

Abstract
Background and Purpose
(−)‐Englerin A (EA) is a potent cytotoxic agent against renal carcinoma cells. It achieves its effects by activation of transient receptor potential canonical (TRPC)4/TRPC1 heteromeric channels. It is also an agonist at channels formed by the related protein, TRPC5. Here, we sought an EA analogue, which might enable a better understanding of these effects of EA.
Experimental Approach
An EA analogue, A54, was synthesized by chemical elaboration of EA. The effects of EA and A54 on the activity of human TRPC4 or TRPC5 channels overexpressed on A498 and HEK 293 cells were investigated, firstly, by measuring intracellular Ca2+ and, secondly, current using whole‐cell patch clamp recordings.
Key Results
A54 had weak or no agonist activity at endogenous TRPC4/TRPC1 channels in A498 cells or TRPC4 or TRPC5 homomeric channels overexpressed in HEK 293 cells. A54 strongly inhibited EA‐mediated activation of TRPC4/TRPC1 or TRPC5 and weakly inhibited activation of TRPC4. Studies of TRPC5 showed that A54 shifted the EA concentration–response curve to the right without changing its slope, consistent with competitive antagonism. In contrast, Gd3+‐activated TRPC5 or sphingosine‐1‐phosphate‐activated TRPC4 channels were not inhibited but potentiated by A54. A54 did not activate TRPC3 channels or affect the activation of these channels by the agonist 1‐oleoyl‐2‐acetyl‐sn‐glycerol.
Conclusions and Implications
This study has revealed a new tool compound for EA and TRPC1/4/5 channel research, which could be useful for characterizing endogenous TRPC1/4/5 channels and understanding EA‐binding sites and their physiological relevance.
Abbreviations
- A498
human renal cell carcinoma cell line 498
- A54
analogue 54
- EA
(−)‐englerin A
- Gd3+
gadolinium ion
- TRPC
transient receptor potential canonical
Introduction
(−)‐Englerin A (EA) is a natural product from Phyllanthus engleri, a south‐east African plant (Wu et al., 2017). It was found to have potent cytotoxic effects against some types of cancer cells cultured from kidney, breast, bone, brain, lung, haematopoietic, prostate and other cancers (Akbulut et al., 2015, Caropreso et al., 2016, Carson et al., 2015, Ratnayake et al., 2009, Sourbier et al., 2013). Subsequent studies demonstrated its in vivo efficacy against xenograft tumours generated in mice using 786‐O and PC3 cancer cell lines, suggesting its potential use as a novel therapeutic strategy (Sourbier et al., 2013). Chemistry studies led to the development of efficient synthesis routes for EA and its derivatives and provided initial insights into structure–activity relationships (Radtke et al., 2011, Willot et al., 2009, Wu et al., 2017).
The transient receptor potential canonical (TRPC) proteins are a subfamily of the TRP superfamily and assemble as homo and heterotetramers to form Ca2+‐permeable non‐selective cationic channels (Abramowitz and Birnbaumer, 2009; Beech, 2013; Damann et al., 2008; Gaunt et al., 2016; Moran et al., 2011; Rubaiy, 2017). They appear to function in two clusters, TRPC1/TRPC4/TRPC5 and TRPC3/TRPC6/TRPC7, which are distinguishable pharmacologically (Akbulut et al., 2015; Bon and Beech, 2013; Maier et al., 2015; Miller et al., 2011; Washburn et al., 2013; Rubaiy et al., 2017a,b).
TRPC1/4/5 channels are potently and strongly activated by EA, whereas TRPC3 and TRPC6 channels are unaffected (Akbulut et al., 2015, Carson et al., 2015, Ludlow et al., 2017, Rubaiy et al., 2017b). EA‐evoked cytotoxicity is TRPC4‐dependent in the human renal cell carcinoma cell line 498 A498 and other cancer cell lines (Carson et al., 2015, Ludlow et al., 2017). TRPC5 is poorly expressed in many cancer cell lines but may be relevant in some cancer cell types (Carson et al., 2015, Ma et al., 2012). The role of TRPC1 is not completely understood, but it has been suggested that it is involved in the death of A498 cells and Hs578T triple‐negative breast cancer cells evoked by EA and has an important role in the control of Na+ versus Ca2+ permeability in these cells (Ludlow et al., 2017). Therefore, ion channels comprising TRPC1/4/5 proteins are an important target of EA and might be a platform for developing novel anticancer agents (Gaunt et al., 2016, Wu et al., 2017). However, obstacles that need to be addressed before it can be considered as a therapeutic strategy include metabolic instability and in vivo toxicity (Carson et al., 2015). A potential route for overcoming these obstacles is a better understanding of the structure–activity relationships of EA at TRPC1/4/5 channels. To this end, we have sought chemical elaboration of EA. Here, we describe the properties of a novel EA analogue and suggest that it is a competitive antagonist with partial agonist (enhancer) capability.
