

Abstract
The cancer-suppressing transcription factor p53 is regulated by a wide variety of cellular factors, including many chaperones. The DNA-binding domain (DBD) of p53 is known to interact with the chaperone Hsp90, but the role of other members of the chaperone network, including co-chaperones such as p23 is unknown. Using a combination of NMR titration, isothermal titration calorimetry, fluorescence anisotropy and native agarose gel electrophoresis, we have identified a direct interaction between the p53 DBD and the Hsp90 co-chaperone p23 that occurs in the absence of Hsp90. The affinity is relatively weak, and largely determined by electrostatic interactions between the acidic C-terminal disordered tail of p23 and the two DNA binding regions of p53 DBD. We show by NMR and native agarose gel electrophoresis that a p53-specific double-stranded DNA sequence competes successfully with p23 for binding to the p53 DBD. The Hsp90-independence of the interaction between p23 and p53 DBD, together with the p23-DNA competition for p53, raise the intriguing possibility that p23, like other small charged proteins, may affect the p53 in hitherto-unknown ways.
Keywords: p53, DNA binding domain (DBD), p23, Hsp90, chaperone, protein-protein interaction
TOC Image
Introduction
The transcription factor and tumor suppressor p53 plays a central role in the cell cycle and functions as an important signaling hub for the cellular response to various stresses.1,2 Mutations in p53 are implicated in the progress of many cancers, and the majority of cancer-causing p53 mutations are found in the central DNA binding domain (DBD) of the 393 amino-acid protein (Figure 1A). The p53 DBD is well folded; transcriptional activity is mediated by binding of this domain to DNA. The remainder of the protein is largely disordered (with the exception of the tetramerization domain in the tetrameric protein); these disordered regions mediate signal-dependent interactions that modulate the function of p53. The p53 DBD consists of a β-sandwich that includes two antiparallel β-sheets and a small β-hairpin, two large loops (L2 (residues 164-194) and L3 (residues 237-250)), and a loop-sheet-helix motif containing loop L1 (residues 113-123), a β-hairpin (residues 124-136) and a long C-terminal helix (H2, residues 278-289) (Figure 1B). The loop-sheet-helix motif and L3 form two DNA binding sites, which contact the major and minor grooves of DNA respectively3,4 L2 includes a short helix H1 that participates in protein-protein interaction between the monomer units of p53 in the structure of the dimer of p53 complexed with DNA.5
Figure 1.
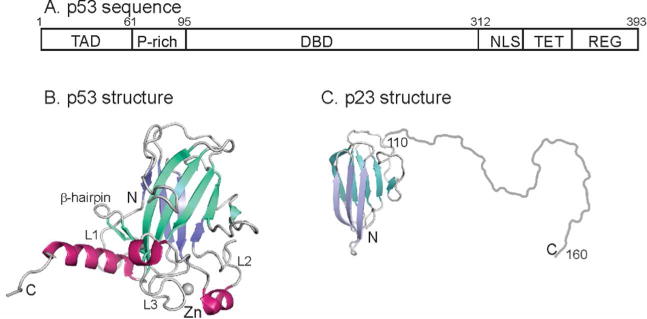
A. Schematic diagram showing the domain structure of human p53. Residues 95-312 comprise the folded DNA-binding domain. TAD – trans-activation domain; P-rich – proline rich domain; DBD – DNA-binding domain; NLS – nuclear localization sequence; TET – tetramerization domain; REG – C-terminal regulatory domain. B. Cartoon representation of the solution structure of the DBD of human p53 (2FEJ4)helices are shown in red, and the strands of the two β-sheets are colored in blue and turquoise. C. Representation of the structure of human p23. The folded domain (residues 1-109) is derived from the crystal structure (1EJF6) and the disordered C-terminus (residues 110-160) is shown schematically as a loop. The strands of the two β-sheets are colored in blue and turquoise.
The interaction of the p53 DBD with the chaperone Hsp90 is important for the stability and activity of p53 in vivo.7-9 A number of previous studies have attempted to define the structural state of the p53 DBD in the presence of Hsp90 - it has variously been described as unfolded,10 folded9 or in a molten-globule state.11 Hsp90, an essential molecular chaperone in eukaryotes, uses a variety of co-chaperones in its interactions with many different proteins. p23, a well-characterized 18 kDa co-chaperone, increases Hsp90 AMP-PNP affinity and reduces the ATPase activity of Hsp90, thereby increasing its affinity for client proteins.12 p23 interacts with the Hsp90 N-terminal and middle domains,13-15 and has been observed as part of the complexes of Hsp90 with various clients, including progesterone receptor, Fes tyrosine kinase and the transcription factor Hsf.16 However, the role of p23 in the interaction of Hsp90 with p53 is unclear. There are very few published results implicating p23 in the p53-Hsp90 interaction, one showing the presence of p23 in the coprecipitation of a p53 temperature-sensitive mutant in murine cells together with Hsp90, Hsp70 and cyclophilin 40,8 and another showing the presence of p23 and p53 in an immunoprecipitation experiment.17 Any effects of p23 on the folding and function of p53 remain unknown.
p23 consists of a N-terminal β-sandwich domain, with two opposing β-sheets, and a long C-terminal unstructured tail (residues 110-160)6 (Figure 1C). While the folded N-terminal domain is responsible for Hsp90 interaction,14 the C-terminal tail is important for its chaperone activity.13,15,18 In addition, p23 has recently been shown to possess other functions, independent of Hsp90. For example, yeast p23 is required for proper Golgi function, ribosome biogenesis and efficient DNA repair.19 p23 has been found localized to the chromatin of hormone-responsive genes, and can inhibit the binding of thyroid hormone receptor (TR) to DNA in vitro, suggesting that it may have a role in transcription, independent of its role as a co-chaperone.20,21
Here, we show by NMR titration, isothermal titration calorimetry (ITC) and native gel electrophoresis that full-length p23 interacts with p53 DBD in the absence of Hsp90. The regions of the two proteins that interact are identified by chemical shift perturbation mapping of full-length and truncated proteins. We find that the primary interaction is between areas near the two DNA-binding sites of p53 and the C-terminal acidic disordered tail of p23. We also show that p23 and DNA compete for p53 binding, suggesting that, as well as playing an important role in the Hsp90-client interaction, p23 could also affect transcription by binding p53 directly, a function reminiscent of its role in the regulation of DNA binding for at least two other proteins, intracellular receptor and Remodeler of Structure of Chromatin (RSC).19,20
Materials and Methods
Protein Expression and Purification
Human p53 DBD (residues 88-312) and full length p23 were expressed and purified as reported previously.11,13,22 The gene for the superstable mutant4 of full-length p53 was cloned into a pET21 expression vector with a 6x His tag and a GB1 fusion protein followed by a TEV cleavage site at the N-terminus. Cells were grown at 37 °C, and protein expression was induced at 18 °C overnight with 0.7mM isopropyl β-D-thiogalactopyranoside (IPTG) and 150 μM ZnSO4 when the absorbance at 600 nm reached 0.7-0.8. Cells were harvested by centrifugation at 4000 × g at 4 °C for 30 min. The cell pellets were resuspended in 40 mM Tris, 1 M NaCl 5 mM DTT. Protease inhibitor (Calbiochem) was added before ultrasonication. DNase I was added to the lysate and after centrifugation the supernatant was applied onto a Ni2+ column (Roche). The eluate at a high imidazole concentration was collected and digested with TEV protease in dialysis at 4 °C overnight. Heparin and Q columns (GE Healthcare) were further used in purification. Truncations of p23 were cloned into pET28 expression vector with a 6x His tag and a SUMO fusion protein at the N-terminus. The protein was expressed under similar conditions as for p53 and purified by a Ni2+ column (Roche), a ULP1 digestion and a Q column (GE Healthcare). GB1-p23 (110-160) or GB1-p23 (120-160) were constructed by cloning the truncated p23 into a pET21 expression vector with a 6x His tag and a GB1 fusion protein at the N-terminus. The protein was expressed under similar conditions as for p53 and purified by a Ni2+ column and a Q column (GE Healthcare).
