

SUMMARY
Constitutive cell-autonomous immunity in metazoans predates interferon-inducible immunity and comprises primordial innate defense. Phagocytes mobilize interferon-inducible responses upon engagement of well-characterized signaling pathways by pathogen-associated molecular patterns (PAMPs). The signals controlling deployment of constitutive cell-autonomous responses during infection have remained elusive. Vita-PAMPs denote microbial viability, signaling the danger of cellular exploitation by intracellular pathogens. We show that cyclic-di-adenosine monophosphate in live Gram-positive bacteria is a vita-PAMP engaging the innate sensor Stimulator of Interferon Genes (STING) to mediate endoplasmic reticulum (ER) stress. Subsequent inactivation of the mechanistic Target of Rapamycin mobilizes autophagy, which sequesters stressed ER membranes, resolves ER stress, and curtails phagocyte death. This vita-PAMP-induced ER-phagy additionally orchestrates an interferon response by localizing ER-resident STING to autophagosomes. Our findings identify stress-mediated ER-phagy as a cell-autonomous response mobilized by STING-dependent sensing of a specific vita-PAMP, and elucidate how innate receptors engage multilayered homeostatic mechanisms to promote immunity and survival after infection.
INTRODUCTION
Self versus non-self discrimination in the innate immune system relies on the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRR), which mobilize signaling pathways whose outcome varies depending on the PAMP/PRR involved (Janeway, 1992). Differences in outcome also reflect PAMP recognition within the context of additional signals associated with the ability of pathogens to replicate and express virulence factors (Vance et al., 2009). Microbial viability comprises a distinct signal in the form of vita-PAMPs, a subset of PAMPs present specifically in live microorganisms and distinct from the classic PAMPs shared by live and dead microorganisms (Blander and Sander, 2012). Vita-PAMPs enable innate immune cells to discriminate between live and dead microorganisms and mobilize highly inflammatory responses only to live microorganisms (Blander and Sander, 2012). One such vita-PAMP is prokaryotic messenger RNA (mRNA), which in context of the classic PAMP lipopolysaccharide from Gram-negative bacteria, activates the NLRP3 inflammasome and augments the type-I interferon (IFN-I) response (Sander et al., 2011). Vita-PAMPs other than bacterial mRNA likely exist, but their identity and the responses they trigger are not defined.
Cell-autonomous immunity relies on both constitutive (pre-existing) and IFN-inducible responses that neutralize microorganisms and protect host tissues from intracellular infection (MacMicking, 2012; Randow et al., 2013). Constitutive cell-autonomous responses are mobilized within the first few hours and depend on pre-formed homeostatic and defense mechanisms (Moretti and Blander, 2017). Interferons induce the expression of potent anti-microbial effector proteins that are deployed to sites of microbial replication (MacMicking, 2012). By controlling the production of IFN-I, compartment-specific PRR, activated upon PAMP detection, dictate the mobilization of IFN-I-inducible cell-autonomous responses (Randow et al., 2013). In contrast, the nature and regulation of constitutive cell-autonomous responses to infection are poorly defined (Moretti and Blander, 2017). Furthermore, a connection between constitutive and IFN-inducible immunity has not been delineated.
Here, we provide evidence for mobilization of a series of integrated constitutive cell-autonomous responses upon engagement of a PRR, Stimulator of Interferon Genes (STING), by cyclic-di-adenosine monophosphate (c-di-AMP), a bacterial second messenger present in live Gram-positive bacteria and known to induce IFN-I production through STING (Corrigan and Grundling, 2013). We identify c-di-AMP as a vita-PAMP whose detection by phagocytes mobilizes STING-dependent endoplasmic reticulum (ER) stress, which protects mice against a Gram-positive infection, and at the cellular level, inactivates the Serine/Threonine kinase mechanistic Target of Rapamycin (mTOR) and induces canonical autophagy. This autophagy resolves ER stress by removing stressed ER membranes, a process termed ER-phagy (Bernales et al., 2007), thereby protecting phagocytes from infection-induced death and localizing STING to autophagosomal membranes as a prelude to IFN-I signaling. Modulation in any step from c-di-AMP availability to STING expression, ER stress, mTORC1 activity, or autophagy, blunts the heightened IFN-I response uniquely triggered by live Gram-positive bacteria. Our work shows that constitutive cell-autonomous responses can be deployed by vita-PAMP engagement of a PRR to preserve cell homeostasis and set the stage for IFN-inducible immunity in a battle of host defense against infection.
RESULTS
Live Gram-positive bacteria elicit canonical autophagy and an augmented IFN-I response
We investigated whether distinct innate responses are triggered by the viability of Gram-positive bacteria. We studied the phagocyte response to avirulent Listeria innocua to decipher the contribution of bacterial viability independently of virulence factors. Macrophages produced greater levels of IFN-I upon stimulation with live compared to dead L. innocua and no detectable IL-1β (Figure 1A). This response differed from that to live avirulent Gram-negative Escherichia coli, which induced both IL-1β secretion associated with NLRP3 inflammasome activation and an augmented IFN-I response (Sander et al., 2011).
Figure 1. Live but not dead Gram-positive bacteria elicit an augmented IFN-I response and induce autophagy irrespective of virulence factors.
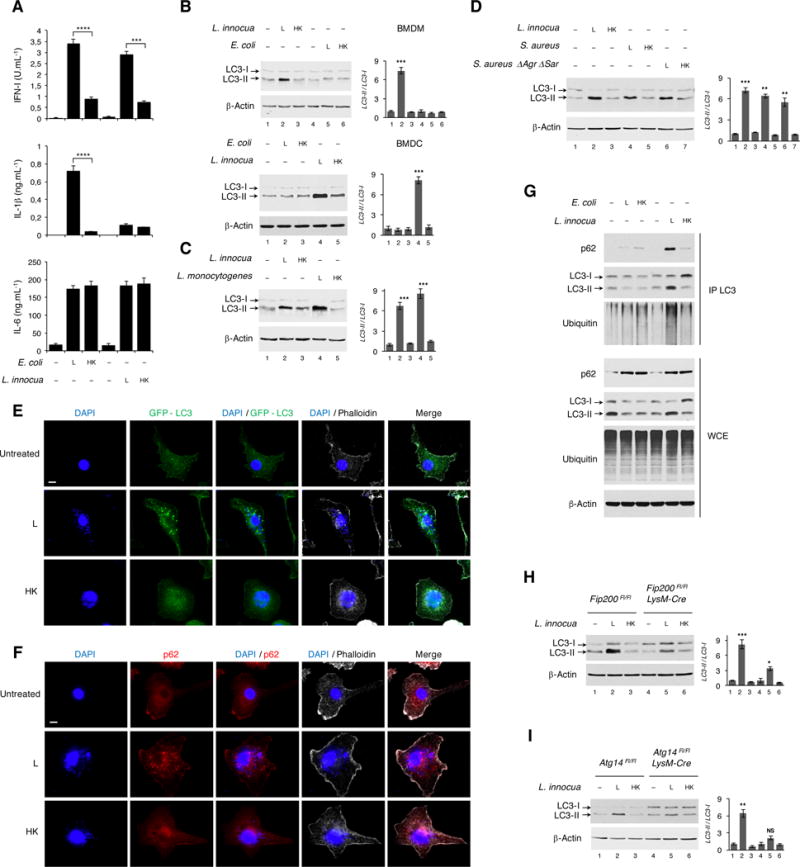
(A) Cytokine production by bone marrow derived macrophages (BMDM) 20 hr post-stimulation with Live (L) or Dead (heat-killed, HK) E. coli or L. innocua as indicated. Type-I IFN (IFN-I) measured by ISRE-Luciferase reporter activity, IL-1β and IL-6 by ELISA.
(B) Western blots (WB) on BMDM or bone marrow derived DC (BMDC) 6 hr post indicated stimuli.
(C and D) WB on BMDM 3 hr post indicated stimuli.
(E and F) Confocal micrographs showing GFP-LC3 transgenic (E) or C57BL/6J wild-type (F) BMDM 3 hr post-stimulation with L or HK L. innocua visualized for GFP or stained for endogenous p62. Scale bar = 10 μm.
(G) WB on immunoprecipitates of endogenous LC3 (IP LC3) and whole cell extracts (WCE) from BMDM 6 hr post indicated stimuli, probed for p62, LC3 and ubiquitin.
(H and I) WB for LC3 on BMDM of indicated genotypes 6 hr post indicated stimuli.
Bar graphs in B-D, H and I show quantification of indicated WB signals. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments.
Autophagy participates in degradation of bacteria that invade the cytoplasm such as Mycobacterium or Salmonella (Huang and Brumell, 2014), and involves LC3-I conjugation to phosphatidylethanolamine to generate LC3-II on autophagosomal membranes (Green and Levine, 2014). Because autophagy is a key regulator of inflammasome activation (Saitoh and Akira, 2016), we investigated its role in differential inflammasome activation by live Gram-negative and Gram-positive bacteria. Lipidated LC3-II levels increased in both macrophages and dendritic cells (DC) in response to live but not dead L. innocua (Figure 1B). Virulent Listeria monocytogenes and Staphylococcus aureus also induced LC3 lipidation compared to their dead counterparts (Figures 1C, 1D and S1A). Virulence factors were dispensable because avirulent ΔAgrΔSar S. aureus also induced LC3 lipidation (Figure 1D). Neither live nor dead E. coli triggered LC3 lipidation (Figure 1B). Two related attenuated strains of Gram-negative Legionella pneumophila also failed to induce LC3 lipidation (Figure S1A).
LC3 lipidation was first observed at 1.5-hours post stimulation with L. innocua, persisted over 6 hours (Figure S1B), and was independent of phagolysosomal activity or dose of bacteria (Figures S1C and S1D). Further characterization using macrophages from GFP-LC3 transgenic mice, showed prominent GFP-LC3 puncta, characteristic of autophagy, only in response to live L. innocua (Figure 1E). No GFP-LC3 puncta were observed to either live or dead E. coli (Figure S1E), consistent with differential LC3 lipidation to L. innocua and not E. coli (Figure 1B). Similar puncta were observed for the autophagy receptor p62 (also known as sequestosome-1 SQSTM1) in response to live L. innocua, confirming the presence of autophagy-related structures (Figure 1F). Immunoprecipitates of endogenous LC3 contained higher levels of p62 and ubiquitinated proteins after stimulation with live L. innocua but not live E. coli or dead forms of these bacteria (Figure 1G). This was despite similar levels of p62 induction by both live and killed bacteria (Figure 1G). Thus, phagocyte detection of live Gram-positive bacteria uniquely induces LC3 lipidation and recruitment of p62 and ubiquitinated proteins to LC3-II, events associated with autophagy.
To decipher whether canonical autophagy or LC3-associated phagocytosis (LAP) (Green et al., 2016) were at play, we tested the response of macrophages lacking components of the pre-initiation ULK kinase complex (containing ULK1/2, Fip200, Atg13 and Atg10) essential for canonical autophagy but dispensable for LAP. Live L. innocua-induced LC3 lipidation was significantly reduced in FIP200-deleted (Figure 1H) and ULK1 deficient macrophages (Figure S1F, and also to other live Gram-positive bacteria). Deletion of ATG14, a member of the class III phosphoinositide 3-kinase (PI3K) complex involved only in canonical autophagy, also impaired LC3 lipidation to live L. innocua (Figure 1I). Notably, DAPI-stained Listeria did not co-localize with LC3 or p62 puncta (Figures 1E and 1F). Altogether, these results show that LC3 lipidation in response to live Gram-positive bacteria reflects canonical autophagy and not LAP.
