

Abstract
In spite of numerous studies on the involvement of fibrinogen in transendothelial migration of leukocytes and thereby inflammation, there is still no clear understanding of which fibrin(ogen) species can stimulate leukocyte transmigration. Although we have previously proposed that interaction of fibrin with the VLDL receptor (VLDLR) promotes leukocyte transmigration, there is no direct experimental evidence for the involvement of fibrin in this process. To address these questions, we performed systematic studies of interaction of VLDLR with fibrinogen, fibrin, and their isolated recombinant BβN- and βN-domains, respectively, and the effect of various fibrin(ogen) species on transendothelial migration of leukocytes. The results obtained revealed that freshly purified fibrinogen does not interact with VLDLR in solution and has practically no effect on leukocyte transmigration. They also indicate that the VLDLR-binding site is cryptic in fibrinogen and becomes accessible upon its adsorption onto a surface or upon its conversion into fibrin. We also found that the D-D:E1 complex and higher molecular mass fibrin degradation products, as well as soluble fibrin and fibrin polymers (clots) anchored to the endothelial monolayer, promote leukocyte transmigration mainly through the VLDL receptor-dependent pathway. Thus, the results of the present study suggest that fibrin degradation products and soluble fibrin that may be present in the circulation in vivo, as well as fibrin clots that may be deposited on the surface of inflamed endothelium, promote leukocyte transmigration. These findings further clarify the molecular mechanisms underlying the fibrin-VLDLR-dependent pathway of leukocyte transmigration and provide an explanation for a possible (patho)physiological role of this pathway.
Keywords: Fibrinogen, Fibrin, VLDL receptor, Endothelium, Leukocyte transmigration
1. Introduction
Fibrinogen is the major plasma protein that plays an important role in hemostasis. Fibrinogen molecule consists of two identical sub-units each of which is composed of three non-identical polypeptide chains, Aα, Bβ, and γ [1]. The N-terminal portions of all six chains linked together by a number of disulfide bonds form the central part of the molecule, which was originally termed the N-terminal disulfide knot (NDSK) [2]. The central part of fibrinogen, also called the E region [3], is released from the molecule upon digestion with plasmin or CNBr in the form of E fragment or NDSK fragment, respectively [1]. Digestion of fibrinogen with plasmin also produces the D fragment derived from two identical terminal D regions of the molecule [1,3]. The N-terminal portions of the Aα and Bβ chains contain fibrinopeptides A and B including amino acid residues 1–16 and 1–14, respectively [1,3]. Upon vascular injury, when the blood coagulation cascade is activated resulting in generation of thrombin, the later converts fibrinogen into fibrin by proteolytic removal of its fibrinopeptides. Fibrin molecules spontaneously polymerize through the interaction between their newly exposed polymerization sites of the central region including knobs “A” and “B” and complementary holes “a” and “b” of the D regions [3]. The resultant fibrin polymer, which is reinforced by covalent crosslinking with factor XIIIa [4], seals damaged vasculature thereby preventing the loss of blood. Crosslinked fibrin polymers (clots) can be dissolved by proteolytic cleavage with plasmin resulting in soluble fibrin degradation products.
Besides its prominent role in hemostasis, fibrinogen participates in various physiological and pathological processes including inflammation. For example, it was shown that fibrinogen is required for efficient inflammatory responses in vivo [5], fibrin deposition contributes to the pathogenesis of intraabdominal abscess formation [6], and fibrin(ogen) mediates acute inflammatory response to implanted biomaterials [7]. Recruitment of leukocytes from the circulation to sites of inflammation is an integral part of the inflammatory response and transendothelial migration of leukocytes is a key step in such recruitment [8–10]. Numerous data suggest that fibrin(ogen) plays a prominent role in leukocyte transmigration and thereby inflammation; however, the proposed mechanisms of fibrin(ogen)-mediated leukocyte transmigration raise a number of questions. For example, it was suggested that binding of fibrinogen to vascular cell receptors mediates a specific pathway of cell-to-cell adhesion by bridging together leukocytes and endothelial cells [11]. Further, it was proposed that such fibrinogen-mediated intercellular bridging, which occurs through the interaction of fibrinogen with the leukocyte receptor Mac-1 and endothelial cell receptor ICAM-1, promotes transendothelial migration of leukocytes [12,13]. However, subsequent studies revealed that Mac-1 binding sites are cryptic in soluble fibrinogen [14,15]. This raises a question of how fibrinogen in the circulation may interact with Mac-1 and promote leukocyte transmigration in the proposed (Mac-1)-fibrinogen-(ICAM-1)-dependent intercellular bridging pathway.
Another suggested mechanism of fibrinogen-dependent leukocyte transmigration involves fibrin-derived products [16]. Specifically, it was shown in the in vitro experiments that fibrin-derived NDSK-II fragment, which corresponds to the central part of the fibrin molecule, promotes leukocyte transmigration and was proposed that NDSK-II bridges leukocytes to the endothelium through the interaction with endothelial receptor VE-cadherin and leukocyte integrin receptor CD11c [16]. It was also suggested that in vivo leukocyte transmigration is stimulated by fibrin degradation product E1 fragment, which is an analog of the NDSK-II fragment [16,17]. Thus, the proposed (CD11c)-E1 fragment-(VE-cadherin)-dependent intercellular bridging pathway of leukocyte transmigration utilizes the ability of the E1 fragment to interact simultaneously with CD11c and VE-cadherin through its N-terminal α chain sequence Gly17-Pro-Arg and β chain sequence 15–42, respectively [16,17]. However, upon fibrin degradation, the E1 fragment is released only in a complex with the D-D fragment (D-D:E1 complex) [18,19] in which the αGly17-Pro-Arg sequences are not accessible. This raises a question of whether such D-D:E1 complex or larger soluble FDP can promote leukocyte transmigration through the proposed (CD11c)-E1 fragment-(VE-cadherin)-dependent bridging pathway.
A third suggested pathway of fibrinogen-dependent leukocyte transmigration involves fibrin. Namely, we found that fibrin interacts with the very low density lipoprotein receptor (VLDL receptor, VLDLR) through a pair of fibrin βN-domains, each consisting of the β chain residues 15–66, and demonstrated that RAP, an antagonist of ligand binding of VLDLR and other low density lipoprotein (LDL) receptor family members, inhibits transendothelial migration of leukocytes [20]. Based on these and some other findings, we proposed that fibrin promotes leukocyte transmigration through a fibrin-VLDLR-dependent pathway [20]. However, to stimulate leukocyte transmigration in our in vitro experiments we used NDSK-II as a simple soluble model of fibrin [20,21], which, in fact, resembles more closely fibrin degradation product E1 fragment. Therefore, there is still no direct experimental evidence that fibrin can stimulate leukocyte transmigration through the proposed fibrin-VLDLR-dependent pathway. Furthermore, a well-established fact that fibrin βN-domains contain secondary polymerization sites (knobs “B”) that interact with the complementary sites (holes ‘b”) of the D regions upon fibrin polymerization [3] raises a question of whether their VLDLR-binding sites are accessible in fibrin polymers. Moreover, our recent study revealed that the VLDLR-binding sites are mainly located in the C-terminal halves of fibrin βN-domains [22], i.e. they may be distant from fibrinopeptides B that are removed upon conversion of fibrinogen into fibrin. This raises another question of whether these sites are accessible for interaction with VLDLR in the fibrinogen molecule.
Thus, in spite of numerous studies on the involvement of fibrin(ogen) in transendothelial migration of leukocytes, there is still no clear evidence that fibrinogen or fibrin can promote leukocyte transmigration. It is also unclear which fibrin degradation product(s) may play a role in this process. To address the questions mentioned above, we performed systematic studies of interaction of fibrinogen, fibrin, and fibrin degradation products with the VLDL receptor, as well as the effect of these fibrin(ogen) species on transendothelial migration of leukocytes.
