

Abstract
Amide bond formation serves as a fundamental reaction in chemistry, and is practically useful for the synthesis of peptides, food additives, and polymers. However, current methods for amide bond formation essentially generate wastes and suffer from poor atom economy under harsh conditions. To solve these issues, we demonstrated an alternative synthesis method for diverse tryptophyl-N-alkylamides by the combination of the first adenylation domain of tyrocidine synthetase 1 with primary or secondary amines as nucleophiles. Moreover, the physiological role of this domain is l-phenylalanine adenylation; however, we revealed that it displayed broad substrate flexibility from mono-substituted tryptophan analogues to even d-tryptophan. To the best of our knowledge, this is the first evidence for an adenylating enzyme-mediated direct amide bond formation via a sequential enzymatic activation of amino acids followed by nucleophilic substitution by general amines. These findings facilitate the design of a promising tool for biocatalytic straightforward amide bond formation with less side products.
Introduction
Amide bonds broadly appear in a vast number of organic compounds, including both biologically active natural products and synthetic chemicals. Thus, amide bond formation can be an important step in the synthesis of pharmaceuticals, bioactive peptides, food additives, flavours, nutrients and polymers. In addition, amide bond formation plays a significant role in synthesising useful compounds for industrial applications1. Various methods for amide bond formation have been developed to date. Typical amide synthesis, such as solid-phase peptide synthesis (SPPS)2, depends on a condensation reagent3 to yield a highly reactive ester for reaction with amines, resulting in amide bond formation. Alternatively, amide bond is also formed by the acylation of amines with carboxylic acid chloride in the presence of a base (Schotten-Baumann reaction)4. These chemical methods are well established and would give good yields; however, they have some underlying drawbacks, such as harmful conditions, toxicity of reagents, and side product formation. SPPS also requires protection and deprotection procedures to avoid unnecessary side reactions, and considering the stoichiometric amount of the condensation reagent, it generates at least an equal amount of side product per product formed1,3. Hence, the requirement for a precisely controlled amide bond formation reaction with lesser steps and improved efficiency remains a serious concern for practical purposes. Catalytic methods like boron-based catalysis or metal-based catalysis, including titanium or zinc complex, are also potential methods for amide production5. However, these reactions need to be carried out under harsh conditions and they suffer from undesirable side reactions. Hence, an efficient, selective, mild, and environment-friendly strategy for a method independent of condensation reagent, which results in less side products, remains in high demand for amide bond formation reactions4.
In nature, the most abundant biological macromolecules linked with amide bonds are peptides and proteins synthesised by a ribosome system in living organisms. Biocatalytic amide bond formation is achieved by various enzymatic or chemoenzymatic methods, i.e. aminolysis reaction by hydrolases6,7, peptide formation between amino acid esters and amino acids by acyltransferases8–10, ligation with two or more unprotected amino acids by ATP-dependent ligases11–16, nucleophilic substitution by transpeptidases17, and hydration of nitrile to amide by nitrile hydratases18. A nonribosomal peptide synthetase (NRPS) is another such method commonly known as a ribosome-independent biological system for secondary metabolite production in bacteria and fungi, and they are composed of huge multidomain modular enzymes19. An adenylation domain (A domain), one of the essential region in NRPS involved in the first step of ATP-driven nonribosomal peptide synthesis, is critical for the selection and incorporation of a specific amino acid into a peptide scaffold, and it activates a carboxy group of the amino acid accompanied by hydrolysis of ATP to AMP and pyrophosphate to form the corresponding aminoacyl-AMP intermediate20.
The truncated A domain of tyrocidine synthetase 1 (TycA) produced in Escherichia coli catalyses the formations of the following dipeptides: l-phenylalanyl-l-phenylalanine, l-phenylalanyl-l-alanine, l-phenylalanyl-l-phenylalaninamide, and l-phenylalanyl-l-leucinamide21. However, this method is applicable only to the combination of amino acids (amide); hence, the products are limited to dipeptides. More recently, acyl-CoA synthetase AcsA from Pseudomonas chlororaphis B23 was identified to catalyse amide bond formation via a thioester bond between short chain fatty acids and l-cysteine22. Likewise, DltA is responsible for transferring d-alanine to a carrier protein, DltC in Bacillus subtilis 168, and it catalyses the synthesis of various dipeptides containing cysteine at C-terminal and some oligopeptides carrying d- or l-cysteine at the second N-terminal position23. In both these biocatalytic cases, thiol-based compounds play a key role in amide bond formation, because a nucleophilic attack is triggered by a thiol group followed by non-enzymatic S- to N- acyl transfer resulting in amide bond formation. To the best of our knowledge, little information is available concerning direct amidation yielding in amide bond formation between the acyl group of amino acids and general amines.