15N-labeled samples were expressed in M9 minimal medium in H2O supplemented with 15NH4SO4 (1 g/l). 13C-labeled samples were expressed in M9 minimal medium in D2O supplemented with 13C-glucose (2 g/l). The expression and purification methods are similar to those used for the unlabeled samples. The unlabeled samples used to perform each of the titrations were protonated.
NMR spectroscopy
NMR titration experiments for protein-protein interaction were performed at 20 °C (15N) or 25 °C (13C) on a Bruker DRX800 or Avance900 spectrometer. All samples were exchanged for NMR into 25mM Tris (pH 7.0), 5 mM DTT in 90% H2O and 10% D2O with 50 mM or 150 mM NaCl. 2 mM AMP-PNP (Roche) was added to the 13C labeled p53 DBD sample for HMQC experiments. Titration was performed with 15N-labeled p53 DBD or p23 at concentrations of 80-150 μM with binding partner at concentration ratios from 1.0:0.0 to 1.0:2.0 or 1.0:2.5. DNA binding was performed with 15N-labeled p53 DBD titrated by a duplex of 20-mer DNA [corresponding to the p21 binding sequence (GAACATGTTCGAACATGTTC)23 containing four p53 binding sites] at concentration ratios (p53 DBD:DNA) of 1.0:0.08 and 1.0:0.2.
All NMR data were processed with NMRPipe24 and analyzed with NMRViewJ.25 Chemical shift perturbation (CSP) was calculated using the formula:
where ΔHN and ΔN are the changes in 1HN and 15N chemical shifts, respectively.
The equilibrium dissociation constants (Kd) were estimated by fitting the observed CSPs to the equation26:
where CSPmax is the CSP at the theoretical saturated condition obtained from the fit; r is the molar ratio of ligand to protein; Cpro is the concentration of initial protein solution; and Clig is the stock concentration of ligand. The Kd value of 15N p53 DBD titrated by p23 is obtained by averaging separate fits to three residues (V272, C277 and G293) of p53 DBD.
1D diffusion ordered spectroscopy (DOSY) experiments were performed at 20 °C on a Bruker Avance900 spectrometer, with diffusion delay of 100 ms and 4.5 ms of gradient lengths. 11 1D DOSY experiments for varying gradient strengths were acquired for each sample with a scan number of 256. The diffusion coefficient was measured by linear fitting of a plot of logarithmical observed intensity versus gradient strengths. The values were calculated by averaging three different peaks.
Native agarose gel electrophoresis
Protein-protein interactions were investigated by native agarose gel electrophoresis following a published protocol:27 a 0.8% agarose gel was run in Tris-glycine (25 mM Tris base, 192 mM glycine) native running buffer (pH 8.3) at 50 V for 0.5-1 hour. The gel was stained by Coomassie blue for 5min then destained for detection. Typically, 50-100 μM sample were mixed with binding partner at concentration ratios from 1.0:0.0 to 1.0:2.0 or 1.0:2.5. DNA binding to p53 DBD was performed by gradient concentration ratios (DNA: p53 DBD) from 1.0:0.0 to 1.0:0.2.
Isothermal Titration Calorimetry (ITC)
Isothermal titration calorimetry was carried out on a VP-ITC 200 instrument (MicroCal, Northampton, MA) at 283K. The samples were exchanged for ITC into a buffer containing 25 mM sodium phosphate (pH 7.0), 20 mM NaCl, 1 mM TCEP, 100 μM PMSF, and 1 μM ZnSO4. Solutions of 100 μM p53 DBD in the cell were titrated by p23. The concentrations of solutions in injection syringe were 1175 μM. The solutions for the cell and the syringe were thoroughly degassed by stirring under vacuum. In total, 21 injections were performed and each injection volume was 2 μl (first and second injection volumes were 1 μl), and spacing between injections was 240 sec.
Fluorescence Anisotropy
Fluorescence anisotropy binding and competition assays were carried out in buffer 50 mM Tris, pH 7.0, 50 mM NaCl, 5 mM DTT at 25 °C on an ISS-PC1 photon-counting steady-state fluorimeter. The 20-mer DNA sequence shown above was labeled with Cy5 dye at the 5′ end. A 5 nM solution of labeled DNA was titrated with p53 DBD in concentration ratios from 1.0:0.0 to 1.0:10.0. The affinity of p53 DBD and DNA was measured by fitting to a standard one-site binding model. The affinity of p23 binding to p53 DBD was determined by a competition method.28,29 5 nM Cy5 dye labeled DNA was first mixed with 110 nM p53 DBD, then the pre-mixed sample was titrated by p23 in concentration ratios (p23:p53 DBD) from 0.0:1.0 to 2000:1.0.
Pull-Down Assay
For in vitro pull-down assays, p53 (88-312) was subcloned into pET21a with a GST fusion at the N-terminus. GST-p53 (88-312) was purified by a GST column followed by a Heparin column (GE Healthcare). 0.2 mg of GST-p53 (88-312) was mixed with 10 μl of GST resin (GE Healthcare) which had been washed with 25 mM Tris, pH 7.0, 50 mM NaCl, 5 mM DTT, 0.1% NP-40. The protein was incubated with resin at room temperature for 2 hours, then the resin was spun down and washed. 0.2 mg Hsp90 and/or p23 was added, and the resin was washed and finally boiled for SDS-PAGE.
Results
Detection of the Interaction Between p53 and p23
The interaction between p53 DBD and p23 was probed by NMR, using a slightly longer construct of p53 DBD, residues 88-312 instead of 94-312 as used previously.11 The longer construct was used to accommodate a possible cation-π interaction reported between R174 and W9123. As 15N labeled p53 DBD was titrated with increasing amounts of unlabeled p23, the 1H-15N HSQC spectrum (Figure 2A, Figure S1) showed changes in cross peak position (for example, for G279 and C277) and intensity (for example, for T211 and G154). The NMR titration was used to obtain a dissociation constant Kd for the interaction of the two proteins. Averaging the Kd values from separate fits for the three residues shown (V272, C277, G293), yielded a Kd for interaction of the two proteins of 8 ± 2 μM (Figure 2B).
Figure 2.
