.09-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk M, Justilien V, Liu J, Hauswirth WW, Lewin AS. Preservation of photoreceptor morphology and function in P23H rats using an allele independent ribozyme. Exp Eye Res. 2007;84:44–52. doi: 10.1016/j.exer.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk MS, Gorbatyuk OS, LaVail MM, Lin JH, Hauswirth WW, Lewin AS. Functional rescue of P23H rhodopsin photoreceptors by gene delivery. Adv Exp Med Biol. 2012;723:191–197. doi: 10.1007/978-1-4614-0631-0_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, Hauswirth WW, Lin JH, Muzyczka N, Lewin AS. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–5966. doi: 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ES, Menz MD, LaVail MM, Flannery JG. Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:1546–1553. [PubMed] [Google Scholar]
- Green ES, Rendahl KG, Zhou S, Ladner M, Coyne M, Srivastava R, Manning WC, Flannery JG. Two animal models of retinal degeneration are rescued by recombinant adeno-associated virus-mediated production of FGF-5 and FGF-18. Mol Ther. 2001;3:507–515. doi: 10.1006/mthe.2001.0289. [DOI] [PubMed] [Google Scholar]
- Grimm C, Wenzel A, Stanescu D, Samardzija M, Hotop S, Groszer M, Naash M, Gassmann M, Reme C. Constitutive overexpression of human erythropoietin protects the mouse retina against induced but not inherited retinal degeneration. J Neurosci. 2004;24:5651–5658. doi: 10.1523/JNEUROSCI.1288-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman GH, Watson RF, Pauer GJ, Bollinger K, Hagstrom SA. Immunocytochemical evidence of Tulp1-dependent outer segment protein transport pathways in photoreceptor cells. Exp Eye Res. 2011;93:658–668. doi: 10.1016/j.exer.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerin K, Gregory-Evans CY, Hodges MD, Moosajee M, Mackay DS, Gregory-Evans K, Flannery JG. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp Eye Res. 2008;87:197–207. doi: 10.1016/j.exer.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471. doi: 10.1016/s0014-4835(02)00332-9. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Grimm C, Simmen BC, Wenzel A, Remé CE. Molecular ophthalmology: an update on animal models for retinal degenerations and dystrophies. Br J Ophthalmol. 2000;84:922–927. doi: 10.1136/bjo.84.8.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom SA, Duyao M, North MA, Li T. Retinal degeneration in tulp1−/− mice: vesicular accumulation in the interphotoreceptor matrix. Invest Ophthalmol Vis Sci. 1999;40:2795–2802. [PubMed] [Google Scholar]
- Hanif AM, Kim MK, Thomas JG, Ciavatta VT, Chrenek M, Hetling JR, Pardue MT. Whole-eye electrical stimulation therapy preserves visual function and structure in P23H-1 rats. Exp Eye Res. 2016;149:75–83. doi: 10.1016/j.exer.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, Mamada H, Tanaka K, Parada LF, Wada K. Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. 2002;22:9228–9236. doi: 10.1523/JNEUROSCI.22-21-09228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckenlively JR, Chang B, Erway LC, Peng C, Hawes NL, Hageman GS, Roderick TH. Mouse model for Usher syndrome: linkage mapping suggests homology to Usher type I reported at human chromosome 11p15. Proc Natl Acad Sci U S A. 1995;92:11100–11104. doi: 10.1073/pnas.92.24.11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombrebueno JR, Tsai MM, Kim HL, De Juan J, Grzywacz NM, Lee EJ. Morphological changes of short-wavelength cones in the developing S334ter-3 transgenic rat. Brain Res. 2010;1321:60–66. doi: 10.1016/j.brainres.2010.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nature Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- Ilia M, Jeffery G. Retinal cell addition and rod production depend on early stages of ocular melanin synthesis. J Comp Neurol. 2000;420:437–444. [PubMed] [Google Scholar]
- Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- Jansen HG, Sanyal S. Development and degeneration of retina in rds mutant mice: electron microscopy. J Comp Neur. 1984;224:71–84. doi: 10.1002/cne.902240107. [DOI] [PubMed] [Google Scholar]
- Jansen HG, Sanyal S, De Grip WJ, Schalken JJ. Development and degeneration of retina in rds mutant mice: ultraimmunohistochemical localization of opsin. Exp Eye Res. 1987;44:347–361. doi: 10.1016/s0014-4835(87)80170-7. [DOI] [PubMed] [Google Scholar]
- Jayaram H, Jones MF, Eastlake K, Cottrill PB, Becker S, Wiseman J, Khaw PT, Limb GA. Transplantation of photoreceptors derived from human Muller glia restore rod function in the P23H rat. Stem Cells Transl Med. 2014;3:323–333. doi: 10.5966/sctm.2013-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RJ. Effects of Antipsychotic Drugs Haloperidol and Clozapine on Visual Responses of Retinal Ganglion Cells in a Rat Model of Retinitis Pigmentosa. J Ocul Pharmacol Ther. 2016;32:685–690. doi: 10.1089/jop.2016.0102. [DOI] [PubMed] [Google Scholar]
- Ji Y, Yu WQ, Eom YS, Bruce F, Craft CM, Grzywacz NM, Lee EJ. The effect of TIMP-1 on the cone mosaic in the retina of the rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;56:352–364. doi: 10.1167/iovs.14-15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Zhu CL, Grzywacz NM, Lee EJ. Rearrangement of the cone mosaic in the retina of the rat model of retinitis pigmentosa. J Comp Neurol. 2012;520:874–888. doi: 10.1002/cne.22800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BW, Marc RE. Retinal remodeling during retinal degeneration. Exp Eye Res. 2005;81:123–137. doi: 10.1016/j.exer.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, Milam AH, LaVail MM, Marc RE. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- Jozwick C, Valter K, Stone J. Reversal of functional loss in the P23H-3 rat retina by management of ambient light. Exp Eye Res. 2006;83:1074–1080. doi: 10.1016/j.exer.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Kaldi I, Dittmar M, Pierce P, Anderson RE. L-NAME protects against acute light damage in albino rats, but not against retinal degeneration in P23H and S334ter transgenic rats. Exp Eye Res. 2003;76:453–461. doi: 10.1016/s0014-4835(02)00334-2. [DOI] [PubMed] [Google Scholar]
- Karli P, Stoeckel ME, Porte A. Dégénérescence des cellules visuelles photoréceptrices et persistance d’une sensibilité de la rétine à la stimulation photique. Observations au microscope électronique. Z Zellforsch. 1965;65:238–252. [PubMed] [Google Scholar]