Here, for the identification of new adenylating enzyme-mediated amide bond formation methods, we have demonstrated l-tryptophyl-N-alkylamide synthesis by enzymatic adenylation of the carboxy group of l-tryptophan followed by nucleophilic substitution with diverse amines.
Results
l-Tryptophyl-N-alkylamides synthesis by TycA-A
We have demonstrated previously that the A domain of TycA (TycA-A) can be used for the synthesis of l-tryptophyl-l-proline24. This finding suggested that the carboxy group of l-tryptophan was first adenylated by TycA-A followed by nucleophilic substitution by the amino group of l-proline. Based on this phenomenon, we speculated that amide bond would be formed directly by activating the carboxy group of the amino acid using an A domain followed by nucleophilic substitution of general amines. This hypothetical reaction mechanism would enable the synthesis of a wide range of amides as well as peptides, and this system should serve as a versatile platform of enzyme-mediated amide bond formation. To investigate the nucleophile specificity of TycA-A, the following nucleophilic species were used in l-tryptophyl-N-alkylamide synthesis: methylamine, dimethylamine, trimethylamine, β-alanine, γ-aminobutyric acid, azetidine, pyrrolidine, piperidine, azepane, azocane, d-proline, cis-4-hydroxy-l-proline, cis-4-hydroxy-d-proline, l-prolinamide, and l-azetidine-2-carboxylic acid. After the reaction, HPLC analysis of the reaction mixture showed that specific peaks corresponding to newly formed products were detected in all amines except for trimethylamine. As speculated, an amide bond was probably formed by the enzymatic adenylation of l-tryptophan and subsequent nucleophilic substitution by amines. Each product was calculated from the amount of decrease in l-tryptophan minus the amount of l-tryptophyl-l-tryptophan generation, and the yield was calculated against the added l-tryptophan concentration (Table 1).
Table 1.
MS detection of amide products formed by activation of l-tryptophan by TycA-A followed by nucleophilic substitution.
| Substrate for TycA-A | Nucleophile | aProduct (mM) | bConversion (%) | Protonated parent ion (m/z) | MS/MS fragment ion (m/z) | |||
|---|---|---|---|---|---|---|---|---|
| cType 1 | dType 2 | |||||||
| l-Tryptophan | (Linear amines) | |||||||
| Methylamine | 0.81 | 8.1 | 218.1302 | 58.0300 | 159.0923 | 87.0552 | 130.0655 | |
| Dimethylamine | 0.81 | 8.1 | 232.0858 | 72.0446 | 159.0915 | 101.0705 | 130.0648 | |
| β-Alanine | 0.96 | 9.6 | 276.1371 | 116.0508 | 159.0930 | 145.0653 | 130.0674 | |
| γ-Aminobutyric acid | 1.16 | 11.6 | 290.1532 | 130.0663 | 159.0934 | ND | ND | |
| (Cyclic amines) | ||||||||
| Azetidine | 0.82 | 8.2 | 244.1457 | 84.0450 | 159.0927 | 113.0713 | 130.0657 | |
| Pyrrolidine | 0.92 | 9.2 | 258.1301 | 98.0598 | 159.0916 | 127.0863 | 130.0646 | |
| Piperidine | 1.40 | 10.4 | 272.1783 | 112.0767 | 159.0930 | 141.1038 | 130.0661 | |
| Azepane | 0.52 | 5.2 | 286.1944 | 126.0927 | 159.0933 | 155.1198 | 130.0663 | |
| Azocane | 1.35 | 13.5 | 300.2090 | 140.1087 | 159.0936 | 169.1350 | 130.0665 | |
| (Cyclic amino acids and derivative) | ||||||||
| d-Proline | 0.46 | 4.6 | 302.1566 | 142.0647 | 159.0917 | 171.0760 | 130.0647 | |
| cis-4-Hydroxy-l-proline | 0.94 | 9.4 | 318.1136 | ND | 159.0910 | 187.0707 | 130.0642 | |
| cis-4-Hydroxy-d-proline | 0.78 | 7.8 | 318.1481 | ND | 159.0934 | 187.0742 | 130.0662 | |
| l-Prolinamide | 0.85 | 8.5 | 301.1682 | ND | 159.0940 | 170.0626 | 130.0666 | |
| l-Azetidine-2-carboxylic acid | 0.78 | 7.8 | 288.1772 | 128.0342 | 159.0918 | 157.0604 | 130.0649 | |
ND, not detected
aCalculated from the amount of decrease in l-tryptophan minus the amount of l-tryptophyl-l-tryptophan generation.
bCalculated against the added l-tryptophan concentration.
cFragment ion generated by C1-C2 cleavage.
dFragment ion generated by C2-C3 cleavage.
