

Abstract
Background and Purpose
Bruton's tyrosine kinase (Btk) is a non‐receptor tyrosine kinase involved in the activation of signalling pathways responsible for cell maturation and viability. Btk has previously been reported to be overexpressed in colon cancers. This kind of cancer is often accompanied by anaemia, which is treated with an erythropoietin supplement. The goal of the present study was to assess the effects of combination therapy with erythropoietin β (Epo) and LFM‐A13 (Btk inhibitor) on colon cancer in in vitro and in vivo models.
Experimental Approach
DLD‐1 and HT‐29 human colon adenocarcinoma cells were cultured with Epo and LFM‐A13. Cell number and viability, and mRNA and protein levels of Epo receptors, Btk and Akt were assessed. Nude mice were inoculated with adenocarcinoma cells and treated with Epo and LFM‐A13.
Key Results
The combination of Epo and LFM‐A13 mostly exerted a synergistic inhibitory effect on colon cancer cell growth. The therapeutic scheme used effectively killed the cancer cells and attenuated the Btk signalling pathways. Epo + LFM‐A13 also prevented the normal process of microtubule assembly during mitosis by down‐regulating the expression of Polo‐like kinase 1. The combination of Epo and LFM‐A13 significantly reduced the growth rate of tumour cells, while it showed high safety profile, inducing no nephrotoxicity, hepatotoxicity or changes in the haematological parameters.
Conclusion and Implications
Epo significantly enhances the antitumour activity of LFM‐A13, indicating that a combination of Epo and LFM‐A13 has potential as an effective therapeutic approach for patients with colorectal cancer.
Abbreviations
- Acl
acalabrutinib
- Btk
Bruton's tyrosine kinase
- DLD‐1
cell line of human colorectal adenocarcinoma
- Epo
erythropoietin
- EpoR
erythropoietin receptor
- Flg
filgrastim
- HCT
haematocrit
- HGB
haemoglobin
- HT‐29
cell line of human colorectal adenocarcinoma
- LFM‐A13
Btk inhibitor
- MCV
mean corpuscular volume
- MYC
a regulator gene that codes for a transcription factor
- NANOG
a transcription factor
- PLK1
Polo‐like kinase 1
- SOX2
a transcription factor
- SPF
specific‐pathogen‐free
- WBC
white blood cells
Introduction
Despite the use of combination therapy in many patients with cancer, satisfactory results are not fully achieved. Tyrosine kinases have become key therapeutic targets for drug development. LFM‐A13 is the first inhibitor of Bruton's tyrosine kinase (Btk), a key signalling molecular complex of receptors on the surface of B cells (Uckun et al., 2002). Btk is also present in monocytes, macrophages and megakaryocytes where it participates in the activation of signalling pathways responsible for processing problems with cell maturation and viability, cytokine production and cellular degranulation (Qiu and Kung, 2000). Overexpression of Btk is associated with an elevated expression of genes with functions related to cell adhesion, the cytoskeletal structure and the extracellular matrix (Kokabee et al., 2015). Btk plays an important role in the development of tumours of B‐cell activating antiapoptotic pathways (Uckun et al., 2002). Increased expression of this kinase was also shown in prostate cancer, where higher levels of Btk positively correlated with the progression of cancer (Guo et al., 2014). Blocking the activity of Btk through the use of already begun treatment (ibrutynib, sorafenib) or currently intensively investigated many kinase inhibitors (GDC0834, CGI‐560, CGI‐1746) effectively inhibits the neoplastic process (Akinleye et al., 2013). It should be emphasized that LFM‐A13 was previously used in haematological malignancies. However, data are emerging suggesting its beneficial use in gastric, lung, prostate and breast cancer (Mahajan et al., 1999; Uckun et al., 2007; Guo et al., 2014; Wang et al., 2016; Peng et al., 2016). This compound promotes apoptosis, acts as an antiproliferative factor and increases the sensitivity of tumour cells to the action of chemotherapeutic agents. It also has a high safety profile, as high doses of LFM‐A13 (20–100 mg·kg−1, i.v.) used in vivo did not induce nephrotoxicity, hepatotoxicity or changes in the blood profile (Uckun et al., 2002).
In light of our previous research and other evidence, erythropoietin (Epo) is regarded as a promoter of the neoplastic process (Yasuda et al., 2003; Hardee et al., 2007; Tankiewicz‐Kwedlo et al., 2016; Tankiewicz‐Kwedlo et al., 2017). Epo, when used to treat anaemia promotes the survival, proliferation and differentiation of the progenitors of erythropoiesis, as well as exerting a pro‐angiogenic and an anti‐apoptotic effect. This action can lead to disease progression and shorten the life span of patients undergoing treatment with Epo (Acs et al., 2001; Dicato et al., 2010).
Initially, the aim of this study was to assess the effects of LFM‐A13 on colon cancer and to find out whether the addition of erythropoietin reduced its anticancer activity. It turned out that Epo, when used with LFM‐A13, did not increase tumour progression but – what was positive and surprising – intensified the antitumor effect of LFM‐A13. Hence, we have focused on explaining this observation.
Herein, we showed that Btk is also expressed in colon cancer cells and that LFM‐A13 when administered in combination with Epo reduces tumour volume in DLD‐1 xenografts more effectively than when LFM‐A13 is administered alone. This is the first time, this novel combined therapy of Epo and LFM‐A13 has been shown to be effective as an anticancer therapy.
Methods
Cell cultures
DLD‐1 (ATCC, Cat# CCL‐221, RRID:CVCL_0248) and HT‐29 (ATCC, Cat# HTB‐38, RRID:CVCL_0320), cell lines of human colorectal adenocarcinoma, were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The characteristics of these cell lines were as presented previously (Tankiewicz‐Kwedlo et al., 2016).
Exogenous erythropoietin, filgrastim, LFM‐A13 and acalabrutinib administration
The colon cancer cell lines DLD‐1 and HT‐29 were incubated with exogenous erythropoietin β at concentrations of 30, 100 IU·mL−1, LFM‐A13 at concentrations of 30, 100 μM, and a combination of these drugs (Epo + LFM‐A13), filgrastim (Flg, human granulocyte colony stimulating factor) at a concentration of 5 μg·kg−1, a combination of Flg + LFM‐A13, acalabrutinib (Acl) at a concentration of 3 μM, and a combination of Epo + Acl or Flg + Acl. Epo (Feldman et al., 2006), LFM‐A13 (Uckun, 2007; Vijayan et al., 2011), Flg (Armstrong et al., 2007) and acalabrutnib (Patel et al., 2017) concentrations were chosen based on data from previously published studies. Epo is normally used at a concentration of 100 IU·mL−1 in these types of experiments (Westenfelder and Baranowski, 2000; Feldman et al., 2006).
Cell count, proliferation and cell cycle analysis
The total number of cells was quantified with an automated cell counter (NucleoCounter® NC3000, Chemometec, Denmark). The proliferation assay was performed as described previously by Tankiewicz‐Kwedlo et al. (2017). The distribution of the cell cycle phases was analysed using flow cytometry. Briefly, DLD‐1 and HT‐29 cells were seeded onto six‐well plates at a density of 2.5 × 105 cells per well and treated with Epo at a concentration of 100 IU·mL−1, LFM‐A13 at a concentration of 100 μM and a combination of these drugs (Epo + LFM‐A13) for 24 h. After the incubation, the cells were harvested and then fixed with 1 mL of 70% ethanol and stored overnight at −20°C. Before analysis, the cells were re‐suspended in PBS, treated with 50 μg·mL−1 of DNase‐free RNase A Solution (Promega) and stained with 100 μg·mL−1 of PI. The FACSCanto II flow cytometer (BD Bioscences Systems) was used to read the fluorescence. To evaluate whether the antiproliferative effects of the combinations of Epo with LFM‐A13 were synergistic, additive or antagonistic, the effects of drug combinations at several constant ratios were evaluated. For each drug (alone or in combination), four independent experiments were performed. Analysis was done using CompuSyn software (http://www.combosyn.com), which allows for automated simulation of synergism and antagonism at all dose and effect levels using the Chou‐Talalay method (Chou and Talalay, 1984).
Quantitative real‐time‐PCR (QRT‐PCR) analysis
Quantitative‐real‐time‐PCR was performed using a standard method described previously (Tankiewicz‐Kwedlo et al., 2016). Primers were designed using PRIMER‐blast software (http://www.ncbi.nlm.nih.gov/tools/primer‐blast). Primer sequences were (5′‐3′ forward, reverse): GATGGGTGGAGTCGCGT, CAGAGTTAAAAGCAGCCCTGG (GAPDH), TCATCCTCGTGGTCATCCTG, GAAGAGGCCTTCAAACTCGC [Erythropoietin receptor (EpoR)], CGACGTGGCTATTGTGAAGG, TTGAGGAGGAAGTAGCGTGG (Akt), CACCTTCCAAGTCCTGGCAT, AATCACTGCGGCCATAGCTT (Btk).
Relative quantification of gene expression was determined by comparing Ct values using the ΔΔC t method according to Livak and Schmittgen (2001). All results were normalized to GAPDH.
Western blot
After stimulation with Epo (up to 100 IU·mL−1), LFM‐A13 (up to 100 μM) and combinations of these agents at 37°C for 5 min (for phosphorylated Btk, phosphorylated Akt) and for 48 h (for phosphorylated EpoR, EpoR, Btk, Akt, phosphorylated PLK1, PLK1, active‐caspase 3, pro‐caspase3), the cells were lysed in NP‐40 [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% Triton X‐100, and a protease inhibitor cocktail (Roche)]. The lysate was centrifuged at 10 000× g for 20 min at 4°C. An aliquot (10 μL) of the supernatant was subjected to electrophoresis in a 10% SDS‐PAGE, followed by transfer to 0.2 μm pore‐size nitrocellulose membrane (Bio‐Rad) according to the method described in the manual accompanying the unit. Blots were blocked for 1 h at room temperature with 5% non‐fat milk (Bio‐Rad, USA) in Tris‐buffered saline, pH 8.0 (Sigma‐Aldrich, USA). The membrane was incubated with mouse monoclonal D‐5 antibody against EpoR (Santa Cruz Biotechnology, Cat# sc‐365662, RRID:AB_10841725), mouse monoclonal Y426 antibody against phospho EpoR (R and D Systems, Cat# MAB6926, RRID:AB_10971652), mouse monoclonal clone 53/Btk against Btk (BD Biosciences, Cat# 611117, RRID:AB_398428), rabbit polyclonal Tyr223 antibody against phospho Btk (Cell Signaling Technology, Cat# 5082P, RRID:AB_10557114), rabbit monoclonal H‐136 antibody against Akt1/2/3 (Santa Cruz Biotechnology, Cat# sc‐8312, RRID:AB_671714), rabbit polyclonal Ser 473 antibody against phospho Akt1/2/3 (Santa Cruz Biotechnology, Cat# sc‐7985 also sc‐7985‐R, RRID:AB_667741), rabbit polyclonal antibody against active caspase‐3 (Abcam, Cat# ab13847, RRID:AB_443014), rabbit polyclonal antibody against caspase‐3 (Abcam, Cat# ab49822, RRID:AB_868673), mouse monoclonal antibody against PLK1 (LifeSpan, Cat# LS‐C63154–200, RRID:AB_1934228), mouse monoclonal Thr210 antibody against phospho PLK1 (BioLegend, Cat# 628901, RRID:AB_439786) or mouse monoclonal antibody against β‐actin (Sigma‐Aldrich, Cat# A2228, RRID:AB_476697) in TBS‐T [20 mM Tris–HCl buffer (pH 7.4) containing 150 mM NaCl and 0.05% Tween 20] overnight. Alkaline phosphatase‐conjugated secondary goat polyclonal antibody against mouse (Sigma‐Aldrich, Cat# A3562, RRID:AB_258091) or secondary goat polyclonal antibody against rabbit (Sigma‐Aldrich, Cat# A3687, RRID:AB_258103) was added at a 1:10 000 dilution in TBS‐T and incubated for 1 h with slow shaking. The nitrocellulose was then washed with TBS‐T (2 × 10 min) and exposed to the Sigma‐Fast BCIP/NBT reagent.