2. Materials and methods
2.1. Proteins, antibodies, and reagents
Human fibrinogen depleted of plasminogen, von Willebrand factor and fibronectin (FIB 3), human plasmin, human thrombin, and HRP-conjugated sheep anti-human fibrinogen antibodies were purchased from Enzyme Research Laboratories. The soluble form of human VLDL receptor, sVLDLR, was prepared using the Drosophila Expression System (Invitrogen) as previously described [23]. The recombinant fibrin(ogen) (Bβ1–66)2 and (β15-66)2 fragments were produced in E. coli and purified as we described earlier [23]. Human receptor-associated protein (RAP) was expressed in E. coli and purified as described in [24]. Anti-VLDL receptor monoclonal antibodies mAb 5F3 and mAb 1H10 were purified from hybridoma supernatants by affinity chromatography on Protein A-Sepharose (Sigma-Aldrich) as we described earlier [21]. Goat secondary anti-mouse polyclonal antibodies conjugated with HRP and HRP substrate SureBlue TMB were from KPL. Calcein AM fluorescent dye and phorbol 12-myristate 13-acetate (PMA) were obtained from BD Biosciences and Promega, respectively, and Gly-Pro-Arg-Pro peptide and N-formyl-Met-Leu-Phe (fMLP) were from Sigma-Aldrich.
2.2. Preparation of acidic fibrin monomer, soluble fibrin, and fibrin(ogen) degradation products
Acidic fibrin monomer was prepared by dissolving non-crosslinked human fibrin in 0.125% acetic acid as described previously [25–27]. To prepare Alexa 488-labeled fibrin monomer, fibrinogen was mixed with Alexa Fluor 488 dye (ThermoFisher Scientific) at 1 to 20 molar ratio, as recommended by the manufacturer, and incubated for 1 h at room temperature. Unconjugated dye was removed on NAP-25 column Sephadex G-25 (GE Healthcare) and Alexa 488-labeled fibrinogen was converted into acidic Alexa 488-labeled fibrin monomer as described above. Soluble fibrin was prepared by rapid dilution of acidic fibrin monomer at 20 μM with Iscove’s modified Dulbecco’s medium (IMDM, Invitrogen) containing fibrinogen-derived D1 fragment to final fibrin concentration of 1.5 μM and final fibrin to D1 molar ratio of 1 to 4.
Fibrinogen-derived D1 and E3 fragments were prepared from plasmin digest of fibrinogen as described earlier [28]. The NDSK-II fragment was prepared from CNBr digest of fibrinogen followed by cleavage of its fibrinopeptides from NDSK with thrombin-agarose as described [29,30]. Fibrin-derived D-D:E1 complex and dimeric D-D fragment were prepared from plasmin digest of crosslinked fibrin using procedures described earlier [28,31]. The high molecular mass fibrin degradation products (HMM-FDP) were prepared from plasmin digest of crosslinked fibrin using the same procedures [28,31] with some modifications. Briefly, plasmin was added to crosslinked fibrin at 1 to 2000 molar ratio in Tris-buffered saline (TBS) containing 10 mM CaCl2 and incubated for 1 h at room temperature. The reaction was terminated by PMSF and aprotinin, and plasmin was removed from the digest on Lys-Sepharose column (GE Healthcare). SDS-PAGE analysis of the early plasmin digest of crosslinked fibrin polymers revealed that the major degradation products were the E1 fragment, D-D dimer, and HMM-FDP (Fig. 1, inset). Subsequent fractionation of this digest by size-exclusion chromatography on Superdex-200 column revealed two major peaks, peak 1 was eluted with the free volume before fibrinogen and peak 2 was eluted between fibrinogen and the D-D fragment (Fig. 1). SDS-PAGE analysis of each of these peaks revealed the presence of HMM-FDP in the first peak and the D-D dimer and E1 fragment in the second peak (Fig. 1, inset). Subsequent analysis of the second peak by native electrophoresis, as we described previously [31], and by re-chromatography on the same column revealed that the D-D dimer and E1 fragment are present exclusively in the form of a non-covalent D-D:E1 complex. This complex was very stable since no dissociation was observed upon its storage in solution at 4 °C for more than a year. In contrast, SDS-PAGE analysis of the prolonged plasmin digest revealed two major terminal fibrin degradation products, the D-D dimer and E3 fragment (not shown) that were purified from the digest as previously described [28,31] and analyzed by SDS-PAGE (Fig. 1, inset).
Fig. 1.
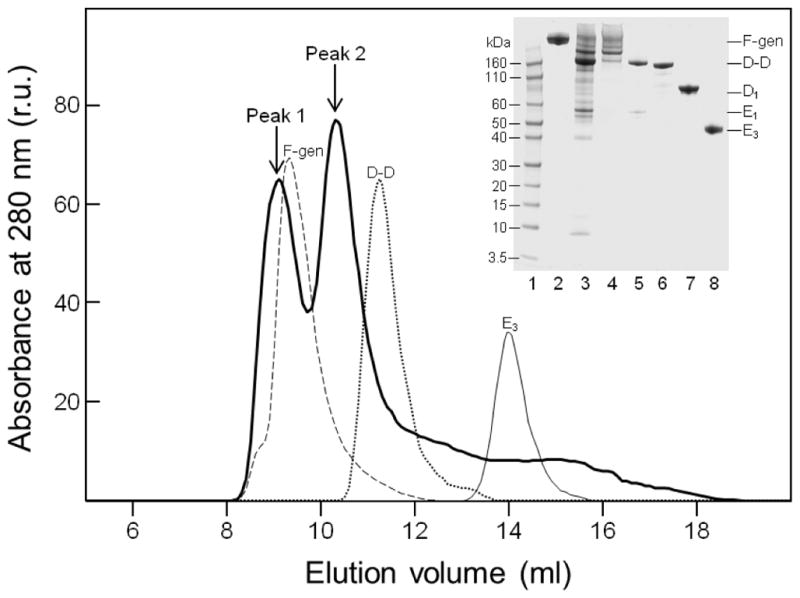
Size-exclusion chromatography and SDS-PAGE analysis of fibrin(ogen) degradation products. Solid thick line shows elution profile of the early plasmin digest of crosslinked fibrin on a Superdex-200 column; elution profiles of fibrinogen (broken line), D-D dimer (dotted line), and E3 fragment (thin line) are shown for comparison. Inset shows SDS-PAGE analysis of the early plasmin digest of crosslinked fibrin (lane 3) and peak 1 and peak 2 fractions (lanes 4 and 5, respectively); the D-D dimer, D1 fragment, and E3 fragment are in lanes 6, 7, and 8, respectively. Novex Sharp Prestained protein markers of the indicated molecular mass and freshly purified fibrinogen are in lanes 1 and 2, respectively.
2.3. Solid-phase binding assay
Binding of sVLDLR to immobilized fibrinogen, fibrin, and the (Bβ1-66)2 and (β15-66)2 fragments was tested using enzyme-linked immunosorbent assay (ELISA), bound sVLDLR was detected with anti-VLDLR mAb 5F3, and the equilibrium dissociation constant (Kd) values were determined as described previously [20–23]. To study binding of fibrinogen and fibrin to immobilized sVLDLR, wells of Immulon 2HB microtiter plates were coated with sVLDLR at 1 μg/mL in 0.1 M Na2CO3, pH 9.5 (coating buffer) overnight at 4 °C. The wells were then blocked with Blocker BSA in TBS (ThermoFisher Scientific) for 1 h at room temperature. Following washing with TBS containing 0.05% Tween 20 and 1 mM CaCl2 (binding buffer), fibrinogen or fibrin, which was solubilized in 5 mM Gly-Pro-Arg-Pro peptide as described earlier [28], each at 1 μM in the binding buffer, were added to the wells and incubated for 1 h at 37 °C. Bound fibrinogen and fibrin were detected by reaction with the HRP-conjugated sheep anti-human fibrinogen antibodies. The peroxidase substrate SureBlue TMB was added and color intensity was measured spectrophotometrically at 450 nm.