Identification of l-tryptophyl-N-alkylamides by MS/MS and NMR spectra
The structures of various amides from l-tryptophan with amines were identified by MS/MS and NMR analyses. The amides produced in the reaction mixture were isolated by HPLC equipped with a fraction collector. Each mass-to-charge ratio (m/z) of protonated ions resulting from products was identical to that of the proposed structures for each l-tryptophyl-N-alkylamide. Further, these structures were identified by assignment of fragment ion peaks obtained from MS/MS spectra to the two cleavage fragments, as expected (Figs 1,S1). We have assigned the structures for various amides deduced from these MS/MS fragments (Table 1).
Figure 1.

Fragmentation pattern of the reaction products.
In addition, l-tryptophylazetidine as a typical cyclic amine and l-tryptophyldimethylamine as a typical linear amine were characterized by NMR (Tables 2,3 Figs S2,S3). The identified structures were in good agreement with the proposed structures as shown in Fig. 2. Consequently, it is uncovered that the nucleophilic substitution by amines had occurred to the carbonyl moiety of l-tryptophyl-AMP intermediate, and then an amide bond formed between l-tryptophan and various amines (Fig. 3).
Table 2.
NMR spectrum data of l-tryptophylazetidine.
| Position no.a | Chemical shift (δ ppm) | ||
|---|---|---|---|
| 13C NMR | 1H NMR (integration, multiplicity, J value [Hz]) | ||
| 1 | 171.1 | — | |
| 2 | 53.1 | 4.20 | (1H, dd, 9.0, 6.0) |
| 3 | 29.3 | 3.36 | (1H, dd, 14.4, 6.0) |
| 3.30 | (1H, dd, 14.4, 9.0) | ||
| 4 | 128.1 | 7.33 | (1H, s) |
| 5 | 109.5 | — | |
| 6 | 129.6 | — | |
| 7 | 120.9 | 7.60 | (1H, ddd, 8.1, 0.9, 0.6) |
| 8 | 125.2 | 7.21 | (1H, ddd, 8.1, 7.2, 0.9) |
| 9 | 122.6 | 7.29 | (1H, ddd, 7.8, 7.2, 0.9) |
| 10 | 115.0 | 7.55 | (1H, ddd, 7.8, 0.9, 0.6) |
| 11 | 139.1 | — | |
| 1′ | 51.6 | 3.94 | (1H, tdd, 9.9, 6.0, 0.6) |
| 3.71 | (1H, tdd, 9.9, 6.0, 1.2) | ||
| 2′ | 17.7 | 2.07 | (1H, dtt, 11.4, 9.6, 6.0) |
| 1.75 | (1H, dtt, 11.4, 9.6, 6.0) | ||
| 3′ | 50.6 | 4.02 | (1H, tdd, 9.6, 6.0, 1.2) |
| 3.11 | (1H, tdd, 9.6, 6.0, 0.6) | ||
aPosition numbers are shown in Fig. 2(a).
Table 3.
NMR spectrum data of l-tryptophyldimethylamine.
| Position no.a | Chemical shift (δ ppm) | ||
|---|---|---|---|
| 13C NMR | 1H NMR (integration, multiplicity, J value [Hz]) | ||
| 1 | 172.1 | — | |
| 2 | 53.8 | 4.73 | (1H, dd, 9.8, 6.0) |
| 3 | 29.3 | 3.39 | (1H, dd, 14.4, 6.0) |
| 3.34 | (1H, dd, 14.4, 9.8) | ||
| 4 | 128.2 | 7.31 | (1H, s) |
| 5 | 109.2 | — | |
| 6 | 129.6 | — | |
| 7 | 120.9 | 7.55 | (1H, ddd, 7.8, 0.9, 0.6) |
| 8 | 125.2 | 7.27 | (1H, ddd, 7.8, 7.2, 0.9) |
| 9 | 122.5 | 7.19 | (1H, ddd, 8.1, 7.2, 0.9) |
| 10 | 115.0 | 7.53 | (1H, ddd, 8.1, 0.9, 0.6) |
| 11 | 139.2 | — | |
| 1′ | 38.6 | 2.81 | (3H, s) |
| 2′ | 39.7 | 2.71 | (3H, s) |
aPosition numbers are shown in Fig. 2(b).