Methods
Cell culture
HEK 293 cells stably expressing human TRPC5 or human TRPC4 [tetracycline‐inducible promoter] were cultured at 37°C in a humidified atmosphere containing 5% CO2/95% air. The HEK 293 cells were from Invitrogen [Paisley, UK; T‐REx™, the tetracycline‐regulated cell lines stably express the tetracyclin repressor], and the American Type Culture Collection number is CRL‐1573. To induce channel expression, tetracyclin (1 μg·mL−1) was added to the cells at least 24 h before the experiment. DMEM‐F12 GlutaMAX (Invitrogen) was used and was supplemented with 10% FBS and 5% penicillin–streptomycin (10 000 U and 10 mg streptomycin mL−1 in 0.9% NaCl, Sigma, Renfrew, UK). The cell culture medium was supplemented with the antibiotics blasticidin (5 μg·mL−1) and phleomycin (400 μg·mL−1, Zeocin) (Invitrogen). The culture medium was changed every 3–4 days until the cells were 70–80% confluent. The cells were used at passages 5 to 20, and the same seeding conditions were maintained between experiments. A‐498 cells were from The Leibniz‐Institute–Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany). These cells were originally established from the kidney carcinoma of a 52‐year‐old man in 1973. A498 cells were cultured in minimum essential Eagle's medium with Earle's balanced salt solution, l‐glutamine, 2.2 g·L−1 NaHCO3 (PAN‐biotech, Aidenbach, Germany) supplemented with 5% sodium pyruvate.
Intracellular calcium measurement
HEK 293 or A498 cells were plated at 90% confluence in microplates (96‐well plates) and incubated overnight at 37°C, 5% CO2. For HEK 293 cells, clear‐bottomed poly‐d‐lysine‐coated black plates (Corning Life Sciences, Lowell, MA, USA) were used, and for A498 cells, clear‐bottomed Nunc plates (Thermo Fisher Scientific, Waltham, MA, USA) were used. The cells were illuminated with alternating 340/380 nm excitation light for fura‐2 fluorescence measurements. Fluorescence emission was measured at 510 nm. In more detail, the cells were incubated for 1 h with fura‐2‐AM (2 μM) in standard bath solution (SBS) at 37°C in the presence of 0.01% pluronic acid. The fura‐2 fluorescence was recorded using a 96‐well fluorescence plate reader at excitation wavelengths of 340 and 380 nm (FlexStation II384, Molecular Devices, Sunnyvale, CA, USA). Ca2+ was indicated by the ratio of the fluorescence (F) emission intensities for the two excitation wavelengths. Measurements were made at room temperature (21 ± 3°C).
Patch‐clamp recording
Conventional whole‐cell configuration was performed under voltage clamp at room temperature using 3–6 MΩ patch pipettes fabricated from borosilicate glass capillaries with an outside diameter of 1 mm and an inside diameter of 0.58 mm (Harvard Apparatus, Holliston, MA, USA). The currents were recorded using an Axopatch 200B amplifier, digitized by a Digidata 1440, and recorded to a computer using pCLAMP10 (Molecular Devices). The data were filtered at 1 kHz and analysed offline using Clampfit software (version 10.2, Molecular Devices) and Origin software version 9.1 (OriginLab, Northampton, MA, USA). The bath solution was SBS, and the pipette solution (intracellular solution) contained 145 mM CsCl, 2 mM MgCl2, 10 mM HEPES, 1 mM EGTA (free acid), 5 mM ATP (sodium salt) and 0.1 mM GTP (sodium salt), titrated to pH 7.2 with CsOH. Cells were plated 24 h earlier on glass coverslips at a low density of 20–30% and stimulated with tetracyclin (1 μg·mL−1).
Chemical synthesis
The synthesis of the ether derivative analogue 54 (A54) (1 in Supporting Information Figure S1) originated from alcohol 2, which is readily accessible in two steps (Supporting information) from a key intermediate of previous englerin syntheses (Radtke et al., 2011, Willot et al., 2009). An iridium catalysed etherification of alcohol 2 gave vinyl ether 3 as 82% yield (Okimoto et al., 2002). Subsequent hydroboration produced the glycol ether 4 as 81% yield (Hoveyda et al., 2011). The primary hydroxyl was protected as silyl ether 5 as 77% yield, followed by hydrogenolytic cleavage of benzyl ether to give alcohol 6. Finally, Yamaguchi esterification with cinnamic acid generated ester 7 (78% yield), which was treated with tetra‐n‐butylammonium fluoride to give the target molecule 1 as 99% yield (Inanaga et al., 1979, Kawanami et al., 1981).
Reagents
(−)‐EA and A54 were stored in aliquots at −80°C. A54 stock and working solutions were prepared similarly to those of (−)‐EA, as previously described (Akbulut et al., 2015). The (−)‐EA analogues were diluted to a final concentration using the SBS containing 0.1% DMSO and 0.01% pluronic acid. The SBS, which was the experimental buffer, contained (mM): NaCl 135, KCl 5, MgCl2 1.2, CaCl2 1.5, glucose 8 and HEPES 10 (pH titrated to 7.4 using NaOH).
