

Abstract
Background and Purpose
We previously showed that nociceptin/orphanin FQ opioid peptide (NOP) receptor agonists attenuate the expression of levodopa‐induced dyskinesia in animal models of Parkinson's disease. We now investigate the efficacy of two novel, potent and chemically distinct NOP receptor agonists, AT‐390 and AT‐403, to improve Parkinsonian disabilities and attenuate dyskinesia development and expression.
Experimental Approach
Binding affinity and functional efficacy of AT‐390 and AT‐403 at the opioid receptors were determined in radioligand displacement assays and in GTPγS binding assays respectively, conducted in CHO cells. Their anti‐Parkinsonian activity was evaluated in 6‐hydroxydopamine hemi‐lesioned rats whereas the anti‐dyskinetic properties were assessed in 6‐hydroxydopamine hemi‐lesioned rats chronically treated with levodopa. The ability of AT‐403 to inhibit the D1 receptor‐induced phosphorylation of striatal ERK was investigated.
Key Results
AT‐390 and AT‐403 selectively improved akinesia at low doses and disrupted global motor activity at higher doses. AT‐403 palliated dyskinesia expression without causing sedation in a narrow therapeutic window, whereas AT‐390 delayed the appearance of abnormal involuntary movements and increased their duration at doses causing sedation. AT‐403 did not prevent the priming to levodopa, although it significantly inhibited dyskinesia on the first day of administration. AT‐403 reduced the ERK phosphorylation induced by SKF38393 in vitro and by levodopa in vivo.
Conclusions and Implications
NOP receptor stimulation can provide significant albeit mild anti‐dyskinetic effect at doses not causing sedation. The therapeutic window, however, varies across compounds. AT‐403 could be a potent and selective tool to investigate the role of NOP receptors in vivo.
Abbreviations
- AIMs
abnormal involuntary movements
- ALO
axial, limb and orolingual
- AT‐390
((1‐(1‐(cis‐4‐isopropylcyclohexyl)piperidin‐4‐yl)‐1H‐indole‐2,3‐diyl)dimethanol)
- AT‐403
(2‐(1‐(1‐((1s,4s)‐4‐isopropylcyclohexyl)piperidin‐4‐yl)‐2‐oxoindolin‐3‐yl)‐N‐methylacetamide)
- L‐DOPA
levodopa
- LID
levodopa‐induced dyskinesia
- MPTP
1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyridine
- MSNs
medium‐sized spiny neurons
- N/OFQ
nociceptin/orphanin FQ
- NOP
nociceptin/orphanin FQ opioid peptide
- 6‐OHDA
6‐hydroxydopamine
- PD
Parkinson's disease
- SNr
substantia nigra reticulata
Introduction
The peptide nociceptin/orphanin FQ (N/OFQ) and the N/OFQ opioid peptide receptor (NOP receptor) (previously known as opioid receptor‐like 1 receptor) constitute a neuropeptide system showing numerous structural and functional analogies with the classical opioid systems (Toll et al., 2016). Nonetheless, the pharmacology of the N/OFQ‐NOP receptor is distinct from those of classical opioid receptor systems, since N/OFQ does not bind classical opioid receptors, and most classical opioid receptor ligands, such as naloxone, do not bind the NOP receptor (Calo et al., 2000; Zaveri, 2016). Because of its widespread distribution in the CNS, the N/OFQ‐NOP receptor system is key in the regulation of multiple central functions, such as sensory nociceptive processing, mood, food intake, pain, reward and locomotion. Moreover, the N/OFQ‐NOP receptor system is involved in various neuropsychiatric disorders such as neuropathic pain (Lin and Ko, 2013), anxiety and depression (Gavioli and Calo, 2013), drug abuse (Cippitelli et al., 2016; Lutfy and Zaveri, 2016) and Parkinson's disease (Marti et al., 2005; Viaro et al., 2008).
As far as Parkinson's disease is concerned, preclinical models disclosed a potential role for NOP receptor agonists as anti‐dyskinetic agents (Marti et al., 2012). Acute central administration of N/OFQ or systemic administration of the small molecule NOP receptor agonist Ro 65‐6570 attenuated the severity of dyskinesia in 6‐hydroxydopamine (6‐OHDA) hemi‐lesioned rats and 1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyridine (MPTP)‐treated macaques chronically treated with levodopa (L‐DOPA). Dyskinesia represents a major complication of L‐DOPA pharmacotherapy of Parkinson's disease (Bastide et al., 2015) and there are currently no drugs that can prevent dyskinesia development when chronically combined with L‐DOPA in de novo patients with Parkinson's disease (Huot et al., 2013; Bastide et al., 2015). Therefore, the identification of anti‐dyskinetic drugs is an unmet clinical need. As the anti‐dyskinetic effect of NOP agonists was observed in rats after acute administration at doses that did not cause hypolocomotion or sedation (Marti et al., 2012), effects of typical NOP receptor agonists (Zaveri, 2016), we hypothesized that NOP agonists may exert specific anti‐dyskinetic effects and be clinically useful in Parkinson's disease patients that have already become dyskinetic due to chronic L‐DOPA therapy. In our first proof‐of‐concept study, we used the NOP agonist Ro 65‐6570, which is only modestly selective for the NOP receptor, as the systemically active agonist to replicate the effects of centrally administered N/OFQ. However, we did not specifically investigate whether N/OFQ or Ro 65‐6570 worsened Parkinsonian disabilities, which is worth investigating in view of the symptomatic effects of NOP receptor antagonists in 6‐OHDA hemi‐lesioned rats (Marti et al., 2004a, 2005, 2008; Volta et al., 2011) and other models of Parkinson's disease (Viaro et al., 2008; Visanji et al., 2008; Volta et al., 2010).
Further, we recently demonstrated that NOP receptor stimulation opposes dopamine D1 receptor signalling in striatum (Olianas et al., 2008; Marti et al., 2012). It is well known that up‐regulation of the D1 receptor signalling cascade in striatal medium‐sized spiny GABAergic neurons (MSNs) projecting to substantia nigra reticulata (SNr) (the so‐called direct pathway) underlies the appearance of dyskinesia (Bastide et al., 2015). In our previous study, we did not assess whether NOP receptor agonists, in addition to preventing expression of L‐DOPA‐induced dyskinesia (LID), a symptomatic effect, also attenuated the process of brain sensitization (priming) that underlies the development of dyskinesia when given chronically in combination with L‐DOPA.
We recently described two high affinity, highly selective NOP receptor agonists, AT‐390 and AT‐403, from two distinct chemical classes, which had greater than 100‐fold selectivity over the μ, δ and κ opioid receptors. We characterized these compounds for their functional activity and found them to be full agonists at the NOP receptor. AT‐403 was recently characterized as a potent non‐peptide surrogate of the natural endogenous peptide ligand N/OFQ, because it was found to have unbiased functional efficacy at the NOP G‐protein as well as the arrestin signalling pathway, like the natural ligand N/OFQ (Ferrari et al., 2017).
Here, we have investigated more fully the anti‐Parkinsonian and anti‐dyskinetic profiles of these two NOP receptor agonists, AT‐403 and AT‐390. In the first series of experiments, AT‐403 and AT‐390 were acutely administered to 6‐OHDA hemi‐lesioned rats to assess whether they improve or worsen Parkinsonian motor deficits. Next, AT‐403 and AT‐390 were acutely administered to 6‐OHDA hemi‐lesioned, L‐DOPA‐primed dyskinetic rats to confirm that NOP receptor stimulation attenuates LID expression. Thirdly, to determine whether NOP receptor stimulation attenuates priming to L‐DOPA and therefore the development of dyskinesia, AT‐403 was chronically administered to 6‐OHDA hemi‐lesioned rats in combination with L‐DOPA for 20 days. Finally, to investigate whether AT‐403 could specifically affect signalling pathways underlying dyskinesia, phosphorylation levels of striatal ERK1 and ERK2, a D1 receptor‐dependent biochemical correlate of LID (Valjent et al., 2005; Santini et al., 2007), were monitored both in vitro and ex vivo.
Methods
Biochemical assays
The binding affinities of AT‐403 and AT‐390 for the opioid receptors were determined in radioligand competition experiments using human opioid receptor‐transfected CHO cells, as previously reported (Adapa and Toll, 1997; Zaveri et al., 2004). [3H]N/OFQ (130 Ci·mmol−1, 0.04 nM) was used as radioligand for the NOP receptor, [3H]U69,593 (120 Ci·mmol−1, 0.2 nM) for the κ receptor, [3H]DAMGO (120 Ci·mmol−1, 0.2 nM) for the μ receptor and [3H]Cl‐DPDPE (120 Ci·mmol−1, 0.2 nM) for the δ receptor. IC50 values were determined from at least six concentrations of test compound and calculated using Graphpad/Prism (ISI, San Diego, CA, USA). Ki values (nM) were derived from the Cheng‐Prusoff equation Ki = IC50/1 + [L]/Kd) where [L] is the concentration of the radioligand.
The functional efficacy of the compounds was determined by their ability to stimulate [35S]GTPγS binding to cell membranes and compared to the standard agonists N/OFQ (NOP), DAMGO (μ), U69,593 (κ) and DPDPE (δ), as previously described (Adapa and Toll, 1997; Zaveri et al., 2004; Spagnolo et al., 2008).
Animals
All animal care and experimental protocols were approved by the Italian Ministry of Health (license no. 170/2013‐B). Adequate measures were taken to minimize animal pain and discomfort. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male Sprague–Dawley rats (150 g, 6 week old; Envigo, S. Pietro al Natisone, Italy) were housed in a standard facility with free access to food (4RF21 standard diet; Mucedola, Settimo Milanese, Milan, Italy) and water and kept under regular lighting conditions (12 h dark/light cycle). Animals were housed in groups of five for a 55 × 33 × 20 cm polycarbonate cage (Tecniplast, Buguggiate, Varese, Italy) with a Scobis Uno bedding (Mucedola, Settimo Milanese, Milan, Italy) and environmental enrichments. At the end of the experiments, rats were killed with an overdose of isoflurane. Six male 2‐month‐old C57Bl6 mice were used for ERK studies in vitro. Mice were housed in a standard facility at Cardiff University, under regular conditions of light (12 h light/dark cycle), with food and water ad libitum. Experimenters were blinded to treatments.