Immunofluorescence
Immunofluorescence was performed as described previously (Tankiewicz‐Kwedlo et al., 2016). The cells were incubated with mouse monoclonal D‐5 antibody against EpoR (Santa Cruz Biotechnology, Cat# sc‐365662, RRID:AB_10841725), rabbit polyclonal antibody against phospho EpoR (Santa Cruz Biotechnology, Cat# sc‐20236‐R, RRID:AB_2098548), rabbit monoclonal H‐136 antibody against Akt1/2/3 (Santa Cruz Biotechnology, Cat# sc‐8312, RRID:AB_671714) and rabbit polyclonal Ser 473 antibody against phospho Akt1/2/3 (Santa Cruz Biotechnology, Cat# sc‐7985 also sc‐7985‐R, RRID:AB_667741) and then incubated with fluorescent (FITC) secondary goat polyclonal antibody against mouse (Sigma‐Aldrich, Cat# F0257, RRID:AB_259378) or fluorescent (FITC) secondary goat polyclonal antibody against rabbit (Sigma‐Aldrich, Cat# F9887, RRID:AB_259816) for 60 min in the dark. After the cells had been washed, the nuclei were stained with Hoechst 33342 (2 lg·mL−1) and analysed using confocal microscopy imaging.
Immunofluorescence and confocal microscopy were used to examine the spindle features of the DLD‐1 cell line treated with Epo, LFM‐A13 and Epo + LFM‐A13, as described by Uckun (2007). The cells were incubated with mouse monoclonal antibodies raised against α‐tubulin (Sigma‐Aldrich, Cat# T9026, RRID:AB_477593), mouse monoclonal antibodies against γ‐tubulin (Sigma‐Aldrich, Cat# T5326, RRID:AB_532292) and fluorescent (FITC) secondary goat polyclonal antibody against the mouse (Sigma‐Aldrich, Cat# F0257, RRID:AB_259378). Cellular DNA was labelled with Hoechst 33342 (ImmunoChemistry technologies, Cat# 639, RRID:AB_2651135). Cells were imaged with a BD Pathway 855 confocal system using a 209 (0.75 NA) objective. Cell populations were analysed for cytoplasmic fluorescence intensity. Images of FITC‐labelled cells were acquired using a 488/10 excitation laser and a 515LP emission laser.
Establishment of a xenograft
All procedures were performed in accordance with the guidelines for animal experiments and the protocol approved by the Local Ethics Committee (129/2015). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Experiments were conducted on male, 4‐week‐old mice weighing 18–20 g, of an inbred strain Cby.Cg‐Foxn1nu/J (RRID:IMSR_JAX:003118, Jackson Laboratory, USA). This strain has a hair follicle defect (homozygous males) and defective development of the thymic epithelium (athymic) and is commonly used for inducing cancer. All animals were kept in SPF cages, with five animals per cage, in a room with a constant temperature and humidity as well as 12 h light cycle. They also had free access to food and water. During the whole experiment, animal welfare was ensured. After a 1 week acclimatization period, the animals were randomized into two groups. The mice in the first group were injected s.c on the dorsal side with 50 μL of suspension containing 1 × 108 DLD‐1 cells in PBS, while the second group of mice were injected with 50 μL of suspension containing 1 × 108 HT‐29 cells in PBS, according to the method described by Shinohara et al. (2012). In order to determine tumour volume, an external caliper was used to measure the greatest longitudinal diameter (length) and the greatest transverse diameter (width). Tumour volumes based on the caliper results were calculated using the modified ellipsoidal formula (Feldman et al., 2009):
According to tumour volume measurements and mouse weight, the mice were allocated into four treatment groups, which received s.c. injections of Epo, LFM‐A13 (i.p.) and simultaneous injections of these agents (Epo + LFM‐A13), as well as the control group that received LFM‐A13 solvent (10% DMSO/PBS; i.p.). In previous studies, we showed that the Epo solvent had no effect on the number of colon cancer cells (Tankiewicz‐Kwedlo et al., 2010), and in accordance with the 3R, we reduced the number of animals used (Törnqvist et al., 2014). When the tumours reached a diameter of about 5 mm, that is, according to the literature, a size suitable to conduct further phases of research, the Epo 600 IU·kg−1 was administered three times a week (Pascual et al., 2013) and LFM‐A13 10 mg·kg−1 was administered twice a day (Uckun, 2007). The treatment was continued for 2 weeks. Animals were killed by an overdose of the combination of ketamine and xylazine administered i.p.
Imaging procedures
To confirm the presence of a tumour, a radiological examination was performed on the 14th day after DLD‐1 cell implantation. All nude mice underwent radiological imaging at 14 days. This radiological examination was performed on clinical Carestream Elite CR Wirtus Plus Systems (Carestream Health, Canada) under the following conditions: molybdenum anode at 23.00 kVp, 12 mAs and molybdenum filter.
Two‐dimensional and colour Doppler ultrasound
Ultrasound with colour Doppler examination was performed using a digital ultrasound system: GE Vivid 7 (General Electric Ultra‐sound System, Milwaukee, WI, USA). The transmitting frequency was set at 7.1 MHz. The ultrasounds were analysed by an experienced radiologist (J.B.) with 11 years of experience in interpreting Doppler ultrasound images.
Histopathology
At the time of necropsy, several tissues (bone marrow, kidney, liver, lung, spleen and tumour) were collected immediately from the mice for histopathological examination. The tissues were fixed in 10% neutral buffered formalin, dehydrated and embedded in paraffin using routine methods. Glass slides with 6‐μm‐thick tissue sections attached were prepared and stained with haematoxylin and eosin.
Immunohistochemistry
Formalin‐fixed, paraffin‐embedded tissue slides were cut on the microtome into sections with a thickness of 6 μm. The slides were deparaffinized in xylenes and hydrated in alcohol. The antigen for the antibody was exposed in a citrate buffer with a pH of 6.0 for 20 min at 97°C, then 20 min at room temperature. Endogenous peroxidase was blocked by incubation with 3% hydrogen peroxide. The sections were incubated with rabbit monoclonal antibody against phospho EpoR (Abcam, Cat# 2585–1, RRID:AB_1580723), rabbit polyclonal antibody against phospho Btk (Thermo Fisher Scientific Cat# 44‐1355G RRID:AB_2533598) and rabbit monoclonal antibody against phospho Akt (Abcam, Cat# ab81283, RRID:AB_2224551) for 2 h at room temperature. Biotinylated anti‐rabbit antibody and avidin conjugated with horseradish peroxidase [ImmPRESS™ HRP Anti‐Rabbit IgG (Peroxidase) Polymer Detection Kit made in Goat (Vector Laboratories Cat# MP‐7451 RRID:AB_2631198)] was used as a detection system. The colour reaction to the peroxidase was performed with DAB (Vector Laboratories, Cat# SK‐4105, RRID:AB_2336520). Protein expression was assessed by two independent pathologists in a quantitative manner based on the percentage of positively stained cells.
Statistical analysis
The number of animals in the group was calculated on the basis of preliminary studies. The power calculation was performed to calculate the sample size. Calculation of sample size revealed that a minimum of 10 samples per group were needed to have a power of 80% and a significance level (two‐tailed) of 0.05 (∆ = 24.96). Shapiro–Wilk's W test of normality was used for data distribution analysis. In all experiments, the mean values for 4–10 assays ± SD or median (minimum – maximum) depending on characteristic distribution was calculated. Multiple group comparisons were performed using one‐way ANOVA, and significant differences between the groups were assessed using the Tukey–Kramer test or non‐parametric Mann–Whitney U‐test. In order to evaluate correlations between the parameters studied, the Pearson correlation coefficient was used. Calculations were performed using GraphPad 6 Prism software (https://www.graphpad.com/scientific‐software/prism, RRID:SCR_002798). The differences were deemed statistically significant when P < 0.05. Quantifications of Western blots and measurements of fluorescence intensity were analysed using Image J 1.50a software (https://imagej.nih.gov, RRID:SCR_003070).
Materials
RPMI‐1640, McCoy's 5a medium, FBS, penicillin and streptomycin were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA). Erythropoietin β (NeoRecormon) was purchased from Roche (Basel, Switzerland). LFM‐A13 [α‐cyano‐b‐hydroxy‐b‐methyl‐N‐(2,5‐dibromophenyl)propenamide] was a product of Tocris (Bristol, UK). Stock solutions of LFM‐A13 were prepared in methanol and stored at −20°C. Due to the poor aqueous solubility of LFM‐A13, this compound was dissolved in 0.01% DMSO in PBS, according to the manufacturer's instructions. A similar quantity of DMSO was added to the control preparations. Acl was purchased from Active Biochem (Bonn, Germany). Filgrastim (Accofil) was a product of Accord Healthcare (Durham, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Erythropoietin–LFM‐A13 combination decreases EpoR expression
Previously, we proved that Epo has varied effects on cells dependent on the presence of the Epo receptor (Tankiewicz‐Kwedlo et al., 2016). Here, we wanted to examine whether the various effects would be maintained after incubation with Epo, LFM‐A13 and both of these compounds together. RT‐PCR analysis showed no change in EpoR mRNA expression upon Epo stimulation or LFM‐A13 treatment. While simultaneous incubation of DLD‐1 and HT‐29 cells with Epo + LFM‐A13 reduced EpoR mRNA expression compared with Epo and LFM‐A13 alone (Figure 1A). Western blot analysis revealed an increase in EpoR levels (measured as phospho/total ratio) after incubation with Epo compared with control cells only in DLD‐1 cells (Figure 1B). Confocal microscopy bio‐imaging of Epo receptors showed their localization in the membrane and cytoplasm of DLD‐1 (Figure 1C, D). It also confirmed differences between these lines in response to Epo (Figure 1B–D). Quantitative analysis of confocal data confirmed a significant increase in p‐Epo receptor levels in DLD‐1 cells after incubation with Epo compared with control cells, as well as compared with cells incubated with LFM‐A13 and Epo + LFM‐A13. LFM‐A13 and Epo + LFM‐A13 did not change the Epo receptor level compared with control levels in both DLD‐1 and HT‐29 cells. These results indicate that the Epo receptor is involved in the mechanism of action of LFM‐A13 alone and when it is combined with Epo.
Figure 1.