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as means ± SEM, and P < 0.05 was considered statistically significant after a two sample test (t‐test) for comparisons of two independent groups or ANOVA (one‐way and post‐test Bonferroni) for multiple groups (post hoc test was only applied when ANOVA gave P < 0.05). Origin 9.1 software was used to analyse the results. In Ca2+ studies, the half maximal inhibitory concentration (IC50) was obtained by normalizing to the maximal response of EA, and the curves were fitted with Origin 9.1 software using y = START + (END − START) * x n/(k n + x n), where y is the Ca2+ concentration (reflected by fura‐2 ratio), x the concentration of the compound, k the IC50 or EC50 and n the number of binding sites per target molecule. The rate of rise of the Ca2+ signal in response to gadolinium ions (Gd3+) was fitted with Origin 9.1 using y = A1 * exp(−x/t1) + y0, where y is Ca2+, x the time, A1 the maximum response, y0 the Ca2+ at the time of Gd3+ application and t1 the time constant. The data in all experiments are represented as n/N, where n indicates independent experiments and N indicates replicates in columns of the 96‐well plate. Statistical significance is indicated by a * (P < 0.05).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Structures of (−)‐englerin A, (−)‐englerin B and analogue 54
EA and its inactive metabolite (−)‐englerin B (EB) (Figure 1A, B) were used to guide the generation of analogues, which might be active at endogenous TRPC4/5 channels. In this article, we show the structure and data for only one of these analogues, A54 (Figure 1C). The synthesis routes and chemical validation are provided in Supporting Information Figure S1.
Figure 1.
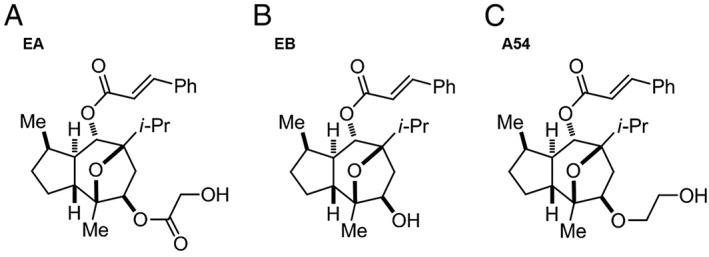
Structures of EA, EB and A54.
A54 lacks agonist activity but inhibits EA‐evoked Ca2+ entry in A498 cells
To test the functionality of EA and its analogues, we first obtained intracellular Ca2+ measurements in the A498 cells, which express endogenous EA‐responsive TRPC4/TRPC1 heteromeric channels (Akbulut et al., 2015, Ludlow et al., 2017). Initially, a single concentration of each compound was tested. As expected, EA was effective and EB ineffective (Figure 2A, B). Like EB, A54 failed to evoke a Ca2+ signal and therefore appeared to be inactive (Figure 2A, B). Nevertheless, we also tested if A54 could affect the response to EA. A498 cells were pre‐incubated with 1 μM A54 for 30 min, and then 100 nM EA was tested in the continuous presence of A54 (Figure 2C). Strikingly, A54 abolished the EA response (Figure 2C). In contrast, EB failed to affect the EA response (Figure 2C). We next constructed a concentration–response curve for A54 against the EA response, pre‐incubating A498 cells with A54 at a range of concentrations (1–1000 nM) for 30 min and then testing EA (Figure 2D). A54 was a potent inhibitor of the EA response, causing 50% inhibition at 62 nM (Figure 2D, E). The data suggest that A54, unlike EB, is an antagonist of EA.
Figure 2.
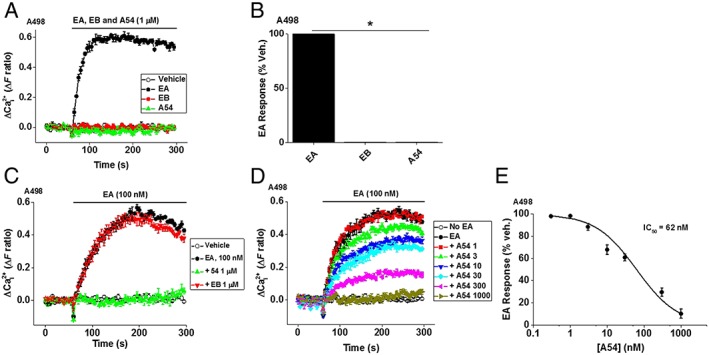
Activity of EA, EB and A54 in A498 cells. (A) Representative Ca2+ measurement data from a single 96‐well plate showing the EA‐evoked Ca2+ entry and lack of effect of EB or A54 in A498 cells. (B) Summary data for experiments of the same type as (A) plotted as a percentage of the maximum EA response and expressed as percentages (n/N = 7/21, one‐way ANOVA and Bonferroni's post‐test). (C) Representative traces showing 1 μM of A54 was sufficient to inhibit activation by EA, whereas 1 μM EB lacked effect. (D) Example of concentration‐dependent inhibition traces of 100 nM EA responses by A54 at increasing concentrations (numbers 1–1000 indicate concentrations in nM). (E) Concentration–response data for inhibition of EA responses by A54 fitted with the Hill equation (IC50 = 62 ± 17 nM, slope = 0.8 ± 0.01) (n/N = 5/15, mean ± SEM). Veh., vehicle.
