

Abstract
Epithelial Na+ channels (ENaCs) are members of the ENaC/degenerin family of ion channels that evolved to respond to extracellular factors. In addition to being expressed in the distal aspects of the nephron, where ENaCs couple the absorption of filtered Na+ to K+ secretion, these channels are found in other epithelia as well as non-epithelial tissues. This review addresses mechanisms by which ENaC activity is regulated by extracellular factors, including proteases, Na+ and shear stress. Other factors, including acidic phospholipids and modification of ENaC cytoplasmic cysteine residues by palmitoylation, enhance channel activity by altering interactions of the channel with the plasma membrane, are also addressed.
Keywords: ENaC, protease, sodium self-inhibition, shear stress, palmitoylation, palmitoyltransferase
The epithelial Na+ channel (ENaC) is expressed in the luminal membrane of principal cells of the aldosterone-sensitive distal nephron (ASDN), where it mediates the reabsorption of filtered Na+ that is coupled to K+ secretion via luminal K+ channels (1; 2). ENaCs are members of the ENaC/Degenerin family of cation-selective channels that are comprised of subunits that have common structural features. These include a large, highly organized extracellular region connected to two transmembrane domains that form the channel pore and where the channel gate resides, and short cytoplasmic amino (N) and carboxyl (C) termini (1; 3). Functional channels are formed by subunits that assemble as trimeric homo- or heteroligomers (4–6). For ENaC, these include the α (or δ), β and γ subunits. The extracellular regions of ENaC subunits and other members of the ENaC/Degenerin family are comprised of distinct domains formed by α helices (termed finger, knuckle and thumb) and β strands (β ball and palm) (1; 3; 4) (Fig. 1). The finger domain is located in the periphery of the extracellular region, and is the least conserved domain in the ENaC/Degenerin family. For example, the α subunit of ENaC and an acid sensing ion channel subunit (ASIC1) have only 8% amino acid identity in the finger domain (7). Mouse ENaC α subunit also has a 73-residue insert that is not present in mouse ASIC1, and this insert contains unique sites relevant for channel activation by proteases (7).
Figure 1.
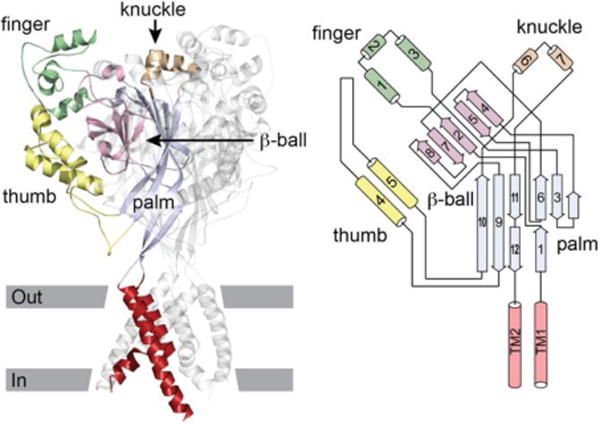
ASIC1 structure. Left, cartoon of ASIC1 trimer. Domains of one subunit highlighted. Approximate location of the membrane is indicated. Right, schematic of an ASIC1 subunit. Cylinders represent helices, arrows represent β strands. The peripheral finger, knuckle and thumb domains are contiguous α helices arising from the central β-ball or palm domains. Modified from (1).
Most ENaC/Degenerin family members are quiescent at rest (1; 8; 9). They are activated by extracellular factors that presumably interact at sites within their extracellular regions, thereby inducing conformation changes that are transmitted to a gate. In contrast, ENaC has evolved as a channel that is constitutively active. This property allows for the bulk transport of Na+ across epithelia. ENaC activity is regulated by both intracellular and extracellular factors, as well as intramembrane lipids that modulate channel open probability. Mechanisms of ENaC regulation by specific factors and lipids are the focus of this review.
ENaCs are expressed in the luminal membranes of a variety of epithelia where they have roles in transepithelial Na+ transport. In the ASDN, ENaCs are modulated by volume regulatory hormones, including aldosterone, angiotensin II, vasopressin and endothelin. Other factors, including luminal flow (or shear stress), ATP, nitric oxide, Na+, H+ and Cl− also alter ENaC activity (1–3). Through its role in reabsorbing filtered Na+, ENaCs are one of several Na+ transporters in the ASDN that regulate total body Na+ content, extracellular fluid volume and blood pressure. While Na+ absorption via ENaC is coupled to K+ secretion in the ASDN, other luminal Na+ transporters in the ASDN, including the Na+,Cl+ co-transporter (NCC) and a Na+-dependent Cl−/HCO3- exchanger (NDCBE) in parallel with a Cl−/HCO3- exchanger (pendrin) facilitate the absorption of both Na+ and Cl− (2; 10–12). This multitude of transporters allows (i) for Na+ absorption to occur in parallel with K+ secretion in states where there is a need for renal K+ excretion, or (ii) for Na+ absorption to occur in parallel with Cl− absorption in states where renal K+ excretion is not required. Recent studies have begun to elucidate mechanisms by which these transporters work in a coordinated manner to absorb Na+ while either absorbing Cl− or secreting K+, as needed. Members of the WNK (with-no-lysine kinases) family, in conjunction with SPAK (Ste20-related proline alanine-rich kinase) and OSR1 (oxidative stress response kinase 1), have important roles in regulating key ASDN Na+ transporters in response to changes in serum [K+], a topic that has been addressed in recent publications (13–16).
ENaCs are also expressed in non-renal tissues where they have roles in modulating blood pressure. For example, ENaCs in lingual epithelia participate in salt taste and influence Na+ ingestion, while ENaCs in the distal colon absorb ingested Na+ (17; 18). Recent studies suggest that ENaC is expressed in specific regions of the CNS (19; 20) and has a role in regulating blood pressure (21). In addition, ENaCs are expressed in both vascular endothelial cells and smooth muscle cells where they are poised to influence vascular tone and pressure-induced myogenic response (22–25).
Extracellular regulators of ENaC
Proteases
The α and γ subunits have imbedded tracts of amino acids that are inhibitory. Proteases activate ENaC by cleaving sites within the finger domains of the α and γ subunits flanking imbedded inhibitory tracts, thereby releasing these tracts and activating the channel (26–28). These inhibitory tracts are apparently not present in other ENaC/Degenerin family members, including β and δ ENaC (3). Proteolytic processing of ENaC subunits by furin occurs as newly formed channels transit through the biosynthetic pathway (28; 29). Furin is a serine protease and member of the family of proprotein convertases that resides primarily in the trans-Golgi network. Furin cleaves the α subunit twice at highly conserved RXXR↓ (Arg-X-X-Arg↓) consensus sites, releasing a 26-residue segment that contains a key 8-residue inhibitory tract (27; 29; 30). Release of this segment has been shown to transition channels from a low to moderate open probability state (26; 31), although there are numerous other factors that also influence channel open probability. In contrast, furin cleaves the γ subunit only once (29). Cleavage of the γ subunit by a number of proteases at sites distal to the furin consensus site releases a segment that contains a key 11-residue inhibitory tract, thereby transitioning channels to a high activity state (26; 32). A growing number of proteases are associated with cleavage of the γ subunit at sites distal to the furin consensus site, including prostasin, kallikrein, neutrophil and pancreatic elastase, transmembrane protease serine 4, matriptase, cathepsin B and S, as well as bacterial derived proteases (26; 33–44). For example, prostasin directs cleavage of the γ subunit at a defined polybasic (RKRK) tract in the finger domain, and a combination of furin and prostasin cleavage releases a ~43-residue peptide containing the 11-mer inhibitory tract (26; 32; 45).
Multiple lines of evidence support the notion that it is release of imbedded inhibitory tracts, rather than cleavage, per se, that is required for channel activation. First, proteolytic activation of ENaC requires cleavage at sites both preceding and following the inhibitory tracts (26; 27; 31). Cleavage at a single site is not sufficient to activate ENaC. Second, mutant channels that lack defined cleavage sites within specific subunits are inactive, whereas mutant channels that lack both defined cleavage sites as well as inhibitory tracts within specific subunits are active, even though these subunits are not cleaved (26; 27). Third, peptides corresponding to the fragments released by proteases inhibit wild type ENaC (24; 26; 27; 30; 32). Fourth, the γ subunit inhibitory tract is removed under physiological conditions when ENaC is activated (40; 45–47).