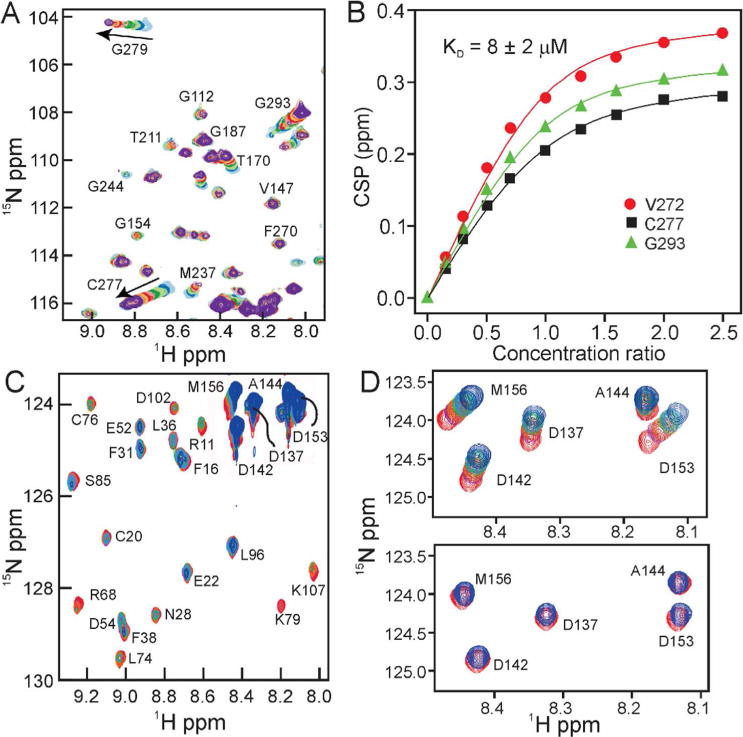
A. Portion of a series of 1H-15N HSQC spectra of 15N-labeled p53(88-312) with increasing mole ratios of p23(1-160). p53:p23 ratios: light blue – 1.0:0.0; dark blue – 1.0:0.17; light green – 1.0:0.3; dark green – 1.0:0.5; pink – 1.0:0.7; red – 1.0:1.0; gold – 1.0:1.3; orange – 1.0:1.6; light purple – 1.0:2.0; purple – 1.0:2.5. The sample is in 25 mM Tris, pH 7.0, 50 mM NaCl, 5 mM DTT, 298K. B. Plots of p53 chemical shift perturbation (CSP) as a function of added p23 for three shifted residues. Solid lines are calculated using the equation shown in Materials and Methods. C. Portion of a series of 1H-15N HSQC spectra of 15N-labeled p23(1-160) with increasing mole ratios of p53(88-312). p23:p53 ratios: red – 1.0:0.0; purple – 1.0:0.5; orange – 1.0:1.0; green – 1.0:1.5; light blue: 1.0:2.0; dark blue: 1.0:2.5. D. Part of the series shown in part C plotted at higher contour level (upper) in 25 mM Tris, pH 7.0, 50 mM NaCl, 5 mM DTT, 298K and (lower) in 25 mM Tris, pH 7.0, 150 mM NaCl, 5 mM DTT, 298K.
Upon titration of 15N labeled p23 with unlabeled p53 DBD, we observe both chemical shift changes and resonance broadening (Figure 2C, Figure S2). An increase of the salt concentration in the buffer from 50mM to 150mM attenuates both the chemical shift change (Figure 2D, Figure S3) and signal broadening (Figure S3), indicating that electrostatic interactions play a major role in the interaction between these two proteins. Addition of full length p53 (residues 1-393) to 15N labeled p23 at 150 mM salt showed similar chemical shift changes and signal broadening (Figure S3C), suggesting that the p53 DBD alone is responsible for the interaction with p23.
Further evidence for the interaction between p53 DBD and p23 is provided by native agarose gel electrophoresis and ITC. The native gel shows that the addition of p23 shifts the band belonging to the p53 DBD in a concentration-dependent way (Figure 3A). ITC also shows that p23 interacts with p53 DBD (Figure 3B), with a Kd of 7 ± 2 μM (ΔH −1.9 ± 100 kcal mol−1; ΔS 17 cal mol−1 deg−1), which is consistent with the value obtained from the NMR titration.
Figure 3.
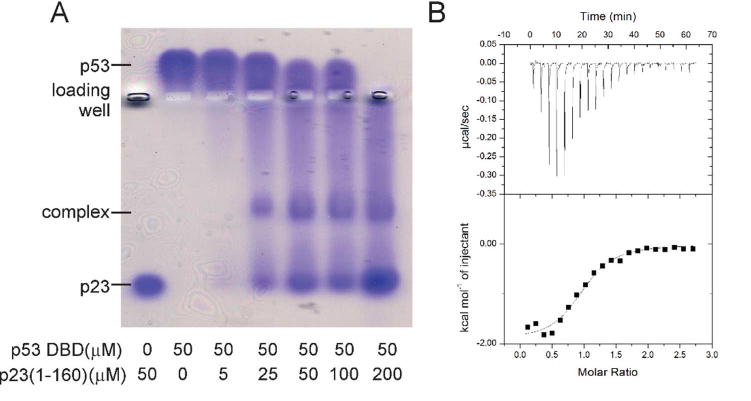
A. Native 0.8% agarose gel run in Tris glycine buffer pH 8.3 and stained with Coomassie blue, showing bands for p23 and p53 and for the complex formed between them. Note that p53(88-312) has a pI of ~9.0 and therefore runs above the loading well. B. Isothermal titration calorimetry of 100 µM p53(88-312) titrated with 2µl of a solution of 1,175 µM p23. Both proteins were dissolved in 25 mM sodium phosphate (pH 7.0), 20 mM NaCl, 1 mM TCEP, 100 µM PMSF, and 1 µM ZnSO4.
We conclude that p23, a co-chaperone for Hsp90, binds directly to the DNA binding domain of p53, independent of the presence of Hsp90, with middle-to-low affinity, likely mediated by electrostatic interactions.
Mapping of binding surfaces for p53 DBD and p23 by NMR
Using published backbone resonance assignments for both p53 DBD30 and p23,13 we have delineated the interacting surfaces by mapping the sequence-dependent intensity decrease and chemical shift perturbation (CSP) observed in the NMR titrations shown in Figures 2, S1 and S2 onto the structures shown in Figure 1. For p53, cross peak intensity decreases are observed with the addition of p23 throughout the sequence (Figure 4A). On the other hand, p53 residues with significant chemical shift change upon addition of p23 are more localized (Figure 4B). The perturbed residues map to two regions of the structure: the most significant CSP is seen for the loop-sheet-helix motif, which includes loop 1 (L1), β-hairpin and helix 2 (H2), and smaller perturbations are seen in a local region near loop 2 (L2) and the zinc-binding loop 3 (L3) (Figure 4C, D). In summary, the CSP results suggest that the loop-sheet-helix motif region is most important for binding p23, with the L2-L3 region as a secondary binding site. The loop-sheet-helix motif and L3 region are the same regions responsible for binding the major groove and minor groove of DNA respectively, indicating that p23 binding may compete with DNA for binding to p53 DBD. The DNA binding sites in p53 have been reported to be involved in interaction with other proteins like Hypoxia inducible factor-1 alpha (HIF-1α), Rad51, Bcl-XL and others,31 suggesting that protein-protein interactions may be important in regulation of DNA binding by p53.