Autophagy controls the augmented IFN-I response to live Gram-positive bacteria
An inverse relationship between autophagy and inflammasome activity has been established (Saitoh and Akira, 2016). We thus tested whether autophagy induction by live Gram-positive bacteria was responsible for the lack of inflammasome activation. As expected, macrophages deleted for Atg7 (involved in the enzymatic catalysis of LC3 lipidation), Beclin-1 (a member of the class III PI3K complex required for vesicle nucleation), or Fip200 showed enhanced IL-1β secretion and pro-caspase-1, pro-caspase-11 and pro-IL-1β cleavage in response to live E. coli, and to a lesser extent to dead E. coli (Figures 2A and S2A). In contrast, we detected no evidence of inflammasome activation by live L. innocua upon deletion of Atg7 or Beclin-1 despite induction of ASC, NLRP3, pro-caspase-1 and pro-IL-1β to levels similar to those by E. coli, and irrespective of bacterial viability (Figure 2A). IL-6 levels were similar in all cases (Figures 2A and S2A). Thus, inability of live L. innocua to activate the inflammasome is not a consequence of the autophagy induced instead.
Figure 2. Autophagy blockade selectively impairs the IFN-I response to live avirulent Gram-positive bacteria.
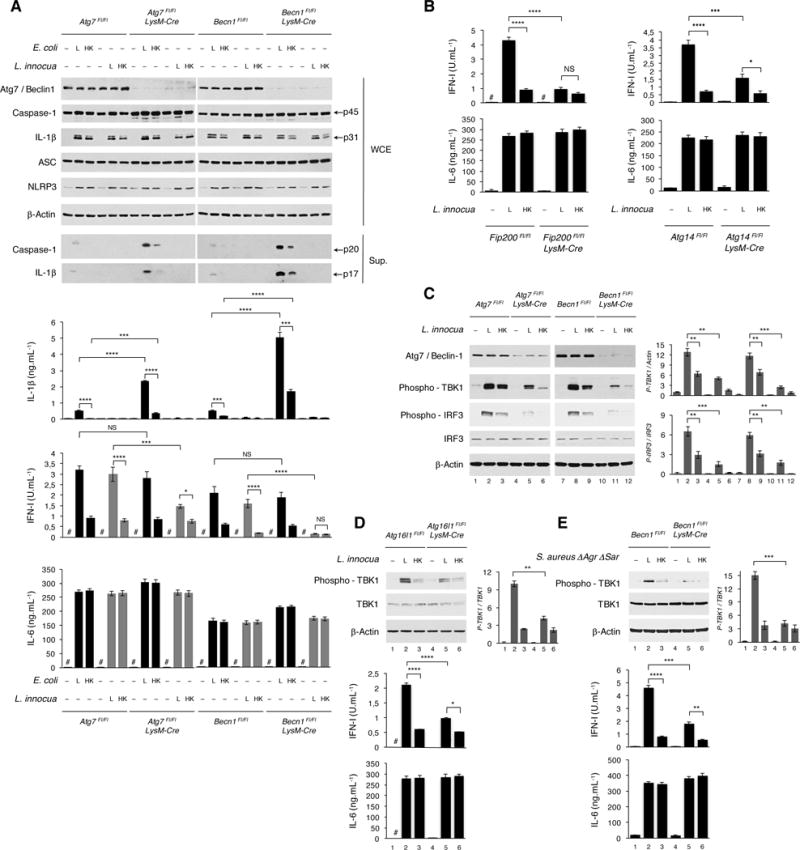
(A) WB on WCE and culture supernatants (sup) from BMDM of indicated genotypes 6 hr (WCE) or 20 hr (sup) post indicated stimuli probed for the proteins indicated (left). Cytokine production (bar graphs) 20 hr post indicated stimuli. Black bars, E. coli. Grey bars, L. innocua.
(B) Cytokine production by BMDM of indicated genotypes 20 hr post indicated stimuli.
(C) WB on BMDM of indicated genotypes 6 hr post indicated stimuli.
(D and E) WB on BMDM of indicated genotypes 6 hr post indicated stimuli. Cytokine production at 20 hr.
In C-E, bar graphs to the right of WB panels show quantification of indicated signals. Cytokine levels measured as in Figure 1A. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments. # = not detected.
We next examined whether the heightened IFN-I response to live Gram-positive bacteria is related to the induction of autophagy (Henault et al., 2012). IFN-I production in response to live L. innocua was significantly reduced in Atg7 and Beclin-1 deficient (Figure 2A), and Fip200 or Atg14 deficient macrophages (Figure 2B). Phosphorylation of TBK1 and IRF3, necessary for the transcriptional activation of IFN-I genes (Honda and Taniguchi, 2006), was higher in response to live compared to dead L. innocua, and was diminished upon deletion of Atg7, Beclin-1, or Atg16L1 (involved at the site of LC3 lipidation for autophagosome elongation) (Figures 2C and 2D). This was concomitant with significant reduction of the IFN-I response to live L. innocua to levels similar to those elicited by dead L. innocua (Figures 2C and 2D). TBK1 activation and IFN-I production to avirulent ΔAgrΔSar S. aureus were also significantly reduced upon Beclin-1 deletion (Figure 2E). Consistent with these results, pharmacological induction of autophagy with Rapamycin significantly augmented IFN-I production upon stimulation with either live or dead L. innocua, while autophagy inhibition with the PI3K inhibitor Wortmannin negated the augmented IFN-I to live bacteria (Figure S2B). Collectively, these results show that canonical autophagy modulates the IFN-I response selectively to live Gram-positive and not Gram-negative bacteria, and this effect does not rely on bacterial virulence factors.
mTOR inactivation by live Gram-positive bacteria promotes autophagy and IFN-I production
To determine the signals controlling the autophagy response to live Gram-positive bacteria, we examined the activity of the nutrient and stress sensor mTORC1 (mechanistic Target of Rapamycin Complex 1), which controls the autophagy pre-initiation ULK kinase complex in response to nutrient availability (Saxton and Sabatini, 2017). We found significant reduction in the phosphorylation of mTOR S2448 uniquely after stimulation of either macrophages or DC with live L. innocua indicating mTORC1 inactivation (Figures 3A and 3B). The phosphorylation of TSC2 at T1462 was also reduced promoting its inhibitory GTPase-activating role within the mTORC1 regulatory complex TSC1/TSC2 (Figures 3A and 3B). Concordant with the inactivation of mTORC1, phosphorylation of the mTORC1 substrates eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and p70 ribosomal protein S6 kinase 1 (p70S6K1) was reduced in response to live compared to dead L. innocua, indicating repression of protein translation (Figures 3A and 3B). Notably, stimulation with E. coli did not modify the levels of phosphorylated TSC2, mTOR, 4EBP1 or p70S6K1 (Figure 3A).
Figure 3. Live but not dead Gram-positive bacteria inactivate mTORC1 to promote autophagy and IFN-I production.
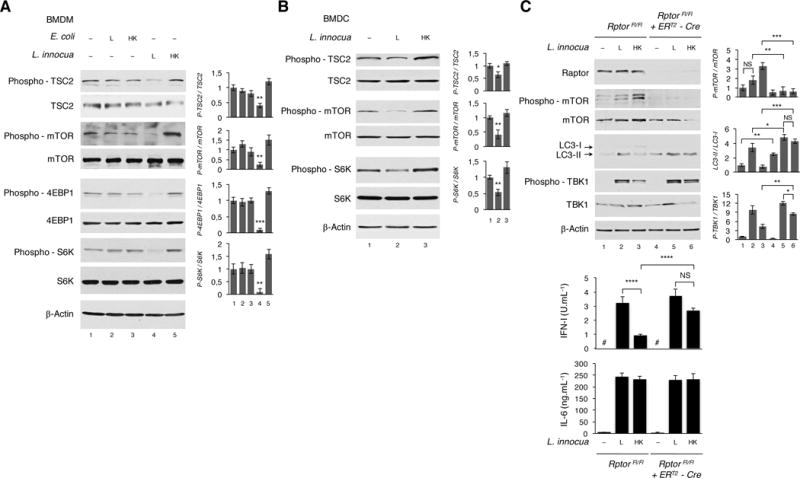
(A and B) WB on BMDM (A) or BMDC (B) 6 hr post indicated stimuli.
(C)RptorFL⁄FL or RptorFL⁄FL ERT2-UbC-Cre BMDM treated with Tamoxifen for 72 hr to induce Cre recombination for Rptor deletion. WB 6 hr and cytokine production 20 hr post indicated stimuli. Cytokine levels measured as in Figure 1A.
WB probed for proteins indicated on left of panels. Bar graphs to right of WB panels show quantification of indicated signals. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments. # = not detected.
To formally test the effect of mTORC1 inactivation on autophagy and the IFN-I response, we specifically inhibited mTORC1 using a genetic approach by inducible deletion of Raptor, an essential component of mTORC1 (Saxton and Sabatini, 2017). Raptor deletion impaired mTOR phosphorylation as expected, and was associated with significantly increased LC3 lipidation, TBK1 phosphorylation and IFN-I production in response to dead L. innocua (Figure 3C). These observations collectively show that mTORC1 activity is critically affected by viability of the bacteria encountered, and orchestrates translation inhibition, autophagy and IFN-I production.
Live Gram-positive bacteria induce ER stress apical to the autophagy and IFN-I response
We considered whether mTORC1 inactivation is part of a broader stress response to bacterial viability that might also involve ER stress. mTORC1 inactivation during nutrient starvation and ER stress due to misfolded protein accumulation – the unfolded protein response (UPR) – both converge on the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), which decreases protein translation while increasing translation of ATF4, the master transcriptional regulator of stress adaptation genes including autophagy genes (Cummings and Lamming, 2016; Saxton and Sabatini, 2017). We observed prominent eIF2α phosphorylation in response to live but not dead L. innocua (Figure 4A). Neither live nor dead E. coli induced eIF2α phosphorylation (Figure 4A).
Figure 4. Live Gram-positive bacteria elicit an apical ER stress response critical for cell-autonomous immunity and organismal defense against infection.