There is evidence of both α and γ ENaC subunit proteolysis in kidneys of animals exposed to a low Na+ diet or aldosterone administration (48–50). However, identifying cleaved subunit fragments of a size consistent with cleavage in the vicinity of the inhibitory tracts does not necessarily demonstrate that two cleavage events have occurred and that the inhibitory tract has been released. This is particularly relevant regarding γ subunit processing, as distinct proteases are required to release the inhibitory tract. Several studies have presented careful analyses of γ subunit molecular weights or have used antibodies directed against the γ subunit inhibitory tract. These studies provided clear evidence that cleavage distal to the 11-mer inhibitory tract has occurred (Carattino, 2014, F1080-7;Uchimura, 2012, F939-43;Svenningsen, 2009, 299-310;Zachar, 2015, 95-106}.
C-terminal α or γ subunit cleavage fragments have been reported that are significantly smaller in size than fragments associated with both channel cleavage and activation, based on studies of channels with mutations at defined cleavage sites (for example, see (47; 51–53). It is not known whether cleavage events that lead to small C-terminal fragments are associated with the release of an inhibitory tract and channel activation. Given the highly organized structure of the extracellular region, where discontinuous stretches of amino acids from the proximal and distal parts of this region come together to form the organized β sheets of the palm and β ball domains (Fig. 1), and where a disulfide bridge spans the region where cleavage has been shown to occur, it is unlikely that large fragments of the extracellular region will be released by limited protease cleavage. For example, TMPRSS4 induces cleavage of the α, β and γ subunits at a site just preceding the second transmembrane domain (TM2) of each subunit. Channels bearing mutations that blocked TMPRSS4 cleavage at sites preceding TM2 are still activated by the protease (53). As recent studies using antibodies against specific ENaC extracellular sites and mass spectrometry analyses of urinary exosomes are identifying human γ subunit fragments that are smaller in size than fragments associated with inhibitory tract release (47; 51; 54), it is important to bear in mind that subunit cleavage does not necessarily equate to channel activation (55).
Numerous in vitro studies have used Xenopus oocytes, cultured epithelial cells, and isolated CCDs to show that proteases activate ENaC (for example, see (26; 29; 33–44; 56; 57)). Furthermore, many in vivo studies have provided correlations between proteolytic processing of ENaC subunits and channel activation (for example, see (45; 46; 48; 49)), or have shown that exogenous proteases activate ENaC in perfused or microdissected tubules (58; 59). However, there are very few in vivo studies where expression of selected proteases has been blocked, providing evidence of a direct role for ENaC proteolysis in channel activation. For example, kallikrein knockout mice exhibited a blunted natriuretic response to amiloride and a reduced amount of a γ subunit cleavage product in kidney cortex (50). However, γ subunit cleavage products were observed with maneuvers to active ENaC (low Na+ diet or aldosterone administration). In addition, these mice had enhanced Na+ absorption in CCDs with no change in transepithelial voltage, suggesting activation of an electroneutral process. Activation of the H+/K+ ATPase and K+ absorption was also noted (60). Prostasin knockout mice have early mortality due to abnormal skin development (61), while a moderate effect on colonic potential difference was observed when prostasin was selectively knocked out in the colon (18). To date, the phenotype of a kidney-specific prostasin knockout has not been described. Given the large, and growing number of identified proteases that can cleave the γ subunit distal to the furin cleavage site and activate ENaC, it is perhaps not surprising that inhibiting expression of one protease would have a limited effect on ENaC activation in vivo.
Proteases often function in cascades that result in an end-effect, as is observed with blood clotting (62), and it is possible that proteolytic cascades may have roles in processing and activating ENaC. In addition, specific endogenous protease inhibitors modulate protease activity and are likely involved in the regulated proteolysis of the γ subunit of ENaC (63–65). To further complicate matters, proteases do not have to be active in order to facilitate channel activation. While prostasin induces γ subunit cleavage and activation, these effects are seen with a prostasin mutant that is catalytically inactive (45; 66). Prostasin co-immunoprecipitates with ENaC, suggesting that it is present within a complex of proteins that are involved in ENaC regulation (45). We found that catalytically inactive prostasin likely functions as a scaffold, recruiting an aprotinin-sensitive serine protease that cleaves and activates ENaC (45).
In addition to channels bearing subunits that have been processed by furin in the biosynthetic pathway, studies in cultured cells suggest that non-cleaved channels reach the plasma membrane (67). The presence of channels with non-cleaved subunits has been observed in plasma membranes of kidney lysates (48; 49; 68). Individual channels with non-cleaved subunits also lack N-glycan processing, suggesting that processing of ENaC subunits by proteases and enzymes involved in N-glycan remodeling/maturation is an all-or-none event within an individual channel complex (67).s At present, it is still unclear how channels bypass these intracellular processing events on route to the plasma membrane. Studies in Fisher rat thyroid cells suggest that increases in intracellular [Na+] enhances the likelihood that non-processed channels reach the plasma membrane (69; 70). This observation is complicated by the fact that these non-processed channels exhibit a dampened sensitivity to cleavage and activation by trypsin, suggesting subunit misfolding.
How do imbedded inhibitory tracts suppress channel activity? Kashlan et al. generated an in silico model of the extracellular region of the α subunit, based largely on homology modeling of the resolved structure of ASIC1 (7). Given the limited homology within the finger domains of ASIC and ENaC subunits, analyses of the effects of an 8-residue inhibitory peptide on specific mutants (replacing residues with Trp) within the finger and thumb domains of the α subunit were used to define distance constraints (7; 71). Inhibitory peptide-ENaC crosslinking studies confirmed key aspects of the in silico model, and suggested that the α subunit inhibitory tract lies within the periphery of the subunit at an interface between the thumb domain and an α-helix in the finger domain (7; 72). Chemically crosslinking the thumb (loop connecting the α4 and α5 helices) and finger (α1 helix) domains stabilized the channel in a low activity state, suggesting that inhibitory tracts bind to and limit the relative mobility of the thumb and finger domains, favoring a low activity state (72).
ENaC activation by proteolysis likely occurs in a variety of clinical conditions, including extracellular volume depletion, states associated with decreased effective arterial volume including heart failure, as well as other states associated with elevated levels of aldosterone (48; 49; 73). Enhanced ENaC proteolysis has been observed in airway cells derived from individuals with cystic fibrosis (64), and may occur in other states of airway or alveolar inflammation. Nephrotic syndrome is characterized by proteinuria, renal Na+ retention and edema, although in some individuals the renin-angiotensin II-aldosterone system is suppressed (74; 75). Studies of nephrotic syndrome in rodents have shown that renal Na+ retention occurs even in the absence of systemic circulating factors that activate renal Na+ transporters. The site of enhanced Na+ retention is the distal nephron, and ENaC has an important role in this process (75–77).
Plasminogen is one of the plasma proteins that is filtered by damaged glomeruli, and is converted to plasmin by urokinase that lines tubular epithelial cells (40). Plasmin can directly cleave the γ subunit distal to its inhibitory tract at a specific site and activate the channel (39; 40). Furthermore, plasmin may interact with other proteases that can cleave and activate ENaC, such as prostasin (78). Whether these proteases act in a cascade to activate ENaC, and whether prostasin functions as a scaffold to recruit plasmin to the channel is unclear. The presence of urinary plasminogen and plasmin has been consistently found in human urine in proteinuric states, including diabetic nephropathy associated with either diabetes mellitus type 1 and type 2, minimal change disease, and preeclampsia (75; 79–82). The extent of proteinuria has been shown to correlate with the extent of albuminuria in different clinical settings, and a significant correlation between urinary plasminogen/plasmin and increases in blood pressure and fluid retention has been observed (79; 82–84). At present, it is unclear whether correlations between urinary plasminogen/plasmin and increases in blood pressure and fluid retention are independent of the effects of albuminuria.
If plasmin-dependent ENaC activation has an important role in Na+ retention in proteinuric states, amiloride could be a useful therapeutic agent as it not only inhibits ENaC, it also inhibits urokinase (85). At present, there is little information regarding the efficacy of amiloride in proteinuric states in humans (86). The risk of hyperkalemia with amiloride use is a concern, as individuals with proteinuria are often receiving other drugs, including angiotensin converting enzyme inhibitors, angiotensin 2 type 1 receptor antagonists, and mineralocorticoid receptor antagonists that also increase the risk of hyperkalemia.