Figure 2.

Structures of l-tryptophyl-N-alkylamide. (a) l-tryptophylazetidine. (b) l-tryptophyldimethylamine.
Figure 3.

Schematic representation of chemoenzymatic amide bond formation using the truncated adenylating enzyme, TycA-A.
Substrate specificity of TycA-A
It was assumed that if TycA-A could accept l-tryptophan analogues, more diverse amides would be synthesised. To investigate the amide synthesis using other substrates, 6-fluorotryptophan, 6-chlorotryptophan, 6-bromotryptophan, and 5-hydroxytryptophan as l-tryptophan analogues were used in place of l-tryptophan. HPLC analysis of the reaction mixtures identified the presence of newly formed product peaks. The chromatograms of the reaction mixture containing 6-fluorotryptophan and amines gave the product peaks at 7.15 min, 5.71 min, and 5.98 min, corresponding to 6-fluorotryptophyl-l-proline, 6-fluorotryptophyldimethylamine, and 6-fluorotryptophylazetidine, respectively (Fig. S4). Similarly, the respective product peaks were observed in case of 6-chlorotryptophan, 6-bromotryptophan, and 5-hydroxytryptophan (Figs S5–S7). As summarized in Table 4, mono-substituted l-tryptophan analogues were also available for adenylation by TycA-A, and the presence of corresponding products could clearly be confirmed.
Table 4.
MS analysis of the ligation products between tryptophan analogs with typical amines.
| Substrate for TycA-A | Nucleophile | ||
|---|---|---|---|
| l-Proline | Azetidine | Dimethylamine | |
| 6-Fluorotryptophan | 320.1475 | 262.1401 | 250.1401 |
| 6-Chlorotryptophan | 336.1170 | 278.1109 | 266.1119 |
| 6-Bromotryptophan | 380.0649 | 322.0614 | 310.0617 |
| 5-Hydroxytryptophan | 318.1503 | 260.1415 | 248.1448 |
Stoichiometry
Time course of l-tryptophan consumption and AMP formation during the synthesis was examined. Although 1.4 mM l-tryptophyl-l-proline was synthesised after a 24-h reaction, little correlation between l-tryptophyl-l-proline formation and AMP formation was found in this reaction (Fig. 4). The major reason for this discrepancy is possibly due to the side reactions. In fact, formation of a smaller amount of l-tryptophyl-l-tryptophan along with l-tryptophyl-l-proline and an unfavourable degradation of ATP to ADP were confirmed in the reaction mixture.
Figure 4.
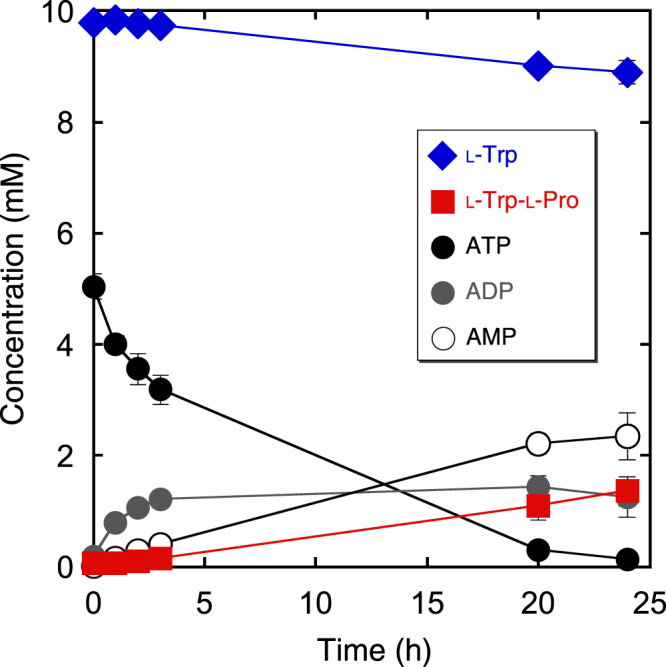
Time course of l-tryptophyl-l-proline synthesis using TycA-A.
Discussion
In this study, we have demonstrated the synthesis of various l-tryptophyl-N-alkylamides by a sequential process as per the following stepwise mechanism: (i) l-tryptophan was enzymatically adenylated by TycA-A to form an l-tryptophyl-AMP intermediate, and (ii) a nucleophilic substitution by an amine to l-tryptophyl-AMP led to an enzyme-independent amide bond formation (Fig. 3).