Figure 4.
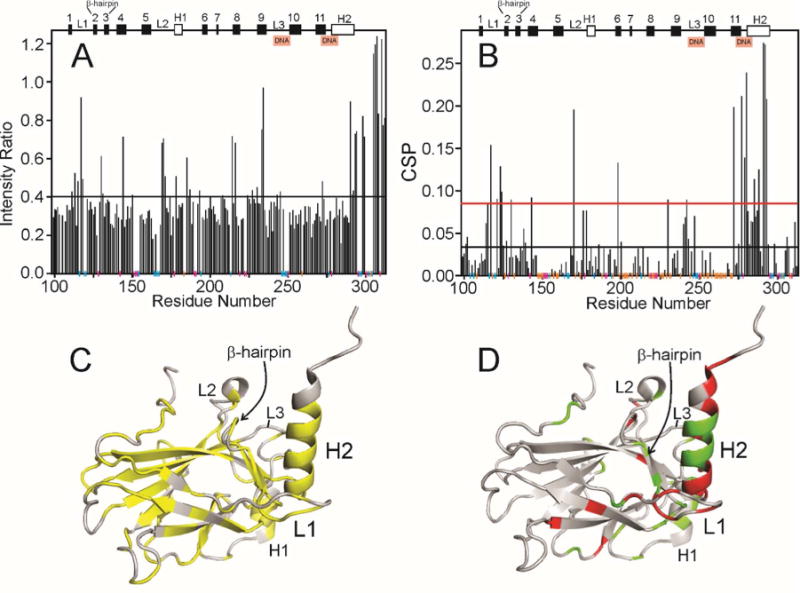
Effect of addition of p23(1-160) to p53 DBD. A. Ratio of the intensity of cross peaks in the 1H-15N HSQC spectrum of 15N-labeled p53(88-312) free (light blue spectrum in Figure 2A) and in the presence of a 1:1 molar ratio of p23(1-160) (red spectrum in Figure 2A). Secondary structure elements are shown at the top of the figure (h indicates helix, otherwise β-strand) and the DNA-binding sites are indicated. Vertical colored lines along the horizontal axis indicate the presence of proline residues (magenta), residues for which cross peaks are not seen or are overlapped in the spectrum of free p53 (blue) and residues for which cross peaks are visible in the free spectrum but not in the 1:1 spectrum (true zero intensity ratio) (orange). The horizontal black line indicates the mean intensity ratio calculated including the orange points but not the blue or red points. B. Chemical shift perturbation of cross peaks in the 1H-15N HSQC spectrum of 15N-labeled p53(88-312) between the free protein (light blue spectrum in Figure 2A) and in the presence of a 1:1 molar ratio of p23(1-160) (red spectrum in Figure 2A) calculated using the formula . Vertical colored lines along the horizontal axis are the same as for Figure 4A. The horizontal black and red lines indicate the mean CSP and the standard deviation above the mean, respectively, calculated including the orange points but not the blue or red points. C. Backbone representation of the structure of p53 DBD (2FEJ4), colored yellow where the intensity ratio shown in Figure 4A lies below the mean value. D. Backbone representation of the structure of p53 DBD (2FEJ4), colored red where the CSP is greater than the mean + standard deviation (above the red line in Figure 4B) and green where mean < value < mean + standard deviation.
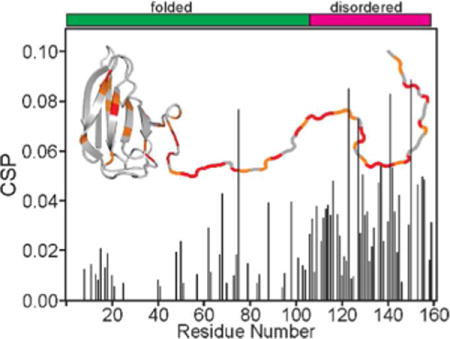
For p23, most of the sequence shows a decrease in cross peak intensity upon interaction with p53 DBD, with the most significant decrease for the N-terminal structured domain (Figure 5A). However, the most significant CSP is observed for the C-terminal disordered sequence, with substantial chemical shift changes observed for only a few resonances of residues in the N-terminal domain (Figure 5B, C). Interestingly, there is a large CSP for C75, the same residue for which a substantial cross peak shift was observed between the 1-119 and 1-160 constructs of free p23.13 This likely reflects contact between the disordered tail and the C75 region in the free protein, a contact that is disrupted by the interaction of p53 DBD with the C-terminal tail. Disruption of contact between the N-and C-terminal parts of p23 may be responsible for much of the perturbation of the resonances of the folded N-terminal domain, since these perturbations appear in the general vicinity of C75 (Figure 5C). Although these results suggest that the C-terminal unstructured region of p23 alone is responsible for binding p53, it remains possible that there is additional involvement of the N-terminal domain.
Figure 5.
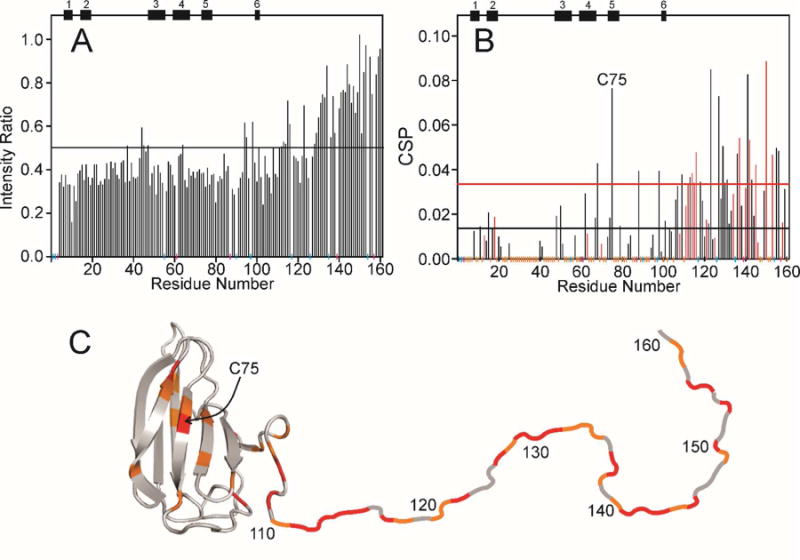
Effect of addition of p53 DBD to p23(1-160). A. Ratio of the intensity of cross peaks in the 1H-15N HSQC spectrum of 15N-labeled p23(1-160) free (red spectrum in Figure 2C) and in the presence of a 1:1 molar ratio of p53(88-312) (dark blue spectrum in Figure 2C). Secondary structure elements are shown at the top of the figure. Vertical colored lines along the horizontal axis are the same as in Figure 4A. The horizontal black line indicates the mean intensity ratio calculated including the orange points but not the blue or red points. B. Chemical shift perturbation of cross peaks in the 1H-15N HSQC spectrum of 15N-labeled p23(1-160) between the free protein (red spectrum in Figure 2C) and in the presence of a 1:1 molar ratio of p53(88-312) (dark blue spectrum in Figure 2C) calculated using the formula used in Figure 4. Red data points indicate acidic residues (Asp or Glu). Vertical colored lines along the horizontal axis are the same as for Figure 4A. The horizontal black and red lines indicate the mean CSP and the standard deviation above the mean, respectively, calculated including the orange points but not the blue or red points. C. Representation of the backbone of p23, where the folded domain (residues 1-109) is derived from the crystal structure (1EJF6) and the disordered C-terminus (residues 110-160) is shown schematically as a loop, colored red where the CSP is greater than the mean + standard deviation (above the red line in Figure 4B) and green where mean < value < mean + standard deviation.