EA‐dependent inhibition of TRPC5 channels by A54
To gain insight into the mechanism of inhibition by A54, we switched to studying HEK 293 cells, which contain tetracyclin‐inducible expression of human TRPC5, generating TRPC5 homomeric channels. These cells exhibit not only TRPC5 activity evoked by EA but also robust, albeit smaller, TRPC5 activity evoked by Gd3+ (Akbulut et al., 2015, Ludlow et al., 2017, Naylor et al., 2016) (Figure 3A). We could, therefore, test A54 against two different modes of channel activity to determine if the effect of A54 had specificity for the EA response. EA evoked a large Ca2+ response in the TRPC5‐expressing cells and, as expected, this effect was abolished by A54 (Figure 3A, B). A54 did not, however, inhibit the Gd3+ response but enhanced its amplitude and rate of onset (Figure 3A, C, D).
Figure 3.
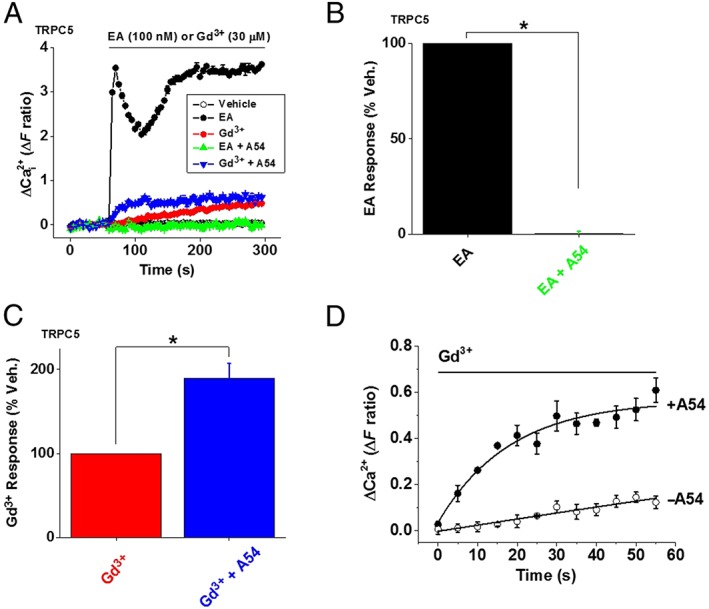
A54 inhibits EA‐ but not Gd3+‐evoked TRPC5 channel activity. Intracellular Ca2+ responses in HEK‐TRPC5 cells to EA (100 nM) or Gd3+ (30 μM) in the absence and presence of 1 μM A54 (A). (B) For EA experiments of the type illustrated in (A), mean ± SEM data normalized to the vehicle (Veh.) control for A54 (n/N = 8/32). (C) For Gd3+ experiments of the type illustrated in (A), mean ± SEM data normalized to the Veh. control for A54 (n/N = 8/32). The time point at which the amplitude of the Gd3+ response was measured was from 150 to 200 s. (D) Activation time course of Gd3+ responses in the absence and presence of A54. The curves were fitted exponential functions with time constants of 780 ± 7.6 s (Gd3+ − A54) and 18.1 ± 3.9 s (Gd3+ + A54) (n/N = 8/32).
To further investigate the dual effect of A54, we used whole‐cell patch clamp recordings. EA activated the TRPC5‐mediated current as expected (Figure 4A, B). Addition of A54 in the continuous presence of EA caused striking inhibition of the EA‐evoked current (Figure 4A–D), and this effect was reversible on washout of A54 (Figure 4A, B). The channels could also be activated by Gd3+, but in this case, A54 further enhanced the current (Figure 5A–D). The data suggest that the inhibitory effect of A54 is EA‐dependent and that, in the absence of EA, A54 has a sharply contrasting positive effect on the Gd3+ response.
Figure 4.
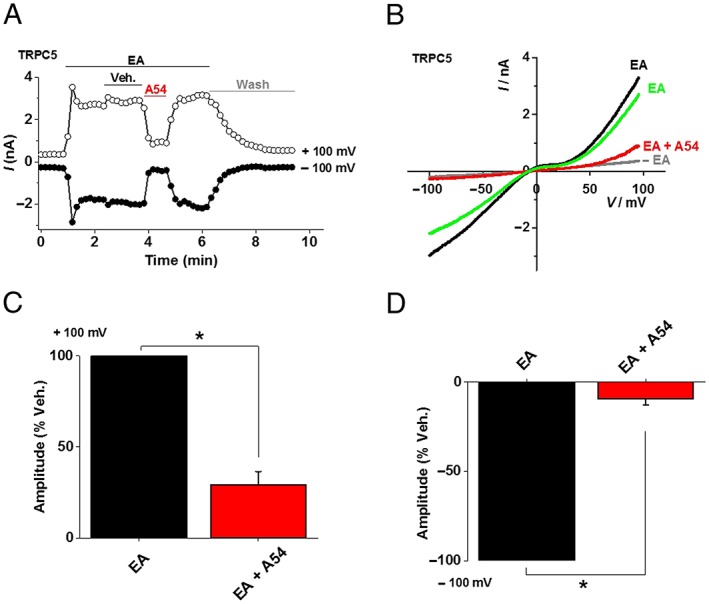
A54 inhibits EA‐evoked current in TRPC5 channels. (A) Example of whole‐cell patch‐clamp data from a TRPC5‐expressing HEK 293 cell showing current sampled at −100 and +100 mV during ramp changes in voltages (all agents were bath applied). EA 10 nM and 1 μM A54 were used. Vehicle (Veh.) was DMSO. Wash indicates washout of EA and A54. (B) Representative current–voltage relationship (I–V) from experiments of the type illustrated in (A). (C, D) Mean percentage inhibition of responses to A54 as illustrated in (A) for (C) +100 mV and (D) −100 mV (n = 5 independent recordings).