- Kaur J, Mencl S, Sahaboglu A, Farinelli P, van Veen T, Zrenner E, Ekstrom P, Paquet-Durand F, Arango-Gonzalez B. Calpain and PARP activation during photoreceptor cell death in P23H and S334ter rhodopsin mutant rats. PLoS One. 2011;6:e22181. doi: 10.1371/journal.pone.0022181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomiets B, Dubus E, Simonutti M, Rosolen S, Sahel JA, Picaud S. Late histological and functional changes in the P23H rat retina after photoreceptor loss. Neurobiol Dis. 2010;38:47–58. doi: 10.1016/j.nbd.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Kolstad KD, Dalkara D, Guerin K, Visel M, Hoffmann N, Schaffer DV, Flannery JG. Changes in adeno-associated virus-mediated gene delivery in retinal degeneration. Hum Gene Ther. 2010;21:571–578. doi: 10.1089/hum.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Sakai T, Komeima K, Kurimoto Y, Ueno S, Nishizawa Y, Usukura J, Fujikado T, Tano Y, Terasaki H. Generation of a transgenic rabbit model of retinal degeneration. Invest Ophthalmol Vis Sci. 2009;50:1371–1377. doi: 10.1167/iovs.08-2863. [DOI] [PubMed] [Google Scholar]
- Kroeger H, Messah C, Ahern K, Gee J, Joseph V, Matthes MT, Yasumura D, Gorbatyuk MS, Chiang WC, LaVail MM, Lin JH. Induction of endoplasmic reticulum stress genes, BiP and chop, in genetic and environmental models of retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53:7590–7599. doi: 10.1167/iovs.12-10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007;81:1345–1351. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- Lau D, McGee LH, Zhou S, Rendahl KG, Manning WC, Escobedo JA, Flannery JG. Retinal degeneration is slowed in transgenic rats by AAV-mediated delivery of FGF-2. Invest Ophthalmol Vis Sci. 2000;41:3622–3633. [PubMed] [Google Scholar]
- LaVail MM. Rod outer segment disc shedding in rat retina: relationship to cyclic lighting. Science. 1976a;194:1071–1074. doi: 10.1126/science.982063. [DOI] [PubMed] [Google Scholar]
- LaVail MM. Survival of some photoreceptor cells in albino rats following long-term exposure to continuous light. Invest Ophthalmol. 1976b;15:64–70. [PubMed] [Google Scholar]
- LaVail MM. Analysis of neurological mutants with inherited retinal degeneration. Invest Ophthalmol Vis Res. 1981;21:638–657. [PubMed] [Google Scholar]
- LaVail MM. Legacy of the RCS rat: Impact of a seminal study on retinal cell biology and retinal degenerative diseases. In: Kolb H, Ripps H, Wu S, editors. Concepts and Challenges in Retinal Biology. A Tribute to John E Dowling. Elsevier; Amsterdam: 2001. pp. 617–627. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Battelle BA. Influence of eye pigmentation and light deprivation on inherited retinal dystrophy in the rat. Exp Eye Res. 1975;21:167–192. doi: 10.1016/0014-4835(75)90080-9. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Blanks JC, Mullen RJ. Retinal degeneration in the pcd cerebellar mutant mouse. I. Light microscopic and autoradiographic analysis. J Comp Neur. 1982;212:217–230. doi: 10.1002/cne.902120302. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Nishikawa S, Duncan JL, Yang H, Matthes MT, Yasumura D, Vollrath D, Overbeek PA, Ash JD, Robinson ML. Sustained delivery of NT-3 from lens fiber cells in transgenic mice reveals specificity of neuroprotection in retinal degenerations. J Comp Neurol. 2008;511:724–735. doi: 10.1002/cne.21858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Sidman M, Rauzin R, Sidman RL. Discrimination of light intensity by rats with inherited retinal degeneration: a behavioral and cytological study. Vision Res. 1974;14:693–702. doi: 10.1016/0042-6989(74)90066-2. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Sidman RL. C57BL/6J mice with inherited retinal degeneration. Arch Ophthalmol. 1974;91:394–400. doi: 10.1001/archopht.1974.03900060406015. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Sidman RL, Gerhardt CO. Congenic strains of RCS rats with inherited retinal dystrophy. J Hered. 1975;66:242–244. doi: 10.1093/oxfordjournals.jhered.a108621. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Unoki K, Yasumura D, Matthes MT, Yancopoulos GD, Steinberg RH. Multiple growth factors, cytokines and neurotrophins rescue photoreceptors from the damaging effects of constant light. Proc Natl Acad Sci USA. 1992;89:11249–11253. doi: 10.1073/pnas.89.23.11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Drenser KA, Flannery JG, Lewin AS, Hauswirth WW. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci USA. 2000;97:11488–11493. doi: 10.1073/pnas.210319397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Lau-Villacorta C, Unoki K, Sung C-H, Steinberg RH. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602. [PubMed] [Google Scholar]
- Lax P, Esquiva G, Altavilla C, Cuenca N. Neuroprotective effects of the cannabinoid agonist HU210 on retinal degeneration. Exp Eye Res. 2014;120:175–185. doi: 10.1016/j.exer.2014.01.019. [DOI] [PubMed] [Google Scholar]
- Lax P, Esquiva G, Fuentes-Broto L, Segura F, Sanchez-Cano A, Cuenca N, Pinilla I. Age-related changes in photosensitive melanopsin-expressing retinal ganglion cells correlate with circadian rhythm impairments in sighted and blind rats. Chronobiol Int. 2016;33:374–391. doi: 10.3109/07420528.2016.1151025. [DOI] [PubMed] [Google Scholar]
- Lee D, Geller S, Walsh N, Valter K, Yasumura D, Matthes M, LaVail MM, Stone J. Photoreceptor degeneration in Pro23His and S334ter transgenic rats. Adv Exp Med Biol. 2003;533:297–302. doi: 10.1007/978-1-4615-0067-4_36. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Ji Y, Zhu CL, Grzywacz NM. Role of Muller cells in cone mosaic rearrangement in a rat model of retinitis pigmentosa. Glia. 2011;59:1107–1117. doi: 10.1002/glia.21183. [DOI] [PubMed] [Google Scholar]
- Leonard KC, Petrin D, Coupland SG, Baker AN, Leonard BC, LaCasse EC, Hauswirth WW, Korneluk RG, Tsilfidis C. XIAP protection of photoreceptors in animal models of retinitis pigmentosa. PLoS ONE. 2007;2:e314. doi: 10.1371/journal.pone.0000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveillard T, Fridlich R, Clerin E, Ait-Ali N, Millet-Puel G, Jaillard C, Yang Y, Zack D, van-Dorsselaer A, Sahel JA. Therapeutic strategy for handling inherited retinal degenerations in a gene-independent manner using rod-derived cone viability factors. C R Biol. 2014;337:207–213. doi: 10.1016/j.crvi.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Lewin AS, Drenser KA, Hauswirth WW, Nishikawa S, Yasumura D, Flannery JG, LaVail MM. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nature Med. 1998;4:967–971. doi: 10.1038/nm0898-967. [DOI] [PubMed] [Google Scholar]