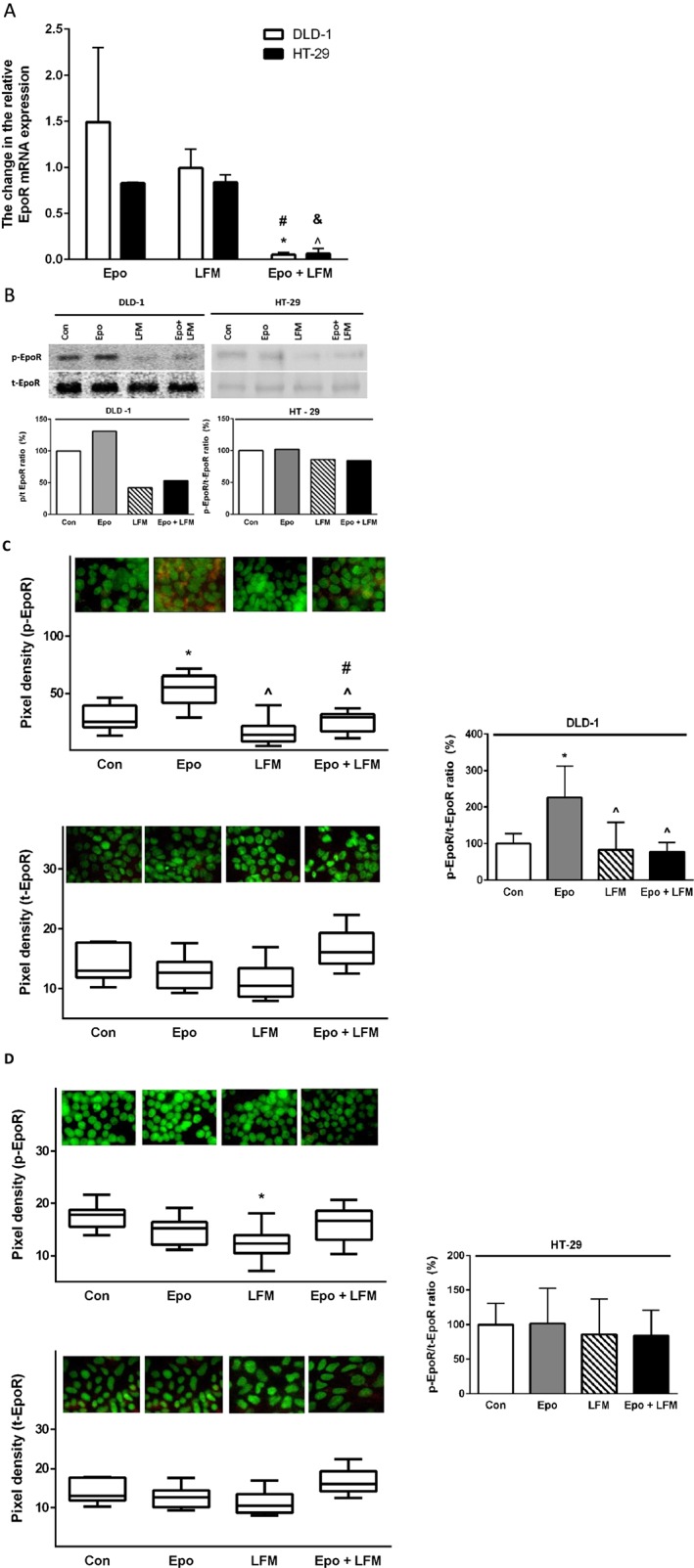
Epo receptor mRNA levels in DLD‐1 and HT‐29 cells treated with Epo, LFM‐A13 (LFM) and their combination. Results are presented as means ± SD, n = 6, ^P < 0.05 (vs. Epo), #P < 0.05 (vs. LFM‐A13) (A). Phosphorylated (p‐EpoR) and total (t‐EpoR) Epo receptor expression as determined by Western blot in DLD‐1 and HT‐29 cells treated with Epo (100 IU·mL−1), LFM‐A13 (100 μM) and their combination for 48 h. Samples used for electrophoresis consisted 20 μg of protein from six pooled independent cell extracts (n = 6). The band staining was quantified by densitometry. Quantitative analysis of the p‐EpoR/t‐EpoR ratio, expressed as a percentage of the control level (B). Phosphorylated and total EpoR expression in DLD‐1 (C) and HT‐29 cells (D); the whole nuclei (Hoechst, green), the recruitment of the receptor (red). Quantitative analysis of confocal data in DLD‐1 and HT‐29. The values given are a percentage of the control, which is 100%. Results are presented as median (minimum – maximum), n = 6. *P < 0.05 (vs. Epo), ^P < 0.05 (vs. Epo), #P < 0.05 (vs. LFM‐A13).
Erythropoietin–LFM‐A13 combination modulates intracellular pathways
To investigate the mechanisms mediating the antiproliferative effects of Epo + LFM‐A13 in DLD‐1 and HT‐29 cells, the levels of intracellular proteins like Btk and Akt were determined by RT‐PCR and Western blots. The incubation of DLD‐1 and HT‐29 cells with Epo + LFM‐A13 down‐regulated Btk mRNA expression compared with LFM‐A13, as well as compared with Epo (Figure 2A). A comparison of the two lines indicated significant differences in Btk mRNA expression in response to Epo. Using Western blots, we observed an increase in Btk expression after Epo stimulation compared with the control in DLD‐1 cells but this was only slight in HT‐29 cells. LFM‐A13 decreased the expression of this kinase in both lines. Moreover, Epo + LFM‐A13 caused the strongest reduction in Btk level (Figure 2B). In these experiments, the most effective concentration (100 IU·mL−1 of Epo and 100 μM of LFM‐A13) was selected for further studies.
Figure 2.
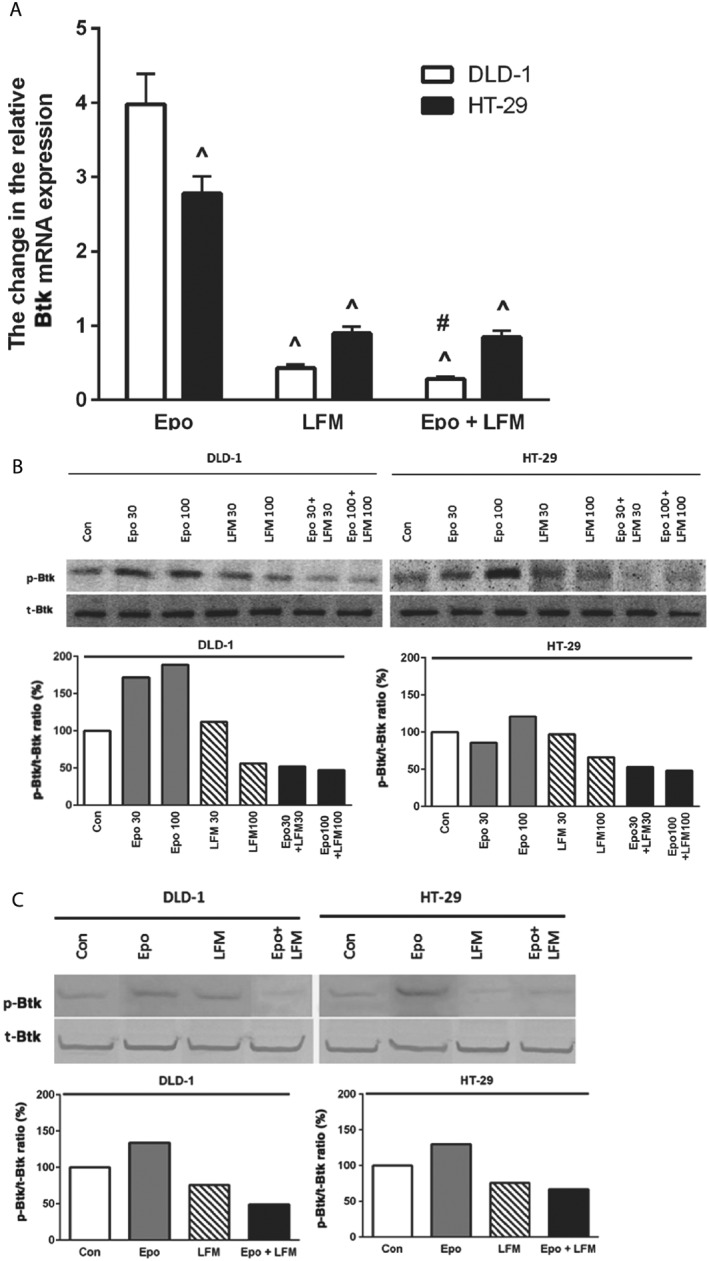
Btk mRNA levels in DLD‐1 and HT‐29 cells treated with Epo, LFM‐A13 (LFM) and their combination. Results are presented as means ± SD, n = 6. ^P < 0.05 (vs. Epo), #P < 0.05 (vs. LFM‐A13) (A). Phosphorylated Btk (p‐Btk) and total Btk (t‐Btk) expression as determined by Western blot in DLD‐1 and HT‐29 cells treated with Epo (30, 100 IU·mL−1), LFM‐A13 (LFM 30, 100 μM) and their combination for 48 h (Btk) and 5 min (p‐Btk). Samples used for electrophoresis consisted of 20 μg of protein from six pooled independent cell extracts (n = 6). Band staining was quantified by densitometry. Quantitative analysis of the p‐Btk/t‐Btk ratio, expressed as a percentage of the control level (B). Phosphorylated Btk (p‐Btk) and total Btk (t‐Btk) expression as determined by Western blot in DLD‐1 and HT‐29 cells treated with the most effective concentration of Epo (100 IU·mL−1), LFM‐A13 (LFM 100 μM) and their combination for 48 h (Btk) and 5 min (p‐Btk). Samples used for electrophoresis consisted of 20 μg of protein from six pooled independent cell extracts (n = 6). Band staining was quantified by densitometry. Quantitative analysis of the p‐Btk/t‐Btk ratio, expressed as a percentage of the control level (C).
We observed that simultaneous treatment with Epo and LFM‐A13 reduced Btk expression both in DLD‐1 and HT‐29 cells (Figure 2B, C).
It has been demonstrated that Akt activation is closely associated with chemoresistance in colon cancer (Zhang et al., 2013). Therefore, in a further step, we assessed Akt expression. It is well known that the Btk kinase family may regulate Akt activity. As in the case of Btk mRNA, we observed a difference in the expression of Akt mRNA in the cell lines examined (Figure 3A). RT‐PCR analysis showed that Epo + LFM‐A13 down‐regulated Akt mRNA expression compared with Epo and LFM‐A13 in DLD‐1 cells. Similar results were observed in the HT‐29 cell line (Figure 3A). Western blots showed that Akt levels were increased after incubation with Epo, and dropped below control values after Epo + LFM‐A13 treatment in both cell lines (Figure 3C). These results were confirmed by confocal microscopy. Incubation of DLD‐1 cells with Epo led to a significant increase in Akt levels compared with control cells, LFM‐A13 and Epo + LFM‐A13. Similar changes, although not so intense, were observed in HT‐29 cells (Figure 3D). Epo treatment increased Akt levels in HT‐29 cells compared with control cells, LFM‐A13 and Epo + LFM‐A13. These results clearly indicate that Epo + LFM‐A13 exhibit antitumor activity by a mechanism dependent on Btk.