We have recently reported the synthesis of various aminoacyl-l-prolines, such as l-tryptophyl-l-proline, l-aspartyl-l-proline, and l-lysyl-l-proline using TycA-A, SurfB2-A, and BacB1-A, respectively24. These adenylating enzyme-catalysed reactions would be achieved by nucleophilic substitution by l-proline to aminoacyl-AMP intermediates. This could mean that if a nucleophilic substitution was actually carried out on an aminoacyl-AMP, an enzyme-independent amide bond formation would be established. We tested this in a reaction by using TycA-A as a model adenylating enzyme with various amines as nucleophiles. Formation of amides could be confirmed for all the reagents tested (Table 1). It is noted that trimethylamine, a tertiary amine, was inert because it was significantly less nucleophilic than primary or secondary amines. In general, tertiary amines are sterically hindered bases that are poor nucleophiles, and thereby it seemed to be hard to enhance the inherent reactivity of an amine in the chemoenzymatic method.
Interestingly, mono-substituted tryptophan analogues and even its d-isomer were accepted as substrates for TycA-A (Table 4), suggesting that TycA-A is a more promiscuous enzyme than previous reports suggested25–28.
In a similar study, investigations on direct peptide bond formation using enzymatic activation of the carboxy group on an amino acid followed by nucleophilic substitution was reported. Nakajima et al. revealed that aminoacyl-tRNA synthetase enables the catalysis of dipeptide formation via an enzymatically formed aminoacyl-AMP-aminoacyl-tRNA synthetase complex, followed by nucleophilic substitution with amino acids29. Dieckmann et al. found that N-phenylalanyl dipeptides are synthesised by TycA-A using l-phenylalanine and some amino acids (amides)21. Our findings are functionally relevant to these reports; however, we also successfully encompassed its applicability beyond peptides to a broad range of general amines.
Previously, Jakubowski had proposed a stimulating enzymatic amide bond formation model involving arginyl-, valyl-, and isoleucyl-tRNA synthetases30 (class I aminoacyl-tRNA synthetases), and lysyl-, aspartyl-, and seryl-tRNA synthetases31 (class II aminoacyl-tRNA synthetases), catalysing the formation of arginylcysteine, valylcysteine, and isoleucylcysteine and lysylcysteine, aspartylcysteine, and serylcysteine, respectively, in a cysteine-dependent manner. More recently, Abe et al. reported a similar enzymatic thiol-based chemistry for amide bond formation using acyl-CoA synthetase22 and d-alanine-activating enzyme DltA23. In this case, a unique enzymatic adenylation of the substrate and subsequent thioester formation by thiol-containing compounds occurred and the resulting highly reactive thioester bond is immediately rearranged to a more stable amide bond by intramolecular S-to-N acyl transfer mechanism, similar to the native chemical ligation in chemical protein synthesis32. This is an important amide-bond forming methodology; however, it is basically limited to the use of aminoacyl thioester chemistry.
The present amide synthesis method described in this study is advantageous for providing diverse aminoacyl-N-alkylamides because of its broader specificity for primary and secondary amines. Importantly, even a d-amino acid was introduced into a dipeptide at the C-terminus position as l-tryptophyl-d-proline. In addition, no extra step is needed, such as CoA addition mediated by phosphopantetheinyl transferase, which is essential to obtain active enzyme of TycA in natural form33.
The present method has some advantages in amide synthesis over conventional method. The major organic synthesis commonly relies on N-(de)protection and condensation to form amino acid amides. These cumbersome procedures require a protecting group, equal amount of condensation reagent and substrate, and solvent. In contrast, the chemoenzymatic synthesis enables direct ligation of an unprotected amino acid with an amine, which is a straightforward strategy and has greater atom economy. Instead, the chemoenzymatic method requires ATP for amino acid activation. To improve productivity of amide compound synthesis, ATP regeneration should be considered using coupling systems. We are currently investigating ATP regeneration with an adenylation domain platform toward catalytic and practical synthesis of a broad spectrum of amide compounds including peptides, which will be reported elsewhere.
We have attempted to measure the kinetic parameters for general amines; however, quantitation was difficult. First, tryptophyl-N-alkylamides, which serves as quantitative standards, were not available from commercial sources. In addition, as shown in stoichiometric analysis (Fig. 4), the decrease in ATP and formation of the amide and AMP did not show any correlation. This is probably due to other possible side reactions, including l-tryptophyl-l-tryptophan formation, caused by competitive substitution by l-tryptophan and unfavourable ATP hydrolysis.