ERK measurement in vitro
Adult male C57Bl6 mice were decapitated after cervical dislocation, and brain slices were freshly prepared according to the protocol described in Marti et al. (2012). The brains were rapidly removed and put on a cool glass plate filled with ice‐cold sucrose‐based dissecting solution (87 mM NaCl, 2.5 mM KCl, 7 mM MgCl2, 1 mM NaH2PO4, 75 mM sucrose, 25 mM NaHCO3, 10 mM D‐glucose, 0.5 mM CaCl2, 2 mM kynurenic acid), carbogenated (95% O2, 5% CO2) and subsequently mounted on the vibratome stage (Vibratome, VT1000S‐Leica Microsystems); 200‐μm‐thick slices were cut and transferred into a brain slice chamber (Brain slice chamber‐BSC1 – Scientific System design Inc., Mississauga, ON, Canada) and allowed to recover for 1 h at 32°C, with a constant perfusion of carbogenated artificial CSF (ACSF: 124 mM NaCl, 5 mM KCl, 1.3 mM MgSO4, 1.2 mM NaH2PO4, 25 mM NaHCO3, 10 mM D‐glucose, 2.4 mM CaCl2). The D1 receptor agonist SKF38393 (100 μM) was applied for 10 min in the presence of AT‐403 (30 nM) or vehicle. After fixation in 4% paraformaldehyde (PFA) for 15 min at room temperature, slices were rinsed three times in PBS and cryoprotected in 30% sucrose solution overnight at 4°C. On the following day, slices were further cut into 18‐μm‐thick slices using a cryostat (Leica CM1850) and mounted onto SuperFrost Plus slides (Thermo Scientific). Immunohistochemistry was performed following the protocol described in Papale et al. (2016): 1 h after blocking in 5% normal goat serum and 0.1% Triton X‐100 solution, slices were incubated overnight at 4°C with anti‐phospho‐p44/42 MAP kinase (Thr202/Tyr204) (1:1000, Cell Signaling Technology cat. #4370 L). Sections were then incubated with biotinylated goat anti‐rabbit IgG (1:200, Vector Laboratories, cat. #BA‐1000) for 2 h at room temperature. Detection of the bound antibodies was carried out using a standard peroxidase‐based method (ABC‐kit, Vectastain, Vector Labs), followed by a 3,3'‐diaminobenzidine (DAB) and H2O2 solution. Images were acquired from the striatum at 40× magnification using a brightfield microscope (Leica Macro/Micro Imaging System), and the number of pERK positive cells in the striatum was counted in each slice.
Unilateral 6‐OHDA lesion
The unilaterally 6‐OHDA lesioned rat (here referred to as hemi‐lesioned) is the most popular and best validated animal model of Parkinson's disease (Schwarting and Huston, 1996; Duty and Jenner, 2011) and was used in our experiments to assess the ability of AT‐403 and AT‐390 to improve Parkinsonian‐like motor deficits. Unilateral lesion of dopaminergic neurons was carried out under isoflurane anaesthesia as previously described (Marti et al., 2005). The neurotoxin 6‐OHDA (8μg free base, dissolved in 0.9% saline solution containing 0.02% ascorbic acid) was stereotaxically injected in the medial forebrain bundle according to the following coordinates from bregma: antero‐posterior = −4.4 mm, medio‐lateral = −1.2 mm, dorso‐ventral = −7.8 mm below dura (Paxinos and Watson, 1986). Animals were pretreated with antibiotics (Synulox™, 50 μL·kg−1, i.p.). The wound was sutured and infiltrated with 2% lidocaine solution (Esteve™). In order to select rats that were successfully hemi‐lesioned, 2 weeks after 6‐OHDA injection, rats were injected with a test dose of d‐amphetamine (5 mg·kg−1, i.p., dissolved in saline), and those performing >7 turns a minute in the direction ipsilateral to the lesion were enrolled in the study. This behaviour has been associated with >95% loss of striatal dopaminergic terminals (Marti et al., 2007) and extracellular dopaminergic levels (Marti et al., 2002).
Behavioural tests
Bar test
The bar test, also known as catalepsy test (Sanberg et al., 1988), measures the ability of the animal to react to an externally imposed position. The right and left forepaws were alternatively placed on three blocks of increasing heights (3, 6 and 9 cm). The immobility time (in seconds) of each forepaw on the blocks was recorded (the cut‐off for each step was set at 20 s).
Drag test
The drag test (Marti et al., 2005), modification of the ‘wheelbarrow test’ (Schallert et al., 1979), measures the ability of the animal to balance its body posture using the forelimbs, in response to backward dragging. Each rat was gently lifted from the abdomen leaving the forepaws on the table and dragged backwards at a constant speed of 20 cm·s−1 for a fixed distance of 1 m. Two different observers counted the number of touches made by each forepaw.
Rotarod test
This test measures the ability of the animal to run on a rotating cylinder and provides different information on a variety of motor parameters such as coordination, balance, muscle tone, gait and motivation to run (Rozas and Labandeira Garcia, 1997). The fixed‐speed rotarod test was employed using a previously validated protocol (Marti et al., 2004b, 2005). Animals were tested starting from 5 rpm, speed was stepwise increased by 5 rpm every 180 s, and total time spent on the rod was calculated.
L‐DOPA treatment and abnormal involuntary movements rating
Rats that successfully performed the amphetamine test were treated for 20 days with L‐DOPA (6 mg·kg−1 + benserazide 12 mg·kg−1, s.c., once daily) to induce abnormal involuntary movements (AIMs), a correlate of LID, as previously described (Mela et al., 2010; Bido et al., 2011; Marti et al., 2012; Mela et al., 2012; Paolone et al., 2015). This represents the best validated model of dyskinesia in rodents (Cenci et al., 1998; Cenci and Lundblad, 2007; Bastide et al., 2015). Rats were observed for 1 min, every 20 min, during the 3 h that followed L‐DOPA injection or until dyskinetic movements ceased. Dyskinetic movements were classified based in their topographical distribution into three subtypes (Cenci et al., 1998; Cenci and Lundblad, 2007): (i) axial AIM, that is, twisted posture or turning of the neck and upper body toward the side contralateral to the lesion; (ii) forelimb AIM, that is, jerky and dystonic movements and/or purposeless grabbing of the forelimb contralateral to the lesion; and (iii) orolingual AIM, that is, orofacial muscle twitching, purposeless masticatory movement and contralateral tongue protrusion. Each AIM subtype was rated on a frequency scale from 0 to 4 (1, occasional; 2, frequent; 3, continuous but interrupted by an external distraction; 4, continuous and not interrupted by an external distraction). In addition, the amplitude of these AIMs was measured on a scale from 0 to 4 based on a previously validated scale (Cenci and Lundblad, 2007). Axial, Limb and Orolingual (ALO) AIMs total value were obtained as the sum of the product between amplitude and frequency of each observation (Cenci and Lundblad, 2007). Therefore, the theoretical maximum ALO AIMs score is 432; to be considered fully dyskinetic, an animal has to score ≥ 100.
Western blot analysis
Dyskinetic rats were treated with saline or AT‐403 (0.1 mg·kg−1), and 15 min later with L‐DOPA (6 mg·kg−1 + benserazide 12 mg·kg−1, s.c.). Thirty minutes after L‐DOPA, rats were anesthetized with isoflurane, killed by decapitation and striata rapidly dissected, frozen on dry ice and stored at −80°C until analysis. Tissues were homogenized in lysis buffer (RIPA buffer, protease inhibitor cocktail and phosphatase inhibitor cocktail) and centrifuged at 18 000 × g at 4°C for 15 min. Supernatants were collected, and protein levels were quantified using the bicinchoninic acid protein assay kit (Thermo Scientific). Thirty micrograms of protein per sample were separated on a 4–12% gradient polyacrylamide precast gels (Bolt® 4–12% Bis‐Tris Plus Gels, Life Technologies) in a Bolt® Mini Gel Tank apparatus (Life Technologies). Proteins were then transferred onto polyvinyldifluoride membrane, blocked for 60 min with 5% non‐fat dry milk in 0.1% Tween20 Tris‐buffered saline and incubated overnight at 4°C with anti‐Thr202/Tyr204‐phosphorylated ERK1/2 (pERK) rabbit monoclonal antibody (Merck Millipore, cat. #05‐797R, 1:1000) or with anti‐ERK1/2 (totERK) rabbit polyclonal antibody (Merck Millipore cat. #06‐182, 1:5000). Membranes were washed, then incubated 1 h at room temperature with horseradish peroxidase‐linked secondary antibodies (Merck Millipore, cat. #12‐348, 1:5000). Immunoreactivity was visualized by enhanced chemiluminescence detection kit (Perkin Elmer), and images were acquired using the ChemiDoc MP System quantified using the ImageLab Software (Bio‐Rad). Membranes were then stripped and re‐probed with rabbit monoclonal anti‐tubulin antibody (Merck Millipore, cat. #04‐1117, 1:50 000). Data were analysed by densitometry, and the optical density of specific pERK or totERK bands was normalized to the corresponding tubulin levels.
Experimental protocols and design
Experiments were performed according to BJP guidelines. Overall, 149 rats and 6 mice were used in this study. Timelines of the different studies are illustrated in Figure 1.
Figure 1.