Extracellular Na+
ENaC not only transports Na+, it is inhibited by extracellular Na+. This phenomena, referred to as Na+ self-inhibition, was first described 40 years ago (87) and represents a reduction in channel open probability in response to external Na+ (31; 88). For selected mutants, a correlation between the magnitude of Na+ self-inhibition and channel open probability has been observed (88). Na+ self-inhibition is mediated by Na+ binding to site(s) in the extracellular region of the channel (89), which drives structural transitions that are transmitted to the channel gate. We and others have identified >90 sites in the different domains of the extracellular regions of mouse or human ENaC where amino acid substitutions altered channel activity, primarily by changing the inhibitory response to external Na+ (31; 71; 88; 90–97). The majority of these mutations likely affect allosteric transitions that occur in response to Na+ binding to the channel.
Urinary [Na+] in ASDN varies considerably under different physiological and pathological conditions. Na+ self-inhibition provides a mechanism for cells in the distal nephron to rapidly tune the rate of Na+ influx according to fluctuations of urinary [Na+]. This regulatory process will tend to limit Na+ absorption in kidney tubules when urinary [Na+] is high. Conversely, low [Na+] relieves ENaCs from Na+ self-inhibition, enhancing Na+ reabsorption and minimizing urinary Na+ losses. In other epithelia, external Na+ concentrations remain high and channels should be inhibited.
There are factors that influence the Na+ self-inhibition response, including cleavage of the α and γ subunits at defined sites in the finger domains with the release of inhibitory tracts (31), extracellular metals (other than Na+) (98), Cl− and protons (99; 100). Proteases that activate ENaC relieve channels of inhibition by external Na+ (28; 31). Non-cleaved channels exhibit a markedly enhanced Na+ self-inhibition response, and very low activity and low open probability at a single channel level when extracellular [Na+] is high. These channels transition to a higher open probability state when extracellular [Na+] is high low (Fig. 2).
Figure 2.
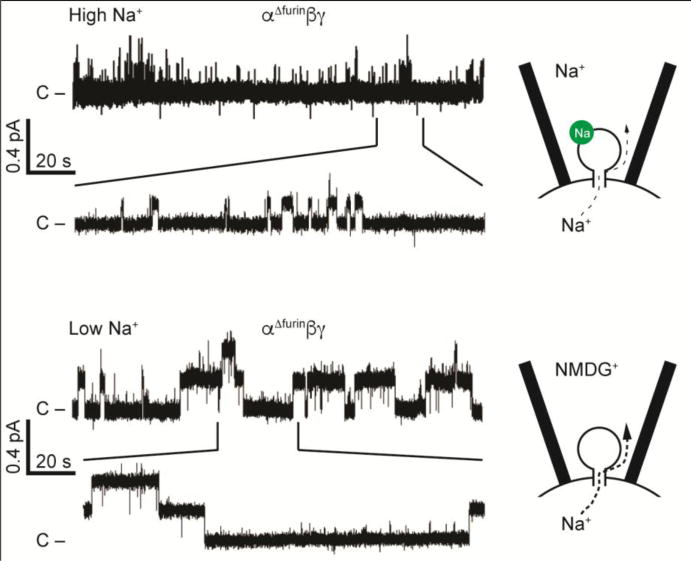
Na+ self-inhibition reduces channel activity by reducing open probability. Top: in the presence of high extracellular Na+ concentrations, Na+ (green circle) binds to a low affinity site in the extracellular domain and drives channels to strongly favor the closed state, restricting Na+ flux. Excised inside-out patch clamp recording shown, with 110 mM Na+ in the pipette, and pipette potential clamped to -100 mV to drive Na+ from the cell into the pipette. Channel mutant shown is not cleaved by furin, retains the α and γ subunit inhibitory tracts, and exhibits a strong Na+ self-inhibition response. Bottom: in the presence of a low Na+ concentration, the inhibitory site is unoccupied, and open probability and Na+ flux are higher. Patch clamp recording was performed similarly to top recording, with NMDG+ replacing Na+ in the pipette. Modified from (31).
Identifying potential Na+ binding sites is challenging, as it is difficult to experimentally distinguish sites involved in Na+ binding from sites involved in subsequent structural transitions that affect the channel gate. It is likely that most of the sites that have been shown to affect Na+ self-inhibition do so by altering structural transitions subsequent to Na+ binding. Na+ self-inhibition is dependent on the cation used to elicit the response, with Na+ being more efficacious than Li+, and K+ and N-methyl-D-glucamine (NMDG) exerting modest or no apparent inhibitory effect (89; 101). We reasoned that introducing mutations at or near an extracellular Na+ binding site should not only affect Na+ self-inhibition, but also the cation selectivity of the response.
ASIC and ENaC subunits have a localized extracellular cluster of acidic residues that could facilitate Na+ binding (4; 7; 89). In studies focusing on sites within the vicinity of this acidic cluster, we found that sites with a loop connecting the β6 and β7 strands of the α subunit, which is adjacent to the α1 helix in the finger domain, are likely involved in Na+ binding. The introduction of mutations at specific sites within this loop affected both the Na+ self-inhibition as well as the cation selectivity of the response (89). Furthermore, crosslinking the β6-β7 loop to the α1 helix reduced channel activity (89).
A growing number of human ENaC non-synonymous variants have been identified. We and other investigators have begun to examine the functional effects of these variants. It is becoming clear that some variants in the extracellular regions of ENaC subunits have a gain-of-function phenotype, reflecting a loss of Na+ self-inhibition. One gain-of-function variant, αW493R, was observed in an individual with atypical cystic fibrosis where only one CFTR allele had a disease-associated mutation (97). For others, either phenotypic information was not available (102), or numbers were too small to correlate phenotype with changes in Na+ self-inhibition (94). Future studies will determine whether ENaC variants in humans or rodents that result in a gain or loss in the Na+ self-inhibition response influence blood pressure, serum [K+], airway mucociliary clearance or other physiological parameters.
Shear Stress
ENaCs are expressed in cells lining structures that that are exposed to varying levels of shear stress. Numerous studies have found that ENaCs are activated by shear stress, and that the extracellular region has a role in sensing shear stress. Xenopus oocytes expressing ENaC respond to an increase in shear stress delivered by a perfusion pipette with a mono-exponential increase in Na+ currents that reaches plateau levels within minutes (103). The increase in current reflects an increase in channel open probability (103; 104). In isolated perfused rabbit tubules, increased tubular flow rates within a physiologically relevant range are accompanied by increases in Na+ absorption (105). As Na+ absorption is tightly coupled to K+ secretion in the ASDN and increases in flow rates enhance K+ secretion via large conductance Ca2+-activated K+ channels (106), it is not surprising that ENaCs are activated by increases in tubular flow rates.
Other groups have suggested that shear stress inhibits ENaC, due to release of ATP from cells that binds to luminal purinergic receptors (107; 108). This results in reduced cellular levels of specific acidic phospholipids and reduced ENaC activity. Shear stress also increases nitric oxide and inhibits ENaC (109). However, measurements of ENaC activity in these studies were performed under conditions where ENaC was not directly exposed to shear stress, with channel activity being assess by cell attached patch clamp or with an Ussing chamber. In contrast, when channel activity is assessed using an outside-out patch clamp configuration application of shear stress increases channel open probability (104). These observations highlight the importance of assessing ENaC activity when channels are directly exposed to shear stress. These observations, taken together, suggest that the response of ENaC to shear stress is dampened by purinergic signaling and by nitric oxide, and should be enhanced by specific purinergic receptor antagonists or by reducing nitric oxide levels.
ENaC is expressed in vascular endothelium and smooth muscle (22–25). Endothelial ENaC activation has been proposed to blunt nitric oxide release and prevent vasodilation (24). This response appears to be flow dependent, suggesting that endothelial ENaC may be functioning as a mechanosensor (110; 111). Vascular smooth muscle ENaC has also been suggested to have a role in the pressure-induced myogenic response (25).