To test whether the N-terminal domain plays a direct role in the interaction, truncations of p23, comprising the N-terminal portion (residues 1-110) and the C-terminal portion (residues 110-160) were used to detect the interaction with p53 DBD. Addition of p23(110-160) to 15N-labeled p53 DBD shows changes in the NMR spectrum identical to those seen with full length p23 (Figure S4A) and attenuation of the chemical shift change is observed upon increasing the salt concentration from 50 mM NaCl to 150 mM NaCl (data not shown). The addition of p23(1-110) does not affect the spectrum of p53 DBD in low salt or high salt conditions (Figure S4B). Consistent results are shown in a native gel, where p23(110-160) shifts p53 DBD, but p23(1-110) does not (Figure 6A). The affinity of the two domains of p23 for p53 DBD was estimated by ITC. The N-terminal domain alone, p23(1-110), shows no binding to the p53 DBD. The ITC measurement was not made for the disordered C-terminal domain alone. Interestingly, the affinity of a construct with a slightly shorter C-terminal tail (residues 120-160) fused at the N-terminus to an unrelated protein, the B1 domain of protein G (GB1), showed a Kd value of 24 ± 15 μM. The NMR titration of 15N-labeled p53 DBD GB1-p23(110-160) (Figure S5) showed almost identical behavior to that of the titration with p23(1-160) (Compare Figure S5 with Figure S1), and gave an identical Kd value (6 ± 4 μM; Figure S5).
Figure 6.
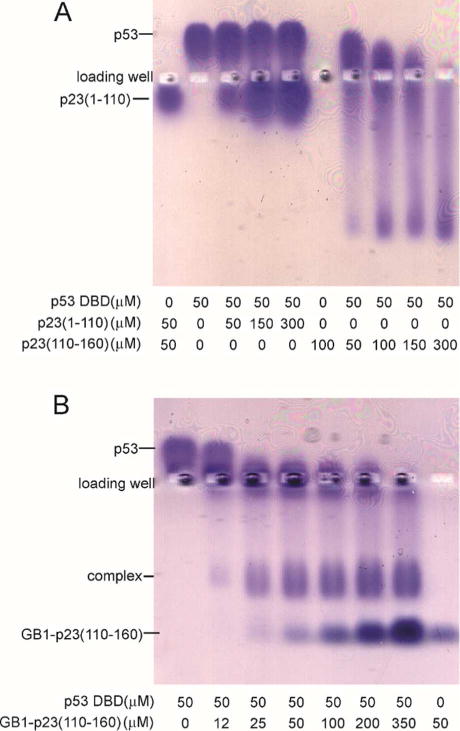
A. Agarose native gel electrophoresis of the interaction of p53 DBD with fragments of p23, residues 1-110 (left lanes) and 110-160 (right lanes). Note that the p23(110-160) construct does not appear on the gel, likely because it is highly negatively charged and does not stain well with Coomassie blue. B. Agarose native gel electrophoresis of the interaction of p53 DBD with a fusion of the B1 domain of protein G (GB1) with residues 110-160 of p23.
The similarity in the affinity of the GB1-p23 fusion to that of the full-length p23 is also demonstrated by a native gel (Figure 6B). No shift of the p53 band is seen in the native gel with GB1 alone (data not shown). These results confirm that the C-terminal disordered tail of p23 is the primary binding site for p53. On a qualitative level the similarity in the behavior of p53 DBD in the native gel in the presence of p23(1-160) (Figure 3A) and in the presence of p23(110-160) (Figure 6A), together with the nearly-identical behavior of p53 DBD in the NMR titrations withp23(1-160) (Figure 2A, S1) and p23(110-160) (Figure S4A) indicates that the presence of the N-terminal structured domain has little effect on the affinity. This conclusion is further confirmed by the similarity of the behavior of p23(1-160), p23(110-160) and the N-terminal GB1 fusion of p23(110-160) in the native gel electrophoresis experiment (Figure 3A, 6A, 6B).
Effect of Hsp90 on the p53-p23 interaction
From the results shown in Figures 2-4, it appears that the co-chaperone p23 and the client protein p53 interact in the absence of Hsp90. To test whether the interaction was modified in the presence of Hsp90, a GST-pulldown experiment was performed with GST-tagged p53 DBD and p23(1-160) and full-length Hsp90 (Figure S6). The results showed weak binding of either Hsp90 or p23 to p53 DBD, and attenuation of Hsp90 binding with both proteins present. Native gel electrophoresis also showed binary interactions of both p23 and Hsp90 with p53, but little influence of Hsp90 on the binary interaction of p23 and p53 (Figure S7). More specific information can be obtained from the NMR spectrum, but an examination by NMR of the effects of Hsp90 on the p23-p53 interaction is problematic because of the size of Hsp90. The effects of the addition of both p23 and Hsp90 on the NMR spectrum of p53 were assessed by comparison of the 1H-13C HMQC spectra of p53 DBD in the presence of one or both of these binding partners. The results (Figure S8) show that p23 and Hsp90 bind independently to p53, and that there is no major synergistic effect of the two binding partners. These observations confirm that the p23-p53 interaction that we observe is independent of Hsp90.
p23 competes with DNA for binding p53
Figure 4B suggests that the primary interaction site of p23 is close to the DNA-binding regions of p53. We used NMR to investigate competition for p53 between p23 and a 20 base-pair fragment of the p21 gene, which contains four pentameric binding sites for p53.3 The 1H-15N HSQC spectrum of 15N-labeled p23 was recorded in the presence of p53 DBD or p53 DBD + DNA. As shown in Figure 5, the addition of p53 DBD to 15N-p23 results in a decrease in the signal intensity, most significantly for the N-terminal domain, while the CSP shows the greatest change in the C-terminal tail. These results have been plotted as blue squares in Figure 7. The results of addition of a mixture of p53 DBD with the 20bp DNA fragment to 15N p23 have been plotted as red circles in Figure 7. The intensity decrease seen for the N-terminal domain of p23 in the presence of p53 is reversed (Figure 7A), and the large CSPs observed for the C-terminal domain disappear as DNA is added (Figure 7B), suggesting that the presence of DNA interferes with the interaction between p23 and p53 DBD.
Figure 7.