Figure 5.

A54 enhances Gd3+‐evoked current in TRPC5 channels. (A) Example of whole‐cell patch‐clamp data from a TRPC5‐expressing HEK 293 cell showing current sampled at −100 and +100 mV during ramp changes in voltages (all agents were bath‐applied). Gd3+ μM and 1 μM A54 were used. Wash indicates washout of Gd3+ and A54. (B) Representative current–voltage relationship (I–V) from experiments of the type illustrated in (A). (C, D) Mean percentage enhancement of responses to A54 as illustrated in (A) for (C) +100 mV and (D) −100 mV (n = 5 independent recordings). Veh., vehicle.
A54 behaves like a competitive antagonist of EA at TRPC5 channels
Because the inhibitory effect of A54 is EA‐dependent, we considered whether it might bind part of the same site as EA and thus compete with EA. To test this hypothesis, we constructed concentration–response curves for EA in the absence of A54 and in the presence of two different concentrations of A54 (10 and 50 nM) (Figure 6A–C). The inhibition by A54 was surmountable, and increasing concentrations of A54 caused parallel rightward shifts in the EA concentration–response curve (Figure 6D). Schild analysis revealed a slope close to unity (Figure 6E). EA responses reached steady state for most concentrations of EA, but the slowness of the response to low concentrations of EA meant that the responses fell short of steady state in some instances. We accepted this weakness because of concern about the health of cells when long exposures to EA were used. The data suggest that A54 is a competitive antagonist of EA at TRPC5 channels.
Figure 6.
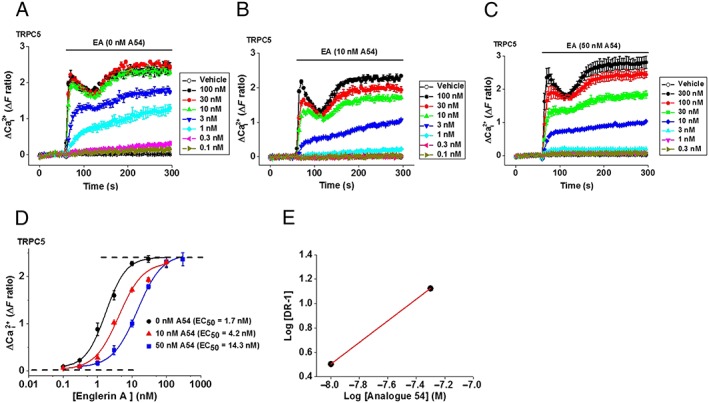
A54 causes parallel rightward shifts of the EA concentration–response curve. (A–C) Example data from HEK‐TRPC5 cells showing the concentration‐dependence of EA responses in the presence of (A) 0, (B) 10 and (C) 50 nM A54. (D) Mean ± SEM concentration–response data for experiments of the type illustrated in (A–C) (n/N = 6/24 for 0 and 50 nM and n/N = 5/16 for 10 nM). EA EC50s were 1.7 nM (0 nM A54), 4.2 nM (10 nM A54) and 14.3 nM (50 nM A54). EA slope values were 1.4 (0 nM A54), 1.3 (10 nM A54) and 1.3 (50 nM A54). (E) Schild plot regression analysis with a slope near 1 (0.9) indicating that A54 is a competitive antagonist (DR in y axis indicates the dose ratio).
A54 is a weak antagonist of EA at TRPC4 channels
We next investigated the effect of A54 on TRPC4 homomeric channels overexpressed in HEK 293 cells. Unexpectedly, A54 was only a mild antagonist of EA at TRPC4 channels (Figure 7A, B), and furthermore, A54 lacked agonist activity on TRPC4 channels (Figure 7C). The data suggest that the EA‐binding site associated with TRPC4 channels does not bind A54 efficiently or that A54 binds tightly but does not compete effectively with EA. These findings contrast with TRPC5 channels and native TRPC4/TRPC1 channels.
Figure 7.