For some mechano-activated channels, intracellular and/or extracellular tethers facilitate the transfer of mechanical forces from the extracellular matrix or intracellular cytoskeleton to the channel gate (112). This is a mechanism used by mechanosenstive channels in C. elegans that are members of the ENaC/Degenerin family and are activated in response to gentle touch (113). Other channels sense deformation or tension within membranes induced by mechanical forces that alter channel gating (112). The response of ENaC to shear stress does not appear to be dependent on changes in membrane properties (114). At present, it is unclear whether tethering ENaC subunits to the extracellular matrix and/or intracellular cytoskeleton is required for mechanosensing. One group has suggested that the extracellular matrix has a role in shear stress activation of ENaC in Xenopus oocytes and endothelium, and that specific α subunit N-glycans facilitate interactions between extracellular matrix and ENaC that are required for flow sensing (111). In contrast, our unpublished work (Kashlan et al.) indicates that ENaC N-glycans are not required for flow sensing. Furthermore, our analyses of channels with mutations within specific domains of the extracellular region suggest that specific domains have roles in ENaC mechanosensing (92; 115; 116)
Intracellular regulators of ENaC
Acidic Phospholipids
Select lipid signaling molecules are well-known regulators of ENaC activity. For example, anionic phospholipids such as PIP2 and PIP3 enhance channel open probability, presumably by binding to cationic sequences within specific ENaC subunits and inducing a conformation change that is transmitted to the channel’s gate (117–120). The CYP-epoxygenase metabolite 11,12-epoxyeicosatrienoic acid (EET) inhibits ENaC by reducing channel open probability (121).
Cys-palmitoylation
Post-translational modification by Cys-palmitoylation is another mechanism by which lipid molecules regulate ENaC (122–124). Cys-palmitoylation is a reversible attachment of palmitate to cytoplasmic Cys residues on proteins adjacent to either a cluster of basic residues, other acyl groups or transmembrane domains (125; 126). This modification can alter (i) the interaction of the palmitoylated domain with membrane lipids or associated proteins, (ii) secondary post-translational modifications such as phosphorylation, (iii) protein structure and function, (iv) membrane trafficking, stability or degradation, or (iv) a combination of these (125–129). Approximately 50 ion channels and/or their modifiers are palmitoylated, where a loss of palmitoylation alters channel function (130).
The β and γ subunits, but not the α subunit, of both mouse and human ENaC are palmitoylated (122; 123; 131). Biochemical analyses identified two sites of cytoplasmic Cys-palmitoylation in each subunit (122; 123). These palmitoylation sites are present in both the N-terminal and C-terminal cytoplasmic domains of the β subunit adjacent to the transmembrane domains, while two palmitoylation sites are localized in the N-terminal cytoplasmic domain of the γ subunit. Mutation of a single palmitoylation site in the β subunit reduced subunit palmitoylation and channel activity to a level seen with a β subunit bearing mutations of both sites (122). Similar results were observed with the γ subunit (123).
Preventing β or γ subunit palmitoylation reduced channel open probability and enhanced the Na+ self-inhibition response, while surface expression and proteolytic processing of channel subunits were unaffected (122–124). In addition, channels lacking γ subunit palmitoylation sites were significantly less active than channels lacking β subunit palmitoylation sites, but their activity was similar to channels lacking both β and γ subunit palmitoylation sites. Altogether, these data suggest that palmitoylation activates ENaC by increasing channel open probability, and that preventing γ subunit palmitoylation has a dominant role in inhibiting ENaC when compared to preventing β subunit palmitoylation (123).
Cys-palmitoylation of ENaC appears to stabilize the open state of the channel. While the structure of the extracellular and transmembrane domains of the α subunit of ENaC have been modeled based on the resolved structure of ASIC1 (1; 4–7), there is limited information regarding the structure of ENaC cytoplasmic domains. The PROF program of cascaded multiple classifiers was used to predict the secondary structure of the cytoplasmic tails of the β and γ subunits in order to understand how Cys-palmitoylation might affect the structure of the pore formed by the transmembrane domains (Fig. 3) (122; 123). There are two predicted α-helices in the N-terminal cytoplasmic domains and a single α-helix within the C-terminal cytoplasmic domains of the β and γ subunits. The C-terminal β subunit palmitoylation site is on a hydrophobic face of a α-helix, where it would be predicted to stabilize the association of the α-helix with the plasma membrane and directly impact the angle of the adjacent transmembrane domain. The N-terminal β subunit palmitoylation site is in a loop between an α-helix and the first transmembrane domain, seven residues from the transmembrane domain. The predicted structure of the γ subunit N-terminal cytoplasmic tail places one palmitoylation site at the terminus of one α-helix and the second site in the adjacent loop 12 residues from the transmembrane domain. The placement of these palmitate residues on the β and γ subunit N-terminal domains is likely to influence the channel pore and gate by altering angles of the first transmembrane domains.
Figure 3.
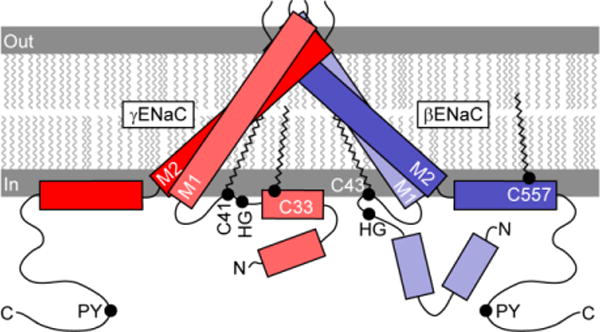
Location of palmitoylation sites on the β and γ subunit. Rectangles represent predicted α-helices in cytoplasmic domains. Rotation, angle and length of first (TM1) and second (TM2) transmembrane domains are based on crystal structure of a related channel (ASIC1), although inter-subunit distances are exaggerated. Palmitoylation of βC43, γC33 and γC41 may tether these sites to the plasma membrane, and may affect the orientation of a His-Gly (HG) motif (132). βC557 is within an amphipathic α-helix. Extracellular domains are not shown.
Previous work has suggested that the palmitoylation sites within the β and γ subunit N-termini are in a region that has an important role in regulating channel gating. These sites are adjacent to a highly conserved His-Gly (HG) motif that is present in the α, β and γ subunit, and where mutations the reduced ENaC activity, likely by reducing channel open probability (132).
While ENaC activity is reduced by blocking its modification by palmitoylation, channel activity and subunit palmitoylation are enhanced by co-expression of ENaC with a palmitoyltransferase (123; 124). Cys-palmitoylation of proteins is catalyzed by a family of 23 palmitoyltransferases in humans and mice (133; 134). These enzymes vary in size, subcellular localization and tissue expression but have a common tetraspanning transmembrane domain and a common Asp-His-His-Cys (DHHC) active site (134; 135). When mouse ENaC was co-expressed with each of the 23 mouse palmitoyltransferases (or DHHCs) in Xenopus oocytes, five were found to significantly enhance ENaC activity (DHHC 1, 2, 3, 7 and 14) (123; 124). Co-immunoprecipitation studies suggest that ENaCs form a complex with the five ENaC-activating DHHCs (124). Transcripts of these five ENaC-activating DHHCs are found in segments of that rat nephron where ENaCs are expressed (136), and in a mouse CCD cell line that expresses endogenous ENaC (124). DHHC 1, 2 and 3 are also expressed in mouse airway epithelia where ENaC has a significant role in regulating surface fluid volume and mucociliary clearance (137).
Summary
There are a growing number of extracellular and intracellular factors that influence ENaC open probability. It is possible that these factors interact in a coordinated manner to regulate channel open probability. For example, the degenerin site is a key residue in the outer vestibule on the channel pore located in the vicinity of the channel gate. Channels with specific mutations at this site (such as βS518K) reside in a high open probability state (27). As discussed above, non-cleaved channels that lack proteolytic sites reside in a very low high open probability state. Non-cleaved channels that also have a degenerin site (βS518K) mutation have intermediate activity, and at a single channel level they rapidly transition between closed and open states (Fig. 4) (27). How the regulatory factors discussed above work in a coordinated manner to modulate channel open probability requires further study using systems where specific factors can be independently altered. Furthermore, new technologies to generate mouse models with specific ENaC mutations will allow investigators to assess the roles of these regulatory factors in vivo.
Figure 4.