2.4. Surface plasmon resonance
Interaction of fibrinogen and the (Bβ1-66)2 and (β15-66)2 fragments with immobilized sVLDLR was studied by surface plasmon resonance (SPR) using the BIAcore 3000 biosensor (Biacore AB) and the Kd values were determined as we described previously [20].
2.5. Cell culture and treatments
Human umbilical vein endothelial cells (HUVECs) purchased from Lonza were cultured in EBM-2 basal medium supplemented with EGM SingleQuot Kit (Lonza), which contained 10% FBS, according to the manufacturer’s instruction and were used at passage 4–6. The HL-60 human promyelocytic cell line (ATCC) was cultured and differentiated to a neutrophil-like lineage as described earlier [20,21,30,32]. Differentiated HL-60 cells were labeled with Calcein AM fluorescent dye in serum-free IMDM as recommended by the manufacturer. All cell cultures were maintained at 37 °C in 5% CO2.
2.6. Leukocyte transendothelial migration assay
Transendothelial migration experiments were performed with 24-well plates containing 8-μm pore size PET membrane inserts (BD Biosciences) as we described earlier [20,21,30]. Briefly, HUVECs were grown to confluence on the insert membranes pre-coated with 0.1% gelatin and serum-starved for 2 h before experiments. Calcein AM-labeled differentiated HL-60 cells were stimulated with PMA. Stimulated HL-60 cells in IMDM containing 1.5 μM NDSK-II, fibrinogen, soluble fibrin, or fibrin-derived species were added on top of HUVEC monolayers. The inserts were placed into the wells containing chemoattractant fMLP, transmigration proceeded for 4 h, and migrated cells were quantified with fluorescence plate reader. The experiments with endothelium-anchored fibrin polymers (clots) were performed in a similar manner. To prepare fibrin clots deposited on endothelial monolayers, acidic fibrin monomer was neutralized by mixing with IMDM to final concentration of 50 nM, the mixture was added to confluent HUVECs and incubated in CO2 incubator for 30 min at 37 °C to allow fibrin to polymerize and anchor to the cells. Then HUVEC monolayers were washed twice with IMDM prior to the use in the experiments.
2.7. Fluorescence microscopy
To confirm the presence of endothelium-anchored fibrin clots on HUVEC monolayer, the inserts with Alexa 488-labeled fibrin clots deposited on HUVECs were washed with Phosphate-buffered saline (PBS) and fixed with solution containing 4% formaldehyde and 5% sucrose in PBS for 20 min at room temperature. The fixed cells were washed with PBS. The images of HUVEC monolayers with anchored Alexa 488-fibrin clots and Calcein AM-labeled differentiated HL-60 cells were captured using EVOS FL Auto microscope (Life Technologies).
2.8. Statistical analysis
Statistical analysis was done using Student’s t-test with a P value of less than 0.05 being considered significant. All statistical analyses were performed in SigmaPlot 13.0 software (Systat Software).
3. Results
3.1. Interaction of fibrinogen and isolated fibrinogen BβN-domains with the VLDL receptor
Our recent finding that the VLDL receptor-binding sites are located mainly within the C-terminal halves of a pair of fibrinogen BβN-domains [22] suggested that these sites may be accessible for VLDLR, i.e. that the later may interact with fibrinogen. To test this suggestion, we studied interaction of soluble extracellular portion of the VLDL receptor (sVLDLR) with fibrinogen and fibrin, and with the recombinant (Bβ1-66)2 and (β15-66)2 fragments representing a pair of fibrinogen BβN-domains and fibrin βN-domains, respectively.
In ELISA, when increasing concentrations of sVLDLR were added to immobilized fibrinogen or fibrin, it exhibited a dose-dependent binding to both species (Fig. 2A). The Kd values were found to be 7.9 and 7.2 nM for fibrinogen and fibrin, respectively. Similarly, when increasing concentrations of sVLDLR were added to immobilized (Bβ1-66)2 or (β15-66)2, sVLDLR bound efficiently to both fragments (Fig. 2B). The Kd values were found to be 7.7 and 5.9 nM for (Bβ1-66)2 and (β15-66)2, respectively, i.e. very close to those determined for fibrinogen and fibrin. The results of these experiments clearly indicate that the VLDL receptor interacts with immobilized fibrinogen and its recombinant BβN-domains.
Fig. 2.

ELISA-detected binding of sVLDLR to immobilized fibrinogen and fibrin (A) and their recombinant (Bβ1-66)2 and (β15-66)2 fragments (B). Increasing concentrations of sVLDLR were incubated with microtiter wells coated with freshly purified fibrinogen or fibrin (A) and with the (Bβ1-66)2 or (β15-66)2 fragments (B), and bound sVLDLR was detected with anti-VLDLR mAb 5F3, as described in Materials and methods. The curves are representative of three independent experiments; error bars represent the standard deviation of triplicate determinations.
In another set of ELISA experiments we tested interaction of fibrinogen and solubilized fibrin with immobilized sVLDLR (Fig. 3). When fibrinogen at 1 μM was added to immobilized sVLDLR, very little binding was observed. In contrast, 1 μM fibrin kept in solution by 5 mM Gly-Pro-Arg-Pro peptide exhibited considerable binding, which was inhibited by RAP, a specific inhibitor of this interaction. This finding was confirmed in SPR experiments in which fibrinogen added at 1 μM exhibited practically no binding to sVLDLR receptor immobilized on the surface of a sensor chip (Fig. 4A). In contrast, the (Bβ1-66)2 and (β15-66)2 fragments both exhibited prominent binding to immobilized sVLDLR. The Kd value for the interaction of (β15-66)2 with sVLDLR was found to be about 4 nM (not shown), i.e. close to that determined by ELISA in this and our previous studies [23]. At the same time, the Kd value for (Bβ1-66)2-sVLDLR interaction was found to be 254 nM (Fig. 4A, inset), i.e. significantly higher than that determined by ELISA. Such a discrepancy may be connected with the fact that the state of binding partners in our ELISA and SPR experiments was different. Namely, in ELISA the (Bβ1-66)2 fragment was immobilized on the surface of microtiter plates and, therefore, had fixed conformation while in SPR it was in solution and, thus, preserved its flexibility. Whatever the reason for such a discrepancy, these results clearly indicate that the VLDLR-binding site is accessible in the (Bβ1-66)2 fragment.
Fig. 3.
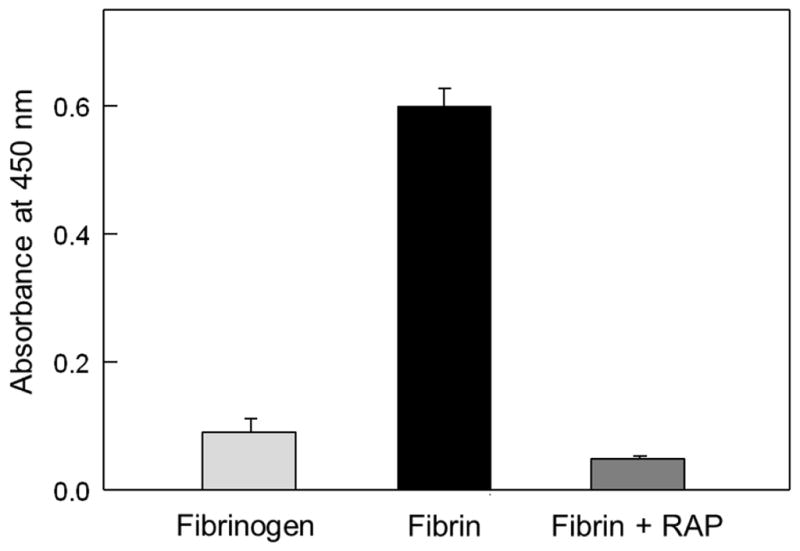
ELISA-detected binding of fibrinogen or solubilized fibrin to immobilized sVLDLR. Freshly purified fibrinogen at 1 μM or 1 μM fibrin solubilized in the presence of 5 mM Gly-Pro-Arg-Pro peptide, without and with 50 nM RAP, was incubated with microtiter wells coated with sVLDLR, and bound fibrinogen or fibrin was detected as described in Materials and methods. The bars are representative of two independent experiments; error bars represent the standard deviation of triplicate determinations.