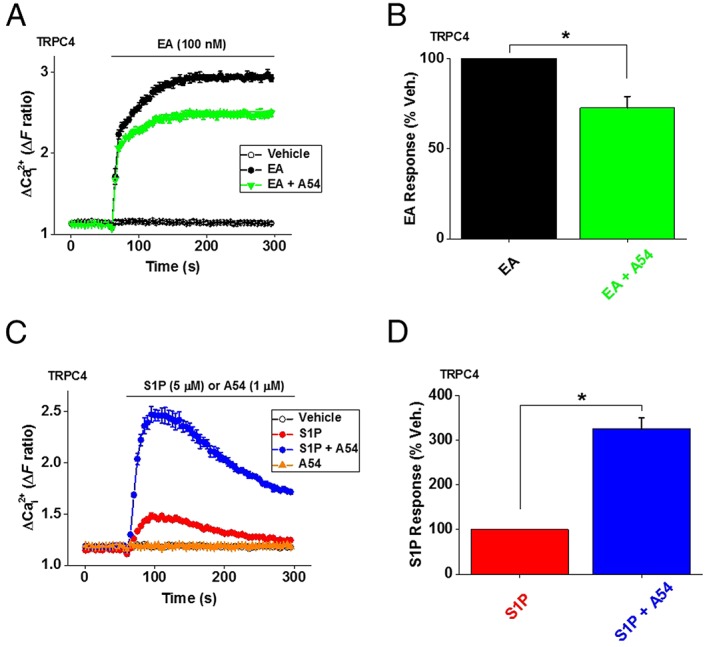
A54 is a striking enhancer of S1P‐evoked TRPC4 channel activity. (A) Intracellular Ca2+ responses in HEK–TRPC4 cells to EA (100 nM) in the absence and presence of 1 μM A54. (B) Mean ± SEM data normalized to the vehicle (Veh.) control for A54 of the type illustrated in (A) (n/N = 5/15 for all data). (C) Intracellular Ca2+ responses in HEK–TRPC4 cells to A54 (1 μM) and S1P (5 μM) in the absence and presence of 1 μM A54. (D) Mean ± SEM of the S1P data normalized to the Veh. control for A54 of the type illustrated in (C); (n/N = 5/15 for all data).
A54 strongly potentiates S1P‐evoked TRPC4 activity
To further investigate the relationship between A54 and TRPC4 channels, we used a physiological agonist, sphingosine 1‐phosphate (S1P), which activates TRPC4 channels via G‐protein signalling. Strikingly, A54 markedly potentiated S1P‐evoked Ca2+ entry through TRPC4 channels (Figure 7C, D). The data suggest that A54 is an enhancer of TRPC4 channel activity evoked by S1P.
A54 has no effect on TRPC3 channels
EA appears to be selective for TRPC1/4/5 channels, lacking effect on 13 other ion channels investigated including TRPC3, a member of the TRPC3/TRPC6/TRPC7 subfamily of TRPC channels (Akbulut et al., 2015). Here, we investigated if there was effect of A54 on TRPC3 homomeric channels overexpressed in HEK 293 cells. A54 (1 μM) did not activate TRPC3 or inhibit or potentiate TRPC3‐mediated Ca2+ entry activated by the TRPC3 agonist 1‐oleoyl‐2‐acetyl‐sn‐glycerol (OAG; Figure 8A, B). The data suggest that A54 has no effect on TRPC3 channels.
Figure 8.
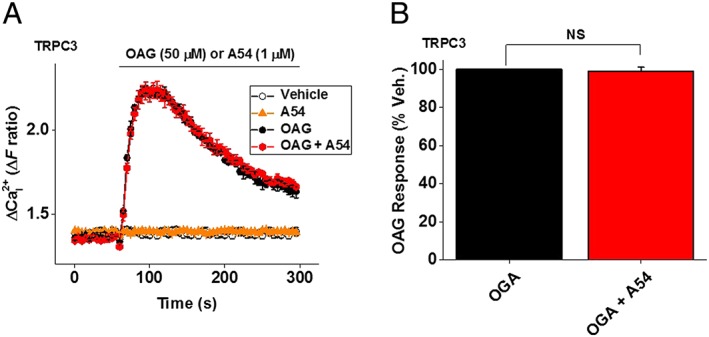
Lack of effect of A54 on TRPC3 channel activity. (A) Intracellular Ca2+ responses to A54 (1 μM) and 1‐oleoyl‐2‐acetyl‐sn‐glycerol (OAG, 50 μM) in the absence and presence of 1 μM A54 in HEK 293 tetracyclin‐positive cell lines stably expressing human TRPC3 (n/N = 6/18). (B) Mean ± SEM data normalized to the vehicle (Veh.) control for A54 of the type illustrated in (A) (n/N = 6/18). NS, not significant.
Discussion and conclusions
The study reveals how subtle chemical elaboration of EA can lead to a compound (A54), which is a potent antagonist of EA at TRPC5 homomeric channels and endogenous TRPC4/TRPC1‐containing channels, yet not a general blocker of the channels. The antagonism had characteristics of surmountable competitive antagonism at TRPC5 channels, suggesting that A54 interacts at the same site as EA but is not able to trigger channel opening. Strikingly, however, A54 was a strong enhancer of TRPC5 channel activity induced by Gd3+ and TRPC4 channel activity induced by S1P. A54 is, therefore, capable of antagonist and enhancer (facilitating) activity; that is, it is not a pure antagonist but retains some agonist‐like (enhancer) capability in the presence of a cofactor (Gd3+ or S1P).