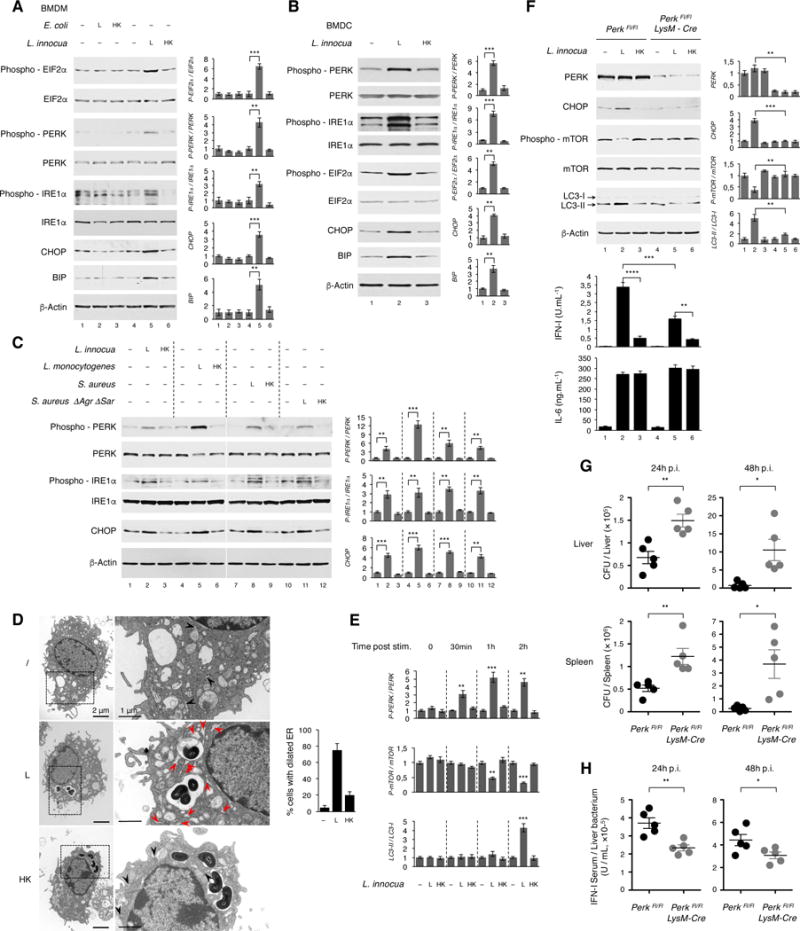
(A-C) WB on BMDM (A, C) or BMDC (B) 4 hr post indicated stimuli.
(D) Electron micrographs showing BMDM 3 hr post stimulation with (L) or (HK) L. innocua. Right panels show enlarged insets. Bar graphs show quantification of dilated ER. Black arrowheads: ER; red arrowheads: dilated ER. Scale bar = 2 μm (left panels) and 1 μm (right panels).
(E) Kinetics of PERK phosphorylation, mTOR inactivation and LC3 lipidation in BMDM at different times post indicated stimuli. Associated WB panels in Figure S3.
(F) WB on BMDM of indicated genotypes 6 hr post indicated stimuli. Cytokine production 20 hr post indicated stimuli.
(G and H) PerkFL⁄FL or PerkFL⁄FL LysM-Cre mice infected intravenously with 3×104 CFU L. monocytogenes. Bacterial burdens in liver and spleen (G) and serum IFN-I (H) 24 or 48 hr following infection. n=5 mice per genotype and per time point.
WB probed for proteins indicated on left of panels. In A-C, E and F (right panels), bar graphs show quantification of indicated WB signals. Cytokine levels in F, H measured as in Figure 1A. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, Student’s t-test, combined results from 3 independent experiments or from n=5 mice ((G) and (H)).
We noted a significant increase in the phosphorylation of the two ER stress sensors PERK and IRE1α, indicative of activation (Hetz, 2012), only in response to live L. innocua (Figure 4A). The pro-apoptotic protein CHOP and the ER chaperone BIP, whose expression are induced by ER stress (Hetz, 2012), also exhibited higher levels in response to live L. innocua (Figure 4A). DC showed similar responses uniquely to live and not killed L. innocua (Figure 4B). The virulent L. monocytogenes and S. aureus, as well as the attenuated ΔAgrΔSar S. aureus, likewise induced PERK and IRE1α phosphorylation as well as CHOP expression when live but not dead (Figure 4C). Moreover, electron microscopy revealed that macrophages stimulated with live but not dead L. innocua exhibited dilated ER, a morphology consistent with ER stress (Figure 4D).
Examining the kinetics of the ER stress response, we observed PERK phosphorylation within 30 minutes following L. innocua infection and preceding mTOR inactivation, which began at 1 hour during peak PERK phosphorylation and overlapped with LC3 lipidation at 2 hours (Figures 4E and S3A). These results suggest an upstream role for ER stress, which is highlighted by concordant mTOR inactivation and LC3 lipidation upon pharmacological induction of ER stress with Tunicamycin or Brefeldin A, and irrespective of concomitant stimulation with bacteria (Figure S3B). As expected, these treatments were toxic to cells over time precluding assessment of IFN-I production. To directly test the relationship between ER stress and the mTORC1-autophagy-IFN-1 response (Figure 3C), we specifically deleted PERK in macrophages and DC without affecting IRE1α expression or activation (Figure S3C). PERK deletion decreased CHOP induction by live L. innocua, as expected (Figure 4F and S3D). Strikingly, PERK deletion also significantly relieved mTOR inactivation and reduced LC3-II and IFN-I levels in response to live L. innocua (Figures 4F and S3D). These results were complementary to our inverse finding that LC3-II and IFN-I levels are induced by dead L. innocua when mTOR is inactivated (Figure 3C). Altogether, these findings show that mTORC1 inactivation and the autophagy and IFN-I response to live L. innocua are mediated by ER stress.
We next tested whether PERK-dependent ER stress controls the organismal innate immune response to Gram-positive bacteria. Specifically deleting PERK in macrophages compromised the ability of mice to clear an infection with L. monocytogenes, as evidenced by significantly increased bacterial burdens in both the liver (a target organ) and spleen relative to control mice (Figure 4G). Macrophage-specific deletion of PERK in mice also lowered serum IFN-I levels post-infection compared to control mice (Figure 4H). These results show that the cell-autonomous ER stress response to live Gram-positive bacteria critically impacts host defense against infection.
ER stress-mediated autophagy resolves ER stress and sustains cell viability
In a reverse set of experiments, we monitored the effects of inhibiting autophagy on ER stress. We noted accumulation of phosphorylated PERK, phosphorylated IRE1α and CHOP in macrophages lacking either Fip200 (Figure 5A), Atg7, Atg16L1, Atg14 or Beclin-1 (Figure S4), under both basal conditions and when stimulated with either live or dead Listeria. Concordantly, induction of canonical autophagy by mTOR inactivation significantly reduced the levels of phosphorylated PERK and almost abrogated CHOP expression (Figure 5B, LC3 lipidation in Figure 3C). These results suggest that autophagy is essential to the resolution of ER stress.
Figure 5. Autophagy blockade sustains ER stress and increases cell death in response to live Gram-positive bacteria.
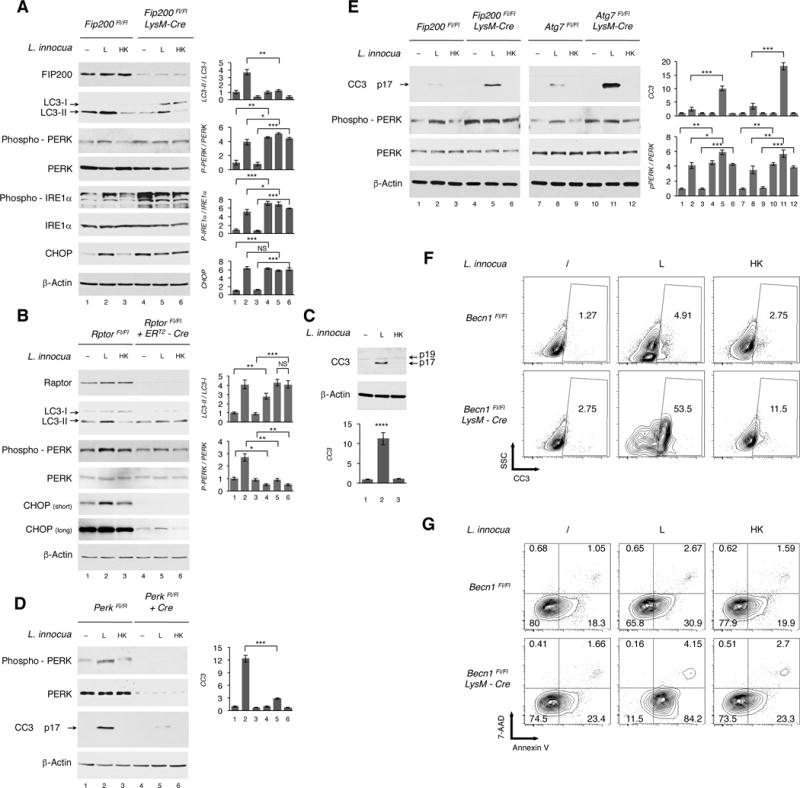
(A) WB on BMDM of indicated genotypes 6 hr post indicated stimuli.
(B) RptorFL⁄FL or RptorFL⁄FL ERT2-UbC-Cre BMDM treated with Tamoxifen as in Figure 3C. WB 4 hr post indicated stimuli.
(C) WB on BMDM 4 hr post indicated stimuli. CC3, cleaved caspase 3.
(D) WB 4 hr post indicated stimuli of PerkFL⁄FL or Perk-deleted PerkFL⁄FL BMDC expressing Cre after lentiviral BM progenitor transduction.
(E) WB on BMDM of indicated genotypes 4 hr post indicated stimuli.
(F and G) Flow cytometry contour plots showing side scatter (SSC) of BMDM staining positively for CC3 (F) or Annexin V and 7-AAD (G) 4 hr post indicated stimuli. % of cells indicated for each quadrant. Analyses represent 3 independent experiments.
WB probed for proteins indicated on left of panels. In A-E, bar graphs show quantification of indicated signals. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from at least 2 independent experiments.
Sustained ER stress induces apoptosis, primarily via the PERK-dependent target CHOP (Tabas and Ron, 2011). Consistent with CHOP induction by live L. innocua (Figure 4) and its accumulation in the absence of autophagy (Figures 5A, S4A and S4B), we detected a prominent cleaved caspase-3 product, indicative of cell death, in the lysates of macrophages treated with live avirulent L. innocua (Figure 5C) or virulent L. monocytogenes (Figure S4D). Notably, PERK deletion in either DC or macrophages strongly reduced caspase-3 cleavage in response to either avirulent or virulent Listeria (Figures 5D and S4D) indicating that infection-induced cell death was PERK-dependent. Sustained ER stress in autophagy-impaired Fip200, Atg7 (Figure 5E), Atg14 and Beclin-1 (Figures S4C and S4D) deleted macrophages was accompanied by increased caspase-3 cleavage in response to live bacteria. Beclin-1 deletion led to a 10-fold increase in the number of macrophages undergoing cleavage of caspase-3 in response to live L. innocua (from 5% to 54%), and to a lesser extent to killed L. innocua and under resting conditions (Figure 5F). The percentage of dying Annexin V+ 7-AAD− macrophages also increased (2.5-fold from 31% to 84%) in response to live L. innocua upon Beclin-1 deletion (Figure 5G). Thus, ER stress-mediated autophagy restores cellular homeostasis by limiting PERK-dependent cell death following infection with Gram-positive bacteria.
ER stress-mediated autophagy sequesters stressed ER membranes
The accumulation of activated ER stress effectors (PERK, IRE1α) and CHOP in the absence of autophagy led us to hypothesize that canonical autophagy relieves ER stress via removal of stressed ER membranes. The ER has been identified as one source for the nucleation of phagophore membranes and elongation of autophagosomes (Bernard and Klionsky, 2013), and parts of the ER can be degraded by autophagy, a process referred to as ER-phagy (Bernales et al., 2007). To test our hypothesis, we fractionated macrophage post-nuclear supernatants on a sucrose density gradient 3-hours post-stimulation with live or dead L. innocua (Figure 6A), and identified a fraction (F5) that was most enriched in LC3-II when macrophages were stimulated with live bacteria (Figure 6B, controls in Figure S5A). Purification of F5 showed enrichment in LC3-II and p62 when macrophages were stimulated with live but not dead L. innocua, but F5 was not enriched for Atg16L1, associated with elongating phagophores/autophagosomes and not mature autophagosomes (Green and Levine, 2014)(Figure 6C). F5 enrichment for LC3-II and p62 was dependent on autophagy (Figure 6D). Thus, F5 purified from macrophages stimulated with live L. innocua is enriched in autophagosomes.