Fig. 4.

SPR-detected interaction of fibrinogen and the (Bβ1-66)2 and (β15-66)2 fragments with sVLDLR. (A) Freshly purified fibrinogen (F-gen) at 1 μM or its recombinant (Bβ1-66)2 fragment at 250 nM were added to immobilized sVLDLR and their association/dissociation was monitored in real time; the association/dissociation of the (β15-66)2 fragment at 25 nM added to sVLDLR is shown for comparison. The inset shows analysis of interaction of the (Bβ1-66)2 fragment with immobilized sVLDLR by SPR. The (Bβ1-66)2 fragment at increasing concentrations, 10, 25, 50, 100, and 250 nM, was added to immobilized sVLDLR and its association/dissociation was monitored in real time. (B) Non-fractionated fibrinogen (F-gen n-f) and freshly purified fibrinogen (F-gen), both at 1 μM, were added to immobilized sVLDLR and their association/dissociation was monitored in real time. The inset shows size-exclusion chromatography of fibrinogen performed on a Superdex-200 column. The fraction of freshly purified monomeric fibrinogen used in all experiments of the present study is shown in gray.
It should be noted that our previous experiments with commercially available fibrinogens from different sources kept either frozen or lyophilized revealed that such fibrinogens contain various amounts of aggregated material which was physiologically active. Therefore, we analyzed fibrinogen used in the present study by size-exclusion chromatography on Superdex-200 column. The analysis revealed the presence of aggregated material that eluted with free volume and appeared as a small left shoulder on the major elution peak (Fig. 4B, inset). Although the amount of aggregates was quite small, non-fractionated fibrinogen exhibited significant binding to immobilized sVLDLR in our preliminary SPR experiments (Fig. 4B, broken curve). To avoid the influence of fibrinogen aggregates on the experimental results, fibrinogen was passed through Superdex-200 column and only freshly prepared samples from the major peak containing only monomeric fibrinogen (Fig. 4B, inset) were used in all experiments described in the present study.
Altogether, the experiments described above indicate that immobilized fibrinogen interacts with the VLDL receptor with high affinity while fibrinogen in solution exhibited practically no interaction with this receptor. In contrast, (Bβ1-66)2 representing isolated fibrinogen BβN-domains interacts with VLDLR being in solution or immobilized. These findings suggest that the VLDLR-binding sites are cryptic in fibrinogen and become available for the interaction only after fibrinogen is aggregated, adsorbed onto a surface, or converted into fibrin.
3.2. Effect of fibrinogen on transendothelial migration of leukocytes
Since our binding experiments suggested that the VLDLR-binding sites are cryptic in fibrinogen, we hypothesized that fibrinogen in solution should not stimulate transendothelial migration of leukocytes. To test this hypothesis, we studied the effect of freshly purified monomeric fibrinogen (Fig. 4B, inset) on leukocyte transmigration using in vitro leukocyte transendothelial migration assay. In this assay, when HUVECs were grown to confluency on the surface of a porous membrane separating lower and upper chambers, Calcein AM-labeled leukocytes (HL-60 cells differentiated into neutrophil-like cells) were added to the upper chamber, and leukocytes migrated to the lower chamber containing chemoattractant fMLP (Fig. 5, control). Note that differentiated HL-60 cells were shown to constitute a valid model system for the analysis of human neutrophil (leukocyte) migration [33]. When leukocytes were added in the presence of fibrin-derived NDSK-II, which was previously shown to stimulate leukocyte transmigration [16,30], the number of migrated leukocytes in the lower chamber was increased by more than two-fold (Fig. 5, NDSK-II). At the same time, when freshly purified fibrinogen was added to leukocytes instead of NDSK-II, only a very small increase (by 17%) was observed (Fig. 5, F-gen). These experiments indicate that contribution of freshly purified fibrinogen to leukocyte transmigration is very small in comparison with that of NDSK-II, in agreement with our hypothesis.
Fig. 5.
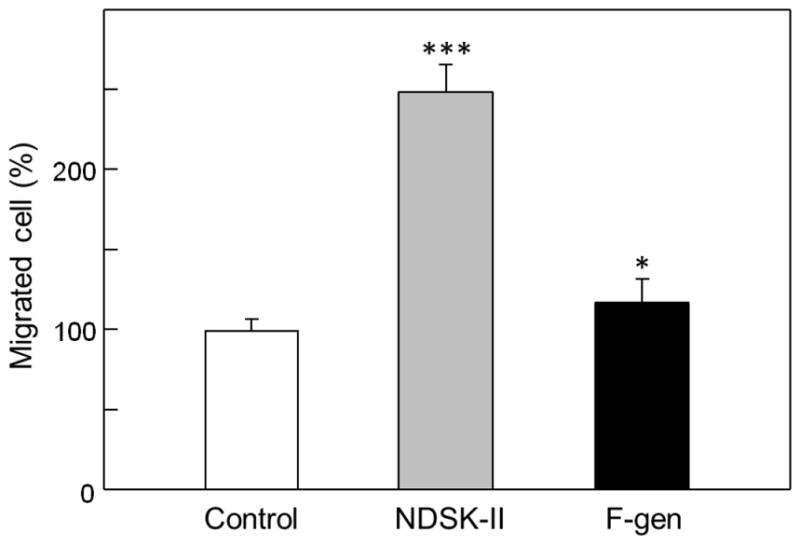
Effect of fibrinogen on transendothelial migration of leukocytes. HUVECs were grown to confluence on gelatin-coated cell culture inserts. Calcein AM-labeled HL-60 cells differentiated into neutrophil-like cells were added to the upper chambers on top of the HUVEC monolayers in the presence of PBS, NDSK-II, or freshly purified fibrinogen (F-gen), each at 1.5 μM. The cells were allowed to migrate into the lower chambers containing chemoattractant fMLP for 4 h at 37°C, collected, and quantified as described in Materials and methods. The results are expressed as percentage of HL-60 cells migrated in the presence of PBS (control). The graph shows combined data obtained from three independent experiments performed in triplicate; error bars denote means ± SD. *P < 0.05; ***P < 0.001.
3.3. Effect of fibrin degradation products on transendothelial migration of leukocytes
It was previously shown that the D-D:E1 complex, in which the D-D and E1 fragments are held together by non-covalent interactions, is the primary soluble fibrin degradation product (FDP) released upon digestion of crosslinked fibrin with plasmin [18]. It is also well known that prolonged plasmin digestion of crosslinked fibrin results in the terminal FDP including the D-D dimer and E3 fragment. These two degradation products, D-D and E3, do not interact with each other due to proteolytic removal of the βN-domains and some other sequences upon degradation of E1 to E3 [34, 35]. Our previous study revealed that the D-D:E1 complex interacts with the immobilized sVLDLR while isolated D-D and E3 fragment do not interact [20]. Based on these findings, we hypothesized that the D-D:E1 complex and larger soluble fibrin degradation products should promote leukocyte transmigration through the VLDLR-dependent pathway. To test this hypothesis, we prepared two fractions from the limited plasmin digest of crosslinked fibrin, the D-D:E1 complex and higher molecular mass fibrin degradation products (HMM-FDP), as described in Materials and methods, and tested their effects on leukocyte transmigration using leukocyte transendothelial migration assay. The experiments revealed that the D-D:E1 complex stimulated leukocyte transmigration to the same extent as control NDSK-II while stimulating effect of HMM-FDP was even higher (Fig. 6). The stimulating effect of D-D:E1 and HMM-FDP was inhibited by RAP indicating the involvement of VLDLR in this process. However, the inhibition was not complete since the 43% difference between RAP-inhibited leukocyte transmigration and control was statistically significant, suggesting involvement of other players/pathways in leukocyte transmigration. Altogether, these experiments clearly indicate that the D-D:E1 complex and higher molecular mass soluble fibrin degradation products both significantly stimulate transendothelial migration of leukocytes mainly through the VLDLR-dependent pathway.