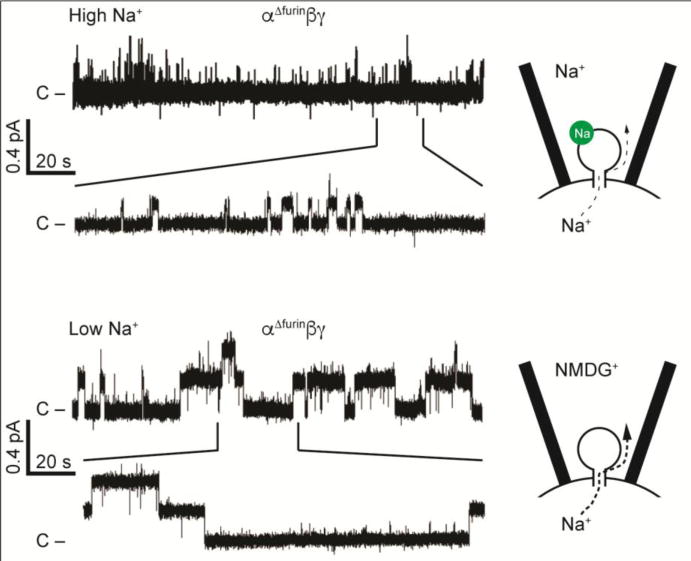
ENaC channels with both a degenerin mutation and mutated furin cleavage sites exhibit an intermediate open probability with frequent switching between open and closed states. Top: Channels with the βS518K degenerin site mutation in the pore have a high open probability and short mean closed times. Bottom: Adding in α and γ subunits with mutated furin cleavage sites that prevent subunit processing by furin reduces open probability, and the channel exhibits both short mean open times and short mean closed times. Modified from (27).
Acknowledgments
Supported by grants DK038470, DK098201 and DK079307
References
- 1.Kashlan OB, Kleyman TR. ENaC structure and function in the wake of a resolved structure of a family member. Am J Physiol Renal Physiol. 2011;301:F684–96. doi: 10.1152/ajprenal.00259.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol. 2015;10:135–46. doi: 10.2215/CJN.05760513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579:95–132. doi: 10.1016/j.gene.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–23. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 5.Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. 2009;460:599–604. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baconguis I, Bohlen CJ, Goehring A, Julius D, Gouaux E. X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na(+)-selective channel. Cell. 2014;156:717–29. doi: 10.1016/j.cell.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kashlan OB, Adelman JL, Okumura S, Blobner BM, Zuzek Z, et al. Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel alpha subunit reveals a mechanism of channel activation by proteases. J Biol Chem. 2011;286:649–60. doi: 10.1074/jbc.M110.167098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002;82:735–67. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 9.Boscardin E, Alijevic O, Hummler E, Frateschi S, Kellenberger S. The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na(+) channel (ENaC): IUPHAR Review 19. Br J Pharmacol. 2016;173:2671–701. doi: 10.1111/bph.13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eladari D, Chambrey R, Picard N, Hadchouel J. Electroneutral absorption of NaCl by the aldosterone-sensitive distal nephron: implication for normal electrolytes homeostasis and blood pressure regulation. Cell Mol Life Sci. 2014;71:2879–95. doi: 10.1007/s00018-014-1585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleyman TR, Satlin LM, Hallows KR. Opening lines of communication in the distal nephron. J Clin Invest. 2013;123:4139–41. doi: 10.1172/JCI71944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinning A, Radionov N, Trepiccione F, Lopez-Cayuqueo KI, Jayat M, et al. Double Knockout of the Na+-Driven Cl-/HCO3- Exchanger and Na+/Cl- Cotransporter Induces Hypokalemia and Volume Depletion. J Am Soc Nephrol. 2017;28:130–9. doi: 10.1681/ASN.2015070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9:2147–63. doi: 10.2215/CJN.05920613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hadchouel J, Ellison DH, Gamba G. Regulation of Renal Electrolyte Transport by WNK and SPAK-OSR1 Kinases. Annu Rev Physiol. 2016;78:367–89. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 15.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016;89:127–34. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, et al. Potassium Sensing by Renal Distal Tubules Requires Kir4.1. J Am Soc Nephrol. 2017 doi: 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandrashekar J, Kuhn C, Oka Y, Yarmolinsky DA, Hummler E, et al. The cells and peripheral representation of sodium taste in mice. Nature. 2010;464:297–301. doi: 10.1038/nature08783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malsure S, Wang Q, Charles RP, Sergi C, Perrier R, et al. Colon-specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J Am Soc Nephrol. 2014;25:1453–64. doi: 10.1681/ASN.2013090936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller RL, Denny GO, Knuepfer MM, Kleyman TR, Jackson EK, et al. Blockade of ENaCs by amiloride induces c-Fos activation of the area postrema. Brain Res. 2015;1601:40–51. doi: 10.1016/j.brainres.2014.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS, Leenen FH. Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1787–97. doi: 10.1152/ajpregu.00063.2005. [DOI] [PubMed] [Google Scholar]
- 21.Leenen FH, Hou X, Wang HW, Ahmad M. Enhanced expression of epithelial sodium channels causes salt-induced hypertension in mice through inhibition of the alpha2-isoform of Na+, K+-ATPase. Physiol Rep. 2015;3:e12383. doi: 10.14814/phy2.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang ZR, Liu HB, Sun YY, Hu QQ, Li YX, et al. Dietary salt blunts vasodilation by stimulating epithelial sodium channels in endothelial cells from salt-sensitive rats. Br J Pharmacol. 2017 doi: 10.1111/bph.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo D, Liang S, Wang S, Tang C, Yao B, et al. Role of epithelial Na+ channels in endothelial function. J Cell Sci. 2016;129:290–7. doi: 10.1242/jcs.168831. [DOI] [PubMed] [Google Scholar]
- 24.Kusche-Vihrog K, Tarjus A, Fels J, Jaisser F. The epithelial Na+ channel: a new player in the vasculature. Curr Opin Nephrol Hypertens. 2014;23:143–8. doi: 10.1097/01.mnh.0000441054.88962.2c. [DOI] [PubMed] [Google Scholar]
- 25.Grifoni SC, Chiposi R, McKey SE, Ryan MJ, Drummond HA. Altered whole kidney blood flow autoregulation in a mouse model of reduced beta-ENaC. Am J Physiol Renal Physiol. 2010;298:F285–92. doi: 10.1152/ajprenal.00496.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, et al. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma-subunit. J Biol Chem. 2007;282:6153–60. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- 27.Carattino MD, Sheng S, Bruns JB, Pilewski JM, Hughey RP, Kleyman TR. The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its alpha subunit. J Biol Chem. 2006;281:18901–7. doi: 10.1074/jbc.M604109200. [DOI] [PubMed] [Google Scholar]
- 28.Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem. 2009;284:20447–51. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, et al. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem. 2004;279:18111–4. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 30.Carattino MD, Passero CJ, Steren CA, Maarouf AB, Pilewski JM, et al. Defining an inhibitory domain in the alpha-subunit of the epithelial sodium channel. Am J Physiol Renal Physiol. 2008;294:F47–52. doi: 10.1152/ajprenal.00399.2007. [DOI] [PubMed] [Google Scholar]
- 31.Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol. 2006;290:F1488–96. doi: 10.1152/ajprenal.00439.2005. [DOI] [PubMed] [Google Scholar]
- 32.Passero CJ, Carattino MD, Kashlan OB, Myerburg MM, Hughey RP, Kleyman TR. Defining an inhibitory domain in the gamma subunit of the epithelial sodium channel. Am J Physiol Renal Physiol. 2010;299:F854–61. doi: 10.1152/ajprenal.00316.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel AB, Chao J, Palmer LG. Tissue kallikrein activation of the epithelial Na channel. Am J Physiol Renal Physiol. 2012;303:F540–50. doi: 10.1152/ajprenal.00133.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adebamiro A, Cheng Y, Rao US, Danahay H, Bridges RJ. A segment of gamma ENaC mediates elastase activation of Na+ transport. J Gen Physiol. 2007;130:611–29. doi: 10.1085/jgp.200709781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol. 2005;288:L813–9. doi: 10.1152/ajplung.00435.2004. [DOI] [PubMed] [Google Scholar]
- 36.Harris M, Firsov D, Vuagniaux G, Stutts MJ, Rossier BC. A novel neutrophil elastase inhibitor prevents elastase activation and surface cleavage of the epithelial sodium channel expressed in Xenopus laevis oocytes. J Biol Chem. 2007;282:58–64. doi: 10.1074/jbc.M605125200. [DOI] [PubMed] [Google Scholar]