Figure 6. Autophagosomes are enriched for ER stress proteins and STING during the phagocyte response to live Gram-positive bacteria.

(A)Schematic for sucrose-density gradient fractionation. BMDM post-nuclear supernatants (PNS) layering is followed by ultracentrifugation, fraction collection, and further purification of fraction 5 (F5) over a Ficoll cushion.
(B and C) WB on sucrose density gradient fractions F1 to F12 probed for LC3 (B) and 20 μg PNS proteins and 2 μg F5 proteins (C) from BMDM 3 hr post indicated stimuli.
(D) WB on 20 μg PNS proteins and 2 μg F5 proteins from BMDM of indicated genotypes 3 hr post stimulation with live L. innocua (L).
(E–H) Analysis of 3 biological replicates of Becn1Fl/Fl (WT#1-3) or or Becn1Fl/Fl LysM-Cre (KO#1-3) F5 by label free quantitation (LFQ) mass spectrometry. Z-scores based on the Log2 transformed LFQ intensity values from samples WT#1-3 and KO#1-3. (E) Heat-maps show hierarchical clustering of Z-scores of 3467 differentially detected proteins from indicated samples (E), among which Endoplasmic Reticulum, Golgi, and Lysosomal proteins are highlighted (F). (G) Scatter plots show the distribution of proteins in (F). Line shows equal distribution between WT and KO samples. Indicated in blue are ER proteins most enriched in WT F5. (H) Functional analyses of the 52 (out of 80) ER proteins abundant in WT compared to KO F5 show proteins involved in Intracellular Trafficking, ER-Golgi Transport and Response to Stress. Also see Figure S5G.
(I) WB on IP LC3 and WCE on BMDM of indicated genotypes 3 hr post indicated stimuli.
WB probed for proteins indicated on left of panels. In C, D, bar graphs show F5/PNS ratio depicting protein enrichment in F5 versus PNS. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, NS = non-significant, Student’s t-test, combining results from at least 2 independent experiments.
Having defined a successful strategy for enriching autophagosomes, we probed for PERK and IRE1α and observed their enrichment selectively in F5 from live and not dead L. innocua-treated macrophages (Figure 6C), and in an autophagy-dependent manner (Figure 6D). This enrichment pattern applied to Calreticulin and ERP72, resident proteins of the ER proper, but not to Golgi protein GM130, the ER Golgi intermediate compartment (ERGIC) markers ERGIC53 and Sec22b, or the lysosomal protein LAMP-1 (Figures 6C and 6D). Analysis of F5 after infection of macrophages with L. monocytogenes led to a similar F5 accumulation of ER proteins in autophagy sufficient and not deficient cells (Figure S5B). Collectively, these results suggest that the ER proper and not ERGIC, Golgi or lysosomes selectively communicates with mature autophagosomes during the response to live Gram-positive bacteria. The molecular mediator of this communication remains unknown, and macrophage deficiency in FAM134B, an ER-resident receptor of the FAM134 reticulon protein family identified as one of the two ER-phagy receptors together with Sec62 (Fumagalli et al., 2016; Khaminets et al., 2015), was not associated with a sustained ER stress response and had no effect on mTOR inactivation, autophagy or IFN-I responses to live L. innocua (Figures S5C and S5D).
Autophagosomes contain ER stress proteins and STING
We next conducted quantitative mass spectrometry on autophagosomal F5 from live L. innocua-stimulated autophagy-sufficient (WT=Becn1Fl/Fl) and autophagy-deficient (KO=Becn1Fl/Fl LysM-Cre) macrophages to obtain an unbiased view of the autophagosome proteome. Hierarchical clustering heat maps highlighted distinct WT versus KO expression patterns of 3467 proteins (Figures 6E and S5E), among which 80 proteins belonged to and/or were associated with the ER, 127 proteins with the Golgi, and 68 proteins with lysosomes (Figure 6F, and respective protein lists in Tables S1 to S3). Notably, and consistent with the absence of co-localization between DAPI-stained Listeria and LC3 or p62 labelled autophagosomes (Figures 1E and 1F), Listeria proteins constituted less than 0.45% of all proteins (Figure S5F).
Importantly, more than 50% of ER proteins (52 out of 80) were more abundant in WT than in KO, while Golgi and lysosomal proteins did not show a preference for either (Figure 6G). Among these, 13% were involved in intracellular trafficking, 10% in ER-Golgi transport, and 23% in the response to stress including proteins involved in ER-associated degradation such as Edem-3, Os9, and Sel-1l whose expression can be controlled by ER stress (Figure 6H and S5G). A subset of mitochondrial proteins also exhibited enrichment in WT versus KO F5 (Figure S5H; protein list Table S4). Among the most enriched ER proteins in WT F5 were UGGT1 (UDP-Glucose glycoprotein glucosyltransferase), P4HA2 (Prolyl4-hydroxylase subunit alpha 2), PTGS2 (Prostaglandin G/H synthase 2), DNAJC3 (Dnaj homolog subfamily C member 3), ALOX5 (Arachidonate 5 lipoxygenase) and RRBP1 (Ribosome binding protein 1) (Figure 6G). Notable in this group was the ER-resident PRR STING (also known as TMEM173), and whose enrichment we could detect by Western blot in autophagy-sufficient compared to autophagy-deficient F5 (Figure 6D).
Immunoprecipitation of endogenous LC3 in Becn1Fl/Fl and Becn1Fl/Fl LysM-Cre macrophages further revealed the co-immunoprecipitation of STING, PERK and p62 with LC3-II in response to live and not killed L. innocua, and dependent on autophagy (Figure 6I). The STING-associated signaling kinase TBK1 also co-immunoprecipitated with LC3-II in response to live L. innocua in an autophagy-dependent manner (Figure 6I), suggesting STING signaling from these autophagosomes. Results from autophagosomal enrichment, mass spectrometry and co-immunoprecipitation collectively indicate that STING, among other select ER trafficking and stress proteins, is within the same subcellular membrane fractions and protein complexes as LC3-II during the response to live L. innocua. Collectively, the data confirm that after infection with Gram-positive bacteria, autophagosomes accumulate ER proteins, among which is the PRR STING and proteins involved in ER stress. These findings are consistent with the notion that autophagy resolves ER stress and restores phagocyte homeostasis by physically removing stressed ER membranes (ER-phagy).
STING engagement by the vita-PAMP c-di-AMP controls the integrated stress response
The multilayered ER stress, mTORC1 inactivation, and autophagy response to live Gram-positive bacteria comprises an integrated stress response that culminates in ER-phagy and IFN-I production. We next sought to determine the bacterial and host signals responsible. The integrated stress response to live L. innocua was notably independent of Toll-like receptor (TLR)2 and TLR9, TLR adaptors TRIF and MyD88, RIG-I and its adaptor MAVS, NOD2 and its effector kinase RIPK2, Syk-dependent PRR, and the type I IFN receptor (IFNAR) (Figures S6A-F). It was also independent of bacterial RNA (Figures 6G and 6H) (Sander et al., 2011) indicating the existence of a vita-PAMP other than prokaryotic mRNA that triggers the distinct cell-autonomous response to live Gram-positive bacteria. The induction of p62 expression was also independent of TLRs and notably IFNAR, consistent with the constitutive nature of the response (Figure S6A).
Because STING was one of the ER-associated proteins enriched in autophagosomes from live bacteria-stimulated macrophages, we turned to the cyclic GMP-AMP synthase (cGAS)-STING pathway, which has been associated with autophagic degradation of Mycobacterium tuberculosis upon cGAS detection of Mycobacterial DNA (Watson et al., 2015). Strikingly, we found that the macrophage response to live and dead bacteria was equivalent in the absence of STING. LC3 lipidation and PERK phosphorylation elicited by live L. innocua were significantly impaired in Sting−/− but not cGas−/− macrophages, while mTOR phosphorylation was restored and even enhanced in Sting−/− but not cGas−/− cells (Figure 7A). Importantly, the increased levels of TBK1 phosphorylation and IFN-I production characteristic of the response to live L. innocua were significantly reduced in Sting−/− macrophages to levels typical of the response to killed bacteria (Figure 7A). Collectively, these data show that while cGAS is not a PRR in the response to L. innocua, STING is essential for mobilizing the integrated stress response to live Gram-positive bacteria.
Figure 7. STING engagement by c-di-AMP controls the integrated stress and IFN-I response.
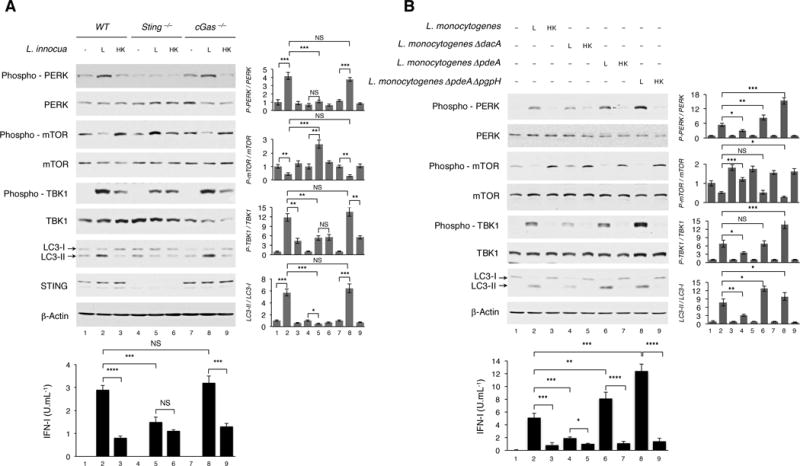
(A and B) WB on BMDM of indicated genotypes 6 hr (A) and 3 hr (B), and IFN-I production 20 hr post indicated stimuli. WB probed for proteins indicated on left of panels. Bar graphs show quantification of indicated WB signals. IFN-I levels measured as in Figure 1A. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments.
Given the dispensable role of cGAS in response to live L. innocua, STING activation in this context could be similar to the manner in which virulent L. monocytogenes bypass cGAS via direct production of the STING agonist c-di-AMP (Burdette and Vance, 2013). Interestingly, c-di-AMP has been classified as the ideal Gram-positive PAMP; instigator of IFN-I responses to L. monocytogenes and immutable for the bacterium (Corrigan and Grundling, 2013). To test whether c-di-AMP serves as the vita-PAMP for Gram-positive bacteria, we made use of a series of well-characterized L. monocytogenes mutants that either accumulate or cannot synthesize c-di-AMP. Strikingly, stimulation with live ΔdacA L. monocytogenes lacking the ability to synthesize c-di-AMP, led to increased mTOR activity, a reduction of phosphorylated PERK and reduced LC3 lipidation as compared to stimulation with wild-type L. monocytogenes (Figure 7B). Concordantly, ΔdacA L. monocytogenes also led to reduced TBK1 activation and IFN-I production. On the contrary, stimulation with ΔpdeA L. monocytogenes, which lack the c-di-AMP phosphodiesterase PdeA responsible for c-di-AMP degradation, and ΔpdeA ΔpgpH L. monocytogenes, which lack both PdeA and another c-di-AMP phosphodiesterase, both lowered mTOR activity and induced higher ER stress and autophagy compared to wild-type L. monocytogenes (Figure 7B). Concordantly, these two strains led to increased TBK1 activity and IFN-I production. Concomitant stimulation of macrophages with c-di-AMP added exogenously with L. innocua significantly changed the macrophage response to killed bacteria manifested by mTOR inactivation and increased PERK phosphorylation, LC3 lipidation, TBK1 activation and IFN-I production (Figure S6I). Exogenous administration of c-di-AMP alone did not induce these changes, and while significantly increasing PERK and TBK1 phosphorylation when added with live L. innocua, it did not lead to further mTOR inactivation or LC3 lipidation likely already at maximal (Figure S6I). Altogether, these results show that c-di-AMP is a vita-PAMP for Gram-positive bacteria and activates STING to mobilize the integrated stress response to live Listeria.