Fig. 6.
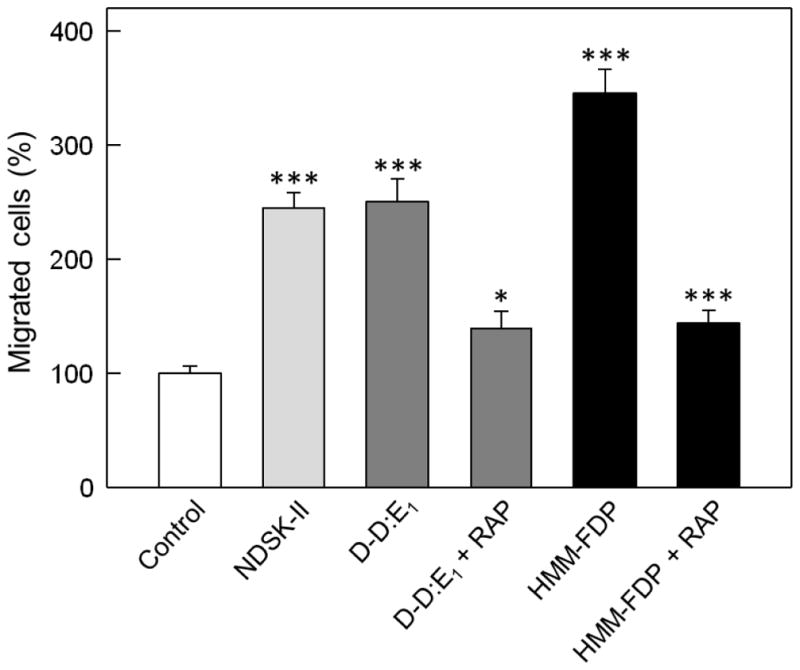
Effect of early fibrin degradation products on transendothelial migration of leukocytes. HUVECs were grown to confluence on gelatin-coated cell culture inserts. Calcein AM-labeled differentiated HL-60 cells were added to the upper chambers on top of the HUVEC monolayers in the presence of PBS or 1.5 μM NDSK-II (controls), or in the presence of 1.5 μM D-D:E1 complex or soluble high molecular mass fibrin degradation products (HMM-FDP) without and with 500 nM RAP. The cells were allowed to migrate into the lower chambers for 4 h at 37°C, collected, and quantified as described in Materials and methods. The results are expressed as percentage of HL-60 cells migrated in the presence of PBS (control). The graph shows combined data from 2 independent experiments; error bars denote means ± SD (n = 6). *P < 0.05; ***P < 0.001.
3.4. Effect of soluble fibrin and endothelium-anchored fibrin polymers on transendothelial migration of leukocytes
Although we proposed earlier that interaction of fibrin with the VLDL receptor promotes leukocyte transmigration, no direct experimental evidence was presented [20]. It was also unclear whether fibrin polymers, in which the βN-domains are involved in polymerization and their VLDLR-binding sites may be inaccessible, can promote leukocyte transmigration. To test the effect of soluble fibrin on leukocyte transmigration, we first solubilized fibrin by incubation of acidic fibrin monomer with fibrinogen D1 fragment as described in Materials and methods. In leukocyte transendothelial migration assay, when added to the upper chamber together with leukocytes, such fibrin stimulated leukocyte transmigration even better than control NDSK-II (Fig. 7, F-sol). This effect was significantly reduced in the presence of mAb 1H10, a monoclonal antibody that was shown to efficiently inhibit fibrin-VLDLR interaction [21], indicating major contribution of this interaction in leukocyte transmigration. At the same time, as in the case with HMM-FDP, the inhibition was not complete suggesting that other players/pathways may be involved in this process.
Fig. 7.
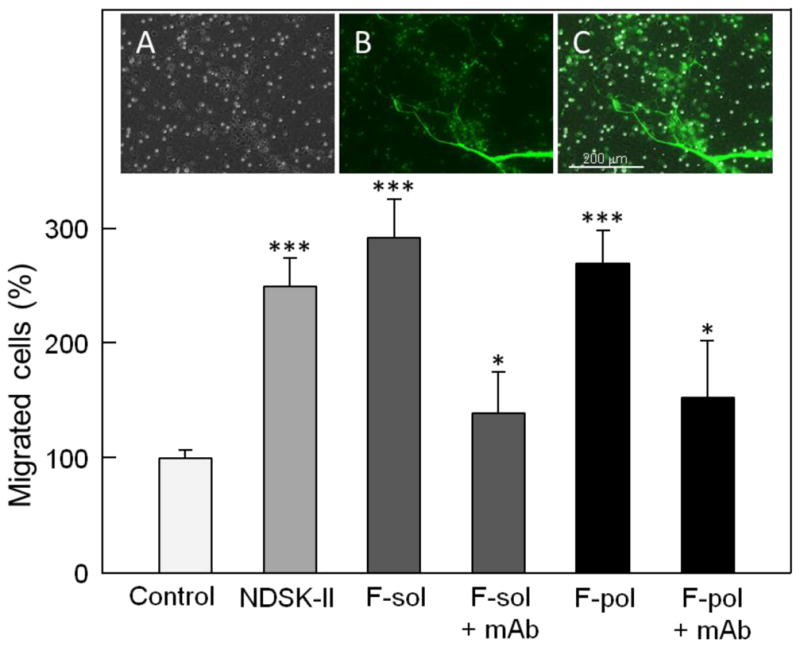
Effect of soluble fibrin and endothelium-anchored fibrin polymers on transendothelial migration of leukocytes. HUVECs were grown to confluence on gelatin-coated cell culture inserts, Calcein AM-labeled differentiated HL-60 cells were added to the upper chambers on top of the HUVEC monolayers in the presence of PBS or 1.5 μM NDSK-II (controls), or 1.5 μM soluble fibrin without (F-sol) and with 0.5 μM mAb 1H10 (F-sol + mAb). Fibrin polymers (F-pol) were deposited (anchored) on the HUVEC monolayers using the procedure described in Materials and methods and tested without or with 0.5 μM mAb 1H10 (F-pol + mAb). The cells were allowed to migrate into the lower chambers containing chemoattractant fMLP for 4 h at 37°C, collected, and quantified as described in Materials and methods. The results are expressed as percentage of HL-60 cells migrated in the presence of PBS (control). The graph shows combined data from 2 independent experiments; error bars denote means ± SD (n = 6). *P < 0.05; ***P < 0.001. The presence of endothelium-anchored fibrin polymers on the HUVEC monolayers was confirmed by fluorescence microscopy as shown in the insets. Inset A shows representative image of membrane insert with bright and dark dots representing membrane pores and differentiated HL-60 cells adherent to the endothelial surface, respectively. Inset B, the same field as in inset A showing endothelium-anchored fibrin polymers labeled with Alexa-488 that appear on the surface as a fiber network colored in green; Calcein AM-labeled differentiated HL-60 cells appear as green dots. Inset C shows merged insets A and B. All images were taken at ×10 magnification using an EVOS FL Auto microscope. Scale bar: 200 μm.
To test the effect of endothelium-anchored fibrin polymers on leukocyte transmigration, fibrin polymers (clots) were deposited on the surface of HUVEC monolayers, as described in Materials and methods. To prove that fibrin polymers were anchored to these monolayers, in parallel experiments Alexa-488-labeled fibrin monomer together with Calcein AM-labeled leukocytes were added to the upper chamber and endothelium-anchored fibrin clots were visualized by fluorescence microscopy (Fig. 7, insets). Our further experiments using leukocyte transmigration assay revealed that non-labeled endothelium-anchored fibrin clots exhibit significant stimulating effect on transendothelial migration of leukocytes, which was comparable with that of NDSK-II (Fig. 7). This effect was significantly reduced in the presence of mAb 1H10. Again, as in the case with HMM-FDP and F-sol, the inhibition was not complete suggesting that other players/pathways may be involved in this process. Altogether, the experiments described above indicate that soluble fibrin and endothelium-anchored fibrin polymers both stimulate leukocyte transmigration mainly through the VLDLR-dependent pathway.