Figure 3.
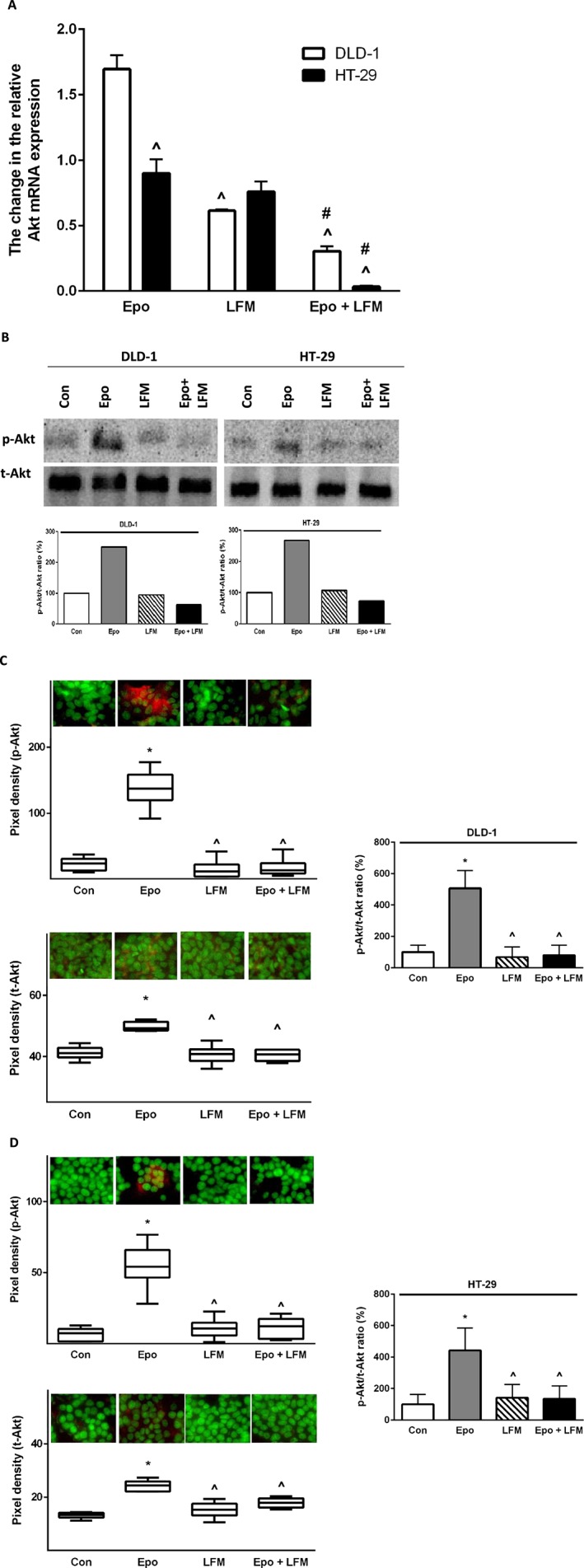
Akt mRNA levels in DLD‐1 and HT‐29 cells after 24 h incubation with Epo (100 IU·mL−1), LFM‐A13 (LFM 100 μM) and their combination. Results are presented as means ± SD, n = 6. ^P < 0.05 (vs. Epo), #P < 0.05 (vs. LFM‐A13) (A). (B) Phosphorylated Akt (p‐Akt) and total Akt (t‐Akt) expression as determined by Western blot in DLD‐1 and HT‐29 cells treated with Epo (100 IU·mL−1), LFM‐A13 (LFM 100 μM) for 5 min (p‐Akt) and 48 h (t‐Akt). Samples used for electrophoresis consisted of 20 μg of protein from six pooled independent cell extracts (n = 6). Band staining was quantified by densitometry. Quantitative analysis of the p‐Akt/t‐Akt ratio, expressed as a percentage of control level (B). Phosphorylated and total Akt expression in DLD‐1 (C) and HT‐29 cells (D); the whole nuclei (Hoechst, green), Akt recruitment (red). Quantitative analysis of confocal data in DLD‐1 and HT‐29. The values given are a percentage of the control which is 100%. Results are presented as median (minimum – maximum), n = 6. *P < 0.05 (vs. Epo), ^P < 0.05 (vs. Epo).
Antimitotic activity of the erythropoietin–LFM‐A13 combination against colon cancer cells
Confocal microscopy analysis demonstrated that LFM‐A13 alone and in conjunction with Epo prevents the normal process of microtubule assembly during mitosis. DLD‐1 and HT‐29‐treated cells developed abnormal monopolar mitotic spindles with highly dense and hyperextended microtubules (Figure 4A). These effects of LFM‐A13 are consistent with the known function of PLK1 in the organization of the spindles during mitosis, which is essential for the proper alignment and segregation of chromosomes. Western blot analysis revealed a down‐regulation of PLK1 induced by LFM‐A13 in both DLD‐1 and HT‐29 cells. A further decrease in PLK1 expression was observed after Epo + LFM‐A13 incubation (Figure 4B).
Figure 4.
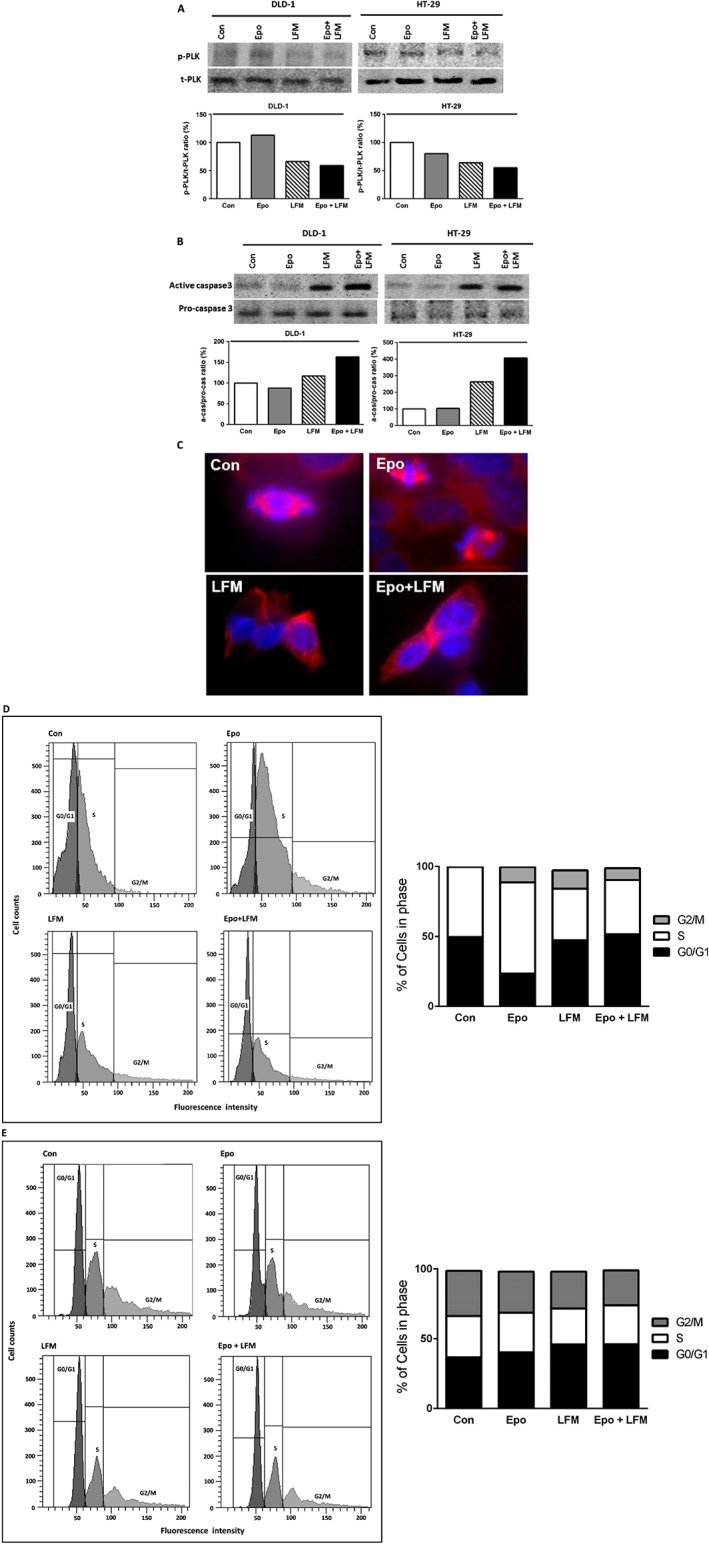
Phosphorylated PLK1 (p‐PLK) and total PLK1 (t‐PLK) expression as determined by Western blot in DLD‐1 and HT‐29 cells treated with Epo (100 IU·mL−1), LFM‐A13 (LFM 100 μM) and their combination for 48 h. Samples used for electrophoresis consisted of 20 μg of protein from six pooled cell extracts (n = 6). The band staining was quantified by densitometry. Bands of phospho‐proteins are normalized to respective total proteins (A). Active and pro‐caspase 3 expression as determined by Western blot in DLD‐1 and HT‐29 cells treated with Epo, LFM‐A13 for 48 h. Samples used for electrophoresis consisted of 20 μg of protein from six pooled independent cell extracts (n = 6). Band staining was quantified by densitometry. Bands of phospho‐proteins were normalized to respective total proteins (B). Effects of LFM‐A13 on spindle assembly in DLD‐1 cells. LFM‐A13‐treated DLD‐1 cells showed aberrant microtubule assembly during mitosis and developed abnormal monopolar mitotic spindles with highly dense and hyperextended microtubules. Orange, γ‐tubulin; red, α‐Tubulin; blue, DNA/chromosomes (C). Effects of Epo, LFM‐A13 and simultaneous use of both compounds (Epo + LFM‐A13) on cell cycle progression of DLD‐1 (D) and HT‐29 cells (E). Cells were cultured in the presence of 100 IU·mL −1of Epo, 100 μM of LFM‐A13 and their combination for 24 h at 37°C and then examined by DNA flow cytometry, as described in the Methods section. Epo + LFM‐A13 treated DLD‐1 and HT‐29 cells show S phase fraction reduction and slight increase in phase G0/G1. Percentages of events at different stages in the cell cycle are shown.
Next, we used flow cytometry to examine the effects of LFM‐A13 alone and in combination with Epo on cell cycle progression of DLD‐1 and HT‐29 cells after 24 h. The G1/S check point is the most critical for control of cell proliferation via intracellular and extracellular signals related to the transportation and integration of molecules into the nucleus (Skotheim et al., 2008). In DLD‐1 cells, the percentage of S phase fraction was reduced from 50% (untreated control cells) to 38.8% following treatment with Epo + LFM‐A13 (Figure 4C). This was accompanied by a concomitant increase in the G0/G1 phase. A similar result was obtained in HT‐29 cells (Figure 4D). We also found that apoptosis induced by Epo + LFM‐A13 was associated with caspase‐3 activation. We confirmed that Epo used together with LFM‐A13 potentiated the antimitotic activity of the latter (Figure 4E).