- 37.Kota P, Garcia-Caballero A, Dang H, Gentzsch M, Stutts MJ, Dokholyan NV. Energetic and structural basis for activation of the epithelial sodium channel by matriptase. Biochemistry. 2012;51:3460–9. doi: 10.1021/bi2014773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Passero CJ, Mueller GM, Myerburg MM, Carattino MD, Hughey RP, Kleyman TR. TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the gamma-subunit distal to the furin cleavage site. Am J Physiol Renal Physiol. 2012;302:F1–8. doi: 10.1152/ajprenal.00330.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem. 2008;283:36586–91. doi: 10.1074/jbc.M805676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20:299–310. doi: 10.1681/ASN.2008040364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan CD, Hobbs C, Sameni M, Sloane BF, Stutts MJ, Tarran R. Cathepsin B contributes to Na+ hyperabsorption in cystic fibrosis airway epithelial cultures. J Physiol. 2014;592:5251–68. doi: 10.1113/jphysiol.2013.267286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haerteis S, Krappitz M, Bertog M, Krappitz A, Baraznenok V, et al. Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch. 2012;464:353–65. doi: 10.1007/s00424-012-1138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alli AA, Song JZ, Al-Khalili O, Bao HF, Ma HP, et al. Cathepsin B is secreted apically from Xenopus 2F3 cells and cleaves the epithelial sodium channel (ENaC) to increase its activity. J Biol Chem. 2012;287:30073–83. doi: 10.1074/jbc.M111.338574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butterworth MB, Zhang L, Heidrich EM, Myerburg MM, Thibodeau PH. Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J Biol Chem. 2012;287:32556–65. doi: 10.1074/jbc.M112.369520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carattino MD, Mueller GM, Palmer LG, Frindt G, Rued AC, et al. Prostasin interacts with the epithelial Na+ channel and facilitates cleavage of the gamma-subunit by a second protease. Am J Physiol Renal Physiol. 2014;307:F1080–7. doi: 10.1152/ajprenal.00157.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uchimura K, Kakizoe Y, Onoue T, Hayata M, Morinaga J, et al. In vivo contribution of serine proteases to the proteolytic activation of gammaENaC in aldosterone-infused rats. Am J Physiol Renal Physiol. 2012;303:F939–43. doi: 10.1152/ajprenal.00705.2011. [DOI] [PubMed] [Google Scholar]
- 47.Zachar RM, Skjodt K, Marcussen N, Walter S, Toft A, et al. The epithelial sodium channel gamma-subunit is processed proteolytically in human kidney. J Am Soc Nephrol. 2015;26:95–106. doi: 10.1681/ASN.2013111173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol. 2009;297:F1249–55. doi: 10.1152/ajprenal.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frindt G, Palmer LG. Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol. 2015;308:F572–8. doi: 10.1152/ajprenal.00585.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Picard N, Eladari D, El Moghrabi S, Planes C, Bourgeois S, et al. Defective ENaC processing and function in tissue kallikrein-deficient mice. J Biol Chem. 2008;283:4602–11. doi: 10.1074/jbc.M705664200. [DOI] [PubMed] [Google Scholar]
- 51.Nielsen MR, Frederiksen-Moller B, Zachar R, Jorgensen JS, Hansen MR, et al. Urine exosomes from healthy and hypertensive pregnancies display elevated level of alpha-subunit and cleaved alpha- and gamma-subunits of the epithelial sodium channel-ENaC. Pflugers Arch. 2017 doi: 10.1007/s00424-017-1977-z. [DOI] [PubMed] [Google Scholar]
- 52.Reihill JA, Walker B, Hamilton RA, Ferguson TE, Elborn JS, et al. Inhibition of Protease-Epithelial Sodium Channel Signaling Improves Mucociliary Function in Cystic Fibrosis Airways. Am J Respir Crit Care Med. 2016;194:701–10. doi: 10.1164/rccm.201511-2216OC. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Caballero A, Dang Y, He H, Stutts MJ. ENaC proteolytic regulation by channel-activating protease 2. J Gen Physiol. 2008;132:521–35. doi: 10.1085/jgp.200810030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi Y, Wang X, Rose KL, MacDonald WH, Zhang B, et al. Activation of the Endogenous Renin-Angiotensin-Aldosterone System or Aldosterone Administration Increases Urinary Exosomal Sodium Channel Excretion. J Am Soc Nephrol. 2016;27:646–56. doi: 10.1681/ASN.2014111137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ray EC, Kleyman TR. Cutting it out: ENaC processing in the human nephron. J Am Soc Nephrol. 2015;26:1–3. doi: 10.1681/ASN.2014060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–10. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 57.Chraibi A, Vallet V, Firsov D, Hess SK, Horisberger JD. Protease modulation of the activity of the epithelial sodium channel expressed in Xenopus oocytes. J Gen Physiol. 1998;111:127–38. doi: 10.1085/jgp.111.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morimoto T, Liu W, Woda C, Carattino MD, Wei Y, et al. Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. Am J Physiol Renal Physiol. 2006;291:F663–9. doi: 10.1152/ajprenal.00514.2005. [DOI] [PubMed] [Google Scholar]
- 59.Nesterov V, Dahlmann A, Bertog M, Korbmacher C. Trypsin can activate the epithelial sodium channel (ENaC) in microdissected mouse distal nephron. Am J Physiol Renal Physiol. 2008;295:F1052–62. doi: 10.1152/ajprenal.00031.2008. [DOI] [PubMed] [Google Scholar]
- 60.El Moghrabi S, Houillier P, Picard N, Sohet F, Wootla B, et al. Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci U S A. 2010;107:13526–31. doi: 10.1073/pnas.0913070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leyvraz C, Charles RP, Rubera I, Guitard M, Rotman S, et al. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170:487–96. doi: 10.1083/jcb.200501038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams RL, Bird RJ. Review article: Coagulation cascade and therapeutics update: relevance to nephrology. Part 1: Overview of coagulation, thrombophilias and history of anticoagulants. Nephrology. 2009;14:462–70. doi: 10.1111/j.1440-1797.2009.01128.x. [DOI] [PubMed] [Google Scholar]
- 63.Wakida N, Kitamura K, Tuyen DG, Maekawa A, Miyoshi T, et al. Inhibition of prostasin-induced ENaC activities by PN-1 and regulation of PN-1 expression by TGF-beta1 and aldosterone. Kidney Int. 2006;70:1432–8. doi: 10.1038/sj.ki.5001787. [DOI] [PubMed] [Google Scholar]
- 64.Myerburg MM, Butterworth MB, McKenna EE, Peters KW, Frizzell RA, et al. Airway surface liquid volume regulates ENaC by altering the serine protease-protease inhibitor balance: a mechanism for sodium hyperabsorption in cystic fibrosis. J Biol Chem. 2006;281:27942–9. doi: 10.1074/jbc.M606449200. [DOI] [PubMed] [Google Scholar]
- 65.Butterworth MB, Zhang L, Liu X, Shanks RM, Thibodeau PH. Modulation of the epithelial sodium channel (ENaC) by bacterial metalloproteases and protease inhibitors. PLoS One. 2014;9:e100313. doi: 10.1371/journal.pone.0100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andreasen D, Vuagniaux G, Fowler-Jaeger N, Hummler E, Rossier BC. Activation of epithelial sodium channels by mouse channel activating proteases (mCAP) expressed in Xenopus oocytes requires catalytic activity of mCAP3 and mCAP2 but not mCAP1. J Am Soc Nephrol. 2006;17:968–76. doi: 10.1681/ASN.2005060637. [DOI] [PubMed] [Google Scholar]
- 67.Hughey RP, Bruns JB, Kinlough CL, Kleyman TR. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem. 2004;279:48491–4. doi: 10.1074/jbc.C400460200. [DOI] [PubMed] [Google Scholar]