To enhance the reactivity, which may be the rate-limiting factor in the overall reaction, a high concentration (200 mM) of amines was found to be favourable for efficient amide bond formation; however, the reaction velocity remained relatively low even at this concentration in comparison to other enzymatic amide or peptide bond forming reactions. This reduced efficiency is possibly due to the lack of the downstream domains. In the NRPS machinery, the adenylation domain, as well as thiolation domain (also referred to as peptidyl carrier protein domain) and condensation domain are indispensable for peptide bond formation, except for a few minor cases. Their absence resulted in both decreased the rate of amide formation and the broad nucleophile specificity. Similarly, in the case of dipeptide synthesis by aminoacyl-tRNA synthetase, the peptide bond was formed between aminoacyl-AMP and various amino acids; however, the reaction rate is extremely low, suggesting the importance of another conformational factor such as tRNA29.
In conclusion, we have revealed a new amide bond formation mechanism involving enzymatic adenylation by TycA-A followed by nucleophilic substitution with primary or secondary amines. Furthermore, TycA-A showed a wide spectrum of substrate flexibility for both mono-substituted tryptophan and d-tryptophan. An improved method for amide bond formation was required, because the current chemical method suffers from low atom economy and results in wastage34. The new method demonstrated here would provide a better approach for a more efficient amide bond formation.
Methods
Reagents
All chemicals were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) or Kanto Chemical Co., Inc. (Tokyo, Japan) unless stated otherwise. Biological materials were used as previously described24.
Preparation of TycA-A
The tycA-A gene was cloned and overexpressed in Escherichia coli as described previously24. The supernatant was used for enzyme purification on the Protino Ni-TED 1000 column (Macherey-Nagel, Düren, Germany) equipped on an ÄKTA prime plus (GE Healthcare), and the eluent was desalted on a PD-10 column (GE Healthcare). The enzyme was purified to apparent homogeneity as judged from sodium dodecyl sulfate-polyacrylamide electrophoresis analysis.
Synthesis of amides by TycA-A
The standard reaction mixture containing 50 mM Tris-HCl buffer (pH 8.0), 10 mM l-tryptophan analogues (acyl donor), 10 mM each of various amines (nucleophile), 20 mM ATP, 20 mM MgSO4, and 0.1 mg/mL TycA-A in a total volume of 100 μL was incubated at 37 °C for up to 24 h. The reaction was terminated by heating at 80 °C for 10 min and it was centrifuged at 20,000× g for 10 min at 4 °C. To assess stoichiometry, the reaction containing 50 mM Tris-HCl buffer (pH 8.0), 10 mM l-tryptophan analogues (acyl donor), 200 mM l-proline (nucleophile), 5 mM ATP, 5 mM MgSO4, and 0.5 mg/mL TycA-A in a total volume of 100 μL was incubated at 37 °C. A small portion of the mixture was withdrawn for the determination of l-tryptophyl-l-proline, ATP, and AMP at each sampling period.
HPLC analysis
To confirm amide formation, the supernatant was analysed by high performance liquid chromatography (HPLC) equipped with an XTerra MS C18 column (Waters, Milford, MA) kept at 40 °C. The separation was carried out using a linear gradient of methanol (0–100%) in 0.1% (v/v) formic acid solvent for 15 min at a flow rate of 1.0 mL/min, and detection was performed at a wavelength of 214 nm. Whenever it was necessary, the reactants were collected by an HPLC system equipped with a fraction collector (model CHF122SB, Advantec, Tokyo, Japan). ATP and AMP were measured as reported previously35.
Structural analysis of various amides
The MS/MS analysis data were acquired with a Triple TOF4600 mass spectrometer (omics analysis LC-MS system; SCIEX, Framingham, MA) by infusion in the positive modes. The conditions were as follows: ion spray voltage floating, 5.5 kV; declustering potential, 80 V; source temperature, 0 °C; desolvation temperature, 500 °C; ion source gas 2, 20 psi; collision energy, 30 V. The data were collected using Analyst TF 1.6 software (SCIEX). NMR spectra were measured with an AVANCE III spectrometer (Bruker, Billerica, MA). For 1H NMR and 13C NMR analysis, the compounds dissolved in D2O containing trimethylsilyl propanoic acid were used as an internal standard, and spectrometer was operated at 600 MHz and 150 MHz, respectively.
Electronic supplementary material
Acknowledgements
This work was partially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant No. 26289318 and 16K14495 (to K.K.), and by the Institute for Fermentation, Osaka (IFO), Japan (to K.K.).