- Li L, Turner JE. Inherited retinal dystrophy in the RCS rat: prevention of photoreceptor degeneration by pigment epithelial cell transplantation. Exp Eye Res. 1988;47:911–917. doi: 10.1016/0014-4835(88)90073-5. [DOI] [PubMed] [Google Scholar]
- Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci USA. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tao W, Luo L, Huang D, Kauper K, Stabila P, LaVail MM, Laties AM, Wen R. CNTF induces regeneration of cone outer segments in a rat model of retinal degeneration. PLoS One. 2010;5:e9495. doi: 10.1371/journal.pone.0009495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang FQ, Aleman TS, Dejneka NS, Dudus L, Fisher KJ, Maguire AM, Jacobson SG, Bennett J. Long-term protection of retinal structure but not function using RAAV.CNTF in animal models of retinitis pigmentosa. Mol Ther. 2001;4:461–472. doi: 10.1006/mthe.2001.0473. [DOI] [PubMed] [Google Scholar]
- Light JG, Fransen JW, Adekunle AN, Adkins A, Pangeni G, Loudin J, Mathieson K, Palanker DV, McCall MA, Pardue MT. Inner retinal preservation in rat models of retinal degeneration implanted with subretinal photovoltaic arrays. Exp Eye Res. 2014;128:34–42. doi: 10.1016/j.exer.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, LaVail MM. Misfolded proteins and retinal dystrophies. Adv Exp Med Biol. 2010;664:115–121. doi: 10.1007/978-1-4419-1399-9_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, LaVail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderholm P, Guyomard JL, Djilas M, Salzmann J, Simonutti M, Sahel JA, Safran AB, Renaud P, Picaud S. Long-term in vivo impedance changes of subretinal microelectrodes implanted in dystrophic P23H rats. Int J Artif Organs. 2013;36:612–619. doi: 10.5301/ijao.5000213. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Peng M, Laties AM, Wen R. Activation of caspase-3 in the retina of transgenic rats with the rhodopsin mutation S334ter during photoreceptor degeneration. J Neurosci. 1999;19:4778–4785. doi: 10.1523/JNEUROSCI.19-12-04778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu TH, Stowe S, Matsushita A, Arikawa K, Naash MI, Williams DS. Defective phototransductive disk membrane morphogenesis in transgenic mice expressing opsin with a mutated N-terminal domain. J Cell Sci. 1997;110(Pt 20):2589–2597. doi: 10.1242/jcs.110.20.2589. [DOI] [PubMed] [Google Scholar]
- Lowe RJ, Duncan JL, Yang H, Donohue-Rolfe KM, Matthes MT, Yasumura D, LaVail MM. Retinal degeneration is slowed by eye pigmentation in P23H but not in S334ter mutant rhodopsin transgenic rats. Invest Ophthalmol Vis Sci. 2005;46 ARVO E-Abstract 3177. [Google Scholar]
- Lu B, Morgans CW, Girman S, Lund R, Wang S. Retinal morphological and functional changes in an animal model of retinitis pigmentosa. Vis Neurosci. 2013;30:77–89. doi: 10.1017/S0952523813000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida S, Chaudhry P, Shinohara T, Singh DP, Reddy VN, Chylack LT, Jr, Sieving PA, Bush RA. Lens epithelium-derived growth factor promotes photoreceptor survival in light-damaged and RCS rats. Invest Ophthalmol Vis Sci. 2001a;42:1087–1095. [PubMed] [Google Scholar]
- Machida S, Chaudhry P, Shinohara T, Singh DP, Reddy VN, Chylack LT, Jr, Sieving PA, Bush RA. Lens epithelium-derived growth factor promotes photoreceptor survival in light-damaged and RCS rats. Invest Ophthalmol Vis Sci. 2001b;42:1087–1095. [PubMed] [Google Scholar]
- Machida S, Kondo M, Jamison JA, Khan NW, Kononen LT, Sugawara T, Bush RA, Sieving PA. P23H rhodopsin transgenic rat: correlation of retinal function with histopathology. Invest Ophthalmol Vis Sci. 2000;41:3200–3209. [PubMed] [Google Scholar]
- Machida S, Raz-Prag D, Fariss RN, Sieving PA, Bush RA. Photopic ERG negative response from amacrine cell signaling in RCS rat retinal degeneration. Invest Ophthalmol Vis Sci. 2008;49:442–452. doi: 10.1167/iovs.07-0291. [DOI] [PubMed] [Google Scholar]
- Maleki S, Gopalakrishnan S, Ghanian Z, Sepehr R, Schmitt H, Eells J, Ranji M. Optical imaging of mitochondrial redox state in rodent model of retinitis pigmentosa. J Biomed Opt. 2013;18:16004. doi: 10.1117/1.JBO.18.1.016004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc R, Pfeiffer R, Jones B. Retinal prosthetics, optogenetics, and chemical photoswitches. ACS Chem Neurosci. 2014;5:895–901. doi: 10.1021/cn5001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RE, Fliesler SJ, Brush RS, Richards MJ, Hopkins SA, Anderson RE. Lipid differences in rod outer segment membranes of rats with P23H and S334ter opsin mutations. Mol Vis. 2005;11:338–346. [PubMed] [Google Scholar]
- Martin RE, Ranchon-Cole I, Brush RS, Williamson CR, Hopkins SA, Li F, Anderson RE. P23H and S334ter opsin mutations: Increasing photoreceptor outer segment n-3 fatty acid content does not affect the course of retinal degeneration. Mol Vis. 2004;10:199–207. [PubMed] [Google Scholar]
- Martinez-Navarrete G, Seiler MJ, Aramant RB, Fernandez-Sanchez L, Pinilla I, Cuenca N. Retinal degeneration in two lines of transgenic S334ter rats. Exp Eye Res. 2011;92:227–237. doi: 10.1016/j.exer.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslim J, Valter K, Egensperger R, Hollander H, Stone J. Tissue oxygen during a critical developmental period controls the death and survival of photoreceptors. Invest Ophthalmol Vis Sci. 1997;38:1667–1677. [PubMed] [Google Scholar]
- McGee Sanftner LH, Abel H, Hauswirth WW, Flannery JG. Glial cell line derived neurotrophic factor delays photoreceptor degeneration in a transgenic rat model of retinitis pigmentosa. Mol Ther. 2001;4:622–629. doi: 10.1006/mthe.2001.0498. [DOI] [PubMed] [Google Scholar]
- McGill TJ, Prusky GT, Douglas RM, Yasumura D, Matthes MT, Lowe RJ, Duncan JL, Yang H, Ahern K, Daniello KM, Silver B, Lavail MM. Discordant anatomical, electrophysiological, and visual behavioral profiles of retinal degeneration in rat models of retinal degenerative disease. Invest Ophthalmol Vis Sci. 2012a;53:6232–6244. doi: 10.1167/iovs.12-9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill TJ, Prusky GT, Luna G, LaVail MM, Fisher SK, Lewis GP. Optomotor and immunohistochemical changes in the juvenile S334ter rat. Exp Eye Res. 2012b;104:65–73. doi: 10.1016/j.exer.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michon JJ, Li ZL, Shioura N, Anderson RJ, Tso MOM. A comparative study of methods of photoreceptor morphometry. Invest Ophthalmol Vis Sci. 1991;32:280–284. [PubMed] [Google Scholar]
- Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Ret Eye Res. 1998;17:175–205. doi: 10.1016/s1350-9462(97)00012-8. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, LaVail MM. Inherited retinal dystrophy: primary defect in pigment epithelium determined with experimental rat chimeras. Science. 1976;192:799–801. doi: 10.1126/science.1265483. [DOI] [PubMed] [Google Scholar]
- Murray SF, Jazayeri A, Matthes MT, Yasumura D, Yang H, Peralta R, Watt A, Freier S, Hung G, Adamson PS, Guo S, Monia BP, LaVail MM, McCaleb ML. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci. 2015;56:6362–6375. doi: 10.1167/iovs.15-16400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naash MI, Peachey NS, Li ZY, Gryczan CC, Goto Y, Blanks J, Milam AH, Ripps H. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant opsin. Invest Ophthalmol Vis Sci. 1996a;37:775–782. [PubMed] [Google Scholar]
- Naash MI, Ripps H, Li S, Goto Y, Peachey NS. Polygenic disease and retinitis pigmentosa: albinism exacerbates photoreceptor degeneration induced by the expression of a mutant opsin in transgenic mice. J Neurosci. 1996b;16:7853–7858. doi: 10.1523/JNEUROSCI.16-24-07853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naash MI, Wu TH, Chakraborty D, Fliesler SJ, Ding XQ, Nour M, Peachey NS, Lem J, Qtaishat N, Al-Ubaidi MR, Ripps H. Retinal abnormalities associated with the G90D mutation in opsin. J Comp Neurol. 2004;478:149–163. doi: 10.1002/cne.20283. [DOI] [PubMed] [Google Scholar]
- Nagai N, Iwata S, Kaji H, Sampei K, Katsukura Y, Onami H, Nishizawa M, Nakazawa T, Mashima Y, Abe T. Protective effects of sustained transscleral unoprostone delivery against retinal degeneration in S334ter rhodopsin mutant rats. J Biomed Mater Res B Appl Biomater. 2016;104:1730–1737. doi: 10.1002/jbm.b.33522. [DOI] [PubMed] [Google Scholar]
- Ng TF, Streilein JW. Light-induced migration of retinal microglia into the subretinal space. Invest Ophthalmol Vis Sci. 2001;42:3301–3310. [PubMed] [Google Scholar]
- Nir I, Harrison JM, Liu C, Wen R. Extended photoreceptor viability by light stress in the RCS rats but not in the opsin P23H mutant rats. Invest Ophthalmol Vis Sci. 2001;42:842–849. [PubMed] [Google Scholar]
- Nir I, Papermaster DS. Immunocytochemical localization of opsin in the inner segment and ciliary plasma membrane of photoreceptors in retinas of rds mutant mice. Invest Ophthalmol Vis Sci. 1986;27:836–840. [PubMed] [Google Scholar]
- Noailles A, Fernandez-Sanchez L, Lax P, Cuenca N. Microglia activation in a model of retinal degeneration and TUDCA neuroprotective effects. J Neuroinflammation. 2014;11:186. doi: 10.1186/s12974-014-0186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noailles A, Maneu V, Campello L, Gomez-Vicente V, Lax P, Cuenca N. Persistent inflammatory state after photoreceptor loss in an animal model of retinal degeneration. Sci Rep. 2016;6:33356. doi: 10.1038/srep33356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohguro H. New drug therapy for retinal degeneration. Nippon Ganka Gakkai Zasshi. 2008;112:7–21. [PubMed] [Google Scholar]
- Olsson JE, Gordon JW, Pawlyk BS, Roof RS, Mukai S, Cowley GS, Berson EL, Dryja TP. Transgenic mice with a rhodopsin mutation (Pro23His): A mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- Organisciak D, Darrow R, Gu X, Barsalou L, Crabb JW. Genetic, age and light mediated effects on crystallin protein expression in the retina. Photochem Photobiol. 2006;82:1088–1096. doi: 10.1562/2005-06-30-RA-599. [DOI] [PubMed] [Google Scholar]
- Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B. Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Invest Ophthalmol Vis Sci. 2003;44:486–492. doi: 10.1167/iovs.02-0708. [DOI] [PubMed] [Google Scholar]
- Organisciak DT, Winkler BS. Retinal light damage: practical and theoretical considerations. In: Osborne N, Chader G, editors. Progress in Retinal and Eye Research. Pergamon Press; Oxford: 1994. pp. 1–29. [Google Scholar]
- Orhan E, Dalkara D, Neuille M, Lechauve C, Michiels C, Picaud S, Leveillard T, Sahel JA, Naash MI, Lavail MM, Zeitz C, Audo I. Genotypic and phenotypic characterization of P23H line 1 rat model. PLoS One. 2015;10:e0127319. doi: 10.1371/journal.pone.0127319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T, Ishiguro S, Hirano S, Baba A, Yamashita T, Tomita H, Nakazawa M. Inhibitory peptide of mitochondrial μ-calpain protects against photoreceptor degeneration in rhodopsin transgenic S334ter and P23H rats. PLoS One. 2013;8:e71650. doi: 10.1371/journal.pone.0071650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt DA, Aguila M, McCulley CH, Bevilacqua D, Mendes HF, Athanasiou D, Novoselov SS, Kanuga N, Munro PM, Coffey PJ, Kalmar B, Greensmith L, Cheetham ME. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014;5:e1236. doi: 10.1038/cddis.2014.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn JS, Li S, Naash MI. Ambient hypoxia reverses retinal vascular attenuation in a transgenic mouse model of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:4007–4013. [PubMed] [Google Scholar]
- Pennesi ME, Nishikawa S, Matthes MT, Yasumura D, LaVail MM. The relationship of photoreceptor degeneration to retinal vascular development and loss in mutant rhodopsin transgenic and RCS rats. Exp Eye Res. 2008;87:561–570. doi: 10.1016/j.exer.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson WM, Flannery JG, Hauswirth WW, Klein RL, Meyer EM, Muzyczka N, Yasumura D, Matthes MT, LaVail MM. Enhanced survival of photoreceptors in P23H mutant rhodopsin transgenic rats by adeno-associated virus (AAV)-mediated delivery of neurotrophic genes. Invest Ophthalmol Vis Sci. 1998;39:S1117. [Google Scholar]
- Picard E, Le Rouzic Q, Oudar A, Berdugo M, El Sanharawi M, Andrieu-Soler C, Naud MC, Jonet L, Latour C, Klein C, Galiacy S, Malecaze F, Coppin H, Roth MP, Jeanny JC, Courtois Y, Behar-Cohen F. Targeting iron-mediated retinal degeneration by local delivery of transferrin. Free Radic Biol Med. 2015;89:1105–1121. doi: 10.1016/j.freeradbiomed.2015.08.018. [DOI] [PubMed] [Google Scholar]
- Pinilla I, Fernandez-Sanchez L, Segura FJ, Sanchez-Cano AI, Tamarit JM, Fuentes-Broto L, Eells JT, Lax P, Cuenca N. Long time remodeling during retinal degeneration evaluated by optical coherence tomography, immunocytochemistry and fundus autofluorescence. Exp Eye Res. 2016;150:122–134. doi: 10.1016/j.exer.2015.10.012. [DOI] [PubMed] [Google Scholar]