The erythropoietin–LFM‐A13 combination has beneficial effects in eliminating colon cancer
In the in vitro study, DLD‐1 cell number decreased after 48 h incubation with Epo + LFM‐A13 compared with the control group, Epo and LFM‐A13 (Figure 5A). Similar results were obtained in HT‐29 cells (Figure 5D). However, Epo did not cause a significant increase in cell number because of the low number or lack of Epo receptors and a stronger effect of LFM‐A13 was observed compared with the control. The addition of Epo to LFM‐A13 intensified the impact of LFM‐A13 on both DLD‐1 (Figure 5A) and HT‐29 cells (Figure 5D). The results indicate that Epo may act as a chemosensitizer.
Figure 5.
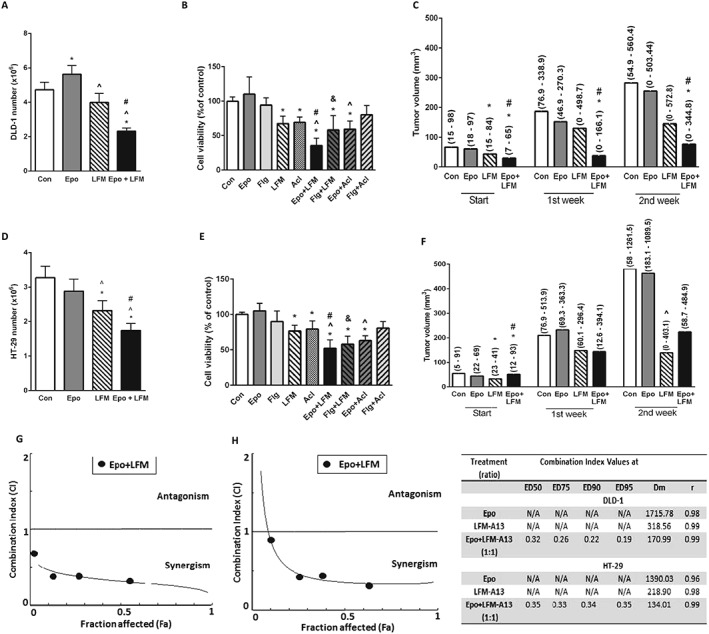
Impact of Epo and LFM‐A13 (LFM) and their combination on human colon models. Number of DLD‐1 (A) and HT‐29 (D) cells after 48 h incubation with Epo, LFM and their combination. Results are presented as means ± SD, n = 6, *P < 0.05 (vs. Con), ^P < 0.05 (vs. Epo), # P < 0.05 (vs. LFM‐A13). Comparison of Epo, filgrastim (Flg), LFM and Acl effects on the viability of DLD‐1 (B) and HT‐29 (E) cells. Results are presented as means ± SD, n = 6, *P < 0.05 (vs. Con), ^P < 0.05 (vs. Epo), # P < 0.05 (vs. LFM‐A13), & P < 0.05 (vs. Flg). The effect of the combined activity of Epo and LFM on tumour volume in DLD‐1 (C) and HT‐29 (F) xenografts. Start – before agent administration, 1, 2 – after first and second week of treatment. The results are presented as median values (minimum – maximum), n = 5–63. Statistical analysis of change in tumour volume in DLD‐1 (C) and HT‐29 (F) xenografts; *P < 0.05 (vs. Con), ^P < 0.05 (vs. Epo), # P < 0.05 (vs. LFM‐A13). The dual combination of Epo and LFM‐A13 in DLD‐1 (G) and HT‐29 (H) at a ratio of 1:1 indicated a mostly synergistic effect; however, the CI value was 0.9 for the lowest concentrations, indicating an additive effect in the HT‐29 line. The table summarizes the Cl values at ED50, ED75, ED90 and ED95 (right). It is worth noting that of all dual combinations, the most potent anticancer agent demonstrated marked synergy at almost all effect levels.
Numerous studies have demonstrated that the Btk inhibitor LFM‐A13 mediates antiproliferative and cytotoxic effects in cultured tumour cells (Uckun et al., 2007; Guo et al., 2014; Harrington et al., 2016). Btk participates in the activation of signalling pathways responsible for the process of cell maturation and viability. Our results confirmed the antiproliferative activity of LFM‐A13 in both DLD‐1 and HT‐29 cells. Similar results were obtained after the cells were treated with the selective Btk inhibitor Acl. We also demonstrated that the addition of Epo to LFM‐A13 or Acl had a greater antiproliferative effect compared with the control or when the latter was used alone (Figure 5B, E). Filgrastim, the recombinant non‐pegylated human granulocyte colony stimulating factor analogue, like Epo, failed to affect cell viability. Simultaneous use of Flg with LFM‐A13 significantly decreased cell proliferation compared with the control and also to LFM‐A13 alone. In turn, this effect was not observed after the incubation of DLD‐1 and HT‐29 cells with Flg and Acl used together. These results suggest that the mechanism of the effect of Epo + LFM‐A13 is dependent on both Btk‐ and JAK2. Epo and LFM‐A13 were tested at increasing concentrations, in four different sets, with the ratio of the two compounds kept constant. For the combinations tested, an additive or mostly synergistic interaction was noted at all effect levels at concentrations of 10 IU·mL−1 of Epo and 10 μM of LFM‐A13. All combination index (CI) values were less than 1, suggesting synergy when the combination ratios of Epo and LFM‐A13 in concentrations of 10 IU·mL−1 of Epo and 10 μM of LFM‐A13 were analysed (Figure 5G, H).
At the beginning of the in vivo experiment, the growth rate of the tumour in DLD‐1 and HT‐29 xenografts was determined. Initial tumour volume was similar and amounted to 83.70 (53.46–298.31) mm3 in DLD‐1 xenografts and 113.12 (51.96–317.47) mm3 in HT‐29 xenografts. However, in the group of HT‐29 xenografts, gains in tumour volume were greater. In the second week, tumour volume increased to 180.11 (54.92–560.40) mm3 in DLD‐1 xenografts and 719.05 (306.14–1261.50) mm3 in HT‐29 xenografts (Supporting Information Figure S1A). After modelling (2 weeks after tumour induction), X‐ray imaging confirmed the presence of subcutaneous soft tissue tumours near the lower limbs of the mouse (Supporting Information Figure S1B, C). Conventional two‐dimensional ultrasonic testing with colour Doppler ultrasound was performed to visualize masses measuring 9.00 × 9.52 mm subcutaneously. The tumour of each mouse was a hypoechogenic solid mass, with intralesional vascularization. Sonography diagnosis of tumours suspected of malignancy is shown in Supporting Information Figure S1D, E.
In DLD‐1 xenografts, tumour development was significantly inhibited in the group receiving Epo + LFM‐A13 compared with the control in the first and second week (Figure 5B, C). Despite the clear difference in tumour size observed in this group at the start compared with the control, the significant decrease in the size of the tumour is worthy of attention. A stronger anticancer effect was obtained after simultaneous administration of Epo with LFM‐A13 in Epo receptor‐positive DLD‐1 xenografts leading to total regression of tumour growth in 4/10 (40.00%) of treated animals. In HT‐29 xenografts, inhibition of tumour development was observed in the group receiving Epo + LFM‐A13 compared with Epo‐treated animals in the second week (Figure 5C, F).
Primary tumour samples (from the control and Epo + LFM‐A13‐treated group) were analysed for p‐EpoR, p‐Btk and p‐Akt expression. In control DLD‐1, eight tumours were strongly positive for p‐EpoR staining 8/8 (100%). Epo + LFM‐A13 samples showed a weak positive staining for p‐EpoR in 5/5 (100%). In control HT‐29, 6/8 (75%) were weakly positive for p‐EpoR, 1/8 (12.50%) was strongly positive and 1/8 (12.50%) was negative. In HT‐29 xenografts, Epo‐LFM‐A13 therapy led to weak positive staining in 7/8 (87.50%) and negative staining in 1/8 (12.50%). p‐EpoR expression was found in the membrane and cytoplasm of cancer cells (Figure 6B).
Figure 6.
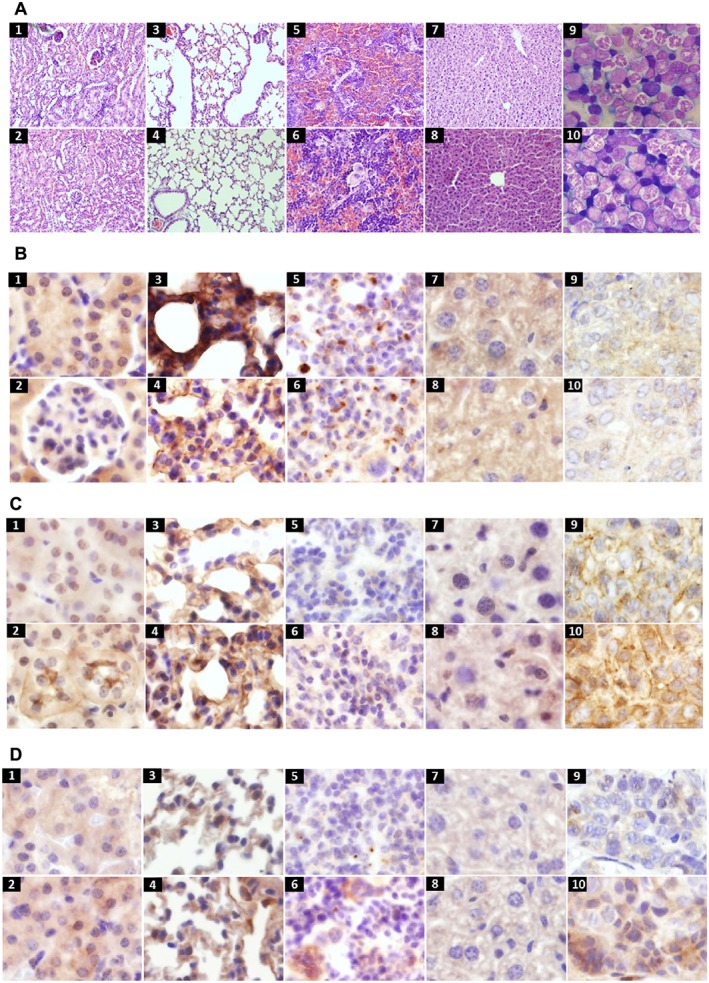
Histological samples from kidney (1, 2), lung (3, 4), spleen (5, 6), liver (7, 8) and bone marrow smears (9, 10) in DLD‐1 (top row) and HT‐29 (bottom row) xenografts 2 weeks after therapy (haematoxylin and eosin, original magnification, 400×) (A). Positive expression of p‐Epo receptors in membrane and cytoplasm of colon cancer xenografts in kidney (1, 2), lung (3, 4), spleen (5, 6), liver (7, 8) and tumour (9,10) in DLD‐1 (top row) and HT‐29 (bottom row) xenografts (original magnification, 400×) (B). Positive expression of p‐Btk in membrane and cytoplasm of colon cancer xenografts, organs in the order as above (original magnification, 400×) (C). Positive expression of p‐Akt in cytoplasm of colon cancer xenografts, organs in the order as above (original magnification, 400×) (D).