4. Discussion
The major findings of the present study are that early high-molecular mass fibrin degradation products (HMM-FDP) and the D-D:E1 complex, as well as soluble fibrin and endothelium-anchored fibrin polymers, promote transendothelial migration of leukocytes mainly through the VLDL receptor-dependent pathway of leukocyte transmigration. Although specific inhibitors of fibrin-VLDLR interaction did not completely inhibit leukocyte transmigration stimulated by these fibrin species suggesting participation of other pathway(s) in this process, their contribution seems to be much lower. These findings provide direct evidence for our previous suggestion that interaction of fibrin with VLDLR may stimulate leukocyte transmigration [20] and identify additional fibrin species that can stimulate leukocyte transmigration through the VLDLR-dependent pathway. They also suggest which fibrin species may be involved in the VLDLR-dependent pathway in vivo and allow us to clarify the (patho)physiological role of this pathway.
Soluble fibrin was prepared by incubation of acidic fibrin monomer with fibrinogen D1 fragment, which at neutral pH interacts with fibrin molecules through its polymerization sites and thus prevents polymerization of fibrin keeping it in solution. Such soluble fibrin actually mimics soluble fibrinogen-fibrin complexes described earlier [36–38] that are present in the circulation in pathological states and hence may stimulate the VLDLR-dependent leukocyte transmigration in vivo. HMM-FDP and D-D:E1 are primary soluble FDP released from crosslinked fibrin at the early stage of fibrinolysis [18] and may also contribute to the VLDLR-dependent pathway of leukocyte transmigration. However, they are rapidly degraded to final FDP, the D-D dimer and E3 fragment, which do not interact with VLDLR [20]. Therefore, their contribution to the VLDLR-dependent pathway may be minor.
As to the endothelium-anchored fibrin polymers, it was shown that the procoagulant activity of inflamed endothelium due to expression of tissue factor results in the deposition of polymerized fibrin (fibrin clots) on the endothelial surface [39–42]. Since endothelium-anchored fibrin polymers mimic fibrin clots deposited on the surface of inflamed endothelium, the results obtained with them may provide a hint on the role of the VLDLR-dependent pathway of leukocyte transmigration in normal and pathological states. Specifically, one can expect that during normal wound healing process fibrin clots on inflamed endothelium should promote leukocyte transmigration to the sites of inflammation through this pathway. In pathological states, for example in the case of myocardial infarction, occlusion of coronary artery whose endothelium contains VLDLR [43] brings fibrin clots in contact with this receptor and their interaction may promote leukocyte transmigration thereby contributing to the pathogenesis of myocardial damage and reperfusion injury.
Another major finding is that the VLDLR-binding site is cryptic in fibrinogen and its exposure in some cases does not require removal of fibrinopeptide A and B, which occurs upon conversion of fibrinogen into fibrin. This is in agreement with our recent finding that the VLDLR-binding site is located mainly in the C-terminal half of fibrin βN-domains [22], i.e. away from the site to be cleaved to release fibrinopeptide B (FpB). The present study revealed that this site is completely exposed in the isolated fibrinogen BβN-domains represented by the recombinant (Bβ1-66)2 fragment and in surface-adsorbed fibrinogen in which fibrin-specific binding sites are usually exposed. Thus, the exposure of the VLDLR-binding site is most probably connected with conformational changes upon conversion of fibrinogen to fibrin, not with the removal of FpB. This is in contrast to the VE-cadherin-binding site, which is located in the N-terminal part of the βN-domains [44] and whose exposure requires FpB removal [45].
The exact mechanism of exposure of the VLDLR-binding site is not clear. However, based on our previous studies one can speculate that it may be connected with the dissociation-association of fibrin(ogen) αC-domains. Indeed, according to the current view, the αC-domains interact intramolecularly with each other and with the central part of the fibrinogen molecule, and FpB located in the N-terminal portions of fibrinogen BβN-domains contributes to this interaction [26,46,47]. Further, upon conversion of fibrinogen into fibrin the αC-domains dissociate from the central part of the molecule and from each other and re-associate intermolecularly forming αC-polymers [26,46,48–50]. Thus, one can hypothesize that the VLDLR-binding sites in fibrinogen are inaccessible for binding to VLDLR due to these intramolecular interactions and their exposure in fibrin is connected with the dissociation of the αC-domains from the central part of the molecule. Similarly, they become accessible upon binding of fibrinogen to a surface, which was shown to involve αC-domains [51]. Further experiments are required to test this hypothesis.
Although the present study focuses mainly on the role of fibrin(ogen) species in the VLDLR-dependent pathway of transendothelial migration of leukocytes, our findings raise some questions about two other pathways of leukocyte transmigration proposed earlier [13,16,17]. First, we found that fibrinogen has only very small stimulating effect on in vitro leukocyte transmigration (Fig. 5), in agreement with the cryptic character of its VLDLR-binding sites mentioned above. At the same time, the previous studies suggested that fibrinogen exhibits significant stimulating effect on leukocyte transmigration [12] through the proposed (Mac-1)-fibrinogen-(ICAM-1)-dependent intercellular bridging pathway [13]. The reason for such discrepancy is unclear, especially taking into account that both the previous [12] and the present studies utilized the same in vitro leukocyte transmigration assay and fibrinogen concentrations used in both studies were comparable. One can only speculate that fibrinogen used in the previous study [12] contained a fraction of active fibrinogen aggregates while in the present study we used freshly prepared monomeric fibrinogen devoid of such aggregates. In agreement with this speculation, it was previously shown that freshly purified fibrinogen in solution does not interact with leukocyte receptor Mac-1; it binds to this receptor only being immobilized onto a surface [14,15]. This suggests that the Mac-1-binding sites are cryptic in the fibrinogen molecule and further confirms our finding that fibrinogen in solution is a poor stimulator of leukocyte transmigration.
Second, the present study revealed that the D-D:E1 complex is able to promote leukocyte transmigration. The D-D:E1 complex, which is the primary soluble FDP released from crosslinked fibrin [18], is very stable in solution, as mentioned in Materials and methods. Such stability is connected with the strong interaction between its individual components. Indeed, it was shown that when purified D-D and E1 are mixed together, they re-associate to form a 1:1 complex [18,31]. Dissociation of this complex occurs only upon its further digestion during which its E1 fragment component is converted to E3 fragment [18,34] in which α17–19 (Gly-Pro-Arg) and β15–53 sequences are proteolitically removed [35]. Thus, the E1 fragment may exist in the circulation only in a complex with the D-D dimer. This raises a question about the proposed bridging role of the E1 fragment in promoting leukocyte transmigration through the interaction with endothelial VE-cadherin and leukocyte CD11c integrin [16,17]. Indeed, if the E1 fragment exists only in a complex with D-D, then in vivo only D-D:E1, not E1, can promote leukocyte transmigration through such bridging mechanism. However, the D-D:E1 complex is kept together through the interaction between complementary polymerization sites containing knobs “A” of E1 and holes “a” of D-D [19]. This means that the α chain Gly17-Pro-Arg sequences corresponding to knobs “A” of E1 are occupied in the complex making impossible interaction of its E1 component with the CD11c integrin, which was shown to occur through this αGly17-Pro-Arg sequence [52]. Thus, the proposed (CD11c)-E1 fragment-(VE-cadherin)-dependent bridging of leukocyte to the endothelium [16,17] cannot occur in vivo through leukocyte CD11c integrin; it should involve a different leukocyte receptor.