DISCUSSION
We have delineated a constitutive cell-autonomous phagocyte response specifically to live and not dead Gram-positive bacteria. It is triggered by the PRR STING and characterized by a multilayered integrated stress response that begins within 30 minutes with manifestations of ER stress, and culminates in ER-phagy and heightened IFN-I production. Our findings comprise the second example of a distinct innate response mobilized uniquely to bacterial viability and irrespective of the expression of bacterial virulence factors. We had previously reported that live but not dead Gram-negative bacteria induce an augmented IFN-I response and NLRP3 inflammasome activation dependent on TRIF, and triggered by the vita-PAMP bacterial mRNA (Sander et al., 2011).
The response we have characterized here to live Gram-positive bacteria is independent of bacterial mRNA leading us to identify c-di-AMP as the vita-PAMP signifying the viability of Gram-positive bacteria. c-di-AMP is dissimilar to host molecules and has already been classified as an ideal PAMP (Whiteley et al., 2015). Many clinically important Gram-positive bacteria including S. aureus, L. monocytogenes, Streptococcus pyogenes, and Mycobacterium tuberculosis produce c-di-AMP, whose synthesis in response to environmental conditions is critical for various bacterial processes (Corrigan and Grundling, 2013). The heightened IFN-I response to bacterial viability is commensurate to the increased threat level signified by vita-PAMP detection. However, it may also incur collateral host damage or trigger a negative feedback loop that tempers inflammation explaining some of the deleterious effects of IFN-I during infections (Stifter and Feng, 2015). Collectively, our findings here and previously (Sander et al., 2011) show that the innate immune system is able to sense bacterial viability in the form of vita-PAMPs and mount a response tailored to the threat live bacteria pose in sterile tissues. They further show that distinct viability-induced innate responses to Gram-positive and Gram-negative bacteria are controlled by different vita-PAMPs and signaling pathways.
The first layer of the STING-mediated cell-autonomous response to live Gram-positive bacteria is ER stress as evidenced by PERK/IRE1α phosphorylation, CHOP/BIP expression, and a dilated ER morphology. The cell-autonomous ER stress response is protective at the organismal level during Listeria infection of mice, as macrophage-specific PERK deletion reduces the serum levels of IFN-1 and impairs the ability to control hepatic and splenic pathogen burdens. Within macrophages, PERK phosphorylation directly correlated with the c-di-AMP levels within the infecting Listeria and was no longer detectable in the absence of STING. Links between ER stress and infection (Moretti and Blander, 2017) as well as chemically-induced ER stress and STING (Liu et al., 2012; Petrasek et al., 2013) have been reported. STING has four transmembrane domains that anchor it to ER membranes (Burdette and Vance, 2013), and the precise mechanism by which STING detection of c-di-AMP leads to the manifestations of ER stress will be important to decipher in future investigations.
The second layer of the response manifests in mTORC1, 4EBP1 and p70S6K1 inactivation (suggesting repression of protein synthesis), and EIF2α activation known to decrease protein translation while promoting the ATF4 pathway of stress adaptation (Rashid et al., 2015; Saxton and Sabatini, 2017). ER stress and mTORC1 activity are interdependent with either synergistic or antagonistic signaling outputs that are context-dependent (Appenzeller-Herzog and Hall, 2012). Our results show that relaying STING-dependent ER stress during infection through PERK triggers mTORC1 inactivation, since selective blockade of either STING or the PERK pathway (keeping IRE1α intact) is sufficient to relieve mTORC1 inactivation in response to live Gram-positive bacteria.
Canonical autophagy constitutes the third layer of the STING-dependent cell-autonomous response to live Gram-positive bacteria, and functions to alleviate ER stress by removing stressed ER membranes (ER-phagy), maintain cellular viability during infection, and mediate a heightened IFN-I response. We provide genetic, biochemical and proteomic evidence for the presence of ER proteins in autophagosome-enriched cellular fractions specifically after stimulation with live bacteria and in a manner dependent on autophagy. The unfolded protein response in yeast triggers autophagy-dependent removal of ER membranes, a process termed ER-phagy (Bernales et al., 2007). The ER-phagy triggered by live Gram-positive bacteria is distinct from the yeast ER-phagy pathway that operates independently of canonical autophagy (Bernales et al., 2007; Schuck et al., 2014), and is likely a branch of ER-stress mediated autophagy that targets the ER (Song et al., 2017). The spectrum of molecules involved in different forms of ER-phagy is not known. Of the known mammalian ER-phagy receptors, FAM134b (Khaminets et al., 2015) is not involved in the ER-phagy response to live Gram-positive bacteria. Whether Sec62 plays a role remains to be determined (Fumagalli et al., 2016).
A correlation has been reported between ER stress and autophagy, presumably explained by the expression of ATG proteins (Kouroku et al., 2007). STING has been linked to autophagy through TBK1-dependent phosphorylation of autophagy receptors, which recruit ubiquitin and LC3 to the Mycobacterium tuberculosis DNA that becomes cytosolic upon phagosomal permeabilization by bacterial virulence factors (Watson et al., 2012). STING activation in this context is by cGAMP produced upon cGAS detection of translocated M. tuberculosis DNA, leading to IFN-I production (Wassermann et al., 2015; Watson et al., 2015). Our findings point to c-di-AMP/STING-dependent ER stress in response to live Gram-positive bacteria apical to mTORC1 inactivation and autophagy. ER stress-mediated autophagy resolves ER stress, rescues cells from dying, and mediates IFN-I production. Thus, STING appears to co-opt ER-phagy, a pre-existing homeostatic cellular response, to link constitutive cell-autonomous responses to IFN-inducible immunity.
Re-localization of STING may be an essential prelude to IFN-I signaling (Burdette and Vance, 2013). Upon activation, STING localizes with the vesicular transport facilitator Sec5 (Ishikawa et al., 2009), exhibits a punctate expression pattern reminiscent of autophagosomes, and gains co-localization with several autophagy proteins such as Atg9a, p62 and LC3 (Saitoh et al., 2009; Watson et al., 2012). The absence of Atg9a (but not of other ATG proteins) enhanced STING signaling in response to double stranded DNA, suggesting Atg9a-dependent regulation of STING signaling independently of the core autophagy process (Saitoh et al., 2009). Negative feedback mechanisms have been proposed for cGAS-dependent activation of STING where autophagy-ULK1-dependent STING phosphorylation turns off STING signaling after its autophagy-dependent relocalization to endosomes (Konno et al., 2013), highlighting the complexity of STING trafficking events. We provide evidence that STING is present on autophagosomal membranes, and both STING and TBK1 interact with autophagy components. Future studies will have to examine how the autophagosomal localization of STING determines its function in IFN-I signaling. One possibility is that autophagosomal membranes are conducive for signaling, perhaps because they contain signaling molecules responsible for the expression of IFN-I.
In summary, our work shows that STING engagement disrupts ER homeostasis and serves as a previously unappreciated mechanism of mobilizing a constitutive cell-autonomous integrated stress response to live Gram-positive bacteria (Model in Movie S1). This stress response is crucial for initiating an appropriate innate immune response and maintaining cell homeostasis in response to bacterial infection. Unlike the classic PAMPs shared between live and dead microorganisms, vita-PAMPs in this context serve a prominent and indispensable role in triggering these responses because they signify the viability of the microorganism and the associated heightened threat to the integrity of the infected cell.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following.
• KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 4EBP1 (Rabbit mAb) | Cell Signaling | 9644 |
| Phospho-4EBP1 T37/46 (Rabbit mAb) | Cell Signaling | 2855 |
| Atg7 (Rabbit mAb) | Cell Signaling | 8558 |
| Atg16L1 (Rabbit mAb) | Cell Signaling | 8089 |
| ASC (Rabbit pAb) | Santa Cruz | sc-22514-R |
| β-Actin (Mouse mAb) | Cell Signaling | 3700 |
| Beclin1 (Rabbit mAb) | Cell Signaling | 3495 |
| BIP (Rabbit mAb) | Cell Signaling | 3177 |
| Calreticulin (Rabbit mAb) | Abcam | ab92516 |
| Caspase-1 p20 (Mouse mAb) | eBiosciences | 14-9832-82 |
| Caspase-11 (Rat mAb) | Sigma Aldrich | C1354 |
| Cleaved Caspase-3 (Rabbit mAb) | Cell Signaling | 9664 |
| Cleaved Caspase-3 Alexa647 (Rabbit mAb) | Cell Signaling | 9602 |
| CHOP (Mouse mAb) | Cell Signaling | 2895 |
| EIF2α (Rabbit mAb) | Cell Signaling | 5324 |
| Phospho-EIF2α (Rabbit mAb) | Cell Signaling | 3398 |
| ERGIC53 (Rabbit pAb) | Sigma Aldrich | E1031 |
| ERP72 (Rabbit mAb) | Cell Signaling | 5033 |
| FIP200 (Rabbit mAb) | Cell Signaling | 12436 |
| GM130 (Rabbit mAb) | Abcam | ab52649 |
| IL-1β (Goat pAb) | R&D | AF-401-NA |
| IRE1α (Rabbit mAb) | Cell Signaling | 3294 |
| Phospho-IRE1α (Rabbit pAb) | Abcam | ab48187 |
| IRF3 (Rabbit mAb) | Cell Signaling | 4302 |
| Phospho-IRF3 S396 (Rabbit mAb) | Cell Signaling | 4947 |
| Lamp1 (Rat mAb) | eBiosciences | 14-1071-82 |
| LC3B (Rabbit pAb) | Abcam | ab48394 |
| mTOR (Rabbit pAb) | Cell Signaling | 2972 |
| Phospho-mTOR S2448 (Rabbit pAb) | Cell Signaling | 2971 |
| NLRP3 (Mouse mAb) | Adipogen | AG-20B-0014-C100 |
| PERK (Rabbit mAb) | Cell Signaling | 3192 |
| Phospho-PERK T980 (Rabbit mAb) | Cell Signaling | 3179 |
| p62 (WB) (Rabbit pAb) | Cell Signaling | 5114 |
| p62 (IF) (Mouse mAb) | Abcam | ab56416 |
| Phospho-p62 (Rat mAb) | Millipore | MABC186 |
| Raptor (Rabbit mAb) | Cell Signaling | 2280 |
| S6K (Rabbit mAb) | Cell Signaling | 2708 |
| Phospho-S6K T389 (Rabbit mAb) | Cell Signaling | 9234 |
| Sec22b (Mouse mAb) | Santa Cruz | sc-101267 |
| STING (Rabbit mAb) | Cell Signaling | 13647 |
| Syntaxin6 (Rabbit mAb) | Cell Signaling | 2869 |
| TBK1 (Rabbit mAb) | Cell Signaling | 3504 |
| Phospho-TBK1 S172 (Rabbit mAb) | Abcam | ab109272 |
| TSC2 (Rabbit mAb) | Cell Signaling | 4308 |
| Phospho-TSC2 T1462 (Rabbit mAb) | Cell Signaling | 3617 |
| Ubiquitin P4D1 (Mouse mAb) | Cell Signaling | 3936 |
| VDAC (Rabbit mAb) | Cell Signaling | 4661 |
| Anti-Mouse Alexa555 (Goat pAb) | Invitrogen | A21424 |
| Bacterial and Virus Strains | ||
| E. coli K12 DH5α thyA− | J. M. Blander | Sander et al., 2011 |
| E. coli RFP thyA− | J. M. Blander | Sander et al., 2011 |
| L. innocua | ATCC | 33091 |
| L. monocytogenes | ATCC | 7644 |
| L. monocytogenes ΔdacA | D. Portnoy | Woodward et al., 2010 |
| L. monocytogenes ΔpdeA | D. Portnoy | Witte et al., 2013 |
| L. monocytogenes ΔpdeA ΔpgpH | J. Woodward | Huynh et al., 2015 |
| L. pneumophila ΔdotA ΔflaA LPO1 | C. Roy | |
| L. pneumophila ΔdotA ΔflaA thyA− LPO2 | R. Vance | Rao et al., 2013 |
| S. aureus | ATCC | 29213 |
| S. aureus ΔAgr ΔSar | L. Stuart | Cheung et al., 2004 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BafilomycinA1 | Sigma Aldrich | B1793 |
| BrefeldinA | Thermo Fisher | 00-4506-51 |
| c-di-AMP | Invivogen | tlrl-nacda |
| Leupeptin | Sigma Aldrich | L2884 |
| Phalloidin Alexa647 | Invitrogen | A22287 |
| Piceatannol | Sigma Aldrich | P0453 |
| Rapamycin | Sigma Aldrich | R0395 |
| Tunicamycin | Sigma Aldrich | T7765 |
| Wortmannin | Sigma Aldrich | W1628 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mutant mice: Perk Fl/Fl (Eif2ak3tm1.2Drc/J) | The Jackson Lab. | Stock 023066 |
| Mutant mice: Perk Fl/Fl LysM-Cre | J. M. Blander | This paper |
• CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, J. Magarian Blander (jmblander@med.cornell.edu).
• EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J mice were purchased from The Jackson Laboratories and bred in-house. Myd88−/− and Trif−/− mice were originally provided by S. Akira. Myd88−/− and Trif−/− mice were interbred to homozygosity to generate Trif−/−Myd88−/− mice, and were provided by R. Medzhitov (Yale University, New Haven CT). Becn1FL/FL mice were obtained from Z. Yue (The Icahn School of Medicine at Mount Sinai, New York, NY) and were bred to LysM-Cre mice (The Jackson Laboratory, Bar Harbor, ME) to obtain Becn1FL/FL LysM-Cre mice. Femurs from Atg7FL/FL and Atg7FL/FL LysM-Cre; Fip200FL/FL and Fip200FL/FL LysM-Cre; Atg14FL/FL and Atg14FL/FL LysM-Cre; Ulk1+/− and Ulk1−/−; as well as GFP-LC3 transgenic mice were provided by D. R. Green (St Jude Children’s Research Hospital, Memphis, TN). Femurs from Atg16l1FL/FL and Atg16l1FL/FL LysM-Cre were provided by K. Cadwell (New York University School of Medicine, New York, NY). Nod2−/− mice and Ripk2−/− bones were provided by G. Yeretssian (formerly at The Icahn School of Medicine at Mount Sinai). RptorFL⁄FL UbiquitinC(UbC)-CreERT2 mice were generated by crossing RptorFL⁄FL mice (Sengupta et al., 2010) with UbC-CreERT2 mice (The Jackson Laboratory, Bar Harbor, ME) where CRE is fused to the estrogen receptor T2 (Covarrubias et al., 2016), in D. Sabatini’s laboratory (Whitehead Institute, Cambridge, MA), in accordance with the guidelines set forth by the Massachusetts Institute of Technology Institutional Animal Care and Use Committee. PerkFL⁄FL were purchased from The Jackson laboratory and bred to LysM-Cre mice to generate PerkFL⁄FL LysM-Cre mice. Femurs from Sting−⁄−and cGas−⁄− mice were provided by Z. J. Chen (University of Texas Southwestern, Dallas, TX). Femurs from Fam134b WT and Fam134b−⁄− were kind gifts from I. Dikic and C. Hübner (Goethe University, Frankfurt, Germany). Mavs−/− mice and femurs from Rig-I wild-type and Rig-I−/− mice were gifts from A. Garcia-Sastre (The Icahn School of Medicine at Mount Sinai).
For generation of bone-marrow derived macrophages and dendritic cells, 6 to 12 weeks old male and female mice were randomly used. For in vivo experiments (see Figure 4G and 4H), n=5 mice (8-10 weeks old) were used in each group. n=5 females were used in each group (PerkFL⁄FL and PerkFL⁄FL LysM-Cre) for the 24 hour time point post infection, and n=5 males were used in each group (PerkFL⁄FL and PerkFL⁄FL LysM-Cre) for the 48 hour time point post infection.
All experiments were approved by the institutional animal care and use committee of both Mount Sinai School of Medicine and Weill Cornell Medicine, and carried out in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ (NIH publication 86–23, revised 1985).
Bone marrow-derived macrophages and dendritic cells
BMDM
Bone marrow-derived macrophages (BMDM) were grown for 7-10 days in RPMI 1640 supplemented with M-CSF and 10% fetal bovine serum (FBS), plus 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 1% MEM non-essential amino acids, and 55 μM β-mercaptoethanol (all Sigma-Aldrich).
BMDC
Bone marrow-derived dendritic cell (BMDC) cultures were grown for 6 days in RPMI 1640 supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF) and 5% fetal bovine serum (FBS), plus 100 U/ml penicillin, 100 μg/ml, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 1% MEM non-essential amino acids, and 55 μM β-mercaptoethanol (all Sigma-Aldrich). BMDM and BMDC were harvested and re-plated 12-16 hours before stimulation with bacteria in appropriate medium at a concentration of 125,000 cells per cm2.
Importantly, the conditions of BMDM and BMDC cultures must be homogenous in between experiments in order to study cell-autonomous stress responses of these cells. Particularly, period of differentiation of the cells, concentration of cells when replating, as well as timelapse between replating the cells and stimulation with bacteria were very important parameters to maintain low stress backgrounds of the different processes studied in this manuscript.
Bacterial strains
Listeria innocua Seeliger (ATCC® 33091™) (Buchrieser et al., 2003) and Listeria monocytogenes strains were purchased from the ATCC, and were grown in Brain Heart Infusion (BHI) Broth (Gibco). Staphylococcus aureus (ATCC®) and Staphylococcus aureus ΔAgrΔSar lacking the global regulators Sar (Staphylococcal accessory regulator) and Agr (Accessory gene regulator) that control synthesis of adhesins, enzymes, and exotoxins (Cheung et al., 2004) (kind gift from L. Stuart, Massachusetts General Hospital) were both grown in BHI Broth (Gibco). Listeria monocytogenes ΔdacA (Woodward et al., 2010) and Listeria monocytogenes ΔpdeA strains (Witte et al., 2013) were kind gifts from D. Portnoy (University of California Berkeley). Listeria monocytogenes ΔpdeAΔpgpH strain was provided by J. Woodward (University of Washington) (Huynh et al., 2015). ΔdacA strain was grown in minimal media (Whiteley et al., 2015), ΔpdeA and ΔpdeAΔpgpH strains were grown in BHI Broth. Escherichia coli K12, strain DH5α was purchased from Invitrogen. Naturally occurring thymidine auxotrophs (thyA−) were selected as previously described (Sander et al., 2011), and were grown in Luria-Bertani (LB) supplemented with 500 μg/ ml Thymidine and 50 μg /ml Trimethoprim (Sigma). Legionella pneumophila ΔdotAΔflaA (LPO1) strain was obtained from C. Roy (Yale University) and the thymidine auxotroph strain Legionella pneumophilaΔdotAΔflaA thyA− (LPO2) was a gift from R. Vance (University of California Berkeley) (Rao et al., 2013). Both were grown on BCYE Plates (Buffered Charcoal Yeast Extract Plates) and BYE media (Buffered Yeast Extract).
• METHOD DETAILS
Bacterial stimulation of phagocytes
BMDM were plated as explained above. Depending on the readouts, different amounts of BMDM were used. For cytokines or IFN-I measurements by ELISA or luciferase, 2.5×105 BMDM per condition were plated 12-16 hours before stimulation with bacteria (one well of a 24 well plate, 2 cm2). For Western blots analysis, and depending on the number of immunoblots planned, at least 2.5×106 BMDM per condition were plated 12-16 hours before stimulation with bacteria (two wells of a 6 well plate, 20 cm2). For separation on sucrose gradient and analysis of autophagosome fraction by Western blot and mass spectrometry, at least 1×108 BMDM per condition plated as above.
For bacterial stimulation of phagocytes, bacteria were grown to mid-log phase (optical density at 600 nm of 0.6) in the appropriate medium, washed three times in PBS, and counted before addition to cells. For heat killing, bacteria were grown to mid-log phase, washed and re-suspended in PBS and subsequently incubated at 60°C for 2 hours before three washes in PBS, counting and addition to cells. All bacteria were added at a ratio of 25 to 1 cell (MOI 25), with the exception of experiments with c-di-AMP mutants of L. monocytogenes where MOI was reduced to 5 in order to avoid hyper-induction of stress responses due to the virulence factors of L. monocytogenes. After addition of live or heat-killed bacteria, cells were briefly centrifuged (1250 rpm, 2 minutes) to synchronize infection. One hour post stimulation with bacteria, antibiotics (cocktail of Penicillin, Streptomycin and Gentamycin) were added to the medium and kept until the end of the experiments.
Western blots
BMDM or BMDC were harvested and washed in cold PBS, then lysed in 50 mM Tris-HCl (pH 7,9), 300 mM NaCl, 1% Triton X-100, supplemented with protease inhibitor and phosphatase inhibitor cocktails (Complete Protease and Phostop – Roche, respectively). Whole cell extracts were cleared by centrifugation at 14,000 rpm for 10 min at 4°C. Protein concentrations were determined using the Bradford method. When indicated, for Western blots on BMDM supernatants, proteins of cell supernatants (prepared in serum-free Opti-MEM) were concentrated using Trichloroacetic acid (TCA) precipitation and washed in cold acetone. Both WCE and TCA precipitate samples were denatured in Laemmli buffer for SDS-PAGE resolution. Proteins were transferred onto a PVDF membrane (Millipore). Membranes were blocked with 7% evaporated milk in PBS 0.2% Tween and were incubated with primary antibodies and peroxidase-conjugated secondary antibodies. Bound antibodies were visualized using the Amersham™ ECL or Pierce® ECL2 detection reagents. All Western blots represent at least 2 independent experiments. All blots were probed for β-actin as a loading control. Grey bar graphs show the densitometric quantification of the indicated signals from independent experiments using ImageJ software. Signal values were reported to β-actin values, and then normalized to unstimulated conditions (set at 1), therefore enabling the pool of several independent experiments for the calculation of the means and s.e.m. shown in the quantification graphs. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments.