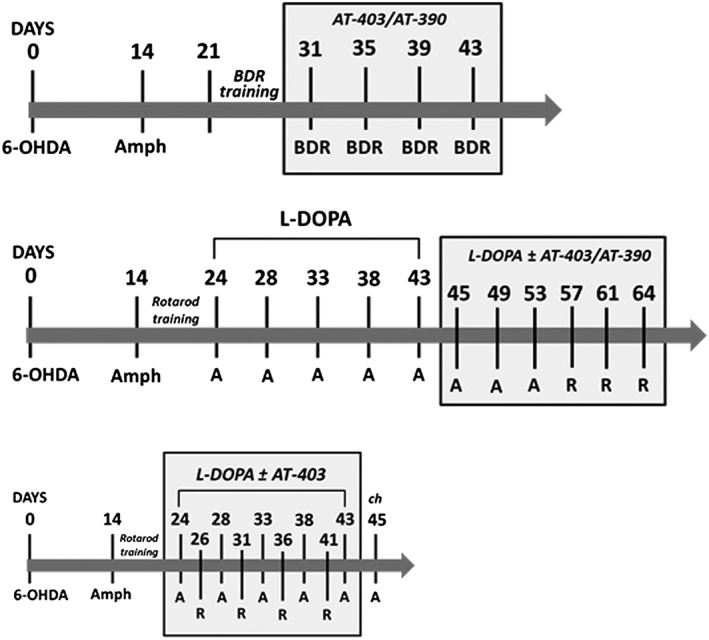
Timelines of studies in 6‐OHDA rats. Timelines of studies on motor disabilities (upper panel), dyskinesia expression (middle panel) and dyskinesia development (lower panel). BDR, bar, drag and rotarod sessions. A, AIMs scoring. R, rotarod sessions.
Motor behaviour in 6‐OHDA hemi‐lesioned, L‐DOPA naïve rats
Fifty‐two (52) rats were hemi‐lesioned with 6‐OHDA, 40 of which passed the amphetamine test after 2 weeks, meeting the selection criteria (Figure 1). Ten days later, these rats were subjected to the bar, drag and rotarod tests repeated in a fixed sequence as a training. When motor performance was reproducible (usually after 7–10 days), rats were randomized and treated with AT‐403 (0.001, 0.01, 0.03, 0.1 and 0.3 mg·kg−1), AT‐390 (0.03, 0.1, 1 and 3 mg·kg−1), the NOP receptor antagonist SB‐612111, AT‐403 + SB‐612111 or vehicle. Rats were tested three to four times, and a 3 day washout was allowed between treatments. Motor activity was assessed 30 and 90 min after AT‐403 administration, or 60, 120 and 180 min after AT‐390 administration, and expressed as absolute values.
LID expression
Seventy‐three (73) rats were hemi‐lesioned with 6‐OHDA, 60 of which passed the selection criteria (Figure 1). Fifty‐two (52) 6‐OHDA hemi‐lesioned rats were chronically treated with L‐DOPA, whereas eight were left untreated as a control for the ERK analysis. At the end of L‐DOPA treatment, 24 fully dyskinetic rats (ALO AIMs score ≥ 100) were randomized to L‐DOPA (6 mg·kg−1 plus benserazide 12 mg·kg−1, s.c.) in combination with AT‐403 (0.03 and 0.1 mg·kg−1), AT‐390 (0.3, 1 and 3 mg·kg−1) or vehicle. Each animal was tested three to four times, with a 3 day washout allowed between treatments. On separate days, motor performance was measured using the rotarod test, both before (OFF L‐DOPA) and 60 min after L‐DOPA administration (ON L‐DOPA), to evaluate whether the potential anti‐dyskinetic effect was associated with an improvement of global motor activity. This time point was chosen based on the ALO AIMs time course, which showed a peak 60–80 min after L‐DOPA administration (Mela et al., 2010, 2012; Bido et al., 2011; Marti et al., 2012; Paolone et al., 2015). Sixteen (16) additional, fully dyskinetic rats were used for ERK analysis and parsed into two groups, one treated with saline plus L‐DOPA and another with 0.1 mg·kg−1AT‐403 plus L‐DOPA.
LID induction
Twenty‐four (24) naïve rats were hemi‐lesioned with 6‐OHDA, 18 of which passed the selection criteria and were treated with L‐DOPA in combination with vehicle (6 mg·kg−1 plus benserazide 12 mg·kg−1, s.c.; n = 9) or AT‐403 (0.03 mg·kg−1, s.c., given 15 min before L‐DOPA; n = 9) for 20 days. ALO AIMs score was performed at days 1, 5, 10, 15 and 20. At the end of the 20 day treatment, all animals were allowed a 24 h washout and then challenged with L‐DOPA (dosage as above) in order to confirm that the anti‐dyskinetic effect was due a reduction of priming, rather than a symptomatic effect. During these 3 weeks, motor activity was assessed in four different occasions (days 3, 8, 14 and 19) with the rotarod test performed both OFF and ON L‐DOPA (60 min after injection).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Motor performance was expressed as absolute values (time on bar or rod, number of steps) and statistically analysed using PRISM 6.0 (GraphPad Software Inc., San Diego, CA, USA) with two‐way repeated measure (RM) ANOVA followed by the Bonferroni test (Figures 2 and 4) or one‐way ANOVA followed by the Newman–Keuls test (Figure 3). ALO AIMs score was expressed as absolute values and statistically analysed with two‐way RM ANOVA followed by the Bonferroni test (time course; Figures 5A, 6A and 7A–E) or one‐way ANOVA followed by the Newman–Keuls test (cumulative AIMs score; Figures 5B and 6B). Rotarod performance in dyskinetic animals was expressed as time on rod (in seconds) and analysed with two‐way ANOVA followed by the Bonferroni test (Figures 5C and 6D). In vitro ERK data (Figure 8A) were analysed using two‐way ANOVA followed by the Bonferroni test, whereas ex vivo ERK data were analysed by one‐way ANOVA followed by the Newman–Keuls test. Statistical significance was set at P < 0.05.
Figure 2.
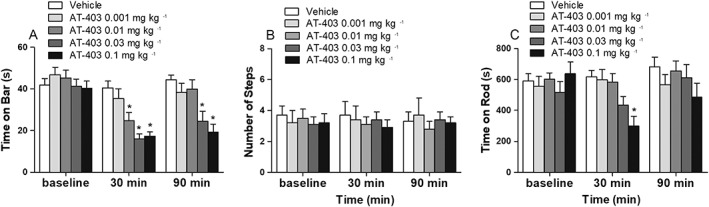
Effect of AT‐403 on Parkinsonian symptoms. Motor activity of 6‐OHDA hemi‐lesioned rats was evaluated in the bar (A), drag (B) and rotarod (C) tests, before and 30 and 90 min after vehicle or AT‐403 administration (0.001, 0.001, 0.03 and 0.1 mg·kg−1, s.c.). Data are expressed as immobility time (in seconds, A), number of steps (B) and time on rod (in seconds, C) and are means ± SEM; n = 14 determinations per group. *P < 0.05, significantly different from vehicle; two‐way RM ANOVA followed by the Bonferroni test for multiple comparisons.
Figure 4.
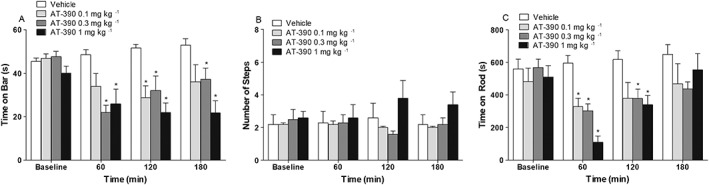
Effect of AT‐390 on Parkinsonian disabilities. Motor activity of 6‐OHDA hemi‐lesioned rats was evaluated in the bar (A), drag (B) and rotarod (C) tests, before and 60, 120 and 180 min after vehicle or AT‐390 administration (0.1, 0.3 and 1 mg·kg−1, s.c.). Data are expressed as immobility time (in seconds, A), number of steps (B) and time on rod (in seconds, C), and are means ± SEM; n = 9 (vehicle, AT‐390 0.3 mg·kg−1, AT‐390 1 mg·kg−1) determinations per group. The effect of AT‐390 0.1 mg·kg−1 was also assessed, although in a lower number of rats (n = 5). *P < 0.05, significantly different from vehicle; two‐way RM ANOVA followed by the Bonferroni test for multiple comparisons.
Figure 3.
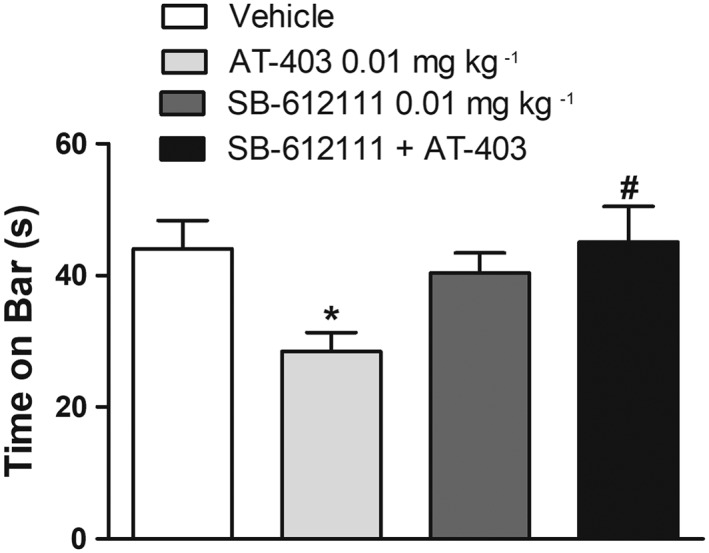
NOP receptor specificity of the AT‐403 effect. The NOP receptor antagonist SB‐612111 (0.01 mg·kg−1, s.c.) was challenged against a low dose of AT‐403 (0.01 mg·kg−1, s.c.) and the effect evaluated in the bar test. Data are expressed as immobility time (in seconds) and are means ± SEM; n = 9 (vehicle, AT‐403, SB‐612111) or n = 8 (AT‐403 + SB‐612111; one data‐set discarded due to experimental loss) determinations per group. *P < 0.05, significantly different from vehicle, #P < 0.05, significantly different from AT‐403; one‐way ANOVA followed by the Newman–Keuls test for multiple comparisons.