- Pinilla I, Lund RD, Sauve Y. Enhanced cone dysfunction in rats homozygous for the P23H rhodopsin mutation. Neurosci Lett. 2005;382:16–21. doi: 10.1016/j.neulet.2005.02.055. [DOI] [PubMed] [Google Scholar]
- Polazzi E, Monti B. Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol. 2010;92:293–315. doi: 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Price BA, Sandoval IM, Chan F, Simons DL, Wu SM, Wensel TG, Wilson JH. Mislocalization and degradation of human P23H-rhodopsin-GFP in a knockin mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52:9728–9736. doi: 10.1167/iovs.11-8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009;12:44–52. doi: 10.1038/nn.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzo C, Xiong W, Cepko CL. Loss of daylight vision in retinal degeneration: are oxidative stress and metabolic dysregulation to blame? J Biol Chem. 2012;287:1642–1648. doi: 10.1074/jbc.R111.304428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu G, Seiler MJ, Mui C, Arai S, Aramant RB, de Juan E, Jr, Sadda S. Photoreceptor differentiation and integration of retinal progenitor cells transplanted into transgenic rats. Exp Eye Res. 2005;80:515–525. doi: 10.1016/j.exer.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Rahmani S, Bogdanowicz L, Thomas J, Hetling JR. Chronic delivery of low-level exogenous current preserves retinal function in pigmented P23H rat. Vision Res. 2013;76:105–113. doi: 10.1016/j.visres.2012.10.016. [DOI] [PubMed] [Google Scholar]
- Ranchon I, LaVail MM, Kotake Y, Anderson RE. Free radical trap phenyl-N-tert-butylnitrone protects against light damage but does not rescue P23H and S334ter rhodopsin transgenic rats from inherited retinal degeneration. J Neurosci. 2003;23:6050–6057. doi: 10.1523/JNEUROSCI.23-14-06050.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport DH, Wong LL, Wood ED, Yasumura D, LaVail MM. Timing and topography of cell genesis in the rat retina. J Comp Neurol. 2004;474:304–324. doi: 10.1002/cne.20134. [DOI] [PubMed] [Google Scholar]
- Rapp LM, Williams TP. A parametric study of retinal light damage in albino and pigmented rats. In: Williams TP, Baker BN, editors. The Effects of Constant Light on Visual Processes. Plenum Press; New York: 1980. pp. 135–159. [Google Scholar]
- Ray A, Sun GJ, Chan L, Grzywacz NM, Weiland J, Lee EJ. Morphological alterations in retinal neurons in the S334ter-line3 transgenic rat. Cell Tissue Res. 2010;339:481–491. doi: 10.1007/s00441-009-0916-5. [DOI] [PubMed] [Google Scholar]
- Raz-Prag D, Zeng Y, Sieving PA, Bush RA. Photoreceptor protection by adeno-associated virus-mediated LEDGF expression in the RCS rat model of retinal degeneration: probing the mechanism. Invest Ophthalmol Vis Sci. 2009;50:3897–3906. doi: 10.1167/iovs.08-3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roddy GW, Rosa RH, Jr, Oh JY, Ylostalo JH, Bartosh TJ, Jr, Choi H, Lee RH, Yasumura D, Ahern K, Nielsen G, Matthes MT, LaVail MM, Prockop DJ. Stanniocalcin-1 rescued photoreceptor degeneration in two rat models of inherited retinal degeneration. Mol Ther. 2012;20:788–797. doi: 10.1038/mt.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque RS, Rosales AA, Jingjing L, Agarwal N, Al-Ubaidi MR. Retina-derived microglial cells induce photoreceptor cell death in vitro. Brain Res. 1999;836:110–119. doi: 10.1016/s0006-8993(99)01625-x. [DOI] [PubMed] [Google Scholar]
- Sagdullaev BT, Aramant RB, Seiler MJ, Woch G, McCall MA. Retinal transplantation-induced recovery of retinotectal visual function in a rodent model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003;44:1686–1695. doi: 10.1167/iovs.02-0615. [DOI] [PubMed] [Google Scholar]
- Sahel JA. Saving cone cells in hereditary rod diseases: a possible role for rod-derived cone viability factor (RdCVF) therapy. Retina. 2005;25:S38–S39. doi: 10.1097/00006982-200512001-00015. [DOI] [PubMed] [Google Scholar]
- Saito Y, Ohguro H, Ohguro I, Sato N, Ishikawa F, Yamazaki H, Metoki T, Ito T, Nakazawa M. Misregulation of rhodopsin phosphorylation and dephosphorylation found in P23H rat retinal degeneration. Clin Ophthalmol. 2008;2:821–828. doi: 10.2147/opth.s4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakami S, Kolesnikov AV, Kefalov VJ, Palczewski K. P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum Mol Genet. 2014;23:1723–1741. doi: 10.1093/hmg/ddt561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan AV, Jacobson SG, Palczewski K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011;286:10551–10567. doi: 10.1074/jbc.M110.209759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzmann J, Linderholm OP, Guyomard JL, Paques M, Simonutti M, Lecchi M, Sommerhalder J, Dubus E, Pelizzone M, Bertrand D, Sahel J, Renaud P, Safran AB, Picaud S. Subretinal electrode implantation in the P23H rat for chronic stimulations. Br J Ophthalmol. 2006;90:1183–1187. doi: 10.1136/bjo.2005.089110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammons J, Gross A. Biochemical analysis of a rhodopsin photoactivatable GFP fusion as a model of G-protein coupled receptor transport. Vision Res. 2013;93:43–48. doi: 10.1016/j.visres.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg MA, Weigel-DiFranco C, Dryja TP, Berson EL. Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1995;36:1934–1942. [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Segura F, Sanchez-Cano A, Jarabo S, Lopez de la Fuente C, Cuenca N, Villegas-Perez MP, Pinilla I. Assessment of Visual and Chromatic Functions in a Rodent Model of Retinal Degeneration. Invest Ophthalmol Vis Sci. 2015;56:6275–6283. doi: 10.1167/iovs.15-17257. [DOI] [PubMed] [Google Scholar]