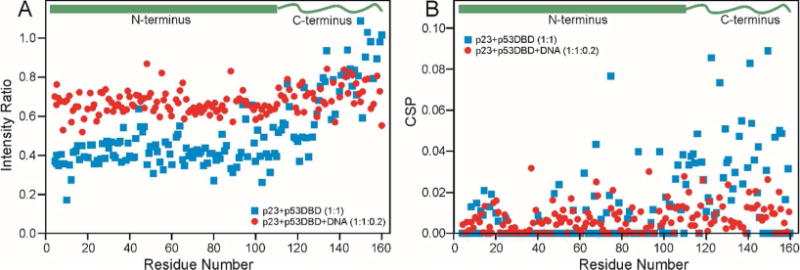
A. The ratio of the intensity of p23 cross peaks in the presence of p53 (concentration ratio 1:1) compared to those of the free protein (blue squares) (same data as Figure 5A). Intensity ratio of p23 cross peaks in the presence of p53 and DNA (concentration ratio p23:p53:DNA of 1:1:0.2) compared to those of the free protein (red circles). A schematic representation of the two domains of p23 is shown at the top. B. CSP of p23 cross peaks from those of the free protein, caused by the presence of p53 (concentration ratio 1:1) (blue squares) (same data as Figure 5B)-r in the presence of p53 and DNA (concentration ratio p23:p53:DNA of 1:1:0.2) (red circles).
Further information on these interactions was provided by using diffusion-ordered spectroscopy (DOSY) to determine the diffusion constant values of p23 in the presence and absence of p53 and DNA. The results are shown in Table 1. The diffusion constants of all of the samples are quite similar, indicating that no major changes in solution viscosity or aggregation state have occurred as a result of the interactions of these proteins. The diffusion constant of p23 decreases after adding p53 DBD, suggesting the formation of a complex between these two proteins. Addition of the 20bp p21 DNA oligonucleotide to the complex results in an increase in the p23 diffusion constant. Addition of p21 DNA alone does not affect the diffusion constant of p23. Since the diffusion constant of the p23:p53 complex is significantly increased in the direction towards that of free p23 after addition of DNA, we conclude that DNA successfully competes with p23 for binding to p53.
Table 1.
Diffusion Constants of p23 with p53 DBD and p53 DBD + DNA
| Sample | ratio | Diffusion Constant (10−7 cm2 s−1) |
|---|---|---|
| 15N p23 | 5.4 ± 0.2 | |
| 15N p23 + p53 DBD | 1:0.5 | 5.1 ± 0.1 |
| 1:1 | 4.2 ± 0.1 | |
| 15N p23 + p53 DBD + DNA | 1:1:0.08 | 4.7 ± 0.1 |
| 1:1:0.2 | 5.0 ± 0.1 | |
| 15N p23 + DNA | 1:0.2 | 5.3 ± 0.2 |
The competition was further characterized by native gel electrophoresis and fluorescence anisotropy decay. The native gel (Figure 8A) shows that both DNA and p23 alone will shift p53 DBD. The addition of increasing amounts of DNA to the p53 DBD-p23 complex results in a shift of the band corresponding to the p53 DBD-p23 complex towards the position of the p53 DBD-DNA complex band, consistent with successful competition by the DNA for binding p53 DBD even in the presence of p23. Fluorescence anisotropy measurements were made with p21 DNA labeled with the fluorescence tag Cy5. Titration of the labeled DNA with p53 DBD results in an increase in the fluorescence anisotropy with a Kd value of 45 ± 7 nM for the four independent p53-binding sites on the DNA, which is consistent with previously-published values.32 The affinity of p23 for p53 DBD in the presence of DNA was measured by titration of the Cy5 labeled p21 DNA-p53 DBD complex with p23. The addition of saturating concentrations of p23 successfully competes for p53 DBD, releasing DNA from p53 DBD and causing the anisotropy value to decrease (Figure 8B). The Kd value for p53 DBD and p23, obtained using a competition fluorescence anisotropy equation,28 is 21 ± 7 μM. This Kd value is consistent with the NMR titration and ITC results (Figure 2A). In summary, the NMR, native gel and fluorescence anisotropy results all show that p23 and DNA compete for binding to p53.
Figure 8.
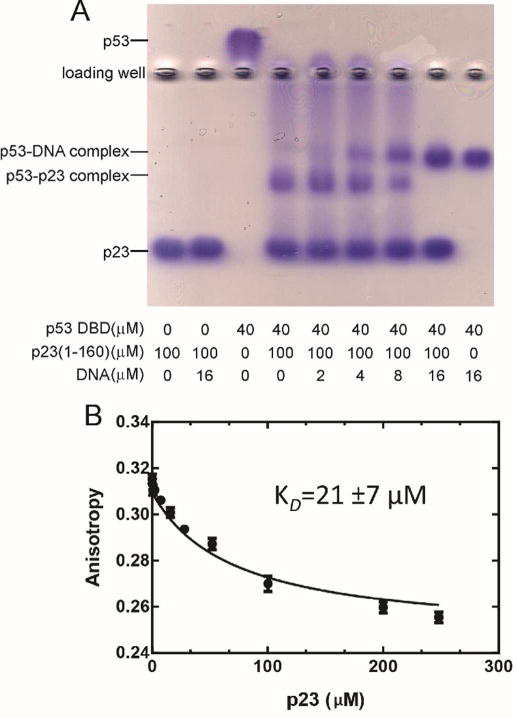
A. Agarose native gel electrophoresis of the interaction of p23(1-160), p53(88-312) and the p21 DNA fragment. B. Fluorescence anisotropy competition of p23(1-160) added to a complex of fluorescently-labeled p21 DNA and p53(88-312).
Discussion
Nature of the Interaction between p53(88-312) and p23(1-160)
The NMR, electrophoresis, ITC and fluorescence data all confirm that an interaction occurs between p53 and p23. This interaction is relatively weak, and highly dependent on the presence of the disordered C-terminal tail of p23: the N-terminal folded domain contributes little if anything to the affinity. Figures 4 and 5 show that both p23 and p53 undergo loss of signal intensity, almost uniformly throughout the protein, when they are mixed. Signal intensity loss has also been observed for p53 in the presence of isolated domains, 2-domain constructs and full-length Hsp90.11 In the p53-Hsp90 system, a variety of spectroscopic and biophysical results were consistent with the formation of a molten globule-like state in p53 as a result of the interaction with Hsp90. Such a structural loosening of p53 could also be occurring in the presence of p23, giving rise to the lowered intensity ratios shown in Figure 4A. p23 also appears to undergo loss of signal intensity upon addition of p53, mostly in the N-terminal folded domain. Interestingly, a similar loss of signal intensity is observed for p23 in the presence of Hsp90 or its domains.13 The most likely source for signal intensity loss is resonance broadening associated with the formation of high-molecular-weight complexes, which will affect the relaxation rates of the ordered regions of the two proteins to a greater extent than the disordered regions. The difference in R2 values between the ordered and disordered components of the complex may also give rise to different relaxation losses during the pulse sequence. It is noticeable from Figure 5A that the C-terminus of p23(1-160) largely retains signal intensity in the presence of p53, seemingly indicating that the C-terminal tail retains its disorder and independence from the large p53-p23 complex. This seems paradoxical in view of the observations in Figure 5B, but likely reflects the presence of disorder in this region within the complex.