The existence and location of a binding site for EA is unknown, but its ability to robustly activate TRPC4 channels in excised membrane patches even in the complete absence of exogenous cofactors is suggestive of an agonist binding site on the channel protein itself (Akbulut et al., 2015). This site is either on the extracellular surface of the channels or only accessible via the outer leaflet of the bilayer, because EA only activates the channels when applied to the outer and not inner surface of the membrane (Akbulut et al., 2015). By implication, the A54 site would also be on the channel itself. However, direct proof is lacking.
The action of EA on TRPC1/4/5 channels is remarkable – it is much stronger and more robust as an agonist than other known activators of the channels. Therefore, there would seem not only to be an EA binding site on the channels but also the very efficient coupling of this binding site to channel activation. Such a site may simply exist by chance and serve no physiological purpose, but it might alternatively serve a purpose, which is not yet recognized. In this latter regard, it will be interesting to test competitive antagonists of EA (e.g. A54) in physiological studies.
As TRPC4 and TRPC5 proteins share 65% amino acid sequence, we were expecting A54 to block the TRPC4 Ca2+ entry evoked by EA. Surprisingly, however, 1 μM A54 had only a modest inhibitory effect (Figure 7) compared with full inhibition at TRPC5 channels (Figures 2 and 3). Moreover, A54 had an enhancing effect in the presence of the physiological agonist S1P, increasing the Ca2+ entry more than threefold (Figure 7). This effect was similar to the enhancing effect of A54 at TRPC5 channels in the presence of Gd3+ (Figures 3 and 5). Such effects might be explained by a conformational change in the channels (induced by S1P or Gd3+), which allows A54 to access an efficacy (agonist) site. Therefore, the EA and A54 data could be explained by the existence of two binding sites, which are in close physical proximity: site 1 allowing binding without efficacy, and site 2 allowing binding and efficacy. In this model, EA would bind sites 1 and 2 (and therefore have direct agonist effect), while A54 would bind not only site 1 in the absence of a cofactor (and therefore antagonize EA) but also site 2 in the presence of the cofactor (and therefore have an enhancing effect).
In conclusion, this study has revealed a new tool compound for EA and TRPC1/4/5 channel research and, to the best of our knowledge, the first known competitive antagonist of EA. This was achieved by minimal structural modification of EA. It is not a perfect antagonist because it has enhancer capability in the presence of a cofactor (Gd3+ at TRPC5 channels and S1P at TRPC4 channels), but it shows impressive proficiency as a surmountable competitive antagonist when used in isolation in the low to middle nmol concentration range. The compound should facilitate studies aimed at better understanding TRPC1/4/5 channel activation, TRPC1/4/5 channel binding sites and the physiological relevance of the action of EA. Because of the in vivo toxicity of EA (Carson et al., 2015), this competitive antagonist might also have use as an antidote to EA poisoning.
Author contributions
H.N.R. performed the research (cell culture, calcium measurements and patch‐clamp electrophysiology) designed the research study, contributed intellectually, analysed the data, generated the figures and wrote the manuscript with D.J.B. M.C. led the chemical synthesis of A54. H.W., B.K., A.C., M.C., T.S., S.H., P.N. and D.J.B. performed or advised on chemical synthesis and usage, generated research funds and ideas, led and coordinated the project or interpreted data.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Chemical synthesis and validation of analogue A54 (1).
{kind=link}
Supporting information
Acknowledgements
This work was supported by the University of Leeds, the Max Planck Society (MPG) and the Lead Discovery Centre (LDC).
Rubaiy, H. N. , Seitz, T. , Hahn, S. , Choidas, A. , Habenberger, P. , Klebl, B. , Dinkel, K. , Nussbaumer, P. , Waldmann, H. , Christmann, M. , and Beech, D. J. (2018) Identification of an (−)‐englerin A analogue, which antagonizes (−)‐englerin A at TRPC1/4/5 channels. British Journal of Pharmacology, 175: 830–839. doi: 10.1111/bph.14128.
Contributor Information
Mathias Christmann, Email: mathias.christmann@fu-berlin.de.
David J Beech, Email: d.j.beech@leeds.ac.uk.