Author Contributions
R.H. conceived the experiments, analyzed the data, and wrote the manuscript. K.H. and S.S. carried out the experiments, analyzed the data, and wrote the manuscript. K.K. supervised the project and improved the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-21408-8.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pattabiraman VR, Bode JW. Rethinking amide bond synthesis. Nature. 2011;480:471–479. doi: 10.1038/nature10702. [DOI] [PubMed] [Google Scholar]
- 2.Merrifield RB. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963;85:2149–2154. doi: 10.1021/ja00897a025. [DOI] [Google Scholar]
- 3.Valeur E, Bradley M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 2009;38:606–631. doi: 10.1039/B701677H. [DOI] [PubMed] [Google Scholar]
- 4.Montalbetti CAGN, Falque V. Amide bond formation and peptide coupling. Tetrahedron. 2005;61:10827–10852. doi: 10.1016/j.tet.2005.08.031. [DOI] [Google Scholar]
- 5.Lundberg H, Tinnis F, Selander N, Adolfsson H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014;43:2714–2742. doi: 10.1039/C3CS60345H. [DOI] [PubMed] [Google Scholar]
- 6.Yazawa K, Numata K. Recent advances in chemoenzymatic peptide syntheses. Molecules. 2014;19:13755–13774. doi: 10.3390/molecules190913755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar D, Bhalla TC. Microbial proteases in peptide synthesis: approaches and applications. Appl. Microbiol. Biotechnol. 2005;68:726–736. doi: 10.1007/s00253-005-0094-7. [DOI] [PubMed] [Google Scholar]
- 8.Yokozeki K, Hara S. A novel and efficient enzymatic method for the production of peptides from unprotected starting materials. J. Biotechnol. 2005;115:211–220. doi: 10.1016/j.jbiotec.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Abe I, Hara S, Yokozeki K. Gene cloning and characterization of alpha-amino acid ester acyl transferase in Empedobacter brevis ATCC14234 and Sphingobacterium siyangensis AJ2458. Biosci. Biotechnol. Biochem. 2011;75:2087–2092. doi: 10.1271/bbb.110181. [DOI] [PubMed] [Google Scholar]
- 10.Hirao Y, Mihara Y, Kira I, Abe I, Yokozeki K. Enzymatic production of L-alanyl-l-glutamine by recombinant E. coli expressing alpha-amino acid ester acyltransferase from Sphingobacterium siyangensis. Biosci. Biotechnol. Biochem. 2013;77:618–623. doi: 10.1271/bbb.120859. [DOI] [PubMed] [Google Scholar]
- 11.Yagasaki M, Hashimoto S. Synthesis and application of dipeptides; current status and perspectives. Appl. Microbiol. Biotechnol. 2008;81:13–22. doi: 10.1007/s00253-008-1590-3. [DOI] [PubMed] [Google Scholar]
- 12.Tabata K, Ikeda H, Hashimoto S. Ywf Ein Bacillus subtilis codes for a novel enzyme, l-amino acid ligase. J. Bacteriol. 2005;187:5195–5202. doi: 10.1128/JB.187.15.5195-5202.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato M, Kirimura K, Kino K. d-Amino acid dipeptide production utilizing d-alanine-d-alanine ligases with novel substrate specificity. J. Biosci. Bioeng. 2005;99:623–628. doi: 10.1263/jbb.99.623. [DOI] [PubMed] [Google Scholar]
- 14.Arai T, Arimura Y, Ishikura S, Kino K. l-Amino acid ligase from Pseudomonas syringae producing tabtoxin can be used for enzymatic synthesis of various functional peptides. Appl. Environ. Microbiol. 2013;79:5023–5029. doi: 10.1128/AEM.01003-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kino K, Arai T, Arimura Y. Poly-alpha-glutamic acid synthesis using a novel catalytic activity of RimK from Escherichia coli K-12. Appl. Environ. Microbiol. 2011;77:2019–2025. doi: 10.1128/AEM.02043-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kino K, Arai T, Tateiwa D. A novel l-amino acid ligase from Bacillus subtilis NBRC3134 catalyzed oligopeptide synthesis. Biosci. Biotechnol. Biochem. 2010;74:129–134. doi: 10.1271/bbb.90649. [DOI] [PubMed] [Google Scholar]
- 17.Buynak JD. Cutting and stitching: the cross-linking of peptidoglycan in the assembly of the bacterial cell wall. ACS Chem. Biol. 2007;2:602–605. doi: 10.1021/cb700182u. [DOI] [PubMed] [Google Scholar]