Information on the specificity of the interaction between p53 and p23 is provided by the NMR chemical shift perturbation. Figure 5B shows that the majority of the CSP occurs in the disordered C-terminus of p23, which is rich in acidic side chains. The marked salt-dependence of the interaction (Figure 2D) indicates that it is primarily electrostatic. Figure 4B shows a general localization of the p23 binding site on p53 in the vicinity of the DNA-binding loops, consistent with an electrostatic interaction mimicking that between the acidic DNA and these areas of p53. Interestingly, the actual DNA contact loops in p53 are not especially rich in lysine and arginine groups; these groups are concentrated on the periphery of the site.5,33
The chemical shift perturbation indicates that the primary binding event is an electrostatic interaction between the C-terminal tail of p23 and the vicinity of the DNA-binding site of p53. The intensity ratios indicate that the folded domain of p23 and the folded p53 DBD influence each other in the complex. For a complex with Kd ~ 7-21 μM, we would not expect that the two component proteins would be so tightly coupled that their combined molecular weight would broaden out the NMR signals, and besides, the effect is concentration-dependent (Figure 2). The intensity ratio of the p23 C-terminus remains high. If the intensity loss was due to resonance broadening from the formation of a high-molecular weight complex, we would expect this region to show the most marked effect, since it constitutes the primary binding site. These observations suggest that the interaction between p53 and p23 is not the formation of a classical protein-protein complex.
Disordered proteins and domains are capable of forming tight and specific complexes with partner proteins, which themselves may be disordered, ordered or partly ordered.34 Crucially, and perhaps counter-intuitively, the complexes themselves may not be fully ordered: complexes with partially ordered or disordered elements have been termed “fuzzy complexes”.35 Although these elements remain disordered in the complex, their presence enhances and in some cases comprises the entirety of the affinity. The behavior of the p23-p53 system is reminiscent of a fuzzy complex. The disordered C-terminus of p23 provides the major affinity, but it apparently remains disordered in the complex. The behavior of the ordered domains indicates that they too may be participating in a fuzzy interaction, possibly through multiple non-specific contacts, although the intensity loss of resonances in the ordered domains could also potentially be explained by differences in the relaxation characteristics of the ordered and disordered domains. The widespread non-specific intensity changes in the ordered domains could also possibly be a result of rapidly changing inter-protein contacts that occur over most of the molecule, a model that has previously been invoked to describe the observations on the p53-Hsp90 complex,11 and could be applied also to the p23-Hsp90 interaction.13 Such a non-specific but relatively high-affinity interaction may well be typical of chaperones with their clients.
p23 interacts close to the DNA-binding surfaces of p53
Information from NMR chemical shifts and native gel electrophoresis show that the p23 interaction sites are close to the DNA-binding surfaces of p53. We further show that DNA competes successfully with p23 for p53 binding. Nucleic acid-binding proteins commonly contain a preponderance of positively-charged amino acids in their DNA-binding sites. By contrast, the DNA-binding surfaces of p53 consist mostly of small non-charged residues, with positive charges at either end. Thus, the major groove binding site has the amino acid sequence Arg-Val-Cys-Ala-Cys-Pro-Gly-Arg, spanning the loop between the last β-strand and the last helix, while the minor groove binding site has the sequence Cys-Met-Gly-Gly-Met-Asn-Arg and forms the C-terminus of a long loop.33 Areas just outside the DNA-binding region are much more positively charged and it is these that appear to be the major sites of p23 interaction. The C-terminal tail of p23 is predominantly acidic. The interaction of these two oppositely-charged regions is consistent with the attenuation of the NMR CSPs in the presence of higher salt concentrations (Figure 2D). Salt concentrations have also been found to influence the hopping and sliding interactions of p53 with DNA, with higher-salt conditions associated with faster diffusion (“hopping”) of the isolated DBD.36
p23 as a Chaperone for p53
Hsp90 is well-known as a chaperone for p53, with the interaction focused on the p53 DBD.7-9 Other interactions of p53, particularly involving disease-related mutant forms, have also been identified as having chaperone-like features. Binding of p53 DBD to a short peptide, CDB3, from a p53 binding protein, was shown to stabilize the structure of unstable p53 mutants, and was termed a “chaperone strategy”37,38. The sequence of the peptide (REDEDEIEW) is strongly reminiscent of that of the C-terminus of p23, raising the possibility that the p23-p53 interaction may play a similar role in stabilizing the p53 DBD before it finds and binds the appropriate DNA sequence.
Weak protein interactions play an indispensable role in cellular function.39,40 That the binding constant between p23 and p53 DBD is weak is not inconsistent with a role for p23 in the regulation of DNA binding by p53. Other well-known p53 binding proteins, such as BCL-xL, have comparable affinities for full length p53 (Kd 1 μM) and p53 DBD (Kd 17 μM) at a salt concentration of 40 mM.41 Like p23, the binding region of BCL-xL overlaps with the DNA-binding site of p53, and charge interactions drive complex formation. The interaction between BCL-xL and p53 was shown to have biological relevance.41 The close proximity of the p23 and DNA binding sites, together with the observation that a specific p53-binding DNA can successfully compete with p23 for p53, suggests that p23, independent of its role as a co-chaperone for Hsp90, could act as a “holdase” for p53 DBD. These observations illustrate the versatility of heterogeneous and relatively non-specific protein-protein interactions, mediated in many cases by disordered regions, in the control of cellular function.
Supplementary Material
Acknowledgments
We thank Xiaoxuan Lyu and F. Wang for performing in-cell assays, Gerard Kroon for help with NMR spectroscopy, Peter Wright for valuable discussions and Euvel Manlapaz for technical assistance. This work was supported by Grant GM113251 from the National Institutes of Health (HJD) and by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (NRF-2014M3A9B6069340) and a grant from the Basic Science Research Program (2016R1D1A1B03931783) (PSJ).
Footnotes
Supplementary Material
Overlaid HSQC spectra for p53 and p23, additional native gels, methyl HMQC TROSY spectra of p53 in the presence and absence of p23.