In the control DLD‐1, tumours were strongly positive for p‐Btk staining in 8/8 (100%). Epo + LFM‐A13 samples indicated weak positive staining for p‐Btk in 5/5 (100%). In control HT‐29, 6/8 (75%) samples were weakly positive for p‐Btk staining, and 2/8 (25%) were medium positive. In HT‐29 xenografts, Epo‐LFM‐A13 therapy led to weak positive p‐Btk staining in 6/8 (75%) and medium positive in 2/8 (25%) samples. p‐Btk expression was found in the membrane and cytoplasm of cancer cells (Figure 6C).
In the case of p‐Akt, 7/8 (87.50%) samples revealed strong positive staining and 1/8 (12.50%) medium positive in DLD‐1 xenografts. Treatment with Epo + LFM‐A13 led to medium positive p‐Akt staining in 4/5 (80%) and 1/5 (20%) were strongly positive. Similar results were observed in HT‐29 xenografts. In the control, 7/8 (87.50%) were weakly positive for p‐Akt staining, and 1/8 (12.50%) was medium positive. Epo‐LFM‐A13 therapy led to medium positive p‐Akt staining in 5/8 (62.50%) and strongly positive in 3/8 (37.50%) samples. p‐Akt expression was mainly found in the cytoplasm of cancer cells (Figure 6D). Immunohistopathological examination of multiple tissues (kidney, lung, spleen and liver) from the Epo + LFM‐A13‐treated group revealed weak positive staining of these proteins.
Lack of toxicity of the combination therapy of Epo and LFM‐A13
The toxicity profile of simultaneous Epo and LFM‐A13 treatment of DLD‐1 and HT‐29 xenografts was examined. Animals were monitored daily for weight, morbidity and mortality. Mice were killed on day 21 to determine the toxicity of Epo (600 IU·kg−1) and LFM‐A13 (10 mg·kg−1) admnistered together by examining their haematological profile and histopathological evaluation of multiple organs (liver, lung, kidney, spleen and bone marrow) for the presence of toxic lesions. None of the treated mice showed signs of morbidity.
Virtually all of the vehicle‐treated as well as Epo + LFM‐A13‐treated mice gained weight during the 21 day observation period. The average weight increased from 22.70 ± 1.92 and 19.35 ± 1.36 g at the start of the experiment to 22.81 ± 2.07 and 20.83 ± 1.04 g during the second week in DLD‐1 and HT‐29 xenografts respectively. No haematological toxicity was observed in the Epo + LFM‐A13‐treated group (Table 1). The morphological characteristics of animals with cancer undergoing therapy were similar to those of untreated animals. A greater response to Epo + LFM‐A13 therapy was observed in the DLD‐1 xenografts. Most of the parameters remained within the normal range in all animals. Two weeks of therapy caused an increase in white blood cell (WBC) and red blood cell (RBC), and a decrease in platelet number in DLD‐1‐xenografts compared with non‐treated animals. Similarly, an increase in the value of haemoglobin (HGB), haematocrit (HCT), mean corpuscular volume (MCV), mean corpuscular haemoglobin and mean corpuscular haemoglobin concentration was observed in DLD‐1 xenografts compared with control.
Table 1.
Haematological and bone marrow values after Epo‐LFM‐A13‐therapy
| DLD‐1 xenografts | HT‐29 xenografts | |||
|---|---|---|---|---|
| Con | Epo + LFM | Con | Epo + LFM | |
| WBC (103 mm−3) | 1.8 (1.4–2.2) | 2.2 (1.8–4)* | 3.5 (2.4–6) | 3.3 (2.3–5.3) |
| RBC (106 mm−3) | 8.9 (8.29–9.61) | 11.4 (11–12.2)* | 8.43 (8.17–11.7) | 10.7 (8.6–12.7)* |
| HGB (g·L−1) | 149 (142–157) | 205 (163–212)* | 150 (143–212) | 190 (151–204)* |
| HCT (%) | 45.1 ± 2.98 | 65.0 ± 2.39* | 42 ± 0.69 | 56.9 ± 7.1* |
| PLT (103 mm−3) | 890.7 ± 57.1 | 746.0 ± 73.6* | 782.4 ± 127.1 | 702.9 ± 30.7 |
| MCV (μm3) | 50 (50–53) | 56.5 (54–58)* | 50.5 (48–53) | 53.5 (50–57)* |
| MCH (pg) | 17 ± 0.31 | 17.8 ± 0.6* | 17.5 ± 0.65 | 17.5 ± 0.85 |
| MCHC (g·L−1) | 316 (308–320) | 340 (322–345)* | 324 (319–348) | 351 (319–358) |
| Proerythroblasts (%) | 0.6 (0–2.2) | 1.5 (0.4–3.6) | 2.3 (1–4) | 2.4 (1–3.4) |
| Basophilic erythroblast (%) | 6.6 (4–10.2) | 5.2 (2.6–8.2) | 5 (4–7.4) | 1.8 (0.8–4)* , # |
| Polychromatic erythroblast (%) | 6.4 (4.2–8.6) | 6.1 (2.2–9.4) | 9 (5.4–12) | 6 (4–8) |
| Orthochromatic erythroblast (%) | 2.8 (2–3.2) | 1.4 (0.4–3) | 5.4 (4.4–8) | 4.4 (4–5.4)# |
| Myeloblast (%) | 0.2 (0–1.6) | 0.2 (0–2) | 0.3 (0–1.2) | 1 (0.6–1.6) |
| Promyelocyte (%) | 1.6 (1.4–3.2) | 3.8 (2.4–5)* | 4.3 (3.4–9) | 3.4 (1.2–6) |
| Neutrophilic myelocyte (%) | 19.2 (14.2–22.2) | 23.6 (21.4–25.2) | 19.2 (18.2–19.6) | 22.4 (19.4–24.2)* |
| Neutrophilic metamyelocyte (%) | 18.4 (15.4–19.8) | 19.4 (17–21) | 14.8 (11.6–16.4) | 16.6 (12.4–21.6) |
| Neutrophilic band (%) | 15 (14.6–20.6) | 17.7 (15.2–19.4) | 15.7 (14.8–17.6) | 15.6 (13.2–18) |
| Neutrophil segmend (%) | 17 (7.8–30.8) | 14 (8.8–19) | 16.4 (13.2–19.4) | 15.2 (13–18.4) |
| Megacariocyte (%) | 0.6 (0.4–1) | 1.3 (0.4–2.4) | 1.3 (0.6–2.4) | 1 (0.4–2.6) |
| Lymphocyte (%) | 3.8 (3.2–4) | 2.9 (2–4) | 2 (1.4–2.6) | 2.8 (1.4–5.8) |
| Monocyte (%) | 0.4 (0.2–0.4) | 1.2 (0–1.8) | 0.8 (0.4–1.2) | 1.2 (0–2.4) |
The results are presented as mean values ± SD or median (minimum – maximum), n = 7–8;
P < 0.05 (vs. Con),
P < 0.05 (DLD‐1 vs. HT‐29).
Slightly smaller changes occurred in HT‐29 xenografts treated with Epo + LFM‐A13. An increase in the value of RBC, HGB, HCT and MCV was found in treated animals compared with untreated mice. These haematological results might suggest the presence of microclots (RBC HGB, HCT increase and PLT decrease); however, these disturbances have not been confirmed by histopathological examination of the parenchymal organs. No myeloid or lymphoid hypoplasia was encountered in the bone marrow of any of the Epo + LFM‐A13‐treated mice (Table 1). There were microerythroblasts in all bone marrow smears. The percentage of erythroid cells was several per cent, and white blood cells ranged from 75 to 80%. In all animals, a moderate number of megakaryocytes producing platelets were present. In DLD‐1 xenografts, an increased percentage of promyelocytes was found. In contrast, in HT‐29 xenografts, a decreased percentage of basophilic erythroblasts and orthochromatic erythroblasts and an increased percentage of neutrophilic myelocyte were found compared with the control group.
Histopathological examination of multiple tissues from the Epo + LFM‐A13‐treated group did not reveal any test article‐related toxic lesions (Figure 6A).
Discussion
In the present study, we showed, for the first time, that the simultaneous use of LFM‐A13 – the reversible inhibitor of Btk – and Epo has a synergistic anticancer effect on colon cancer cells both in in vitro as well as in vivo. This combination therapy could constitute a new scheme for colon cancer treatment. Herein, we reported that LFM‐A13 inhibits colon cancer cell growth. Moreover, the combination of Epo and LFM‐A13 is more effective than LFM‐A13 alone in both DLD‐1 and HT‐29 cells. Stronger inhibitory effects were observed in DLD‐1 than in HT‐29 cells, which seems to be associated with the higher expression of Epo receptors in DLD‐1 cells. Using real‐time RT‐PCR and confocal microscopy, we confirmed the existence of Epo receptors in DLD‐1 but little/no expression in HT‐29 cells.
An increasing body of experimental and clinical data in recent years supports a major role of Btk not only in B cell malignancies (Hendriks et al., 2014) but also in other solid tumours, including breast (Eifert et al., 2013), ovarian (Zucha et al., 2015) and prostate cancer (Guo et al., 2014). Btk has also previously been reported to be overexpressed in prostate cancer, which correlated with cancer grades (Guo et al., 2014). An up‐regulation of Btk is associated with low overall survival in ovarian cancer. It has also been shown that ovarian cancer cells, which highly express Btk, were cisplatin‐resistant (Zucha et al., 2015). The involvement of Btk in colon cancer development and progression is still not completely understood. There is only one report that indicates that Btk is abundantly expressed in colon cancer cell lines and tumour tissues (Grassilli et al., 2016). Grassilli et al. revealed that Btk, via p65Btk expression, is a novel and powerful oncogene acting downstream of the RAS/MAPK pathway and suggest that targeting it may be a promising therapeutic approach.
We confirmed the expression of Btk in both of the colon carcinoma cell lines analysed. Exposure to Epo resulted in increased expression of Btk in both cell lines, which is inconsistent with the observations of Schmidt et al. (2004). They demonstrated that Epo‐induced signal transduction is inhibited in cells lacking Btk. Herein, we observed that incubation with LFM‐A13 was the reason for the decreased expression of Btk (slighter in HT‐29), and a stronger down‐regulation of this kinase was obtained after the simultaneous use of Epo and LFM‐A13. Furthermore, similar results were obtained after combined administration of Epo with another Btk inhibitor – the highly selective aclacalabrutinib. It was previously shown that knockdown/blocking of Btk expression selectively inhibits the proliferation of prostate cancer cells (Guo et al., 2014), gastric cancer cells (Wang et al., 2016) and leukaemic cells (Cheng et al., 2014). This is in line with our results because treatment of DLD‐1 and HT‐29 with LFM‐A13 or Acl resulted in decreased Btk expression and hence inhibition of the proliferation of these cells. Further stronger antiproliferative effects were observed after simultaneous use of Epo with LFM‐A13 and Acl in both DLD‐1 and HT‐29 cells. Therefore, Epo appears to be a chemosensitizer and showed a synergistic antiproliferative effect with LFM‐A13. The dual combination of Epo and LFM‐A13 at a ratio of 1:1 mostly exerted a synergistic effect; however, the CI value was 0.9 for the lowest concentrations, indicating an additive effect in the HT‐29 cell line (Figure 5H). It is worth noting that of all the dual combinations, the most potent anticancer agent demonstrated marked synergy at almost all effect levels. Interestingly, the combination of Epo and LFM‐A13 was not antagonistic but was mostly synergistic, suggesting that these two compounds may have different mechanisms of action.