- Seiler MJ, Aramant RB, Jones MK, Ferguson DL, Bryda EC, Keirstead HS. A new immunodeficient pigmented retinal degenerate rat strain to study transplantation of human cells without immunosuppression. Graefes Arch Clin Exp Ophthalmol. 2014;252:1079–1092. doi: 10.1007/s00417-014-2638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler MJ, Aramant RB, Thomas BB, Peng Q, Sadda SR, Keirstead HS. Visual restoration and transplant connectivity in degenerate rats implanted with retinal progenitor sheets. Eur J Neurosci. 2010a;31:508–520. doi: 10.1111/j.1460-9568.2010.07085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler MJ, Jones BW, Aramant RB, Yang PB, Keirstead HS, Marc RE. Computational molecular phenotyping of retinal sheet transplants to rats with retinal degeneration. Eur J Neurosci. 2012;35:1692–1704. doi: 10.1111/j.1460-9568.2012.08078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler MJ, Lin RE, McLelland BT, Mathur A, Lin B, Sigman J, De Guzman AT, Kitzes LM, Aramant RB, Thomas BB. Vision Recovery and Connectivity by Fetal Retinal Sheet Transplantation in an Immunodeficient Retinal Degenerate Rat Model. Invest Ophthalmol Vis Sci. 2017;58:614–630. doi: 10.1167/iovs.15-19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler MJ, Rao B, Aramant RB, Yu L, Wang Q, Kitayama E, Pham S, Yan F, Chen Z, Keirstead HS. Three-dimensional optical coherence tomography imaging of retinal sheet implants in live rats. J Neurosci Methods. 2010b;188:250–257. doi: 10.1016/j.jneumeth.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler MJ, Sagdullaev BT, Woch G, Thomas BB, Aramant RB. Transsynaptic virus tracing from host brain to subretinal transplants. Eur J Neurosci. 2005;21:161–172. doi: 10.1111/j.1460-9568.2004.03851.x. [DOI] [PubMed] [Google Scholar]
- Seiler MJ, Thomas BB, Chen Z, Arai S, Chadalavada S, Mahoney MJ, Sadda SR, Aramant RB. BDNF-treated retinal progenitor sheets transplanted to degenerate rats: improved restoration of visual function. Exp Eye Res. 2008a;86:92–104. doi: 10.1016/j.exer.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler MJ, Thomas BB, Chen Z, Wu R, Sadda SR, Aramant RB. Retinal transplants restore visual responses: trans-synaptic tracing from visually responsive sites labels transplant neurons. Eur J Neurosci. 2008b;28:208–220. doi: 10.1111/j.1460-9568.2008.06279.x. [DOI] [PubMed] [Google Scholar]
- Sekirnjak C, Hulse C, Jepson LH, Hottowy P, Sher A, Dabrowski W, Litke AM, Chichilnisky EJ. Loss of responses to visual but not electrical stimulation in ganglion cells of rats with severe photoreceptor degeneration. J Neurophysiol. 2009;102:3260–3269. doi: 10.1152/jn.00663.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirnjak C, Jepson LH, Hottowy P, Sher A, Dabrowski W, Litke AM, Chichilnisky EJ. Changes in physiological properties of rat ganglion cells during retinal degeneration. J Neurophysiol. 2011;105:2560–2571. doi: 10.1152/jn.01061.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JA, Kim HS, Vargas A, Yu WQ, Eom YS, Craft CM, Lee EJ. Inhibition of Matrix Metalloproteinase 9 Enhances Rod Survival in the S334ter-line3 Retinitis Pigmentosa Model. PLoS One. 2016;11:e0167102. doi: 10.1371/journal.pone.0167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde V, Pitale PM, Howse W, Gorbatyuk O, Gorbatyuk M. Neuronatin is a stress-responsive protein of rod photoreceptors. Neuroscience. 2016;328:1–8. doi: 10.1016/j.neuroscience.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde VM, Sizova OS, Lin JH, LaVail MM, Gorbatyuk MS. ER stress in retinal degeneration in S334ter Rho rats. PLoS One. 2012;7:e33266. doi: 10.1371/journal.pone.0033266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidman RL. Histogenesis of the mouse eye studied with thymidine [3H] In: Smelser G, editor. The Structure of the Eye. Academic Press; New York: 1961. pp. 497–506. [Google Scholar]
- Sizova OS, Shinde VM, Lenox AR, Gorbatyuk MS. Modulation of cellular signaling pathways in P23H rhodopsin photoreceptors. Cell Signal. 2014;26:665–672. doi: 10.1016/j.cellsig.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB. C57BL/6J-vit/vit mouse model of retinal degeneration: light microscopic analysis and evaluation of rhodopsin levels. Exp Eye Res. 1992;55:903–910. doi: 10.1016/0014-4835(92)90017-m. [DOI] [PubMed] [Google Scholar]
- Song Y, Zhao L, Tao W, Laties AM, Luo Z, Wen R. Photoreceptor protection by cardiotrophin-1 in transgenic rats with the rhodopsin mutation s334ter. Invest Ophthalmol Vis Sci. 2003;44:4069–4075. doi: 10.1167/iovs.02-1130. [DOI] [PubMed] [Google Scholar]
- Sotoca JV, Alvarado JC, Fuentes-Santamaria V, Martinez-Galan JR, Caminos E. Hearing impairment in the P23H-1 retinal degeneration rat model. Front Neurosci. 2014;8:297. doi: 10.3389/fnins.2014.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg RH, Flannery JG, Naash M, Oh P, Matthes MT, Yasumura D, Lau-Villacorta C, Chen J, LaVail MM. Transgenic rat models of inherited retinal degeneration caused by mutant opsin genes. Invest Ophthalmol Vis Sci. 1996;37:S698. [Google Scholar]
- Stiles M, Qi H, Sun E, Tan J, Porter H, Allegood J, Chalfant CE, Yasumura D, Matthes MT, LaVail MM, Mandal NA. Sphingolipid profile alters in retinal dystrophic P23H-1 rats and systemic FTY720 can delay retinal degeneration. J Lipid Res. 2016;57:818–831. doi: 10.1194/jlr.M063719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl K, Zrenner E. Electronic approaches to restitute vision in patients with neurodegenerative diseases of the retina. Ophthalmic Res. 2013;50:215–220. doi: 10.1159/000354424. [DOI] [PubMed] [Google Scholar]
- Stone J, Maslim J, Valter-Kocsi K, Mervin K, Bowers F, Chu Y, Barnett N, Provis J, Lewis G, Fisher SK, Bisti S, Gargini C, Cervetto L, Merin S, Peer J. Mechanisms of photoreceptor death and survival in mammalian retina. Prog Retin Eye Res. 1999;18:689–735. doi: 10.1016/s1350-9462(98)00032-9. [DOI] [PubMed] [Google Scholar]
- Strauss O, Stumpff F, Mergler S, Wienrich M, Wiederholt M. The Royal College of Surgeons rat: an animal model for inherited retinal degeneration with a still unknown genetic defect. Acta Anat. 1998;162:101–111. doi: 10.1159/000046474. [DOI] [PubMed] [Google Scholar]
- Stuck MW, Conley SM, Naash MI. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog Retin Eye Res. 2016;52:47–63. doi: 10.1016/j.preteyeres.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Davenport CM, Hennessey JC, Maumenee IH, Jacobson SG, Heckenlively JR, Nowakowski R, Fishman G, Gouras P, Nathans J. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:6481–6485. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung C-H, Makino C, Baylor D, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung C-H, Tai AW. Rhodopsin trafficking and its role in retinal dystrophies. Int Rev Cytol. 2000;195:215–265. doi: 10.1016/s0074-7696(08)62706-0. [DOI] [PubMed] [Google Scholar]
- Tamai M, Chader GJ. The early appearance of disc shedding in the rat retina. Invest Ophthalmol Vis Sci. 1979;18:913–917. [PubMed] [Google Scholar]
- Tan E, Wang Q, Quiambao AB, Xu X, Qtaishat NM, Peachey NS, Lem J, Fliesler SJ, Pepperberg DR, Naash MI, Al-Ubaidi MR. The relationship between opsin overexpression and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2001;42:589–600. [PubMed] [Google Scholar]
- Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2017;101:25–30. doi: 10.1136/bjophthalmol-2016-308823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Wen R, Goddard MB, Sherman SD, O’Rourke PJ, Stabila PF, Bell WJ, Dean BJ, Kauper KA, Budz VA, Tsiaras WG, Acland GM, Pearce-Kelling S, Laties AM, Aguirre GD. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43:3292–3298. [PubMed] [Google Scholar]
- Tessitore A, Parisi F, Denti MA, Allocca M, Di Vicino U, Domenici L, Bozzoni I, Auricchio A. Preferential silencing of a common dominant rhodopsin mutation does not inhibit retinal degeneration in a transgenic model. Mol Ther. 2006;14:692–699. doi: 10.1016/j.ymthe.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Thanos S. Sick photoreceptors attract activated microglia from the ganglion cell layer: a model to study the inflammatory cascades in rats with inherited retinal dystrophy. Brain Res. 1992;588:21–28. doi: 10.1016/0006-8993(92)91340-k. [DOI] [PubMed] [Google Scholar]
- Thomas BB, Arai S, Ikai Y, Qiu G, Chen Z, Aramant RB, Sadda SR, Seiler MJ. Retinal transplants evaluated by optical coherence tomography in photoreceptor degenerate rats. J Neurosci Methods. 2006 doi: 10.1016/j.jneumeth.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Thomas BB, Samant DM, Seiler MJ, Aramant RB, Sheikholeslami S, Zhang K, Chen Z, Sadda SR. Behavioral evaluation of visual function of rats using a visual discrimination apparatus. J Neurosci Methods. 2007a;162:84–90. doi: 10.1016/j.jneumeth.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BB, Seiler MJ, Aramant RB, Samant D, Qiu G, Vyas N, Arai S, Chen Z, Sadda SR. Visual functional effects of constant blue light in a retinal degenerate rat model. Photochem Photobiol. 2007b;83:759–765. doi: 10.1562/2006-09-19-RA-1044. [DOI] [PubMed] [Google Scholar]
- Thomas BB, Seiler MJ, Sadda SR, Aramant RB. Superior colliculus responses to light - preserved by transplantation in a slow degeneration rat model. Exp Eye Res. 2004a;79:29–39. doi: 10.1016/j.exer.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Thomas BB, Seiler MJ, Sadda SR, Coffey PJ, Aramant RB. Optokinetic test to evaluate visual acuity of each eye independently. J Neurosci Methods. 2004b;138:7–13. doi: 10.1016/j.jneumeth.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Thomas BB, Shi D, Khine K, Kim LA, Sadda SR. Modulatory influence of stimulus parameters on optokinetic head-tracking response. Neurosci Lett. 2010;479:92–96. doi: 10.1016/j.neulet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Ali RR, Banin E, Branham KE, Flannery JG, Gamm DM, Hauswirth WW, Heckenlively JR, Iannaccone A, Jayasundera KT, Khan NW, Molday RS, Pennesi ME, Reh TA, Weleber RG, Zacks DN, Monaciano C. Advancing therapeutic strategies for inherited retinal degeneration: recommendations from the Monaciano Symposium. Invest Ophthalmol Vis Sci. 2015;56:918–931. doi: 10.1167/iovs.14-16049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, McHenry CL, Li Y, Richards JE, Othman MI, Schwinger E, Vollrath D, Jacobson SG, Gal A. Retinal dystrophy due to paternal isodisomy for chromosome 1 or chromosome 2, with homoallelism for mutations in RPE65 or MERTK, respectively. Am J Hum Genet. 2002;70:224–229. doi: 10.1086/338455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani I, Banfi S, Simonelli F, Surace EM, Auricchio A. Gene therapy of inherited retinal degenerations: prospects and challenges. Hum Gene Ther. 2015;26:193–200. doi: 10.1089/hum.2015.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani I, Puppo A, Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. 2014;43:108–128. doi: 10.1016/j.preteyeres.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverso V, Bush RA, Sieving PA, Deretic D. Retinal cAMP levels during the progression of retinal degeneration in rhodopsin P23H and S334ter transgenic rats. Invest Ophthalmol Vis Sci. 2002;43:1655–1661. [PubMed] [Google Scholar]
- Treisman JE, Morabito MA, Barnstable CJ. Opsin expression in the rat retina is developmentally regulated by transcriptional activation. Mol Cell Biol. 1988;8:1570–1579. doi: 10.1128/mcb.8.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usukura J, Bok D. Changes in the localization and content of opsin during retinal development in the rds mutant mouse: immunocytochemistry and immunoassay. Exp Eye Res. 1987;45:501–515. doi: 10.1016/s0014-4835(87)80061-1. [DOI] [PubMed] [Google Scholar]
- Valter K, Kirk DK, Stone J. The potential of ambient light restriction to restore function to the degenerating P23H-3 rat retina. Adv Exp Med Biol. 2008;613:193–199. doi: 10.1007/978-0-387-74904-4_22. [DOI] [PubMed] [Google Scholar]
- Valter K, Kirk DK, Stone J. Optimising the structure and function of the adult P23H-3 retina by light management in the juvenile and adult. Exp Eye Res. 2009;89:1003–1011. doi: 10.1016/j.exer.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Vargas A, Kim HS, Baral E, Yu WQ, Craft CM, Lee EJ. Protective effect of clusterin on rod photoreceptor in rat model of retinitis pigmentosa. PLoS One. 2017;12:e0182389. doi: 10.1371/journal.pone.0182389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasireddy V, Chavali VR, Joseph VT, Kadam R, Lin JH, Jamison JA, Kompella UB, Reddy GB, Ayyagari R. Rescue of photoreceptor degeneration by curcumin in transgenic rats with P23H rhodopsin mutation. PLoS One. 2011;6:e21193. doi: 10.1371/journal.pone.0021193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan DK, Coulibaly SF, Darrow RM, Organisciak DT. A morphometric study of light-induced damage in transgenic rat models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003;44:848–855. doi: 10.1167/iovs.02-0709. [DOI] [PubMed] [Google Scholar]