Materials
AT‐403 (Ferrari et al., 2017) and AT‐390 were synthesized at Astraea Therapeutics (Mountain View, CA, USA), according to methods reported by Zaveri et al. (2017). These compounds were dissolved in 1% CH3COOH 1 M and 3% DMSO in water. Radioligands [3H]N/OFQ, [3H]DAMGO and [3H]DPDPE were obtained from the National Institute of Drug Abuse Drug Supply Program. [3H]U69593 and [35S]GTPγS was purchased from Perkin Elmer. L‐DOPA, benserazide, 6‐OHDA hydrobromide and d‐amphetamine sulphate were purchased from Tocris Bioscience (Bristol, UK). L‐DOPA, benserazide and d‐amphetamine sulphate were dissolved in saline, and 6‐OHDA was dissolved in 0.02% ascorbic acid in saline.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/18 (Alexander et al., 2017a, 2017b).
Results
Receptor and GTPγS binding
AT‐403 and AT‐390 showed nanomolar affinity for the NOP receptor expressed in CHO cells (Ki of 1.1 and 0.9 nM, respectively) and significantly lower affinity for the other opioid receptors (Table 1). The selectivity ratios for NOP over classical opioid receptors were as follows: AT‐390, 59‐fold (μ), 125‐fold (δ) and 95‐fold (κ); AT‐403, 89‐fold (μ), 3700‐fold (δ) and 1420‐fold (κ). Both compounds were characterized for their functional activity in the [35S]GTPγS binding assay using a range of concentrations up to 10 μM. In this assay, AT‐403 and AT‐390 mimicked the stimulatory effects of N/OFQ at the NOP receptor, showing similar efficacy (110 and 104%, respectively) and slightly lower potency (see EC50 values in Table 1), suggesting that they are full agonists at the NOP receptor. Higher concentrations of AT‐403 and AT‐390 appeared to stimulate the μ opioid receptor, although with significantly lower potency (overall <60%). Potency selectivity ratios of AT‐403 and AT‐390 for NOP over the μ receptor were 33‐fold and 10‐fold, respectively, and maximal stimulation was 33 and 54% of positive controls respectively (Table 1).
Table 1.
Receptor binding assays (upper panel) and [35S]GTPγS functional activity (lower panel) of NOP agonists in membranes of CHO cells stably expressing the human recombinant NOP and classical μ, δ and κ opioid receptors
| Binding (K i nM) | ||||
|---|---|---|---|---|
| NOP | μ | δ | κ | |
| AT‐390 | 0.9 ± 0.32 | 53.13 ± 16.5 | 113.15 ± 9.8 | 85.28 ± 19.8 |
| AT‐403 | 1.13 ± 0.13 | 97.94 ± 15.0 | 4074.3 ± 17.3 | 1563.74 ± 203.9 |
| [35S]GTPγS assay | ||||||||
|---|---|---|---|---|---|---|---|---|
| NOP | μ | δ | κ | |||||
| EC50 | % Stim | EC50 | % Stim | EC50 | % Stim | EC50 | % Stim | |
| AT‐390 | 15.20 ± 0.4 | 110.1 ± 11.4 | 143.80 ± 0.6 | 54.3 ± 9.4 | 3847.0 ± 73 | 61.3 ± 6 | 534.3 ± 147.2 | 19.25 ± 1.3 |
| AT‐403 | 6.3 ± 1.42 | 104.6 ± 1.2 | 206.4 ± 78.9 | 33.5 ± 14.6 | – | – | – | – |
Experiments were performed as previously described (references in text). Values represent average ± SEM for three experiments conducted in triplicate. Standard ligands in binding and [35S]GTPγS experiments were N/OFQ (NOP), DAMGO (μ), DPDPE (δ) and U69,593 (κ). Affinity values (Ki, nM) were 0.12 ± 0.01 for N/OFQ, 2.96 ± 0.54 for DAMGO, 1.11 ± 0.07 for DPDPE and 1.05 ± 0.02 for U69,593. Efficacy (EC50 nM) of standard ligands in the [35S]GTPγS was 3.6 ± 0.7 for N/OFQ, 32.6 ± 4.06 for DAMGO, 8.98 ± 2.31 for DPDPE and 60.14 ± 7.45 for U69,593. Agonist stimulation by AT‐390 orAT‐403 is expressed as a percentage of that of the standard agonist (set to 100).
Effect on Parkinsonian symptoms
Previous studies showed that i.c.v. injection of N/OFQ has a biphasic effect on motor behaviour in naïve animals. To investigate whether a highly selective NOP receptor agonist could affect Parkinsonian‐like symptoms, AT‐403 was administered systemically (s.c.) in the 0.001–0.3 mg·kg−1 dose‐range, and motor activity evaluated by the bar, drag and rotarod tests 30 and 90 min after injection. After preliminary screening, we observed that rats (n = 3) treated with AT‐403 0.3 mg·kg−1 were unable to perform for 1 h after drug administration, due to profound, though reversible, sedation/hypo‐locomotion. Therefore, the study was continued with the lower doses.
In the bar test (Figure 2A), ANOVA showed a significant effect of treatment (F4,2 = 5.99), time (F2,130 = 57.33) and time × treatment interaction (F8,130 = 6.07) at the contralateral paw. Post hoc analysis revealed that AT‐403 caused a transient reduction (30 min time point) of the time on bar at the contralateral paw at 0.01 mg·kg−1, showing more profound and long‐lasting effects at the higher doses (Figure 2A). AT‐403 did not affect stepping activity at the contralateral paw (Figure 2B). In the rotarod test (Figure 2C), ANOVA did not show an overall effect of treatment (F4,2 = 1.45), but a significant effect of time (F2,130 = 6.34) and time × treatment interaction (F8,130 = 3.08). Post hoc analysis revealed a significant decrease of rotarod performance with the highest dose of AT‐403 (0.1 mg·kg−1) at 30 min after administration.
To investigate the NOP selectivity of AT‐403, the effects of systemic administration of AT‐403 (0.01 mg·kg−1, s.c.) in combination with the NOP receptor antagonist SB‐612111 were evaluated in the bar test (Figure 3). SB‐612111 was administered at a dose (0.01 mg·kg−1, s.c.) ineffective per se on motor behaviour (Marti et al., 2013). ANOVA showed a significant effect of treatment (F3,31 = 3.77) (Figure 3). Post hoc analysis revealed that AT‐403 reduced the time on bar and SB‐612111, ineffective by itself, prevented such effects of AT‐403.
To confirm that NOP receptor stimulation dually modulated Parkinsonian disabilities, behavioural observations were extended up to 180 min after AT‐390 injection (Figure 4). In a dose‐finding experiment, a profound and long‐lasting sedation and hypo‐locomotion was observed with 3 mg·kg−1 AT‐390. Animals were unable to perform for at least 2 h after drug administration and, therefore, the study was continued with lower doses. Very much like AT‐403, AT‐390 improved the immobility time at the contralateral paw (Figure 4A; treatment F3,3 = 5.39; time F3,84 = 11.09; time × treatment interaction F9,84 = 4.59). A transient reduction of immobility time was observed at 120 min after injection of 0.1 mg·kg−1 AT‐390, whereas a sustained (up to 180 min) inhibition was observed with both 0.3 and 1 mg·kg−1 AT‐390, with the effect of the former showing a tendency to fade over time. In the drag test, no significant effect was observed at the contralateral paw (Figure 4B). Finally, in the rotarod test (Figure 4C), transient inhibition was detected at 60 min with all doses (treatment F3,3 = 4.07; time F3,84 = 19.65; time × treatment interaction F9,84 = 5.54). The effect was particularly dramatic with 1 mg·kg−1 AT‐390. A milder, though significant, inhibition was still observed at 2 h with 0.3 and 1 mg·kg−1 AT‐390, whereas the effects had disappeared 3 h after treatment.
Effects on AIMs expression
To investigate whether NOP receptor stimulation prevents the expression of AIMs, AT‐403 and AT‐390 were administered 15 min before an L‐DOPA challenge in dyskinetic rats (Figure 5). AT‐403 caused a delay in LID appearance without significantly affecting the overall duration of the response to L‐DOPA (treatment F2,8 = 7.92; time F8,352 = 47.47; time ×treatment interaction F16,352 = 15.17; Figure 5A). ANOVA on itemized cumulative AIMs (Figure 5B) showed that AT‐403 at 0.1 mg·kg−1 markedly inhibited all AIM subtypes whereas AT‐403 at 0.03 mg·kg−1 cause a mild and selective inhibition of limb AIMs (Figure 5B). The beneficial effect of 0.1 mg·kg−1 AT‐403 was accompanied by overt sedation or hypo‐locomotion within the first hour after compound administration. This was also evident when rats were challenged on the rotarod. Indeed, control animals displayed dramatic reduction (75%) of the rotarod performance at peak dyskinesia (Figure 5C), whereas animals treated with 0.03 mg·kg−1 AT‐403 were significantly less impaired (55% reduction). Animals treated with 0.1 mg·kg−1 AT‐403 instead were inhibited as much as controls, indicating that the absence of dyskinetic movements was not associated with better rotarod performance.
Figure 5.