- 68.Frindt G, Ergonul Z, Palmer LG. Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol. 2008;131:617–27. doi: 10.1085/jgp.200809989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Knight KK, Wentzlaff DM, Snyder PM. Intracellular sodium regulates proteolytic activation of the epithelial sodium channel. J Biol Chem. 2008;283:27477–82. doi: 10.1074/jbc.M804176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heidrich E, Carattino MD, Hughey RP, Pilewski JM, Kleyman TR, Myerburg MM. Intracellular Na+ regulates epithelial Na+ channel maturation. J Biol Chem. 2015;290:11569–77. doi: 10.1074/jbc.M115.640763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kashlan OB, Boyd CR, Argyropoulos C, Okumura S, Hughey RP, et al. Allosteric inhibition of the epithelial Na+ channel through peptide binding at peripheral finger and thumb domains. J Biol Chem. 2010;285:35216–23. doi: 10.1074/jbc.M110.167064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kashlan OB, Kleyman TR. Epithelial Na(+) channel regulation by cytoplasmic and extracellular factors. Experimental cell research. 2012;318:1011–9. doi: 10.1016/j.yexcr.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng H, Liu X, Sharma NM, Li Y, Pliquett RU, Patel KP. Urinary Proteolytic Activation of Renal Epithelial Na+ Channels in Chronic Heart Failure. Hypertension. 2016;67:197–205. doi: 10.1161/HYPERTENSIONAHA.115.05838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meltzer JI, Keim HJ, Laragh JH, Sealey JE, Jan KM, Chien S. Nephrotic syndrome: vasoconstriction and hypervolemic types indicated by renin-sodium profiling. Ann Intern Med. 1979;91:688–96. doi: 10.7326/0003-4819-91-5-688. [DOI] [PubMed] [Google Scholar]
- 75.Ray EC, Rondon-Berrios H, Boyd CR, Kleyman TR. Sodium retention and volume expansion in nephrotic syndrome: implications for hypertension. Adv Chronic Kidney Dis. 2015;22:179–84. doi: 10.1053/j.ackd.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ichikawa I, Rennke HG, Hoyer JR, Badr KF, Schor N, et al. Role for intrarenal mechanisms in the impaired salt excretion of experimental nephrotic syndrome. J Clin Invest. 1983;71:91–103. doi: 10.1172/JCI110756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deschenes G, Wittner M, Stefano A, Jounier S, Doucet A. Collecting duct is a site of sodium retention in PAN nephrosis: a rationale for amiloride therapy. J Am Soc Nephrol. 2001;12:598–601. doi: 10.1681/ASN.V123598. [DOI] [PubMed] [Google Scholar]
- 78.Svenningsen P, Uhrenholt TR, Palarasah Y, Skjodt K, Jensen BL, Skott O. Prostasin-dependent activation of epithelial Na+ channels by low plasmin concentrations. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1733–41. doi: 10.1152/ajpregu.00321.2009. [DOI] [PubMed] [Google Scholar]
- 79.Andersen H, Friis UG, Hansen PB, Svenningsen P, Henriksen JE, Jensen BL. Diabetic nephropathy is associated with increased urine excretion of proteases plasmin, prostasin and urokinase and activation of amiloride-sensitive current in collecting duct cells. Nephrol Dial Transplant. 2015;30:781–9. doi: 10.1093/ndt/gfu402. [DOI] [PubMed] [Google Scholar]
- 80.Andersen RF, Buhl KB, Jensen BL, Svenningsen P, Friis UG, et al. Remission of nephrotic syndrome diminishes urinary plasmin content and abolishes activation of ENaC. Pediatr Nephrol. 2013;28:1227–34. doi: 10.1007/s00467-013-2439-2. [DOI] [PubMed] [Google Scholar]
- 81.Buhl KB, Friis UG, Svenningsen P, Gulaveerasingam A, Ovesen P, et al. Urinary plasmin activates collecting duct ENaC current in preeclampsia. Hypertension. 2012;60:1346–51. doi: 10.1161/HYPERTENSIONAHA.112.198879. [DOI] [PubMed] [Google Scholar]
- 82.Buhl KB, Oxlund CS, Friis UG, Svenningsen P, Bistrup C, et al. Plasmin in urine from patients with type 2 diabetes and treatment-resistant hypertension activates ENaC in vitro. J Hypertens. 2014;32:1672–7. doi: 10.1097/HJH.0000000000000216. [DOI] [PubMed] [Google Scholar]
- 83.Oxlund CS, Buhl KB, Jacobsen IA, Hansen MR, Gram J, et al. Amiloride lowers blood pressure and attenuates urine plasminogen activation in patients with treatment-resistant hypertension. J Am Soc Hypertens. 2014;8:872–81. doi: 10.1016/j.jash.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 84.Schork A, Woern M, Kalbacher H, Voelter W, Nacken R, et al. Association of Plasminuria with Overhydration in Patients with CKD. Clin J Am Soc Nephrol. 2016;11:761–9. doi: 10.2215/CJN.12261115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vassalli JD, Belin D. Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett. 1987;214:187–91. doi: 10.1016/0014-5793(87)80039-x. [DOI] [PubMed] [Google Scholar]
- 86.Andersen H, Hansen PB, Bistrup C, Nielsen F, Henriksen JE, Jensen BL. Significant natriuretic and antihypertensive action of the epithelial sodium channel blocker amiloride in diabetic patients with and without nephropathy. J Hypertens. 2016;34:1621–9. doi: 10.1097/HJH.0000000000000967. [DOI] [PubMed] [Google Scholar]
- 87.Fuchs W, Larsen EH, Lindemann B. Current-voltage curve of sodium channels and concentration dependence of sodium permeability in frog skin. J Physiol. 1977;267:137–66. doi: 10.1113/jphysiol.1977.sp011805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maarouf AB, Sheng N, Chen J, Winarski KL, Okumura S, et al. Novel determinants of epithelial sodium channel gating within extracellular thumb domains. J Biol Chem. 2009;284:7756–65. doi: 10.1074/jbc.M807060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kashlan OB, Blobner BM, Zuzek Z, Tolino M, Kleyman TR. Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J Biol Chem. 2015;290:568–76. doi: 10.1074/jbc.M114.606152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sheng S, Bruns JB, Kleyman TR. Extracellular histidine residues crucial for Na+ self-inhibition of epithelial Na+ channels. J Biol Chem. 2004;279:9743–9. doi: 10.1074/jbc.M311952200. [DOI] [PubMed] [Google Scholar]
- 91.Sheng S, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem. 2007 doi: 10.1074/jbc.M611761200. [DOI] [PubMed] [Google Scholar]
- 92.Shi S, Blobner BM, Kashlan OB, Kleyman TR. Extracellular finger domain modulates the response of the epithelial sodium channel to shear stress. J Biol Chem. 2012;287:15439–44. doi: 10.1074/jbc.M112.346551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Winarski KL, Sheng N, Chen J, Kleyman TR, Sheng S. Extracellular allosteric regulatory subdomain within the gamma subunit of the epithelial Na+ channel. J Biol Chem. 2010;285:26088–96. doi: 10.1074/jbc.M110.149963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ray EC, Chen J, Kelly TN, He J, Hamm LL, et al. Human epithelial Na+ channel missense variants identified in the GenSalt study alter channel activity. Am J Physiol Renal Physiol. 2016;311:F908–F14. doi: 10.1152/ajprenal.00426.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Edelheit O, Ben-Shahar R, Dascal N, Hanukoglu A, Hanukoglu I. Conserved charged residues at the surface and interface of epithelial sodium channel subunits--roles in cell surface expression and the sodium self-inhibition response. FEBS J. 2014;281:2097–111. doi: 10.1111/febs.12765. [DOI] [PubMed] [Google Scholar]
- 96.Collier DM, Snyder PM. Identification of epithelial Na+ channel (ENaC) intersubunit Cl- inhibitory residues suggests a trimeric alpha gamma beta channel architecture. J Biol Chem. 2011;286:6027–32. doi: 10.1074/jbc.M110.198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rauh R, Diakov A, Tzschoppe A, Korbmacher J, Azad AK, et al. A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J Physiol. 2010;588:1211–25. doi: 10.1113/jphysiol.2009.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sheng S, Perry CJ, Kleyman TR. Extracellular Zn2+ activates epithelial Na+ channels by eliminating Na+ self-inhibition. J Biol Chem. 2004;279:31687–96. doi: 10.1074/jbc.M405224200. [DOI] [PubMed] [Google Scholar]
- 99.Collier DM, Snyder PM. Extracellular protons regulate human ENaC by modulating Na+ self-inhibition. J Biol Chem. 2009;284:792–8. doi: 10.1074/jbc.M806954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Collier DM, Snyder PM. Extracellular chloride regulates the epithelial sodium channel. J Biol Chem. 2009;284:29320–5. doi: 10.1074/jbc.M109.046771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bize V, Horisberger JD. Sodium self-inhibition of human epithelial sodium channel: selectivity and affinity of the extracellular sodium sensing site. Am J Physiol Renal Physiol. 2007;293:F1137–46. doi: 10.1152/ajprenal.00100.2007. [DOI] [PubMed] [Google Scholar]