- 18.Kato Y, Ooi R, Asano Y. Isolation and characterization of a bacterium possessing a novel aldoxime-dehydration activity and nitrile-degrading enzymes. Arch. Microbiol. 1998;170:85–90. doi: 10.1007/s002030050618. [DOI] [PubMed] [Google Scholar]
- 19.Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem. Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 20.Schmelz S, Naismith JH. Adenylate-forming enzymes. Curr. Opin. Struct. Biol. 2009;19:666–671. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dieckmann R, Neuhof T, Pavela-Vrancic M, von Dohren H. Dipeptide synthesis by an isolated adenylate-forming domain of non-ribosomal peptide synthetases (NRPS) FEBS Lett. 2001;498:42–45. doi: 10.1016/S0014-5793(01)02471-1. [DOI] [PubMed] [Google Scholar]
- 22.Abe T, Hashimoto Y, Hosaka H, Tomita-Yokotani K, Kobayashi M. Discovery of amide (peptide) bond synthetic activity in Acyl-CoA synthetase. J. Biol. Chem. 2008;283:11312–11321. doi: 10.1074/jbc.M709654200. [DOI] [PubMed] [Google Scholar]
- 23.Abe T, et al. Peptide bond synthesis by a mechanism involving an enzymatic reaction and a subsequent chemical reaction. J. Biol. Chem. 2016;291:1735–1750. doi: 10.1074/jbc.M115.700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara R, Suzuki R, Kino K. Hydroxamate-based colorimetric assay to assess amide bond formation by adenylation domain of nonribosomal peptide synthetases. Anal. Biochem. 2015;477:89–91. doi: 10.1016/j.ab.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Schaffer ML, Otten LG. Substrate flexibility of the adenylation reaction in the Tyrocidine non-ribosomal peptide synthetase. J. Mol. Catal. B-Enzym. 2009;59:140–144. doi: 10.1016/j.molcatb.2009.02.004. [DOI] [Google Scholar]
- 26.Dieckmann R, Lee YO, van Liempt H, von Dohren H, Kleinkauf H. Expression of an active adenylate-forming domain of peptide synthetases corresponding to acyl-CoA-synthetases. FEBS Lett. 1995;357:212–216. doi: 10.1016/0014-5793(94)01342-X. [DOI] [PubMed] [Google Scholar]
- 27.Pfeifer E, Pavela-Vrancic M, von Dohren H, Kleinkauf H. Characterization of tyrocidine synthetase 1 (TY1): requirement of posttranslational modification for peptide biosynthesis. Biochemistry. 1995;34:7450–7459. doi: 10.1021/bi00022a019. [DOI] [PubMed] [Google Scholar]
- 28.Pavela-Vrancic M, Dieckmann R, Dohren HV, Kleinkauf H. Editing of non-cognate aminoacyl adenylates by peptide synthetases. Biochem. J. 1999;342:715–719. doi: 10.1042/bj3420715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakajima H, et al. Dipeptide synthesis catalyzed by aminoacyl-tRNA synthetases from Bacillus stearothermophilus. Int. J. Pept. Protein Res. 1986;28:179–185. doi: 10.1111/j.1399-3011.1986.tb03245.x. [DOI] [PubMed] [Google Scholar]
- 30.Jakubowski H. Synthesis of cysteine-containing dipeptides by aminoacyl-tRNA synthetases. Nucleic Acids Res. 1995;23:4608–4615. doi: 10.1093/nar/23.22.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jakubowski H. Aminoacyl thioester chemistry of class II aminoacyl-tRNA synthetases. Biochemistry. 1997;36:11077–11085. doi: 10.1021/bi970589n. [DOI] [PubMed] [Google Scholar]
- 32.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 33.Lambalot RH, et al. A new enzyme superfamily - the phosphopantetheinyl transferases. Chem. Biol. 1996;3:923–936. doi: 10.1016/S1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 34.Constable DJC, et al. Key green chemistry research areas - a perspective from pharmaceutical manufacturers. Green Chem. 2007;9:411–420. doi: 10.1039/B703488C. [DOI] [Google Scholar]
- 35.Sato M, Masuda Y, Kirimura K, Kino K. Thermostable ATP regeneration system using polyphosphate kinase from Thermosynechococcus elongatus BP-1 for d-amino acid dipeptide synthesis. J. Biosci. Bioeng. 2007;103:179–184. doi: 10.1263/jbb.103.179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.