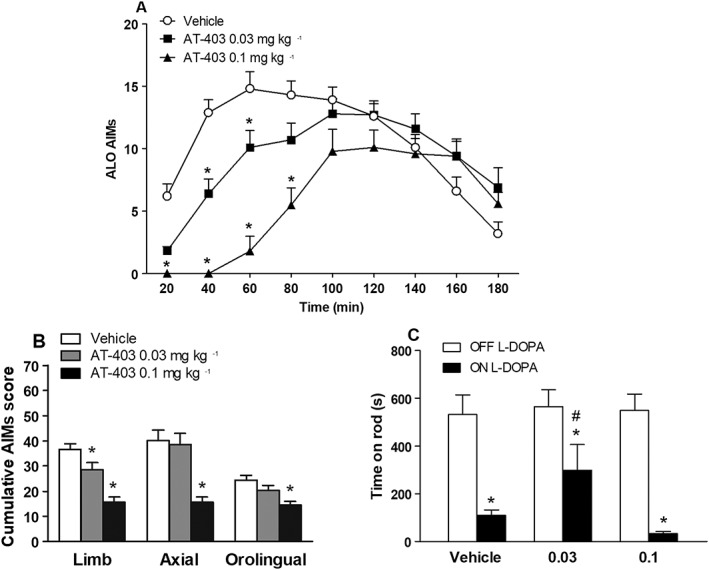
AT‐403 attenuated the expression of L‐DOPA‐induced dyskinesia. ALO AIMs were scored in 6‐OHDA hemi‐lesioned dyskinetic rats following challenge with L‐DOPA (6 mg·kg−1 plus benserazide 12 mg·kg−1, s.c., n = 16) combined with vehicle (s.c., n = 16), AT‐403 0.03 mg·kg−1 (s.c., n = 15; one data point was discarded as the animal did not respond to L‐DOPA) or AT‐403 0.1 mg·kg−1 (s.c., n = 16). Data (means ± SEM) are expressed as ALO AIMs score for each time point in absolute values (A) or as separate ALO AIMs scores over the 3 h observation period (i.e. cumulative scores calculated as the sum of scores at each time point; B). On a separate day, treatments were replicated in the same animals, and rotarod performance (time on rod in seconds, n = 10 determinations per group) was evaluated before and 60 min after drug administration (C). *P < 0.05, significantly different from vehicle. #P < 0.05, different from vehicle ON L‐DOPA. Statistical analysis was performed by two‐way RM (A) or two‐way (C) ANOVA followed by the Bonferroni test for multiple comparisons or one‐way ANOVA followed by the Newman–Keuls test for multiple comparisons (B).
AT‐390 significantly modified AIMs time course, although in a quite unexpected way (Figure 6A). Although 0.3 mg·kg−1 AT‐390 did not significantly affect the response to L‐DOPA, 1 mg·kg−1 AT‐390 delayed the appearance of AIMs and prolonged AIMs duration up to 240 min after L‐DOPA injection, such that the overall response to L‐DOPA was quantitatively unchanged (treatment F2,13 = 0.14) whereas time (F13,260 = 51.43) and the interaction of time × treatment (F26,260 = 9.34) were significant (Figure 6A). To confirm this profile, we injected a higher dose of AT‐390 (3 mg·kg−1) in a small number of rats (n = 4, Figure 6C). The effect was particularly dramatic as a further rightward shift in the time course was observed, with longer delay and duration of the AIMs response. As observed for AT‐403, a clear sedative or hypo‐locomotive effect was associated with the delayed response to 1 mg·kg−1 AT‐390. Indeed, even though AIMs were significantly reduced at 60 min after L‐DOPA injection, rotarod performance was markedly impaired (90%), similar to controls (Figure 6D).
Figure 6.
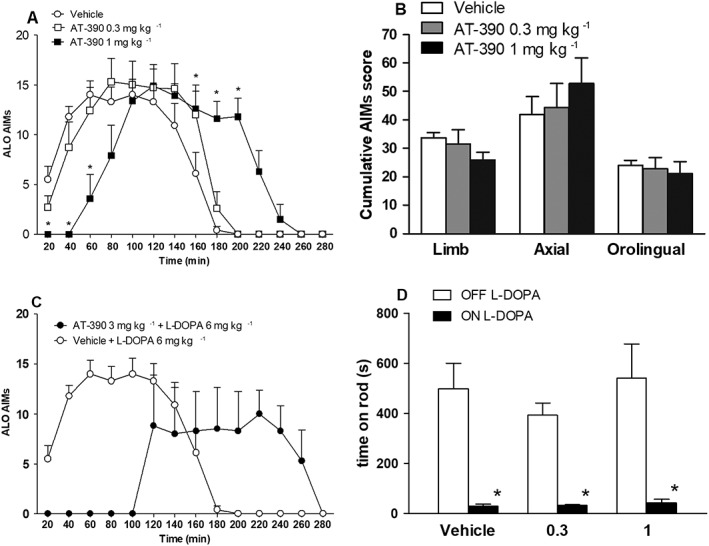
AT‐390 delayed and prolonged the expression of L‐DOPA‐induced dyskinesia. ALO AIMs were scored in 6‐OHDA hemi‐lesioned dyskinetic rats following challenge with L‐DOPA (6 mg·kg−1 plus benserazide 12 mg·kg−1, s.c.) combined with vehicle or AT‐390 (0.3 or 1 mg·kg−1, s.c., n = 8 determinations per group). The effect of AT‐390 3 mg·kg−1 was evaluated separately in a small number of rats (n = 4). Data (means ± SEM) are expressed as ALO AIMs score for each time point in absolute values (A, C) or as separate ALO AIMs scores over the 3 h observation period (i.e. cumulative scores calculated as the sum of scores at each time point; B). On a separate day, treatments were replicated in the same animals, and rotarod performance was evaluated (as time on rod in seconds; n = 6 determinations per group) before and 60 min after drug administration (D). *P < 0.05, significantly different from L‐DOPA + vehicle. Statistical analysis was performed by two‐way RM (A) or two‐way (D) ANOVA followed by the Bonferroni test for multiple comparisons or one‐way ANOVA followed by the Newman–Keuls test for multiple comparisons (B).
Effect on AIMs development (priming protocol)
The experiments in dyskinetic animals revealed that only AT‐403 (0.03 mg·kg−1) provided an acute, albeit mild, anti‐dyskinetic effect, without causing primary sedation or hypolocomotion. We therefore tested whether this dose could attenuate AIMs development when chronically combined with L‐DOPA.
Animals treated with L‐DOPA alone developed severe AIMs within the first week of treatment (Figure 7A). Then, dyskinesia scores remained maximal until the end of the 20 day period. AT‐403 did not confer overall protection against AIMs development, although AT‐403‐treated rats showed a tendency to be less dyskinetic than controls throughout the study. Nonetheless, a significant attenuation of dyskinesia was observed at first administration. On day 1; treatment (F1,8 = 2.55) was not significant; but time (F8,128 = 8.99) and time × treatment interaction (F8,128 = 2.51) were significantly affected (Figure 7B). This effect, however, disappeared in subsequent experimental sessions (Figure 7C–E).
Figure 7.
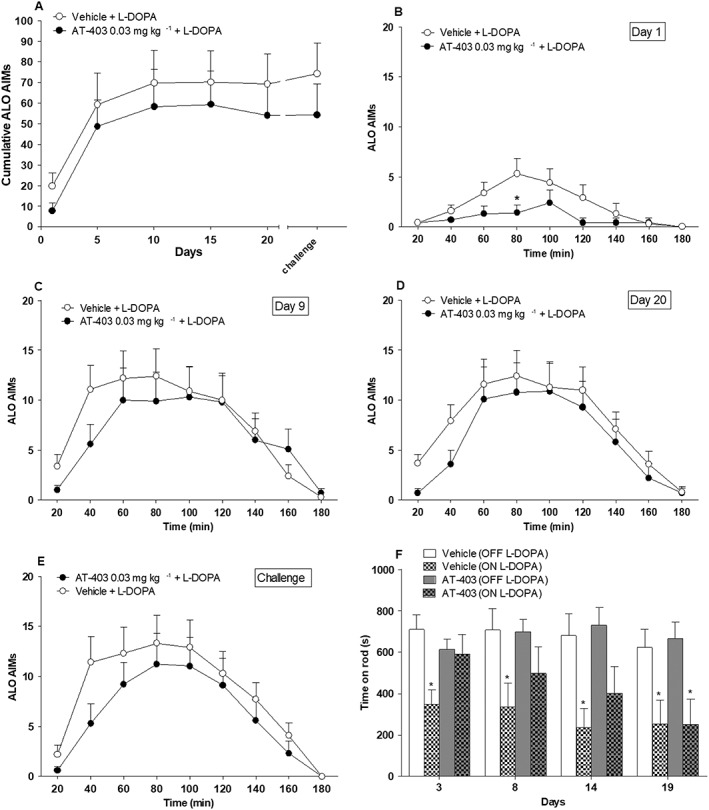
Chronic treatment with AT‐403 significantly inhibited LID at day 1, however, without interfering with its overall development. ALO AIMs were scored in 6‐OHDA hemi‐lesioned rats chronically treated for 20 days with L‐DOPA (6 mg·kg−1 plus benserazide 12 mg·kg−1, s.c.) combined with vehicle or AT‐403 0.03 mg·kg−1 (s.c., n = 9 rats per group). The time course of AIMs development over the 20 day period (A) and the AIMs response separately at days 1 (B), 9 (C), 20 (D) and 21 (challenge, E) are shown. In days different from AIMs scoring, rotarod performance was evaluated (as time on rod in seconds) before and 60 min after drug administration (F). *P < 0.05, significantly different from vehicle; two‐way RM ANOVA followed by the Bonferroni test for multiple comparisons.
At days 3, 8, 14 and 19, rotarod activity was evaluated at 60 min after L‐DOPA administration, with significant effects of treatment (F3,3 = 5.89) and time (F3,96 = 3.14) but not for the time × treatment interaction (F9,96 = 0.62) (Figure 7F). ANOVA revealed that rats treated with L‐DOPA alone poorly performed on the rotarod from the first through the last session, whereas animals treated with 0.03 mg·kg−1 AT‐403 were significantly impaired only at day 19.