In conclusion, the present study revealed that the VLDLR-binding sites are cryptic in the fibrinogen molecule and fibrinogen in solution is a very poor stimulator of leukocyte transmigration, in agreement with the current view that fibrinogen is rather inert in the circulation. This site is exposed only upon conversion of fibrinogen into fibrin or its adsorption onto a surface, most probably due to conformational changes caused by these processes. The study also revealed that the D-D:E1 complex and higher molecular mass fibrin degradation products, as well as soluble fibrin and fibrin polymers anchored to the endothelium (fibrin clots), promote leukocyte transmigration mainly through the VLDLR-dependent pathway. These findings further clarify the molecular mechanisms underlying fibrin-induced VLDLR-dependent pathway of transendothelial migration of leukocytes and raise some questions about other proposed mechanisms of leukocyte transmigration. They also provide an explanation for a possible (patho)physiological role of the VLDLR-dependent pathway.
Highlights.
The VLDL receptor-dependent pathway of leukocyte transmigration plays an important role in inflammation.
The present study identified fibrin species that stimulate leukocyte transmigration through this pathway.
The species include early fibrin degradation products, soluble fibrin, and fibrin clots deposited on the endothelium.
These findings clarify the molecular mechanism and (patho)physiological role of this pathway.
Acknowledgments
We thank Dr. A. Belkin for his help in planning and performing some experiments and fruitful discussions. This work was supported by the National Institutes of Health grant HL 056051 to L. Medved.
Abbreviations
- VLDLR
very low density lipoprotein receptor
- FDP
fibrin degradation products
- HMM-FDP
high molecular mass fibrin degradation products
- FpB
fibrinopeptide B
- ELISA
enzyme-linked immunosorbent assay
- SPR
surface plasmon resonance
- RAP
receptor-associated protein
Footnotes
Conflicts of interest
All authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Henschen A, McDonagh J. Fibrinogen, fibrin and factor XIII. In: Zwaal RFA, Hemker HC, editors. Blood Coagulation. Elsevier Science Publishers; Amsterdam: 1986. pp. 171–241. [Google Scholar]
- 2.Blomback B, Blomback M, Henschen A, Hessel B, Iwanaga S, Woods KR. N-terminal disulphide knot of human fibrinogen. Nature. 1968;218:130–134. doi: 10.1038/218130a0. [DOI] [PubMed] [Google Scholar]
- 3.Medved L, Weisel JW on behalf of Fibrinogen and Factor XIII Subcommittee of Scientific Standardization Committee of International Society on Thrombosis and Haemostasis. Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost. 2009;7:355–359. doi: 10.1111/j.1538-7836.2008.03242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 5.Colvin RB, Johnson RA, Mihm MC, Jr, Dvorak HF. Role of the clotting system in cell-mediated hypersensitivity. I. Fibrin deposition in delayed skin reactions in man. J Exp Med. 1973;138:686–698. doi: 10.1084/jem.138.3.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McRitchie DI, Girotti MJ, Glynn MF, Goldberg JM, Rotstein OD. Effect of systemic fibrinogen depletion on intraabdominal abscess formation. J Lab Clin Med. 1991;118:48–55. [PubMed] [Google Scholar]
- 7.Tang L, Eaton JW. Fibrin(ogen) mediates acute inflammatory responses to biomaterials. J Exp Med. 1993;178:2147–2156. doi: 10.1084/jem.178.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 9.Johnson-Léger C, Aurrand-Lions M, Imhof BA. The parting of the endothelium: miracle. or simply a junctional affair? J Cell Sci. 2000;113:921–933. doi: 10.1242/jcs.113.6.921. [DOI] [PubMed] [Google Scholar]
- 10.Aurrand-Lions M, Johnson-Léger C, Imhof BA. Role of interendothelial adhesion molecules in the control of vascular functions. Vascul Pharmacol. 2002;39:239–246. doi: 10.1016/s1537-1891(03)00012-0. [DOI] [PubMed] [Google Scholar]
- 11.Languino LR, Plescia J, Duperray A, Brian AA, Plow EF, Geltosky JE, Altieri DC. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell. 1993;73:1423–1434. doi: 10.1016/0092-8674(93)90367-y. [DOI] [PubMed] [Google Scholar]
- 12.Languino LR, Duperray A, Joganic KJ, Fornaro M, Thornton GB, Altieri DC. Regulation of leukocyte-endothelium interaction and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc Natl Acad Sci U S A. 1995;92:1505–1509. doi: 10.1073/pnas.92.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altieri DC. Regulation of leukocyte-endothelium interaction by fibrinogen. Thromb Haemost. 1999;82:781–786. [PubMed] [Google Scholar]
- 14.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated unmasking of the cryptic binding site for integrin αMβ2 in the γC-domain of fibrinogen. Biochemistry. 2002;41:12942–12951. doi: 10.1021/bi026324c. [DOI] [PubMed] [Google Scholar]
- 15.Yakovlev S, Zhang L, Ugarova T, Medved L. Interaction of fibrin(ogen) with leukocyte receptor αMβ2 (Mac-1): further characterization and identification of a novel binding region within the central domain of the fibrinogen γ-module. Biochemistry. 2005;44:617–626. doi: 10.1021/bi048266w. [DOI] [PubMed] [Google Scholar]
- 16.Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, Groger M, Wolff K, Zacharowski K. The fibrin-derived peptide Bβ15–42 protects the myocardium against ischemia-reperfusion injury. Nat Med. 2005;11:298–304. doi: 10.1038/nm1198. [DOI] [PubMed] [Google Scholar]
- 17.Zacharowski K, Zacharowski P, Reingruber S, Petzelbauer P. Fibrin(ogen) and its fragments in the pathophysiology and treatment of myocardial infarction. J Mol Med. 2006;84:469–477. doi: 10.1007/s00109-006-0051-7. [DOI] [PubMed] [Google Scholar]
- 18.Olexa SA, Budzynski AZ. Primary soluble plasmic degradation product of human cross-linked fibrin. Isolation and stoichiometry of the (DD)E complex. Biochemistry. 1979;18:991–995. doi: 10.1021/bi00573a009. [DOI] [PubMed] [Google Scholar]
- 19.Moskowitz KA, Budzynski AZ. The (DD)E complex is maintained by a composite fibrin polymerization site. Biochemistry. 1994;33:12937–12944. doi: 10.1021/bi00248a001. [DOI] [PubMed] [Google Scholar]
- 20.Yakovlev S, Mikhailenko I, Cao C, Zhang L, Strickland DK, Medved L. Identification of VLDLR as a novel endothelial cell receptor for fibrin that modulates fibrin-dependent transendothelial migration of leukocytes. Blood. 2012;119:637–644. doi: 10.1182/blood-2011-09-382580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yakovlev S, Belkin AM, Chen L, Cao C, Zhang L, Strickland DK, Medved L. Anti-VLDL receptor monoclonal antibodies inhibit fibrin-VLDL receptor interaction and reduce fibrin-dependent leukocyte transmigration. Thromb Haemost. 2016;116:1122–1130. doi: 10.1160/TH16-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yakovlev S, Medved L. Interaction of fibrin with the very low-density lipoprotein (VLDL) receptor: further characterization and localization of the VLDL receptor-binding site in fibrin βN-domains. Biochemistry. 2017;56:2518–2528. doi: 10.1021/acs.biochem.7b00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yakovlev S, Medved L. Interaction of fibrin with the very low density lipoprotein receptor: Further characterization and localization of the fibrin-binding site. Biochemistry. 2015;54:4751–4761. doi: 10.1021/acs.biochem.5b00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams SE, Ashcom JD, Argraves WS, Strickland DK. A novel mechanism for controlling the activity of α2-macroglobulin receptor/low density lipoprotein receptor-related protein. Multiple regulatory sites for 39-kDa receptor-associated protein. J Biol Chem. 1992;267:9035–9040. [PubMed] [Google Scholar]