We presume that an increase in the number of LFM‐A13/Acl target proteins (p‐Btk) may enhance its antiproliferative activity and explain the stronger effects of the combined therapy tested. Moreover, LFM‐A13 efficiently inhibits Epo‐induced phosphorylation of the Epo receptors, JAK2, thus breaking the intracellular signalling pathway, which also may explain the results obtained (van den Akker et al., 2004). This hypothesis seems to confirm the results from our experiment using another haematopoietic growth factor – filgrastim (Flg). Simultaneous administration of Flg with LFM‐A13, but not with Acl, significantly reduced the proliferation of DLD‐1 and HT‐29 cells. The involvement of JAK2 in the mechanism of action of Flg could be inhibited by LFM‐A13 but not by the highly selective Btk inhibitor Acl. However, a more detailed role of JAK 2 in the mechanism of action of LFM‐A13 will be the object of our further investigations.
The Btk family kinases are closely involved in the interaction with the cellular apoptotic machinery and appear to be major regulators of apoptosis. Btk is an upstream activator of multiple antiapoptotic signalling molecules and networks, including the phosphatidylinositol‐3‐kinase/Akt pathway which is one of the strongest intracellular prosurvival signalling systems (Novero et al., 2014). It was previously demonstrated that Btk is required for the phosphorylation of Akt in B lymphocytes (Craxton et al., 1999). Another study has shown that an elevated expression of Btk in myeloma cells leads to Akt/WNT/b‐catenin–dependent up‐regulation of the key stem genes (octamer‐binding transcription factor 4, SOX2, NANOG and MYC) and enhanced self‐renewal (Yang et al., 2015). In addition, simultaneous use of Epo and LFM‐A13 even more strongly down‐regulates the Btk and without an effect on Akt signalling pathways, thereby much more effectively inducing apoptosis in DLD‐1 and HT‐29 cells compared with LFM‐A13 alone. Our experiments, carried out using the Western blot technique, revealed that LFM‐A13, by up‐regulating caspase‐3, increased the intensity of apoptosis and resulted in the inhibition of DLD‐1 and HT‐29 cell proliferation. Adding Epo to LFM‐A13 further enhanced these effects and induced a high level of apoptosis. It has been previously noted that inhibition of Btk using LFM‐A13 results in increased apoptosis in BT474 and MCF‐7 breast cancer cells (Kokabee et al., 2015). Moreover, Herman et al. (2011) demonstrated dose‐ and time‐dependent cytotoxicity of ibrutinib, a Btk inhibitor, in CLL through the caspase‐3‐dependent apoptotic pathway.
Our molecular studies are in line with previous observations and confirm that LFM‐A13 acts as a dual‐function inhibitor of Btk and PLK (Uckun et al., 2007). There are several reports of PLK overexpression in cancer cells including colorectal cancer (Reagan‐Shaw and Ahmad, 2005; Kanaji et al., 2006; Han et al., 2012). PLK is an M‐phase specific protein kinase which regulates the activation of the Cdc2‐cyclin B complex/mitosis promoting factor that regulates the maturation and function of centrosomes and spindles, chromosome segregation and initiation of anaphase by regulation of the anaphase‐promoting complex (Uckun, 2007). Therefore in our study, LFM‐A13 alone and also with Epo induced cell cycle arrest, prevented the normal process of microtubule assembly and caused gross mitotic aberrations in DLD‐1 and HT‐29 cells. Similar results were obtained by Kumar, who showed that LFM‐A13 inhibits bipolar spindle assembly formation in human cancer cell lines, including breast cancer and glioblastoma (Kumar and Kim, 2015).
Furthermore, we provide evidence that the therapeutic scheme used is effective not only in in vitro but also in vivo. The use of in vivo animal models to assess the potency of a treatment is generally preferred over in vitro test systems, since animal model assays have the ability to directly measure a product's functional activity (Stroncek et al., 2007). Therefore, our combination therapy was investigated in mouse models. Four‐week‐old mice Cby.Cg‐Foxn1nu/J were inoculated with DLD‐1 and HT‐29 colon cancer cells and treated with Epo and LFM‐A13 on their own or as a combination. We examined dynamic tumour growth over 2 weeks and found that HT‐29 xenograft tumours were larger than DLD‐1 ones but responded similarly to the Epo‐LFM‐A13 treatment.
LFM‐A13 at a nontoxic, low dose level of 10 mg·kg−1 caused a reduction of tumour burden. The anticancer activity of LFM‐A13 has been previously documented in the MMTVneu transgenic mouse model of HER2 positive breast cancer, in which LFM‐A13 delayed tumour progression (Uckun et al., 2007). Moreover, Epo in combination with LFM‐A13 markedly enhanced the anticancer activity of LFM‐A13 in DLD‐1 xenografts. During the first and second week in DLD‐1 xenografts, we noted a significant decrease in tumour volume in the Epo + LFM‐A13‐treated group compared with the control. Our in vivo studies confirmed the stronger anticancer effect of the simultaneous administration of Epo with LFM‐A13 in EpoR‐positive DLD‐1 xenografts, leading to total regression of tumour growth in 3/8 (37.50%) of the treated animals. This result accords with the ideal strategy to cure cancer, to kill all tumour cells (Eastman, 2017). LFM‐A13 was more potent when it was administered in combination with Epo than when administered separately.
The toxicity profile of the combined Epo and LFM‐A13 therapy use in mice was also examined. Virtually all of the vehicle‐treated as well as the Epo + LFM‐A13‐treated mice gained weight during the 21 day observation period. The haematological profiles of these mice were similar to those of vehicle‐treated control mice. We found that the number of platelets in the DLD‐1 xenografts was decreased compared with the control. This is in line with Rushworth et al., who reported that ibrutinib (a Btk inhibitor), at a concentration that causes cytotoxicity in lymphoproliferative disorders, significantly inhibited platelet aggregation primarily in response to collagen and ADP in blood samples obtained from patients with chronic lymphocytic leukaemia and mantle‐cell lymphoma (Rushworth et al., 2013). No haematological toxicity in the Epo + LFM‐A13‐treated group was observed. Histopathological examination of multiple tissues from the Epo + LFM‐A13‐treated group did not reveal any test article‐related toxic lesions. These results are consistent with other in vivo studies, in which high doses of LFM‐A13 (20–100 mg·kg−1 i.v.) did not induce nephrotoxicity, hepatotoxicity or changes in the blood chemistry profile. It is worth noting the beneficial effect of adding Epo, which not only enhanced the antitumor effect of LFM‐A13 but also improved the haematological profile.
Immunohistochemistry staining of p‐EpoR, p‐Btk and p‐Akt revealed a down‐regulation in the expression of these proteins only in tumour samples after Epo + LFM‐A13 treatment compared with the control. Evaluation of multiple organs (liver, lungs, kidneys and spleen) indicated similar expression of p‐EpoR, p‐Btk and p‐Akt compared with the results obtained in the control sample.
Taken together, this study provides unprecedented evidence that the simultaneous administration of Epo and LFM‐A13 significantly intensifies the anticancer activity of LFM‐A13. We have come to the conclusion that Epo, as an adjunct to LFM‐A13, has synergistic inhibitory effects on human colon cancer cell growth, indicating that this therapeutic scheme may improve treatment outcome as well as the quality of life in cancer patients. Thus, the outcomes of this study indicate that Epo and LFM‐A13 should be considered as a combination therapy for colorectal cancer.
Author contributions
A.T.K. developed the study concept, carried out in vitro and in vivo studies, statistical analysis of the results and interpretation of data for the work and drafted the manuscript. J.M.H. developed the study concept, carried out in vitro and in vivo studies and drafted the manuscript. T.D. carried out RT‐PCR research. K.P. supervised over RT‐PCR research and interpreted of data for the work. M.R. carried out bone marrow analysis. A.P. carried out immunohistochemical studies. A.S. carried out confocal imaging. T.K. carried out statistical analysis of the results. A.K. examined the spindle features. D.P. supervised over the entire study, interpreted of data for the work and provided critical revisions. All authors read and approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Growth rate of colon tumour in DLD‐1 and HT‐29 xerographs (A). 0 – beginning of observation, when the tumour was approx. 5.00 x 5.00 mm, 1, 2 – after the first and second week. The results are presented as median (minimum – maximum), n = 10. *P < 0.05 (0 vs. 1 and 0 vs. 2 in DLD‐1 xenografts); #P < 0.05 (1 vs. 2 in HT‐29 xenografts). ^P < 0.05 (0 vs. 1 and 0 vs. 2 in HT‐29 xenografts). X‐ray images taken of a DLD‐1 xenograft 14 days after subcutaneous injection of tumour cells. The primary tumour is indicated by the arrow (B, C). The conventional two‐dimensional ultrasonic testing with colour Doppler ultrasound was performed to visualize masses measuring 9.00 x 9.52 mm subcutaneously (D, E).
Acknowledgements
The research project was funded by grants nos N/ST/ZB/17/001/2228 and N/ST/MN/17/001/2233 financed by the Medical University of Bialystok, Poland, grants from the Leading National Science Centre in Bialystok no. 122/KNOW/2015 and National Science Centre, Poland, no. DEC‐2017/01/X/NZ5/00362. This study was conducted using equipment purchased by the Medical University of Bialystok as part of OP DEP 2007–2013, Priority Axis I.3, contract no. POPW.01.03.00‐20‐022/09. Medical University of Bialystok, no. N/ST/ZB/17/001/2228 to Anna Tankiewicz‐Kwedlo. Medical University of Bialystok, no. N/ST/MN/17/001/2233 to Justyna Magdalena Hermanowicz. Leading National Science Centre in Bialystok, no. 122/KNOW/2015 to Tomasz Kamiński.
Tankiewicz‐Kwedlo, A. , Hermanowicz, J. M. , Domaniewski, T. , Pawlak, K. , Rusak, M. , Pryczynicz, A. , Surazynski, A. , Kaminski, T. , Kazberuk, A. , and Pawlak, D. (2018) Simultaneous use of erythropoietin and LFM‐A13 as a new therapeutic approach for colorectal cancer. British Journal of Pharmacology, 175: 743–762. doi: 10.1111/bph.14099.