Effects on ERK phosphorylation
Activation of D1 receptors increases pERK levels in striatal MSNs (Valjent et al., 2005), and elevated pERK levels are associated with LID (Pavon et al., 2006; Santini et al., 2007). We therefore investigated whether AT‐403 could reduce D1 receptor agonist‐stimulated pERK levels in striatum. In the first set of experiments (Figure 8A), application of the D1 receptor agonist SKF38393 (100 μM) to striatal slices of naïve mice caused an approximately fourfold increase in the number of pERK immunoreactive cells. As further shown in Figure 8A, AT‐403 alone tended to reduce basal pERK levels but, when co‐applied with SKF38393, clearly prevented its effect (AT‐403 F1,44 = 29.20; SKF38393 F1,44 = 43.30; interaction F1,44 = 29.61). We therefore investigated whether AT‐403 could also be effective in vivo (Figure 8B, C; Supporting Information Figure S1). 6‐OHDA dyskinetic rats were pretreated with saline or AT‐403 0.1 mg·kg−1 and 15 min later, challenged with L‐DOPA. Dyskinetic animals showed a 40% increase of pERK levels in the ipsilateral compared to the contralateral striatum whereas AT‐403‐treated dyskinetic and 6‐OHDA hemi‐lesioned rats showed similar levels in both striata (Figure 8B; F5,42 = 5.96). Indeed, pERK levels at the ipsilateral side were normalized in AT‐403‐treated dyskinetic rats compared to dyskinetic controls and 6‐OHDA hemi‐lesioned rats. Interestingly, statistical analysis also revealed about 30% reduction in pERK levels at the contralateral side in AT‐403‐treated dyskinetic rats compared to 6‐OHDA hemi‐lesioned rats. AT‐403 did not affect total ERK levels (Figure 8C), showing that the effect was specific for the phosphorylated form of the protein.
Figure 8.
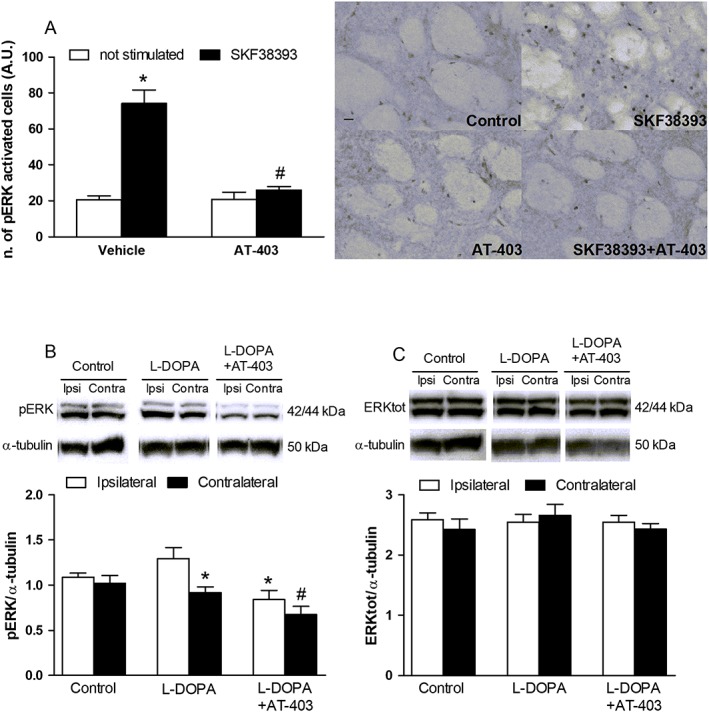
AT‐403 inhibited D1 receptor‐stimulated ERK signalling in striatum. Number of ERK‐positive cells in striatal slices of naïve mice following simultaneous application of SKF38393 and AT‐403 (30 nM) (A) and representative microphotographs of treated slices. Data are mean ± SEM of n = 12 slices per group, obtained from six mice and pooled together. *P < 0.05, significantly different from not stimulated vehicle; #P < 0.05, significantly different from SKF38393 alone; two‐way ANOVA followed by the Bonferroni post hoc test. Western blot representative images (upper panel) and quantification (lower panel) of pERK (B) and total ERK (C) in the striatum of 6‐OHDA hemi‐lesioned, L‐DOPA‐naïve or L‐DOPA primed, dyskinetic rats. Dyskinetic rats were treated with 0.1 mg·kg−1 AT‐403 or vehicle and, 15 min later, challenged with L‐DOPA. Data are means ± SEM of n = 8 rats per group. *P < 0.05, significantly different from the ipsilateral side of L‐DOPA treated rats; #P < 0.05, significantly different from the contralateral side of control rats; one‐way ANOVA followed by the Newman–Keuls post hoc test.
Discussion
AT‐403 and AT‐390 are novel low MW NOP receptor agonists with affinity, potency and selectivity for the NOP receptor better than those of our previously used NOP agonist Ro 65‐6570 (Marti et al., 2012), and in line with most potent and selective NOP receptor agonists described thus far (Zaveri, 2016). In vivo, both AT‐403 and AT‐390 significantly improved Parkinsonian‐like akinesia at low doses and, at higher doses, disrupted motor coordination and global behaviour. AT‐403 was remarkably potent, 10‐fold more than AT‐390, causing behavioural effects at doses as low as 0.01 mg·kg−1, and overt sedation at 0.3 mg·kg−1. Various studies have shown that genetic deletion or pharmacological blockade of the NOP receptor improves motor activity in naïve mice, rats and nonhuman primates (Candeletti and Ferri, 2000; Marti et al., 2004b, 2008; Viaro et al., 2008; Rizzi et al., 2011) suggesting that endogenous N/OFQ tonically inhibits motor behaviour. Motor inhibition is also typically observed when intermediate‐to‐high doses of the natural agonist N/OFQ are exogenously administered. However, exogenous N/OFQ is capable of promoting movement when given at low doses (Florin et al., 1996; Kuzmin et al., 2004; Marti et al., 2009; Viaro et al., 2013), an effect that originates from changes of primary motor cortex activity (Marti et al., 2009). The biphasic effect on motor function produced by AT‐403 and AT‐390 is similar to that of the NOP receptor agonist SCH 655842 in mice, where low doses stimulated and higher doses inhibited, the total distance travelled (Lu et al., 2011). Nonetheless, not all low MW NOP receptor agonists exhibit this profile, since Ro 64‐6198 caused only inhibition (Kuzmin et al., 2004). The finding that low doses of AT‐403 and AT‐390 reduced the immobility time in the catalepsy test and that this effect persisted also at doses inhibiting rotarod performance would indicate a specific control operated by NOP receptors on neuronal circuits regulating time to initiate movement, that is, akinesia. Indeed, the effect is clearly mediated by NOP receptors, as it was reversed by SB‐612111, at doses per se ineffective on motor function. The reason why AT‐403 and AT‐390 did not also affect stepping activity or rotarod performance at these low doses may be due to plastic changes of N/OFQ transmission in the Parkinsonian brain. In particular, as N/OFQ tone is reduced in some areas (e.g. striatum) and elevated in others (e.g. SNr), we speculate that AT‐403 and AT‐390 may target specific subpopulations of NOP receptors that are not saturated by endogenous N/OFQ or that are even up‐regulated, perhaps as a compensatory response following the drop of N/OFQ levels (Marti et al., 2012). Alternatively, we must consider the possibility that the reduction of immobility time may reflect changes in non‐motor functions. Indeed in rats, NOP receptor agonists exert anxiolysis at low, non‐sedative doses (Jenck et al., 2000; Varty et al., 2008; Lu et al., 2011). However, the finding that Ro 65‐6570 ameliorated axial symptoms in MPTP‐treated non‐human primates (Marti et al., 2012) seems to confirm the view that NOP receptor stimulation can exert positive motor effects, even under Parkinsonian conditions, in different animal species.
We previously reported that the poorly NOP‐selective (10‐fold over μ receptor) low MW NOP agonist Ro 65‐6570, reduced LID expression without causing primary hypo‐locomotion (Marti et al., 2012). The present study confirms that NOP receptor stimulation counteracts the emergence of dyskinesia. Nonetheless, only for AT‐403 it was possible to find a (narrow) therapeutic window where inhibition of LID was not associated with sedation or hypo‐locomotion. In fact, the anti‐dyskinetic effect of 0.03 mg·kg−1 AT‐403 was associated with improvement of rotarod performance on L‐DOPA, as previously shown for Ro 65‐6570, and as expected from drugs that reduce dyskinetic movements thereby improving motor coordination (Marti et al., 2012). We cannot prove whether the anti‐dyskinetic and the sedative effects are temporally spaced aspects of a common behavioural response or if they are independent phenomena. In support of the latter, however, we showed that N/OFQ injection in the striatum, that is, a brain area not specifically involved in attention control, inhibited AIMs expression (Marti et al., 2012). Moreover, we found that AT‐403 was capable of inhibiting D1 receptor‐stimulated expression of ERK, a biochemical fingerprint of LID, in vitro and ex vivo, suggesting that the anti‐dyskinetic effect of NOP agonists specifically relies on the NOP receptor ability to negatively interfere with D1 receptor signalling in direct pathway striatal MSNs neurons (Marti et al., 2012). The finding that pERK levels at the contralateral side were reduced in AT‐403‐treated rats compared to 6‐OHDA hemi‐lesioned rats is consistent with this view. This raises the possibility that the anti‐dyskinetic and sedative and hypo‐locomotive have different neuroanatomical substrates and can be pharmacologically separated. In fact, not all NOP agonists tested thus far displayed the same thresholds for anti‐dyskinetic and sedative responses. For instance, Ro 65‐6570 exerted motor inhibition at doses (1 mg·kg−1) higher than those attenuating dyskinesia (0.01 mg·kg−1) whereas for AT‐390, the two responses were superimposable. The width of the therapeutic window may thus be compound‐dependent and relate to several pharmacodynamic or pharmacokinetic considerations, among which selectivity or activity against different populations of NOP receptors (or NOP receptor‐regulated motor pathways), different brain penetrance in brain areas mediating the two effects, or different off‐targets. Moreover, recent studies pointed out that Ro 65‐6570 is a G protein biased ligand, being less efficient in activating the β‐arrestin2 pathway than N/OFQ (Ferrari et al., 2016) or AT‐403 (Ferrari et al., 2017). A different efficacy towards the Gi and β‐arrestin2 pathways may thus result in a dissociation between the anti‐dyskinetic and sedative effects.