Immunoprecipitation of endogenous LC3
BMDM were harvested and washed in cold PBS, then lysed in 50 mM Tris-HCl (pH 7,9), 150 mM NaCl, 0.5% Triton X-100 (alternatively 0.25% Triton X-100 for STING co-immunoprecipitation experiment), supplemented with protease inhibitor and phosphatase inhibitor cocktails (Complete Protease and Phostop – Roche, respectively). Portions of cleared whole cell extracts were pre-adsorbed on Protein G-agarose beads for 30 minutes (Sigma Aldrich) before incubation with anti-LC3 antibody for 2 hours at 4°C, and final binding on Protein G-agarose beads for 30 minutes.
Infection of mice
L. monocytogenes were grown until an optical density at 600 nm of 0.1 in BHI medium and washed several times in cold PBS. PerkFL⁄FL or PerkFL⁄FL LysM-Cre mice were infected intravenously with 3×104 CFU L. monocytogenes in PBS (injected volume of 200 μL). At 24 and 48 hours post infection, blood was collected and serum was isolated for quantification of serum type I IFN. Mice were sacrificed 24 or 48 hours following infection. Livers and spleens were harvested, weighed and a portion of the organs was mechanically homogenized in 10 mL Ultrapure water containing 0.2% NP-40. Homogenates were subjected to serial dilutions that were plated on BHI-agar (Gibco) plates. Bacterial CFU grown after a 24-hour incubation at 37°C were then counted.
Cytokine ELISA
Supernatants from cultured BMMs or BMDCs were collected at 20 hours after stimulation or at the times indicated. Enzyme-linked immunosorbent assay (ELISA) capture/detection antibody pairs used for IL-6 or IL-1β were as follows: IL-6, MP5-20F3/MP5-32C11 (BDPharmingen); IL-1β, B12/rabbit polyclonal antibody (eBioscience). All ELISA antibodies were used at 2 μg/mL capture and 0.5 μg/mL detection, with the exception of IL-6 capture, which was used at 1 μg/ml. Detection antibodies were biotinylated and labeled by streptavidin-conjugated horseradish peroxidase (HRP), and visualized by the addition 3,3′, 5,5′-tetramethylbenzidine solution (TMB, KPL). Color development was stopped with TMB-Stop Solution (KPL). Recombinant cytokines served as standards and were purchased from Peprotech. Absorbances at 450 nm were measured on a microplate reader (VersaMax, Molecular Devices). Cytokine supernatant concentrations were calculated by extrapolating absorbance values from standard curves where known concentrations were plotted against absorbance using SoftMax Pro 5 software.
Type I IFN measurements
Type-I IFN production was measured in culture supernatants or mouse sera using the ISRE-Luciferase reporter cell line, a kind gift from Bruce Beutler (University of Texas Southwestern). Supernatants or mouse sera were incubated on ISRE-Luciferase cells for 8 hours before lysis of cells in Passive lysis buffer and detection of luciferase luminescence using the Luciferase assay system (Promega). Recombinant IFN-β was used for standard curve determination.
Antibodies
See also Key Resources Table. Antibodies for Western Blot were obtained from: 1) Cell Signaling Technology: anti-Atg7 (#8558), anti-Atg16L1 (#8089), anti-β-Actin (#3700), anti-Beclin1 (#3495), anti-BIP (#3177), anti-CC3 (#9664), anti-CHOP (#2895), anti-EIF2α (#5324), anti-Phospho-EIF2α (#3398), anti-ERP72 (#5033), anti-FIP200 (#12436), anti-IRE1α (#329), anti-IRF3 (#4302), anti-Phospho-IRF3 S396 (#4947), anti-mTOR (#2972), anti-Phospho-mTOR S2448 (#2971), anti-PERK (#3192), anti-Phospho-PERK T980 (#3179), anti-p62 (#5114), anti-Raptor (#2280), anti-S6K (#2708), anti-Phospho-S6K T389 (#9234), anti-STING (#13647), anti-TBK1 (#3504), anti-TSC2 (#4308), anti-Phospho-TSC2 T1462 (#3617), anti-UbP4D1 (#3936), anti-4EBP1 (#9644), and anti-Phospho-4EBP1 T37/46 (#2855). 2) From Abcam: anti-Calreticulin (ab92516), anti-GM130 (ab52649), anti-Phospho-IRE1α S724 (ab48187), anti-LC3B (ab48394, also for immunoprecipitation), and anti-Phospho-TBK1 S172 (ab109272). 3) From eBiosciences: anti-Caspase-1 p20 (#14-9832-82), and anti-Lamp1 (#14-1071-82). 4) From Santa Cruz Technologies: anti-ASC (#sc-22514-R), and anti-Sec22b (#sc-101267). 5) From Sigma Aldrich: anti-Caspase-11 (#C1354), and anti-ERGIC53 (#E1031). 6) From Adipogen: anti-NLRP3 (AG-20B-0014-C100). 7) From Millipore: anti-Phospho-p62 S403 (MABC186). 8) From Research & Development: anti-IL-1β (AF-401-NA).
For flow cytometry, anti-CC3-Alexa647 was used from Cell Signaling Technology (#9602).
Chemical treatments of macrophages
To inhibit lysosomal degradation, cells were treated with 50 μM Leupeptin or 1 μM Bafilomycin A1. To inhibit autophagy, cells were treated with 10 μM Wortmannin, an inhibitor of Class III PI(3)K. To inhibit mTOR, Rapamycin was added at 250nM final concentration. All the above treatments were applied 2 hours before bacterial stimulation and maintained until the end of the experiments. To induce ER stress, cells were treated either with 4 μg/mL Tunicamycin or 1 μg/mL BrefeldinA starting from 1 hour before bacterial stimulation and until the end of the experiments.
To induce Cre expression and delete Raptor, RptorFL⁄FL and RptorFL⁄FL ERT2-UbC-Cre BMDM were treated with 1 μg/mL Tamoxifen after 6 days of differentiation. Tamoxifen was kept in the culture for 72 hours until stimulation of BMDM with L. innocua.
Lentiviral transduction
293T cells were transfected with empty or Cre-encoding lentiviral vector with puromycin resistance (Cre-IRES-Puro, Addgene #30205) together with GAG, REV and Env-expressing vectors. PerkFL⁄FL bone marrow progenitors cultured into DC were transduced on day 1 with virion-containing 293T cells supernatants two days before puromycin selection (day 3). BMDC were replated for bacterial stimulation on day 6.
Cell death analysis by flow cytometry
BMDM were harvested 4 hours after stimulation with bacteria. For Caspase-3 cleavage (CC3) analysis, cells were fixed and permeabilized before CC3 labeling. For Annexin V/7AAD analyses, cells were directly stained using the Annexin V-PE/7AAD Apoptosis Detection kit (BD Pharmingen), and analyzed by flow cytometry (FACSCalibur, BD).
Sucrose density gradient fractionation
200 to 400 million BMDM per condition were washed and harvested in cold PBS before resuspension in homogenization buffer HB-Sucrose buffer (250 mM sucrose, 0.5 mM EGTA, and 20 mM HEPES/KOH). Cells were disrupted by 10 strong strokes in a Dura Grind stainless-steel homogenizer (Wheaton Scientific) in homogenization buffer (HB) to prepare Post-nuclear supernatants (PNS). PNS were adjusted to 39% sucrose in HB and layered in a sucrose-density gradient as depicted in Figure 6A (respective layers: 65%, 55%, 39% (PNS), 32.5% and 10%). After ultracentrifugation for 1 hour at 28400 rpm, fractions were collected and submitted to Western blot analyses. F5 fractions were further re-suspended in HB and cleared over a 15% Ficoll cushion by ultracentrifugation for 15 minutes at 28400 rpm. Purified F5 fractions were collected in lysis buffer (50 mM Tris-HCl (pH 7,9), 300 mM NaCl, 1% Triton X-100) before Western blot analysis or quantitative Mass Spectrometry.
Label-free quantitative mass spectrometry
Samples were reduced with 0.02 M dithiothreitol (pH 8) for 1 hour at 57°C and subsequently alkylated with 0.05M iodoacetamide (pH 8) for 45 minutes in the dark at room temperature. Then the samples were loaded into a NuPAGE® 4-12% Bis-Tris Gel 1.0 mm (Life Technologies Corporation) and ran for 35 minutes at 200 V. The gel was stained using using GelCode Blue Stain Reagent (Thermo Scientific), gel lanes were divided into 3 sections, and cut into 1 × 1 mm pieces.
Protein gel sections were destained using 50% methanol in 100mM ammonium bicarbonate (pH = 8) (v:v). After at least 5 buffer changes of 15 minutes each, the gel pieces were dehydrated using acetonitrile and dried in a speedvac. Then 300ng trypsin (Promega) was added and the gel pieces were covered with 100mM ammonium bicarbonate. The digestion was allowed to proceed overnight with gentle shaking at room temperature. The resulting peptide mixture was extracted from the gel pieces as previously described (Cotto-Rios et al., 2012). The desalted peptide mixture was concentrated in a SpeedVac concentrator and reconstituted in 0.5% acetic acid.
An aliquot of the sample was loaded onto an Acclaim PepMap 100 precolumn (75μm × 2cm, C18, 3μm, 100Å, Thermo Scientific) equilibrated with solvent A (2% acetonitrile, 0.5% acetic acid) in-line with an EASY-Spray, PepMap column (75μm × 50cm, C18, 2μm, 100Å Thermo Scientific) with a 5μm emitter using the autosampler of an EASY-nLC 1000 (Thermo Scientific). Peptides were gradient eluted into a Fusion Orbitrap mass spectrometer (Thermo Scientific) using a 120-minute linear gradient from 2% to 40% solvent B (95% acetonitrile, 0.5% acetic acid), followed by a 20-minute linear gradient from 40% to 100% solvent B. Solvent B is held at 100% for another 15 minutes for column wash. MS1 spectra were acquired with a resolution of 120,000 (@m/z 400), an AGC target of 4e5, with a maximum ion time of 50ms and a scan range of 400 to 1,500 m/z. Following each MS1, data-dependent low resolution HCD MS2 spectra were acquired using the universal data dependent acquisition method that varies maximal inject time based on the number and intensities of precursor ions. MS2 spectra for precursors of charge states 2 – 5 were collected for a duty cycle of 2 seconds using the following instrument parameters: rapid scan with ion trap detection, one microscan, 2 m/z isolation window, 30s dynamic exclusion list, and Normalized Collision Energy of 27.
MS2 spectra were searched against a Mus musculus uniprot database and proteins were quantified using the MaxQuant software suite (Cox et al., 2014; Luber et al., 2010). For the first search the peptide tolerance was set to 20 ppm and for the main search peptide tolerance was 4.5 ppm. Trypsin specific cleavage was selected with 2 missed cleavages. A PSM FDR of 1% and a Protein FDR of 1% was selected for identification. Label free quantitation was performed with a LFQ minimum ratio of 2 and allowing for unique and razor peptides to be used for quantitation. Matching between runs was allowed with a 0.7 min match window and a 20-minute alignment time window. Carbamidomethylation of Cys was added as a static modification. Oxidation of methionine, deamidation of asparagine and glutamine, and acetylation of the protein N-terminus were the allowed variable modifications.
Results were filtered to remove any proteins that were not identified in at least all three replicates of either wildtype or knock out samples. LFQ intensity values were log2 transformed and all missing values were replaced with values from the normal distribution. Z-scores for all values were calculated, hierarchical clustering was performed and heat maps were generated.