References
- 1.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 2.Soussi T, Legros Y, Lubin R, Ory K, Schlichtholz B. Multifactorial analysis of p53 alteration in human cancer: a review. Int J Cancer. 1994;57:1–9. doi: 10.1002/ijc.2910570102. [DOI] [PubMed] [Google Scholar]
- 3.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 4.Cañadillas JMP, Tidow H, Freund SMV, Rutherford TJ, Ang HC, Fersht AR. Solution structure of p53 core domain: Structural basis for its instability. Proc Natl Acad Sci USA. 2006;103:2109–2114. doi: 10.1073/pnas.0510941103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho WC, Fitzgerald MX, Marmorstein R. Structure of the p53 Core Domain Dimer Bound to DNA. J Biol Chem. 2006;281:20494–20502. doi: 10.1074/jbc.M603634200. [DOI] [PubMed] [Google Scholar]
- 6.Weaver AJ, Sullivan WP, Felts SJ, Owen BA, Toft DO. Crystal structure and activity of human p23, a heat shock protein 90 co-chaperone. J Biol Chem. 2000;275:23045–23052. doi: 10.1074/jbc.M003410200. [DOI] [PubMed] [Google Scholar]
- 7.Blagosklonny MV, Toretsky J, Bohen S, Neckers L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci USA. 1996;93:8379–8383. doi: 10.1073/pnas.93.16.8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitesell L, Sutphin PD, Pulcini EJ, Martinez JD, Cook PH. The physical association of multiple molecular chaperone proteins with mutant p53 is altered by geldanamycin, an hsp90-binding agent. Mol Cell Biol. 1998;18:1517–1524. doi: 10.1128/mcb.18.3.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Müller L, Schaupp A, Walerych D, Wegele H, Büchner J. Hsp90 regulates the activity of wild type p53 under physiological and elevated temperatures. J Biol Chem. 2004;279:48846–48854. doi: 10.1074/jbc.M407687200. [DOI] [PubMed] [Google Scholar]
- 10.Rüdiger S, Freund SM, Veprintsev DB, Fersht AR. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc Natl Acad Sci USA. 2002;99:11085–11090. doi: 10.1073/pnas.132393699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park SJ, Borin BN, Martinez-Yamout MA, Dyson HJ. The client protein p53 forms a molten globule-like state in the presence of Hsp90. Nat Struct Mol Biol. 2011;18:537–541. doi: 10.1038/nsmb.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLaughlin SH, Sobott F, Yao ZP, Zhang W, Nielsen PR, Grossmann JG, Laue ED, Robinson CV, Jackson SE. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J Mol Biol. 2006;356:746–758. doi: 10.1016/j.jmb.2005.11.085. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Yamout MA, Venkitakrishnan RP, Preece NE, Kroon G, Wright PE, Dyson HJ. Localization of sites of interaction between p23 and Hsp90 in solution. J Biol Chem. 2006;281:14457–14464. doi: 10.1074/jbc.M601759200. [DOI] [PubMed] [Google Scholar]
- 14.Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karagoz GE, Duarte AM, Ippel H, Uetrecht C, Sinnige T, van RM, Hausmann J, Heck AJ, Boelens R, Rüdiger SG. N-terminal domain of human Hsp90 triggers binding to the cochaperone p23. Proc Natl Acad Sci USA. 2011;108:580–585. doi: 10.1073/pnas.1011867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felts SJ, Toft DO. p23, a simple protein with complex activities. Cell Stress Chaperones. 2003;8:108–113. doi: 10.1379/1466-1268(2003)008<0108:paspwc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dasgupta G, Momand J. Geldanamycin prevents nuclear translocation of mutant p53. Exp Cell Res. 1997;237:29–37. doi: 10.1006/excr.1997.3766. [DOI] [PubMed] [Google Scholar]
- 18.Weikl T, Abelmann K, Buchner J. An unstructured C-terminal region of the Hsp90 co-chaperone p23 is important for its chaperone function. J Mol Biol. 1999;293:685–691. doi: 10.1006/jmbi.1999.3172. [DOI] [PubMed] [Google Scholar]
- 19.Echtenkamp FJ, Zelin E, Oxelmark E, Woo JI, Andrews BJ, Garabedian M, Freeman BC. Global Functional Map of the p23 Molecular Chaperone Reveals an Extensive Cellular Network. Mol Cell. 2011;43:229–241. doi: 10.1016/j.molcel.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeman BC, Felts SJ, Toft DO, Yamamoto KR. The p23 molecular chaperones act at a late step in intracellular receptor action to differentially affect ligand efficacies. Genes Devel. 2000;14:422–434. [PMC free article] [PubMed] [Google Scholar]
- 21.Freeman BC, Yamamoto KR. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science. 2002;296:2232–2235. doi: 10.1126/science.1073051. [DOI] [PubMed] [Google Scholar]
- 22.Park SJ, Kostic M, Dyson HJ. Dynamic interaction of Hsp90 with its client protein p53. J Mol Biol. 2011;411:158–173. doi: 10.1016/j.jmb.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bista M, Freund SM, Fersht AR. Domain-domain interactions in full-length p53 and a specific DNA complex probed by methyl NMR spectroscopy. Proc Natl Acad Sci USA. 2012;109:15752–15756. doi: 10.1073/pnas.1214176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delaglio F, Grzesiek S, Vuister GW, Guang Z, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 26.French M, Swanson K, Shih SC, Radhakrishnan I, Hicke L. Identification and characterization of modular domains that bind ubiquitin. Methods Enzymol. 2005;399:135–157. doi: 10.1016/S0076-6879(05)99009-5. [DOI] [PubMed] [Google Scholar]
- 27.Kim R. Native agarose gel electrophoresis of multiprotein complexes. Cold Spring Harb Protoc. 2011;2011:884–887. doi: 10.1101/pdb.prot4558. [DOI] [PubMed] [Google Scholar]
- 28.Lee CW, Ferreon JC, Ferreon AC, Arai M, Wright PE. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc Natl Acad Sci USA. 2010;107:19290–19295. doi: 10.1073/pnas.1013078107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roehrl MHA, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein–protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 30.Wong KB, DeDecker BS, Freund SM, Proctor MR, Bycroft M, Fersht AR. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc Natl Acad Sci USA. 1999;96:8438–8442. doi: 10.1073/pnas.96.15.8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez-Fernandez MR, Sot B. The relevance of protein-protein interactions for p53 function: the CPE contribution. Prot Eng Des Sel. 2011;24:41–51. doi: 10.1093/protein/gzq074. [DOI] [PubMed] [Google Scholar]
- 32.Nikolova PV, Henckel J, Lane DP, Fersht AR. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc Natl Acad Sci USA. 1998;95:14675–14680. doi: 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malecka KA, Ho WC, Marmorstein R. Crystal structure of a p53 core tetramer bound to DNA. Oncogene. 2009;28:325–333. doi: 10.1038/onc.2008.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 35.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single molecule characterization of p53 search on DNA. Proc Natl Acad Sci USA. 2011;108:563–568. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedler A, Hansson LO, Veprintsev DB, Freund SMV, Rippin TM, Nikolova PV, Proctor MR, Rüdiger S, Fersht AR. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc Natl Acad Sci USA. 2002;99:937–942. doi: 10.1073/pnas.241629998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedler A, Veprintsev DB, Rutherford T, von Glos KI, Fersht AR. Binding of Rad51 and other peptide sequences to a promiscuous, highly electrostatic binding site in p53. J Biol Chem. 2005;280:8051–8059. doi: 10.1074/jbc.M411176200. [DOI] [PubMed] [Google Scholar]
- 39.Vaynberg J, Fukuda T, Chen K, Vinogradova O, Velyvis A, Tu Y, Ng L, Wu C, Qin J. Structure of an Ultraweak Protein-Protein Complex and Its Crucial Role in Regulation of Cell Morphology and Motility. Mol Cell. 2005;17:513–523. doi: 10.1016/j.molcel.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 40.Acuner Ozbabacan SE, Engin HB, Gursoy A, Keskin O. Transient protein–protein interactions. Protein Engineering, Design and Selection. 2011;24:635–648. doi: 10.1093/protein/gzr025. [DOI] [PubMed] [Google Scholar]
- 41.Follis AV, Llambi F, Ou L, Baran K, Green DR, Kriwacki RW. The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat Struct Mol Biol. 2014;21:535–543. doi: 10.1038/nsmb.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.