The ability of AT‐390 to prolong AIMs duration was surprising. This effect was dose‐dependent and probably unrelated to the mechanisms underlying the delay in AIMs appearance as it was not shared by AT‐403. Interestingly, Ro 65‐6570 potentiated cocaine‐induced locomotion. Both L‐DOPA and cocaine are capable of elevating striatal dopamine levels, and indirect evidence that NOP agonists improve dopaminergic transmission has been presented. In fact, the motor‐stimulating effect of N/OFQ is blocked by a D2 receptor antagonist or by genetic removal of the D2 receptor (Florin et al., 1996; Viaro et al., 2013). Therefore, the prolongation of L‐DOPA effect might rely on a prolonged dopamine output. Interestingly, we previously showed that NOP antagonists can also promote movement by elevating D2 receptor transmission (Viaro et al., 2013) and are capable of potentiating L‐DOPA therapeutic effects, at the cost of inducing dyskinesia (Visanji et al., 2008; Marti et al., 2012). Therefore, low level stimulation of NOP receptors and NOP receptor blockade appear to share similar mechanisms and motor effects (Viaro et al., 2013). However, prolongation of AIMs duration was not previously reported with Ro 65‐6570 or N/OFQ itself (Marti et al., 2012). Therefore, alternative explanations should be taken into account, for instance that AT‐390 stimulates a specific subpopulation of NOP receptors, inhibits enzymic degradation of synaptic dopamine (newly formed from L‐DOPA) or even L‐DOPA itself, or has off‐target effects. Nonetheless, the ability of AT‐390 to prolong L‐DOPA action deserves further investigation. In fact, the prolongation of L‐DOPA action might help minimize long‐term side effects of the drug, such as dyskinesia and motor fluctuations, which are due to oscillations of L‐DOPA plasma levels and, consequently, brain levels of dopamine.
Chronic administration of 0.03 mg·kg−1 AT‐403 failed to affect overall LID development, suggesting that NOP receptor stimulation with such low dose of AT‐403 does not prevent the sensitization (i.e. priming) of striatal MSNs to L‐DOPA. Nonetheless, AT‐403 significantly reduced AIMs appearance at first administration and, consistently, spared rotarod performance ON L‐DOPA. Thus, we might speculate that the inability of AT‐403 to prevent the priming to L‐DOPA is due to development of NOP receptor desensitization. Indeed, receptor tolerance to the analgesic effect can develop after prolonged exposure to NOP agonists (Khroyan et al., 2007; Micheli et al., 2015). If this were the case, an NOP receptor partial agonist might exert a prolonged efficacy, due to its lower propensity to desensitize the receptor. In addition, such a molecule might provide additional symptomatic benefit due to its ability to attenuate overactive N/OFQ transmission in SNr, which appears to contribute to motor deficit in Parkinsonism models (Marti et al., 2005, 2012). However, we should note that other behavioural responses to NOP receptor agonists, such as the anxiolytic effect of Ro 64‐6198 (Dautzenberg et al., 2001) or the protective effect on alcohol drinking of MT‐7716 (Ciccocioppo et al., 2014) do not undergo tolerance after repeated administration. Alternatively, we can speculate that the modulation operated by such a low dose of AT‐403 is insufficient to fully counteract LID development, being effective only when the process is in its initial phase. In this respect, as the anti‐dyskinetic effect of AT‐403 might rely on a negative NOP‐D1 receptor functional interaction in direct pathway striatal MSNs, higher AT‐403 doses (e.g. 0.1 mg·kg−1) would prove more effective. However, these doses are accompanied by severe sedation, and whether repeated administration would increase the therapeutic window, having a different effect on the thresholds of the anti‐dyskinetic and the sedative actions, cannot be predicted.
In conclusion, the novel low MW NOP receptor agonists, AT‐390 and AT‐403, were used to investigate the effect of NOP receptor stimulation on Parkinsonian‐like symptoms and LID in 6‐OHDA hemi‐lesioned rats. Both compounds selectively improved akinesia at low doses and disrupted motor coordination and general behaviour at higher doses, confirming that NOP receptor stimulation biphasically regulates motor function. AT‐403 also provided a specific, albeit mild, therapeutic effect in protocols of LID expression and development, which was associated with inhibition of a biochemical marker of D1 receptor‐induced activation of striatal MSNs. These data confirm that, beyond their well characterized anti‐reward, anxiolytic and analgesic effects (Zaveri, 2016), NOP receptor agonists may be therapeutically useful to attenuate dyskinesia in L‐DOPA‐treated, Parkinsonian patients (Marti et al., 2012). Nonetheless, further studies are needed to pin down the neurobiological substrates of the anti‐dyskinetic and sedative and hypo‐locomotive effects in order to develop NOP receptor agonists with an optimal therapeutic window.
Author contributions
L.A. and C.A.P. performed surgery, L‐DOPA treatments and AIMs scoring; L.A., S.N. and M.F. carried out motor tests. B.V.J. and M.E.M. synthesized AT‐403 and AT‐390, and W.E.P. performed binding and GTPγS experiments. I.M. and S.F. carried out ERK studies in slices, and D.M. carried out Western blot analysis. R.B. drafted the manuscript; N.T.Z. and M.M. conceived the study and drafted the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Uncropped representative blots of Figure 8B‐C.
Acknowledgements
This study was supported by a grant (Dyskinesia Challenge 2013 Program) from the Michael J Fox Foundation for Parkinson's research (to M.M. and N.T.Z.) and National Institutes of Health grant R01DA027811 (to N.T.Z.).
Arcuri, L. , Novello, S. , Frassineti, M. , Mercatelli, D. , Pisanò, C. A. , Morella, I. , Fasano, S. , Journigan, B. V. , Meyer, M. E. , Polgar, W. E. , Brambilla, R. , Zaveri, N. T. , and Morari, M. (2018) Anti‐Parkinsonian and anti‐dyskinetic profiles of two novel potent and selective nociceptin/orphanin FQ receptor agonists. British Journal of Pharmacology, 175: 782–796. doi: 10.1111/bph.14123.
References
- Adapa ID, Toll L (1997). Relationship between binding affinity and functional activity of nociceptin/orphanin FQ. Neuropeptides 31: 403–408. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastide MF, Meissner WG, Picconi B, Fasano S, Fernagut PO, Feyder M et al (2015). Pathophysiology of L‐dopa‐induced motor and non‐motor complications in Parkinson's disease. Prog Neurobiol 132: 96–168. [DOI] [PubMed] [Google Scholar]
- Bido S, Marti M, Morari M (2011). Amantadine attenuates levodopa‐induced dyskinesia in mice and rats preventing the accompanying rise in nigral GABA levels. J Neurochem 118: 1043–1055. [DOI] [PubMed] [Google Scholar]
- Calo G, Guerrini R, Rizzi A, Salvadori S, Regoli D (2000). Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br J Pharmacol 129: 1261–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candeletti S, Ferri S (2000). Effects of an antisense oligonucleotide to pronociceptin and long‐term prevention of morphine actions by nociceptin. Peptides 21: 1119–1124. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Bjorklund A (1998). L‐DOPA‐induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin‐ and glutamic acid decarboxylase mRNA. Eur J Neurosci 10: 2694–2706. [PubMed] [Google Scholar]
- Cenci MA, Lundblad M (2007). Ratings of L‐DOPA‐induced dyskinesia in the unilateral 6‐OHDA lesion model of Parkinson's disease in rats and mice. Curr Protoc Neurosci Chapter 9: Unit 9 25. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Stopponi S, Economidou D, Kuriyama M, Kinoshita H, Heilig M et al (2014). Chronic treatment with novel brain‐penetrating selective NOP receptor agonist MT‐7716 reduces alcohol drinking and seeking in the rat. Neuropsychopharmacology 39: 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cippitelli A, Schoch J, Debevec G, Brunori G, Zaveri NT, Toll L (2016). A key role for the N/OFQ‐NOP receptor system in modulating nicotine taking in a model of nicotine and alcohol co‐administration. Sci Rep 6: 26594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautzenberg FM, Wichmann J, Higelin J, Py‐Lang G, Kratzeisen C, Malherbe P et al (2001). Pharmacological characterization of the novel nonpeptide orphanin FQ/nociceptin receptor agonist Ro 64‐6198: rapid and reversible desensitization of the ORL1 receptor in vitro and lack of tolerance in vivo . J Pharmacol Exp Ther 298: 812–819. [PubMed] [Google Scholar]
- Duty S, Jenner P (2011). Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 164: 1357–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Cerlesi MC, Malfacini D, Asth L, Gavioli EC, Journigan BV et al (2016). In vitro functional characterization of novel nociceptin/orphanin FQ receptor agonists in recombinant and native preparations. Eur J Pharmacol 793: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Malfacini D, Journigan BV, Bird MF, Trapella C, Guerrini R et al (2017). In vitro pharmacological characterization of a novel unbiased NOP receptor‐selective nonpeptide agonist AT‐403. Pharmacol Res Perspect 5: e00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin S, Suaudeau C, Meunier JC, Costentin J (1996). Nociceptin stimulates locomotion and exploratory behaviour in mice. Eur J Pharmacol 317: 9–13. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Calo G (2013). Nociceptin/orphanin FQ receptor antagonists as innovative antidepressant drugs. Pharmacol Ther 140: 10–25. [DOI] [PubMed] [Google Scholar]
- Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM (2013). The pharmacology of L‐DOPA‐induced dyskinesia in Parkinson's disease. Pharmacol Rev 65: 171–222. [DOI] [PubMed] [Google Scholar]