- 102.Chen J, Kleyman TR, Sheng S. Gain-of-function variant of the human epithelial sodium channel. Am J Physiol Renal Physiol. 2013;304:F207–13. doi: 10.1152/ajprenal.00563.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carattino MD, Sheng S, Kleyman TR. Epithelial Na+ channels are activated by laminar shear stress. J Biol Chem. 2004;279:4120–6. doi: 10.1074/jbc.M311783200. [DOI] [PubMed] [Google Scholar]
- 104.Althaus M, Bogdan R, Clauss WG, Fronius M. Mechano-sensitivity of epithelial sodium channels (ENaCs): laminar shear stress increases ion channel open probability. FASEB J. 2007;21:2389–99. doi: 10.1096/fj.06-7694com. [DOI] [PubMed] [Google Scholar]
- 105.Satlin LM, Sheng S, Woda CB, Kleyman TR. Epithelial Na(+) channels are regulated by flow. Am J Physiol Renal Physiol. 2001;280:F1010–8. doi: 10.1152/ajprenal.2001.280.6.F1010. [DOI] [PubMed] [Google Scholar]
- 106.Woda CB, Bragin A, Kleyman TR, Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol. 2001;280:F786–93. doi: 10.1152/ajprenal.2001.280.5.F786. [DOI] [PubMed] [Google Scholar]
- 107.Bugaj V, Sansom SC, Wen D, Hatcher LI, Stockand JD, Mironova E. Flow-sensitive K+-coupled ATP secretion modulates activity of the epithelial Na+ channel in the distal nephron. J Biol Chem. 2012;287:38552–8. doi: 10.1074/jbc.M112.408476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tarran R, Trout L, Donaldson SH, Boucher RC. Soluble mediators, not cilia, determine airway surface liquid volume in normal and cystic fibrosis superficial airway epithelia. J Gen Physiol. 2006;127:591–604. doi: 10.1085/jgp.200509468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hyndman KA, Bugaj V, Mironova E, Stockand JD, Pollock JS. NOS1-dependent negative feedback regulation of the epithelial sodium channel in the collecting duct. Am J Physiol Renal Physiol. 2015;308:F244–51. doi: 10.1152/ajprenal.00596.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, et al. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ Res. 2016;118:935–43. doi: 10.1161/CIRCRESAHA.115.308269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Knoepp F, Ashley Z, Barth D, Kazantseva M, Szczesniak PP, et al. Mechanical activation of epithelial Na+ channel relies on an interdependent activity of the extracellular matrix and extracellular N-glycans of αENaC. bioRxiv 2017 [Google Scholar]
- 112.Hamill OP, Martinac B. Molecular basis of mechanotransduction in living cells. Physiol Rev. 2001;81:685–740. doi: 10.1152/physrev.2001.81.2.685. [DOI] [PubMed] [Google Scholar]
- 113.Syntichaki P, Tavernarakis N. Genetic models of mechanotransduction: the nematode Caenorhabditis elegans. Physiol Rev. 2004;84:1097–153. doi: 10.1152/physrev.00043.2003. [DOI] [PubMed] [Google Scholar]
- 114.Carattino MD, Liu W, Hill WG, Satlin LM, Kleyman TR. Lack of a role of membrane-protein interactions in flow-dependent activation of ENaC. Am J Physiol Renal Physiol. 2007;293:F316–24. doi: 10.1152/ajprenal.00455.2006. [DOI] [PubMed] [Google Scholar]
- 115.Shi S, Carattino MD, Kleyman TR. Role of the wrist domain in the response of the epithelial sodium channel to external stimuli. J Biol Chem. 2012;287:44027–35. doi: 10.1074/jbc.M112.421743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shi S, Ghosh DD, Okumura S, Carattino MD, Kashlan OB, et al. Base of the thumb domain modulates epithelial sodium channel gating. J Biol Chem. 2011;286:14753–61. doi: 10.1074/jbc.M110.191734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yue G, Malik B, Eaton DC. Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J Biol Chem. 2002;277:11965–9. doi: 10.1074/jbc.M108951200. [DOI] [PubMed] [Google Scholar]
- 118.Ma HP, Eaton DC. Acute regulation of epithelial sodium channel by anionic phospholipids. J Am Soc Nephrol. 2005;16:3182–7. doi: 10.1681/ASN.2005040434. [DOI] [PubMed] [Google Scholar]
- 119.Pochynyuk O, Staruschenko A, Tong Q, Medina J, Stockand JD. Identification of a functional phosphatidylinositol 3,4,5-trisphosphate binding site in the epithelial Na+ channel. J Biol Chem. 2005;280:37565–71. doi: 10.1074/jbc.M509071200. [DOI] [PubMed] [Google Scholar]
- 120.Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens. 2008;17:533–40. doi: 10.1097/MNH.0b013e328308fff3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, et al. Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol. 2004;124:719–27. doi: 10.1085/jgp.200409140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mueller GM, Maarouf AB, Kinlough CL, Sheng N, Kashlan OB, et al. Cys palmitoylation of the beta subunit modulates gating of the epithelial sodium channel. J Biol Chem. 2010;285:30453–62. doi: 10.1074/jbc.M110.151845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mukherjee A, Mueller GM, Kinlough CL, Sheng N, Wang Z, et al. Cysteine palmitoylation of the gamma subunit has a dominant role in modulating activity of the epithelial sodium channel. J Biol Chem. 2014;289:14351–9. doi: 10.1074/jbc.M113.526020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mukherjee A, Wang Z, Kinlough CL, Poland PA, Marciszyn AL, et al. Specific Palmitoyltransferases Associate with and Activate the Epithelial Sodium Channel. J Biol Chem. 2017;292:4152–63. doi: 10.1074/jbc.M117.776146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nadolski MJ, Linder ME. Protein lipidation. FEBS J. 2007;274:5202–10. doi: 10.1111/j.1742-4658.2007.06056.x. [DOI] [PubMed] [Google Scholar]
- 126.Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 127.Shipston MJ. Ion channel regulation by protein palmitoylation. J Biol Chem. 2011;286:8709–16. doi: 10.1074/jbc.R110.210005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yeste-Velasco M, Linder ME, Lu YJ. Protein S-palmitoylation and cancer. Biochim Biophys Acta. 2015;1856:107–20. doi: 10.1016/j.bbcan.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 129.Chamberlain LH, Shipston MJ. The physiology of protein S-acylation. Physiol Rev. 2015;95:341–76. doi: 10.1152/physrev.00032.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shipston MJ. Ion channel regulation by protein S-acylation. J Gen Physiol. 2014;143:659–78. doi: 10.1085/jgp.201411176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mueller GM, Yan W, Copelovitch L, Jarman S, Wang Z, et al. Multiple residues in the distal C terminus of the alpha-subunit have roles in modulating human epithelial sodium channel activity. Am J Physiol Renal Physiol. 2012;303:F220–8. doi: 10.1152/ajprenal.00493.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, et al. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J. 1997;16:899–907. doi: 10.1093/emboj/16.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Korycka J, Lach A, Heger E, Boguslawska DM, Wolny M, et al. Human DHHC proteins: a spotlight on the hidden player of palmitoylation. Eur J Cell Biol. 2012;91:107–17. doi: 10.1016/j.ejcb.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 134.Tsutsumi R, Fukata Y, Fukata M. Discovery of protein-palmitoylating enzymes. Pflugers Arch. 2008;456:1199–206. doi: 10.1007/s00424-008-0465-x. [DOI] [PubMed] [Google Scholar]
- 135.Ohno Y, Kihara A, Sano T, Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim Biophys Acta. 2006;1761:474–83. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 136.Lee JW, Chou CL, Knepper MA. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes. J Am Soc Nephrol. 2015;26:2669–77. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–5. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]