- 25.Pozdnjakova TM, Musjalkovskaja AA, Ugarova TP, Protvin DD, Kotsjuruba VN. On the properties of fibrin monomer prepared from fibrin clot with acetic acid. Thromb Res. 1979;16:283–288. doi: 10.1016/0049-3848(79)90292-5. [DOI] [PubMed] [Google Scholar]
- 26.Veklich YI, Gorkun OV, Medved LV, Nieuwenhuizen W, Weisel JW. Carboxyl-terminal portions of the α chains of fibrinogen and fibrin. Localization by electron microscopy and the effects of isolated αC fragments on polymerization. J Biol Chem. 1993;268:13577–13585. [PubMed] [Google Scholar]
- 27.Gorkun OV, Veklich YI, Weisel JW, Lord ST. The conversion of fibrinogen to fibrin: recombinant fibrinogen typifies plasma fibrinogen. Blood. 1997;89:4407–4414. [PubMed] [Google Scholar]
- 28.Yakovlev S, Gorlatov S, Ingham K, Medved L. Interaction of fibrin(ogen) with heparin: further characterization and localization of the heparin-binding site. Biochemistry. 2003;42:7709–7716. doi: 10.1021/bi0344073. [DOI] [PubMed] [Google Scholar]
- 29.Bach TL, Barsigian C, Yaen CH, Martinez J. Endothelial cell VE-cadherin functions as a receptor for the β15–42 sequence of fibrin. J Biol Chem. 1998;273:30719–30728. doi: 10.1074/jbc.273.46.30719. [DOI] [PubMed] [Google Scholar]
- 30.Yakovlev S, Gao Y, Cao C, Chen L, Strickland DK, Zhang L, Medved L. Interaction of fibrin with VE-cadherin and anti-inflammatory effect of fibrin-derived fragments. J Thromb Haemost. 2011;9:1847–1855. doi: 10.1111/j.1538-7836.2011.04438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yakovlev S, Makogonenko E, Kurochkina N, Nieuwenhuizen W, Ingham K, Medved L. Conversion of fibrinogen to fibrin: mechanism of exposure of tPA- and plasminogen-binding sites. Biochemistry. 2000;39:15730–15741. doi: 10.1021/bi001847a. [DOI] [PubMed] [Google Scholar]
- 32.Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci U S A. 1978;75:2458–2462. doi: 10.1073/pnas.75.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hauert AB, Martinelli S, Marone C, Niggli V. Differentiated HL-60 cells are a valid model system for the analysis of human neutrophil migration and chemotaxis. Int J Biochem Cell Biol. 2002;34:838–854. doi: 10.1016/s1357-2725(02)00010-9. [DOI] [PubMed] [Google Scholar]
- 34.Olexa SA, Budzynski AZ. Binding phenomena of isolated unique plasmic degradation products of human cross-linked fibrin. J Biol Chem. 1979;254:4925–4932. [PubMed] [Google Scholar]
- 35.Olexa SA, Budzynski AZ, Doolittle RF, Cottrell BA, Greene TC. Structure of fragment E species from human cross-linked fibrin. Biochemistry. 1981;20:6139–6145. doi: 10.1021/bi00524a035. [DOI] [PubMed] [Google Scholar]
- 36.Sherman LA, Harwig S, Lee J. In vitro formation and in vivo clearance of fibrinogen: fibrin complexes. J Lab Clin Med. 1975;86:100–111. [PubMed] [Google Scholar]
- 37.Graeff H, Hafter R, Von Hugo R. On soluble fibrinogen-fibrin complexes. Thromb Res. 1979;16:575–576. doi: 10.1016/0049-3848(79)90106-3. [DOI] [PubMed] [Google Scholar]
- 38.Nakahara K, Kazahaya Y, Shintani Y, Yamazumi K, Eguchi Y, Koga S, Wada H, Matsuda M. Measurement of soluble fibrin monomer-fibrinogen complex in plasmas derived from patients with various underlying clinical situations. Thromb Haemost. 2003;89:832–836. [PubMed] [Google Scholar]
- 39.Kirchhofer D, Sakariassen KS, Clozel M, Tschopp TB, Hadváry P, Nemerson Y, Baumgartner HR. Relationship between tissue factor expression and deposition of fibrin, platelets, and leukocytes on cultured endothelial cells under venous blood flow conditions. Blood. 2003;81:2050–2058. [PubMed] [Google Scholar]
- 40.Kirchhofer D, Tschopp TB, Hadváry P, Baumgartner HR. Endothelial cells stimulated with tumor necrosis factor-α express varying amounts of tissue factor resulting in inhomogenous fibrin deposition in a native blood flow system. Effects of thrombin inhibitors. J Clin Invest. 1994;93:2073–2083. doi: 10.1172/JCI117202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell RA, Overmyer KA, Bagnell CR, Wolberg AS. Cellular procoagulant activity dictates clot structure and stability as a function of distance from the cell surface. Arterioscler Thromb Vasc Biol. 2008;28:2247–2254. doi: 10.1161/ATVBAHA.108.176008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campbell RA, Overmyer KA, Selzman CH, Sheridan BC, Wolberg AS. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114:4886–4896. doi: 10.1182/blood-2009-06-228940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wyne KL, Pathak K, Seabra MC, Hobbs HH. Expression of the VLDL receptor in endothelial cells. Arterioscler Thromb Vasc Biol. 1996;16:407–415. doi: 10.1161/01.atv.16.3.407. [DOI] [PubMed] [Google Scholar]
- 44.Gorlatov S, Medved L. Interaction of fibrin(ogen) with the endothelial cell receptor VE-cadherin: mapping of the receptor-binding site in the NH2-terminal portions of the fibrin β chains. Biochemistry. 2002;41:4107–4116. doi: 10.1021/bi0160314. [DOI] [PubMed] [Google Scholar]
- 45.Martinez J, Ferber A, Bach TL, Yaen CH. Interaction of fibrin with VE-cadherin. Ann N Y Acad Sci. 2001;936:386–405. doi: 10.1111/j.1749-6632.2001.tb03524.x. [DOI] [PubMed] [Google Scholar]
- 46.Weisel JW, Medved L. The structure and function of the αC domains of fibrinogen. Ann NY Acad Sci. 2001;936:312–327. doi: 10.1111/j.1749-6632.2001.tb03517.x. [DOI] [PubMed] [Google Scholar]
- 47.Litvinov RI, Yakovlev S, Tsurupa G, Gorkun OV, Medved L, Weisel JW. Direct evidence for specific interactions of the fibrinogen αC-domains with the central E region and with each other. Biochemistry. 2007;46:9133–9142. doi: 10.1021/bi700944j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Medved LV, Gorkun OV, Manyakov VF, Belitser VA. The role of fibrinogen αC-domains in the fibrin assembly process. FEBS Lett. 1985;181:109–112. doi: 10.1016/0014-5793(85)81123-6. [DOI] [PubMed] [Google Scholar]
- 49.Gorkun OV, Veklich YI, Medved LV, Henschen AH, Weisel JW. Role of the αC domains of fibrin in clot formation. Biochemistry. 1994;33:6986–6997. doi: 10.1021/bi00188a031. [DOI] [PubMed] [Google Scholar]
- 50.Tsurupa G, Pechik I, Litvinov RI, Hantgan RR, Tjandra N, Weisel JW, Medved L. On the mechanism of αC polymer formation in fibrin. Biochemistry. 2012;51:2526–2538. doi: 10.1021/bi2017848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clarke ML, Wang J, Chen Z. Conformational changes of fibrinogen after adsorption. J Phys Chem B. 2005;109:22027–22035. doi: 10.1021/jp054456k. [DOI] [PubMed] [Google Scholar]
- 52.Loike JD, Sodeik B, Cao L, Leucona S, Weitz JI, Detmers PA, Wright SD, Silverstein SC. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the Aα chain of fibrinogen. Proc Natl Acad Sci U S A. 1991;88:1044–1048. doi: 10.1073/pnas.88.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]