- Jenck F, Ouagazzal AM, Pauly‐Evers M, Moreau JL (2000). OrphaninFQ: role in behavioral fear responses and vulnerability to stress? Mol Psychiatry 5: 572–574. [DOI] [PubMed] [Google Scholar]
- Khroyan TV, Zaveri NT, Polgar WE, Orduna J, Olsen C, Jiang F et al (2007). SR 16435 [1‐(1‐(bicyclo[3.3.1]nonan‐9‐yl)piperidin‐4‐yl)indolin‐2‐one], a novel mixed nociceptin/orphanin FQ/mu‐opioid receptor partial agonist: analgesic and rewarding properties in mice. J Pharmacol Exp Ther 320: 934–943. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin A, Sandin J, Terenius L, Ogren SO (2004). Evidence in locomotion test for the functional heterogeneity of ORL‐1 receptors. Br J Pharmacol 141: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AP, Ko MC (2013). The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chem Nerosci 4: 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SX, Higgins GA, Hodgson RA, Hyde LA, Del Vecchio RA, Guthrie DH et al (2011). The anxiolytic‐like profile of the nociceptin receptor agonist, endo‐8‐[bis(2‐chlorophenyl)methyl]‐3‐phenyl‐8‐azabicyclo[3.2.1]octane‐3‐carboxami de (SCH 655842): comparison of efficacy and side effects across rodent species. Eur J Pharmacol 661: 63–71. [DOI] [PubMed] [Google Scholar]
- Lutfy K, Zaveri NT (2016). The Nociceptin Receptor as an Emerging Molecular Target for Cocaine Addiction. Prog Mol Biol Transl Sci 137: 149–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Guerrini R, Beani L, Bianchi C, Morari M (2002). Nociceptin/orphanin FQ receptors modulate glutamate extracellular levels in the substantia nigra pars reticulata. A microdialysis study in the awake freely moving rat. Neuroscience 112: 153–160. [DOI] [PubMed] [Google Scholar]
- Marti M, Mela F, Budri M, Volta M, Malfacini D, Molinari S et al (2013). Acute and chronic antiparkinsonian effects of the novel nociceptin/orphanin FQ receptor antagonist NiK‐21273 in comparison with SB‐612111. Br J Pharmacol 168: 863–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Mela F, Fantin M, Zucchini S, Brown JM, Witta J et al (2005). Blockade of nociceptin/orphanin FQ transmission attenuates symptoms and neurodegeneration associated with Parkinson's disease. J Neurosci 25: 9591–9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Mela F, Guerrini R, Calo G, Bianchi C, Morari M (2004a). Blockade of nociceptin/orphanin FQ transmission in rat substantia nigra reverses haloperidol‐induced akinesia and normalizes nigral glutamate release. J Neurochem 91: 1501–1504. [DOI] [PubMed] [Google Scholar]
- Marti M, Mela F, Veronesi C, Guerrini R, Salvadori S, Federici M et al (2004b). Blockade of nociceptin/orphanin FQ receptor signaling in rat substantia nigra pars reticulata stimulates nigrostriatal dopaminergic transmission and motor behavior. J Neurosci 24: 6659–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Rodi D, Li Q, Guerrini R, Fasano S, Morella I et al (2012). Nociceptin/orphanin FQ receptor agonists attenuate L‐DOPA‐induced dyskinesias. J Neurosci 32: 16106–16119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Trapella C, Morari M (2008). The novel nociceptin/orphanin FQ receptor antagonist Trap‐101 alleviates experimental parkinsonism through inhibition of the nigro‐thalamic pathway: positive interaction with L‐DOPA. J Neurochem 107: 1683–1696. [DOI] [PubMed] [Google Scholar]
- Marti M, Trapella C, Viaro R, Morari M (2007). The nociceptin/orphanin FQ receptor antagonist J‐113397 and L‐DOPA additively attenuate experimental parkinsonism through overinhibition of the nigrothalamic pathway. J Neurosci 27: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Viaro R, Guerrini R, Franchi G, Morari M (2009). Nociceptin/orphanin FQ modulates motor behavior and primary motor cortex output through receptors located in substantia nigra reticulata. Neuropsychopharmacology 34: 341–355. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mela F, Marti M, Bido S, Cenci MA, Morari M (2012). In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to L‐DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol Dis 45: 573–582. [DOI] [PubMed] [Google Scholar]
- Mela F, Millan MJ, Brocco M, Morari M (2010). The selective D(3) receptor antagonist, S33084, improves parkinsonian‐like motor dysfunction but does not affect L‐DOPA‐induced dyskinesia in 6‐hydroxydopamine hemi‐lesioned rats. Neuropharmacology 58: 528–536. [DOI] [PubMed] [Google Scholar]
- Micheli L, Di Cesare Mannelli L, Guerrini R, Trapella C, Zanardelli M, Ciccocioppo R et al (2015). Acute and subchronic antinociceptive effects of nociceptin/orphanin FQ receptor agonists infused by intrathecal route in rats. Eur J Pharmacol 754: 73–81. [DOI] [PubMed] [Google Scholar]
- Olianas MC, Dedoni S, Boi M, Onali P (2008). Activation of nociceptin/orphanin FQ‐NOP receptor system inhibits tyrosine hydroxylase phosphorylation, dopamine synthesis, and dopamine D(1) receptor signaling in rat nucleus accumbens and dorsal striatum. J Neurochem 107: 544–556. [DOI] [PubMed] [Google Scholar]
- Paolone G, Brugnoli A, Arcuri L, Mercatelli D, Morari M (2015). Eltoprazine prevents levodopa‐induced dyskinesias by reducing striatal glutamate and direct pathway activity. Mov Disord 30: 1728–1738. [DOI] [PubMed] [Google Scholar]
- Papale A, Morella IM, Indrigo MT, Bernardi RE, Marrone L, Marchisella F et al (2016). Impairment of cocaine‐mediated behaviours in mice by clinically relevant Ras‐ERK inhibitors. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon N, Martin AB, Mendialdua A, Moratalla R (2006). ERK phosphorylation and FosB expression are associated with L‐DOPA‐induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry 59: 64–74. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986). The Rat Brain in Stereotaxic Coordinates, 2nd edn. Academic Press: Sydney; Orlando. [Google Scholar]
- Rizzi A, Molinari S, Marti M, Marzola G, Calo G (2011). Nociceptin/orphanin FQ receptor knockout rats: in vitro and in vivo studies. Neuropharmacology 60: 572–579. [DOI] [PubMed] [Google Scholar]
- Rozas G, Labandeira Garcia JL (1997). Drug‐free evaluation of rat models of parkinsonism and nigral grafts using a new automated rotarod test. Brain Res 749: 188–199. [DOI] [PubMed] [Google Scholar]
- Sanberg PR, Bunsey MD, Giordano M, Norman AB (1988). The catalepsy test: its ups and downs. Behav Neurosci 102: 748–759. [DOI] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA et al (2007). Critical involvement of cAMP/DARPP‐32 and extracellular signal‐regulated protein kinase signaling in L‐DOPA‐induced dyskinesia. J Neurosci 27: 6995–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T, De Ryck M, Whishaw IQ, Ramirez VD, Teitelbaum P (1979). Excessive bracing reactions and their control by atropine and L‐DOPA in an animal analog of Parkinsonism. Exp Neurol 64: 33–43. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP (1996). The unilateral 6‐hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol 50: 275–331. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo B, Calo G, Polgar WE, Jiang F, Olsen CM, Berzetei‐Gurske I et al (2008). Activities of mixed NOP and mu‐opioid receptor ligands. Br J Pharmacol 153: 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Bruchas MR, Calo G, Cox BM, Zaveri NT (2016). Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol Rev 68: 419–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC et al (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A 102: 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varty GB, Lu SX, Morgan CA, Cohen‐Williams ME, Hodgson RA, Smith‐Torhan A et al (2008). The anxiolytic‐like effects of the novel, orally active nociceptin opioid receptor agonist 8‐[bis(2‐methylphenyl)methyl]‐3‐phenyl‐8‐azabicyclo[3.2.1]octan‐3‐ol (SCH 221510). J Pharmacol Exp Ther 326: 672–682. [DOI] [PubMed] [Google Scholar]
- Viaro R, Calcagno M, Marti M, Borrelli E, Morari M (2013). Pharmacological and genetic evidence for pre‐ and postsynaptic D2 receptor involvement in motor responses to nociceptin/orphanin FQ receptor ligands. Neuropharmacology 72: 126–138. [DOI] [PubMed] [Google Scholar]
- Viaro R, Sanchez‐Pernaute R, Marti M, Trapella C, Isacson O, Morari M (2008). Nociceptin/orphanin FQ receptor blockade attenuates MPTP‐induced parkinsonism. Neurobiol Dis 30: 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji NP, de Bie RM, Johnston TH, McCreary AC, Brotchie JM, Fox SH (2008). The nociceptin/orphanin FQ (NOP) receptor antagonist J‐113397 enhances the effects of levodopa in the MPTP‐lesioned nonhuman primate model of Parkinson's disease. Mov Disord 23: 1922–1925. [DOI] [PubMed] [Google Scholar]
- Volta M, Mabrouk OS, Bido S, Marti M, Morari M (2010). Further evidence for an involvement of nociceptin/orphanin FQ in the pathophysiology of Parkinson's disease: a behavioral and neurochemical study in reserpinized mice. J Neurochem 115: 1543–1555. [DOI] [PubMed] [Google Scholar]
- Volta M, Viaro R, Trapella C, Marti M, Morari M (2011). Dopamine‐nociceptin/orphanin FQ interactions in the substantia nigra reticulata of hemiparkinsonian rats: involvement of D2/D3 receptors and impact on nigro‐thalamic neurons and motor activity. Exp Neurol 228: 126–137. [DOI] [PubMed] [Google Scholar]
- Zaveri NT (2016). Nociceptin opioid receptor (NOP) as a therapeutic target: progress in translation from preclinical research to clinical utility. J Med Chem 59: 7011–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri NT, Jiang F, Olsen CM, Deschamps JR, Parrish D, Polgar W et al (2004). A novel series of piperidin‐4‐yl‐1,3‐dihydroindol‐2‐ones as agonist and antagonist ligands at the nociceptin receptor. J Med Chem 47: 2973–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri N T, Meyer M M, Journigan BV, Yasuda D (2017). Piperidinyl nociceptin receptor compounds. International Patent publication number: WO 2017/096323 A1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Uncropped representative blots of Figure 8B‐C.