References
- Acs G, Acs P, Beckwith SM, Pitts RL, Clements E, Wong K et al (2001). Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 61: 3561–3565. [PubMed] [Google Scholar]
- Akinleye A, Chen Y, Mukhi N, Song Y, Liu D (2013). Ibrutinib and novel Btk inhibitors in clinical development. J Hematol Oncol 6: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Akker E, van Dijk TB, Schmidt U, Felida L, Beug H, Löwenberg B et al (2004). The BTK inhibitor LFM‐A13 is a potent inhibitor of Jak2 kinase activity. Biol Chem 385: 409–413. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The concise guide to PHARMACOLOGY 2017/18: catalytic receptors. Br J Pharmacol 174 (Suppl 1): S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The concise guide to PHARMACOLOGY 2017/18: enzymes. Br J Pharmacol 174 (Suppl 1): S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DK, Bookman MA, McGuire W, Bristow RE, Schilder JM (2007). Gynecologic Oncology Group. A phase I study of paclitaxel, topotecan, cisplatin and Filgrastim in patients with newly diagnosed advanced ovarian epithelial malignancies: a Gynecologic Oncology Group study. Gynecol Oncol 105: 667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Ma J, Guo A, Lu P, Leonard JP, Coleman M et al (2014). BTK inhibition targets in vivo CLL proliferation through its effects on B‐cell receptor signaling activity. Leukemia 28: 649–657. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P (1984). Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55. [DOI] [PubMed] [Google Scholar]
- Craxton A, Jiang A, Kurosaki T, Clark EA (1999). Syk and Bruton's tyrosine kinase are required for B cell antigen receptor‐mediated activation of the kinase Akt. J Biol Chem 274: 30644–30650. [DOI] [PubMed] [Google Scholar]
- Dicato M, Plawny L, Diederich M (2010). Anemia in cancer. Ann Oncol 21: 167–172. [DOI] [PubMed] [Google Scholar]
- Eastman A (2017). Improving anticancer drug development begins with cell culture: misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 8: 8854–8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eifert C, Wang X, Kokabee L, Kourtidis A, Jain R, Gerdes MJ et al (2013). A novel isoform of the B cell tyrosine kinase BTK protects breast cancer cells from apoptosis. Genes Chromosomes Cancer 52: 961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman L, Wang Y, Rhim JS, Bhattacharya N, Loda M, Sytkowski AJ (2006). Erythropoietin stimulates growth and STAT5 phosphorylation in human prostate epithelial and prostate cancer cells. Prostate 66: 135–145. [DOI] [PubMed] [Google Scholar]
- Feldman JP, Goldwasser R, Mark S, Schwartz J, Orion I (2009). A mathematical model for tumor volume evaluation using two‐dimensions. JAQM 4: 455–462. [Google Scholar]
- Grassilli E, Pisano F, Cialdella A, Bonomo S, Missaglia C, Cerrito MG et al (2016). A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS‐mediated transformation. Oncogene 35: 4368–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Liu R, Bhardwaj G, Yang JC, Changou C, Ma AH et al (2014). Targeting BTK/Etk of prostate cancer cells by a novel dual inhibitor. Cell Death Dis 5: e1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DP, Zhu QL, Cui JT, Wang PX, Qu S, Cao QF et al (2012). Polo‐like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Med Sci Monit 18: 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardee ME, Cao Y, Fu P, Jiang X, Zhao Y, Rabbani ZN et al (2007). Erythropoietin blockade inhibits the induction of tumor angiogenesis and progression. PLoS One 2: e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington BK, Gardner HL, Izumi R, Hamdy A, Rothbaum W, Coombes KR et al (2016). Preclinical evaluation of the novel BTK inhibitor acalabrutinib in canine models of B‐cell non‐Hodgkin lymphoma. PLoS One 11 (7): e0159607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks RW, Yuvaraj S, Kil LP (2014). Targeting Bruton's tyrosine kinase in B cel malignancies. Nat Rev Cancer 14: 219–232. [DOI] [PubMed] [Google Scholar]
- Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S et al (2011). Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI‐32765. Blood 117: 6287–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaji S, Saito H, Tsujitani S, Matsumoto S, Tatebe S, Kondo A et al (2006). Expression of polo‐like kinase 1 (PLK1) protein predicts the survival of patients with gastric carcinoma. Oncology 70: 126–133. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokabee L, Wang X, Sevinsky CJ, Wang WL, Cheu L, Chittur SV et al (2015). Bruton's tyrosine kinase is a potential therapeutic target in prostate cancer. Cancer Biol Ther 16: 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Kim J (2015). PLK‐1 targeted inhibitors and their potential against tumorigenesis. Biomed Res Int 2015: 705745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Mahajan S, Ghosh S, Sudbeck EA, Zheng Y, Downs S, Hupke M et al (1999). Rational design and synthesis of a novel anti‐leukemic agent targeting Bruton's tyrosine kinase (BTK), LFM‐A13 [alpha‐cyano beta‐hydroxy‐beta‐methyl‐N‐(2,5‐dibromophenyl) propenamide]. J Biol Chem 274: 9587–9599. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novero A, Ravella PM, Chen Y, Dous G, Liu D (2014). Ibrutinib for B cell malignancies. Exp Hematol Oncol 3: 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Bohle B, Alonso S, Mayol X, Salvans S, Grande L et al (2013). Preoperative administration of erythropoietin stimulates tumor recurrence after surgical excision of colon cancer in mice by a vascular endothelial growth factor‐independent mechanism. J Surg Res 183: 270–277. [DOI] [PubMed] [Google Scholar]
- Patel V, Balakrishnan K, Bibikova E, Ayres M, Keating MJ, Wierda WG et al (2017). Comparison of acalabrutinib, a selective Bruton tyrosine kinase inhibitor, with ibrutinib in chronic lymphocytic leukemia cells. Clin Cancer Res 23: 3734–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Wang Q, Liu H, Ye M, Wu X, Guo L (2016). EPHA3 regulates the multidrug resistance of small cell lung cancer via the PI3K/BMX/STAT3 signaling pathway. Tumour Biol 37: 11959–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Kung HJ (2000). Signaling network of the BTK family kinases. Oncogene 19: 5651–5661. [DOI] [PubMed] [Google Scholar]
- Reagan‐Shaw S, Ahmad N (2005). Polo‐like kinase (Plk) as a target for prostate cancer management. IUBMB Life 57: 677–682. [DOI] [PubMed] [Google Scholar]
- Rushworth SA, MacEwan DJ, Bowles KM (2013). Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 369: 1277–1278. [DOI] [PubMed] [Google Scholar]
- Schmidt U, van den Akker E, Parren‐van Amelsvoort M, Litos G, de Bruijn M, Gutiérrez L et al (2004). BTK is required for an efficient response to erythropoietin and for SCF‐controlled protection against TRAIL in erythroid progenitors. J Exp Med 199: 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara N, Tsuduki T, Ito J, Honma T, Kijima R, Sugawara S et al (2012). Jacaric acid, a linolenic acid isomer with a conjugated triene system, has a strong antitumor effect in vitro and in vivo . Biochim Biophys Acta 1821: 980–988. [DOI] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, Cross FR (2008). Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroncek DF, Jin P, Wang E, Jett B (2007). Potency analysis of cellular therapies: the emerging role of molecular assays. J Transl Med 5: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankiewicz‐Kwedlo A, Pawlak D, Domaniewski T, Buczko W (2010). Effect of erythropoietin, 5‐fluorouracil and SN‐38 on the growth of DLD‐1 cells. Pharmacol Rep 62: 926–937. [DOI] [PubMed] [Google Scholar]
- Tankiewicz‐Kwedlo A, Hermanowicz J, Surażynski A, Rożkiewicz D, Pryczynicz A, Domaniewski T et al (2016). Erythropoietin accelerates tumor growth through increase of erythropoietin receptor (EpoR) as well as by the stimulation of angiogenesis in DLD‐1 and Ht‐29 xenografts. Mol Cell Biochem 421: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankiewicz‐Kwedlo A, Hermanowicz JM, Surazynski A, Kwedlo W, Rozkiewicz D, Pawlak K et al (2017). Erythropoietin enhances the cytotoxic effect of hydrogen peroxide on colon cancer cells. Curr Pharm Biotechnol 18: 127–137. [DOI] [PubMed] [Google Scholar]
- Törnqvist E, Annas A, Granath B, Jalkesten E, Cotgreave I, Öberg M (2014). Strategic focus on 3R principles reveals major reductions in the use of animals in pharmaceutical toxicity testing. PLoS ONE 9: e101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uckun FM (2007). Chemosensitizing anti‐cancer activity of LFM‐A13, a leflunomide metabolite analog targeting polo‐like kinases. Cell Cycle 6: 3021–3026. [DOI] [PubMed] [Google Scholar]
- Uckun FM, Zheng Y, Cetkovic‐Cvrlje M, Vassilev A, Lisowski E, Waurzyniak B et al (2002). In vivo pharmacokinetic features, toxicity profile, and chemosensitizing activity of alpha‐cyano‐beta‐hydroxy‐beta‐methyl‐N‐(2,5‐dibromophenyl)propenamide (LFM‐A13), a novel antileukemic agent targeting Bruton's tyrosine kinase. Clin Cancer Res 8: 1224–1233. [PubMed] [Google Scholar]
- Uckun FM, Dibirdik I, Qazi S, Vassilev A, Ma H, Mao C et al (2007). Anti‐breast cancer activity of LFM‐A13, a potent inhibitor of Polo‐like kinase (PLK). Bioorg Med Chem 15: 800–814. [DOI] [PubMed] [Google Scholar]
- Vijayan V, Baumgart‐Vogt E, Naidu S, Qian G, Immenschuh S (2011). Bruton's tyrosine kinase is required for TLR‐dependent heme oxygenase‐1 gene activation via Nrf2 in macrophages. J Immunol 187: 817–827. [DOI] [PubMed] [Google Scholar]
- Wang JD, Chen XY, Ji KW, Tao F (2016). Targeting BTK with ibrutinib inhibit gastric carcinoma cells growth. Am J Transl Res 8: 3003–3012. [PMC free article] [PubMed] [Google Scholar]
- Westenfelder C, Baranowski RL (2000). Erythropoietin stimulates proliferation of human renal carcinoma cells. Kidney Int 58: 647–657. [DOI] [PubMed] [Google Scholar]
- Yang Y, Shi J, Gu Z, Salama ME, Das S, Wendlandt E et al (2015). Bruton tyrosine kinase is a therapeutic target in stem‐like cells from multiple myeloma. Cancer Res 75: 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Fujita Y, Matsuo T, Koinuma S, Hara S, Tazaki A et al (2003). Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 24: 1021–1029. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang L‐L, Shen L, Xu X‐M, Yu H‐G (2013). Regulation of AKT gene expression by cisplatin. Oncol Lett 5: 756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucha MA, Wu AT, Lee WH, Wang LS, Lin WW, Yuan CC et al (2015). Bruton's tyrosine kinase (BTK) inhibitor ibrutinib suppresses stem‐like traits in ovarian cancer. Oncotarget 6: 13255–13268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Growth rate of colon tumour in DLD‐1 and HT‐29 xerographs (A). 0 – beginning of observation, when the tumour was approx. 5.00 x 5.00 mm, 1, 2 – after the first and second week. The results are presented as median (minimum – maximum), n = 10. *P < 0.05 (0 vs. 1 and 0 vs. 2 in DLD‐1 xenografts); #P < 0.05 (1 vs. 2 in HT‐29 xenografts). ^P < 0.05 (0 vs. 1 and 0 vs. 2 in HT‐29 xenografts). X‐ray images taken of a DLD‐1 xenograft 14 days after subcutaneous injection of tumour cells. The primary tumour is indicated by the arrow (B, C). The conventional two‐dimensional ultrasonic testing with colour Doppler ultrasound was performed to visualize masses measuring 9.00 x 9.52 mm subcutaneously (D, E).