- Veleri S, Lazar CH, Chang B, Sieving PA, Banin E, Swaroop A. Biology and therapy of inherited retinal degenerative disease: insights from mouse models. Dis Model Mech. 2015;8:109–129. doi: 10.1242/dmm.017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey KA, Greferath U, Aplin FP, Jobling AI, Phipps JA, Ho T, De Iongh RU, Fletcher EL. Adenosine triphosphate-induced photoreceptor death and retinal remodeling in rats. J Comp Neurol. 2014;522:2928–2950. doi: 10.1002/cne.23558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viringipurampeer IA, Metcalfe AL, Bashar AE, Sivak O, Yanai A, Mohammadi Z, Moritz OL, Gregory-Evans CY, Gregory-Evans K. NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Hum Mol Genet. 2016;25:1501–1516. doi: 10.1093/hmg/ddw029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollrath D, Feng W, Duncan JL, Yasumura D, D’Cruz PM, Chappelow A, Matthes MT, Kay MA, LaVail MM. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci U S A. 2001;98:12584–12589. doi: 10.1073/pnas.221364198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh N, van Driel D, Lee D, Stone J. Multiple vulnerability of photoreceptors to mesopic ambient light in the P23H transgenic rat. Brain Res. 2004;1013:194–203. doi: 10.1016/j.brainres.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Wang M, Lam TT, Tso MOM, Naash MI. Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci. 1997;14:55–62. doi: 10.1017/s0952523800008750. [DOI] [PubMed] [Google Scholar]
- Wen R, Tao W, Li Y, Sieving PA. CNTF and retina. Prog Retin Eye Res. 2012;31:136–151. doi: 10.1016/j.preteyeres.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- White MP, Gorrin GM, Mullen RJ, LaVail MM. Retinal degeneration in the nervous mutant mouse. II. Electron microscopic analysis. J Comp Neur. 1993;333:182–198. doi: 10.1002/cne.903330205. [DOI] [PubMed] [Google Scholar]
- Wong LL, Pye QN, Chen L, Seal S, McGinnis JF. Defining the catalytic activity of nanoceria in the P23H-1 rat, a photoreceptor degeneration model. PLoS One. 2015;10:e0121977. doi: 10.1371/journal.pone.0121977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Li Y, Huang D, Wang Z, Luo L, Song Y, Zhao L, Wen R. Oncostatin M protects rod and cone photoreceptors and promotes regeneration of cone outer segment in a rat model of retinal degeneration. PLoS One. 2011;6:e18282. doi: 10.1371/journal.pone.0018282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai A, Hafeli UO, Metcalfe AL, Soema P, Addo L, Gregory-Evans CY, Po K, Shan X, Moritz OL, Gregory-Evans K. Focused magnetic stem cell targeting to the retina using superparamagnetic iron oxide nanoparticles. Cell Transplant. 2012;21:1137–1148. doi: 10.3727/096368911X627435. [DOI] [PubMed] [Google Scholar]
- Yang PB, Seiler MJ, Aramant RB, Yan F, Mahoney MJ, Kitzes LM, Keirstead HS. Trophic factors GDNF and BDNF improve function of retinal sheet transplants. Exp Eye Res. 2010a;91:727–738. doi: 10.1016/j.exer.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Justus S, Li Y, Tsang SH. BEST1: the Best Target for Gene and Cell Therapies. Mol Ther. 2015;23:1805–1809. doi: 10.1038/mt.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mohand-Said S, Danan A, Simonutti M, Fontaine V, Clerin E, Picaud S, Leveillard T, Sahel JA. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol Ther. 2009;17:787–795. doi: 10.1038/mt.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mohand-Said S, Leveillard T, Fontaine V, Simonutti M, Sahel JA. Transplantation of photoreceptor and total neural retina preserves cone function in P23H rhodopsin transgenic rat. PLoS One. 2010b;5:e13469. doi: 10.1371/journal.pone.0013469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW. Cell proliferation during postnatal development of the retina in the mouse. Dev Brain Res. 1985;21:229–239. doi: 10.1016/0165-3806(85)90211-1. [DOI] [PubMed] [Google Scholar]
- Yu DY, Cringle S, Valter K, Walsh N, Lee D, Stone J. Photoreceptor death, trophic factor expression, retinal oxygen status, and photoreceptor function in the P23H rat. Invest Ophthalmol Vis Sci. 2004;45:2013–2019. doi: 10.1167/iovs.03-0845. [DOI] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ. Retinal degeneration and local oxygen metabolism. Exp Eye Res. 2005;80:745–751. doi: 10.1016/j.exer.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Yu TY, Acosta ML, Ready S, Cheong YL, Kalloniatis M. Light exposure causes functional changes in the retina: increased photoreceptor cation channel permeability, photoreceptor apoptosis, and altered retinal metabolic function. J Neurochem. 2007;103:714–724. doi: 10.1111/j.1471-4159.2007.04766.x. [DOI] [PubMed] [Google Scholar]
- Yu WQ, Eom YS, Shin JA, Nair D, Grzywacz SX, Grzywacz NM, Craft CM, Lee EJ. Reshaping the Cone-Mosaic in a Rat Model of Retinitis Pigmentosa: Modulatory Role of ZO-1 Expression in DL-Alpha-Aminoadipic Acid Reshaping. PLoS One. 2016;11:e0151668. doi: 10.1371/journal.pone.0151668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WQ, Grzywacz NM, Lee EJ, Field GD. Cell type-specific changes in retinal ganglion cell function induced by rod death and cone reorganization in rats. J Neurophysiol. 2017;118:434–454. doi: 10.1152/jn.00826.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, Fariss RN, Wong WT. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia. 2016;64:1479–1491. doi: 10.1002/glia.23016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbin M. Cell-Based Therapy for Degenerative Retinal Disease. Trends Mol Med. 2016;22:115–134. doi: 10.1016/j.molmed.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Zarbin MA, Arlow T, Ritch R. Regenerative nanomedicine for vision restoration. Mayo Clin Proc. 2013;88:1480–1490. doi: 10.1016/j.mayocp.2013.05.025. [DOI] [PubMed] [Google Scholar]
- Zeiss CJ, Johnson EA. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Invest Ophthalmol Vis Sci. 2004;45:971–976. doi: 10.1167/iovs.03-0301. [DOI] [PubMed] [Google Scholar]
- Zhang C, Shen JK, Lam TT, Zeng HY, Chiang SK, Yang F, Tso MO. Activation of microglia and chemokines in light-induced retinal degeneration. Mol Vis. 2005;11:887–895. [PubMed] [Google Scholar]
- Zhang Y, Rauch U, Perez MT. Accumulation of neurocan, a brain chondroitin sulfate proteoglycan, in association with the retinal vasculature in RCS rats. Invest Ophthalmol Vis Sci. 2003;44:1252–1261. doi: 10.1167/iovs.02-0450. [DOI] [PubMed] [Google Scholar]
- Zhu CL, Ji Y, Lee EJ, Grzywacz NM. Spatiotemporal pattern of rod degeneration in the S334ter-line-3 rat model of retinitis pigmentosa. Cell Tissue Res. 2013a;351:29–40. doi: 10.1007/s00441-012-1522-5. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Misra S, Nivison-Smith L, Acosta ML, Fletcher EL, Kalloniatis M. Mapping cation entry in photoreceptors and inner retinal neurons during early degeneration in the P23H-3 rat retina. Vis Neurosci. 2013b;30:65–75. doi: 10.1017/S0952523813000047. [DOI] [PubMed] [Google Scholar]
- Zulliger R, Conley SM, Naash MI. Non-viral therapeutic approaches to ocular diseases: An overview and future directions. J Control Release. 2015;219:471–487. doi: 10.1016/j.jconrel.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]