References
- Abramowitz J, Birnbaumer L (2009). Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J 23: 297–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbulut Y, Gaunt HJ, Muraki K, Ludlow MJ, Amer MS, Bruns A et al (2015). (−)‐Englerin A is a potent and selective activator of TRPC4 and TRPC5 calcium channels. Angew Chem Int Ed Engl 54: 3787–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ (2013). Characteristics of transient receptor potential canonical calcium‐permeable channels and their relevance to vascular physiology and disease. Circ J 77: 570–579. [DOI] [PubMed] [Google Scholar]
- Bon RS, Beech DJ (2013). In pursuit of small molecule chemistry for calcium‐permeable non‐selective TRPC channels – mirage or pot of gold? Br J Pharmacol 170: 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caropreso V, Darvishi E, Turbyville TJ, Ratnayake R, Grohar PJ, Mcmahon JB et al (2016). Englerin A inhibits EWS‐FLI1 DNA binding in Ewing sarcoma cells. J Biol Chem 291: 10058–10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson C, Raman P, Tullai J, Xu L, Henault M, Thomas E et al (2015). Englerin A agonizes the TRPC4/C5 cation channels to inhibit tumor cell line proliferation. PLoS One 10: e0127498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damann N, Voets T, Nilius B (2008). TRPs in our senses. Curr Biol 18: R880–R889. [DOI] [PubMed] [Google Scholar]
- Gaunt HJ, Vasudev NS, Beech DJ (2016). Transient receptor potential canonical 4 and 5 proteins as targets in cancer therapeutics. Eur Biophys J 45: 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoveyda HR, Marsault E, Gagnon R, Mathieu AP, Vezina M, Landry A et al (2011). Optimization of the potency and pharmacokinetic properties of a macrocyclic ghrelin receptor agonist (part I): development of ulimorelin (TZP‐101) from hit to clinic. J Med Chem 54: 8305–8320. [DOI] [PubMed] [Google Scholar]
- Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M (1979). A rapid esterification by means of mixed anhydride and its application to large‐ring lactonization. Bull Chem Soc Jpn 52: 1989–1993. [Google Scholar]
- Kawanami Y, Dainobu Y, Inanaga J, Katsuki T, Yamaguchi M (1981). Synthesis of thiol esters by carboxylic trichlorobenzoic anhydrides. Bull Chem Soc Jpn 54: 943–944. [Google Scholar]
- Ludlow MJ, Gaunt HJ, Rubaiy HN, Musialowski KE, Blythe NM, Vasudev NS et al (2017). (−)‐Englerin A‐evoked cytotoxicity is mediated by Na+ influx and counteracted by Na+/K+‐ATPase. J Biol Chem 292: 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Cai Y, He D, Zou C, Zhang P, Lo CY et al (2012). Transient receptor potential channel TRPC5 is essential for P‐glycoprotein induction in drug‐resistant cancer cells. Proc Natl Acad Sci U S A 109: 16282–16287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Follmann M, Hessler G, Kleemann HW, Hachtel S, Fuchs B et al (2015). Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol‐sensitive TRPC cation channels. Br J Pharmacol 172: 3650–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Wu M, Xu J, Weaver D, Li M, Zhu MX (2011). High‐throughput screening of TRPC channel ligands using cell‐based assays In: Zhu MX. (ed). TRP Channels: Boca Raton (FL), CRC Press/Taylor & Francis. [PubMed] [Google Scholar]
- Moran MM, Mcalexander MA, Biro T, Szallasi A (2011). Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov 10: 601–620. [DOI] [PubMed] [Google Scholar]
- Naylor J, Minard A, Gaunt HJ, Amer MS, Wilson LA, Migliore M et al (2016). Natural and synthetic flavonoid modulation of TRPC5 channels. Br J Pharmacol 173: 562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto Y, Sakaguchi S, Ishii Y (2002). Development of a highly efficient catalytic method for synthesis of vinyl ethers. J Am Chem Soc 124: 1590–1591. [DOI] [PubMed] [Google Scholar]
- Radtke L, Willot M, Sun H, Ziegler S, Sauerland S, Strohmann C et al (2011). Total synthesis and biological evaluation of (−)‐englerin A and B: synthesis of analogues with improved activity profile. Angew Chem Int Ed Engl 50: 3998–4002. [DOI] [PubMed] [Google Scholar]
- Ratnayake R, Covell D, Ransom TT, Gustafson KR, Beutler JA (2009). Englerin A, a selective inhibitor of renal cancer cell growth, from Phyllanthus engleri . Org Lett 11: 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubaiy HN (2017). A short guide to electrophysiology and ion channels. Journal of Pharmacy and Pharmaceutical Sciences 20: 48–67. [DOI] [PubMed] [Google Scholar]
- Rubaiy HN, Ludlow MJ, Bon RS, Beech DJ (2017a). Pico145 – powerful new tool for TRPC1/4/5 channels. Channels (Austin) 11: 362–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubaiy HN, Ludlow MJ, Henrot M, Gaunt HJ, Miteva K, Cheung SY et al (2017b). Picomolar, selective and subtype specific small‐molecule inhibition of TRPC1/4/5 channels. J Biol Chem 292: 8158–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourbier C, Scroggins BT, Ratnayake R, Prince TL, Lee S, Lee MJ et al (2013). Englerin A stimulates PKCθ to inhibit insulin signaling and to simultaneously activate HSF1: pharmacologically induced synthetic lethality. Cancer Cell 23: 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn DG, Holt DA, Dodson J, Mcatee JJ, Terrell LR, Barton L et al (2013). The discovery of potent blockers of the canonical transient receptor channels, TRPC3 and TRPC6, based on an anilino‐thiazole pharmacophore. Bioorg Med Chem Lett 23: 4979–4984. [DOI] [PubMed] [Google Scholar]
- Willot M, Radtke L, Konning D, Frohlich R, Gessner VH, Strohmann C et al (2009). Total synthesis and absolute configuration of the guaiane sesquiterpene englerin A. Angew Chem Int Ed Engl 48: 9105–9108. [DOI] [PubMed] [Google Scholar]
- Wu Z, Zhao S, Fash DM, Li Z, Chain WJ, Beutler JA (2017). Englerins: a comprehensive review. J Nat Prod 80: 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Chemical synthesis and validation of analogue A54 (1).
Supporting information