• QUANTIFICATION AND STATISTICAL ANALYSES
For statistical analyses of all quantifications (measurement of cytokine production and densitometric quantification of Western blots), Student’s t-tests were performed combining results from independent experiments as indicated in Figure legends. For statistical analyses of in vivo experiments (Figures 4G and 4H), Student’s t-tests were performed (n=5 mice per group). Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, # = not detected.
Supplementary Material
(A–D, and F) WB on C57BL/6J wild-type (A–C) and Ulk−/+ or Ulk−/− (F) BMDM at 6 hr (A, C, F) and at different time points (B) post indicated stimuli. (C) BMDM pre-treatment with 50 μM Leupeptin and 1 μM Bafilomycin A1.
(E)Confocal micrographs on BMDM 3 hr post indicated stimuli visualized for GFP-LC3 puncta. Scale bar = 10 μm.
WB probed for proteins indicated on left of panels. LC3-II/LC3-I densitometric ratios indicated numerically in A, C, D, and F, and as bar graphs in B.
(A)WB on concentrated culture supernatants (conc. sup.) and WCE from BMDM of indicated genotypes at 20 and 6 hr, respectively, post indicated stimuli. WB probed for proteins indicated on left of panels. Bar graphs show cytokine levels measured by ELISA 20 hr post indicated stimuli.
(B)IFN-I levels in supernatants of BMDM 20 hr post indicated stimuli. BMDM pre-treatment with dimethyl sulfoxide (DMSO) vehicle, 10 μM Wortmannin or 250nM Rapamycin.
Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments. # = not detected.
(A–C) WB on BMDM at different time points (A), at 6 hr (B, C) post indicated stimuli. WB quantifications in A shown in Figure 4E. In B, BMDM pre-treatment with 4 μg/mL Tunicamycin or 1 μg/mL Brefeldin A, and bar graphs show quantification of indicated WB signals. All BMDM wild-type except of indicated genotypes in C.
(D)WB 4 hr and cytokine production 20 hr post indicated stimuli of PerkFL⁄FL or Perk-deleted PerkFL⁄FL BMDC expressing Cre after lentiviral BM progenitor transduction.
WB probed for proteins indicated on left of panels. Error bars, mean ± s.e.m. ***P≤0.001, Student’s t-test, combining results from at least two independent experiments.
(A–D) WB on BMDM of indicated genotypes 6 hr (A–C) and 4 hr (D) post indicated stimuli.
WB probed for proteins indicated on left of panels. Bar graphs show quantification of indicated signals. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments.
(A) WB on sucrose density gradient fractions F1 to F12 from BMDM of indicated genotypes 3 hr post stimulation with live L. innocua. No genotype inherent differences in fractionation of ER proteins Calreticulin and ERP72, Golgi proteins GM130 and Syntaxin 6, or the mitochondrial protein VDAC.
(B) WB on 20 μg PNS proteins and 2 μg F5 proteins from BMDM of indicated genotypes 3 hr post stimulation with L. monocytogenes.
(C and D) WB on BMDM (C) or BMDC (D) of C57BL/6J wild-type (WT) and Fam134b−/− genotypes 6 hr post indicated stimuli. Bar graphs show IFN-I production 20 hr post indicated stimuli.
(E–H) 3 independent replicates of the Becn1Fl/Fl (WT#1-3) or Becn1Fl/Fl LysM-Cre (KO#1-3) F5 fractions analyzed by label free quantitation (LFQ) mass spectrometry. (E) The average numbers of total or exclusive proteins identified in either WT, KO or both WT and KO samples. Of 3467 proteins identified, 2840 were common to WT and KO F5 showing a global qualitative similarity. (F) The percentage of proteins from Mus musculus genome or Listeria innocua genome in each sample. (G) Detailed information on the ER proteins shown in Figure 6H that are more abundant in WT versus KO F5. (H) Heat-maps showing hierarchical clustering of proteins from Mitochondria (135 proteins, Z-scores generated based on the Log2 transformed LFQ intensity values from samples WT#1-3 and KO#1-3). Scatter plot show the distribution of 135 proteins from Mitochondria, based on the Log2 LFQ average intensity for each selected protein. Line shows the equal distribution between WT and KO samples.
(A–G, and I) WB on BMDM of indicated genotypes 6 hr post indicated stimuli. F, BMDM pre-treatment with DMSO vehicle or 50 μM Piceatannol to inhibit Syk-dependent PRRs such as Dectin 1 or Fc receptors. G and I, IFN-I production 20 hr post indicated stimuli. G, addition of 10 μg/mL total RNA (RNAtot) purified from L. innocua failed to augment IFN-I production in response to HK L. innocua. I, c-di-AMP at 10 and 30 μM added to BMDM concomitantly with bacteria.
(H) IL-1β production 20 hr post stimulation of BMDM with L or HK E. coli. Addition of 10 μg/mL L. innocua RNAtot enabled IL-1β secretion in response to dead E. coli showing no inherent immune stimulatory differences between RNA from Gram-positive or Gram-negative bacteria.
WB probed for proteins indicated on left of panels. In C, E, G, LC3-II/LC3-I densitometric ratios are indicated to quantify LC3 lipidation. In D and I, bar graphs show densitometric quantification of indicated WB signals. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, NS = non-significant, Student’s t-test, combining results from independent experiments.
Examination of the innate immune response to live or dead gram-positive bacteria revealed a heightened IFN-I response induced specifically by live bacteria. The path to this IFN-I response involves unique and pre-existing cell autonomous responses mobilized quickly within 30 minutes and only in response to live bacteria. After phagocytosis of live bacteria, the second messenger, cyclic di-adenosine monophosphate, gains access to the cytosol and activates the ER-resident transmembrane pattern recognition receptor STING. Detection of c-di-AMP by STING, best known for its induction of an IFN-I response, leads to the initiation of an integrated ER stress response characterized by the phosphorylation of ER proteins PERK and IRE1α. The metabolic sensor mTORC1 detects these cellular perturbations releasing its suppression of autophagy. Upon induction of autophagy, autophagosomes sequester membranes from the stressed ER. This ER-phagy response to live bacteria serves to resolve ER stress and relocate STING from ER membranes to autophagosomal membranes, an event critical for the augmented IFN-I response. Notably, this elaborate cell-autonomous response is mobilized specifically by c-di-AMP, the second member of vita-PAMPs, and not a classic PAMP like peptidoglycan for example shared by either live or dead gram-positive bacteria. The model illustrates PRR-dependent mobilization of preformed constitutive cell-autonomous responses upstream of IFN-I-inducible immunity.
Acknowledgments
We are grateful to I. Dikic, D.M. Sabatini, C. Hübner, D. Portnoy, C. Roy, R. Vance, J. Woodward, B. Beutler, A. Garcia-Sastre, Z. Yue, and K. Cadwell for bacterial strains, cells and mice, R. Gordon for electron microscopy, L. Campisi for flow cytometry analyses, Blander lab members and P. Nair-Gupta for discussions, C. Brou, P. Chastagner, A. Israël, D. Filipescu, and D. Kachaner, M. Blander and S.J. Blander for advice and support. This work was supported by Icahn School of Medicine at Mount Sinai (ISMMS) seed funds to J.M.B. Support for J.Mo. in part by the Philippe Foundation; J.Ma. by NIH Intramural Research Program 1ZIAES10328601; D.W.L was supported by NIH/NIA AG041765 (K99/R00 Pathway to Independence Award) and has received funding from, and is a scientific advisory board member of, Delos Pharmaceuticals, which seeks to develop novel, selective mTOR inhibitors for the treatment of various diseases; T.H. by NIH AI102964 and 119763; G.Y. while at ISMMS by The Leona M. and Harry B. Helmsley Charitable Trust (the article does not represent the work, views, or opinions of Helmsley); D.R.G. by NIH AI040646 and a Distinguished Investigator Award from the Lupus Research Alliance; J.M.B. and her laboratory by NIH (DK072201, DK111862, AI095245 and AI123284, AI127658), the Burroughs Wellcome Fund, and Leukemia & Lymphoma Society, the mass spectrometer by NIH/ORIP grant S10OD010582.
Footnotes
AUTHOR CONTRIBUTIONS
J.Mo. performed all experiments and data analyses. J.Mo. and J.M.B. designed experiments and wrote the manuscript. D.W.L. and G.Y. helped with manuscript editing. S.R. conducted experiments at early stages. J.R.C and B.U. performed mass spectrometry and related analyses with J.Mo. D.B. and G.Y. conducted infections of mice and related analyses. D.W.L., J.Ma., Z.J.C., T.H., and D.R.G. provided reagents and mice. J.M.B. designed movie, and conceived of the study.
Supplemental Information includes six figures, four tables and a Movie, and can be found with this article online.
References
- Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274–282. doi: 10.1016/j.tcb.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–287. doi: 10.4161/auto.3930. [DOI] [PubMed] [Google Scholar]
- Bernard A, Klionsky DJ. Autophagosome formation: tracing the source. Dev Cell. 2013;25:116–117. doi: 10.1016/j.devcel.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol. 2012;12:215–225. doi: 10.1038/nri3167. [DOI] [PubMed] [Google Scholar]
- Buchrieser C, Rusniok C, Kunst F, Cossart P, Glaser P, Listeria C. Comparison of the genome sequences of Listeria monocytogenes and Listeria innocua: clues for evolution and pathogenicity. FEMS Immunol Med Microbiol. 2003;35:207–213. doi: 10.1016/S0928-8244(02)00448-0. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- Cheung AL, Bayer AS, Zhang G, Gresham H, Xiong YQ. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol Med Microbiol. 2004;40:1–9. doi: 10.1016/S0928-8244(03)00309-2. [DOI] [PubMed] [Google Scholar]
- Corrigan RM, Grundling A. Cyclic di-AMP: another second messenger enters the fray. Nat Rev Microbiol. 2013;11:513–524. doi: 10.1038/nrmicro3069. [DOI] [PubMed] [Google Scholar]
- Cotto-Rios XM, Bekes M, Chapman J, Ueberheide B, Huang TT. Deubiquitinases as a signaling target of oxidative stress. Cell Rep. 2012;2:1475–1484. doi: 10.1016/j.celrep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 2016;5 doi: 10.7554/eLife.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings NE, Lamming DW. Regulation of metabolic health and aging by nutrient-sensitive signaling pathways. Mol Cell Endocrinol. 2016 doi: 10.1016/j.mce.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Solda T, et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol. 2016;18:1173–1184. doi: 10.1038/ncb3423. [DOI] [PubMed] [Google Scholar]
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016;23:915–926. doi: 10.1038/cdd.2015.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37:986–997. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014;12:101–114. doi: 10.1038/nrmicro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TN, Luo S, Pensinger D, Sauer JD, Tong L, Woodward JJ. An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc Natl Acad Sci U S A. 2015;112:E747–756. doi: 10.1073/pnas.1416485112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA., Jr The immune system evolved to discriminate infectious nonself from noninfectious self. Immunology today. 1992;13:11–16. doi: 10.1016/0167-5699(92)90198-G. [DOI] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- Liu YP, Zeng L, Tian A, Bomkamp A, Rivera D, Gutman D, Barber GN, Olson JK, Smith JA. Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J Immunol. 2012;189:4630–4639. doi: 10.4049/jimmunol.1102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, Akira S, Wiegand M, Hochrein H, O’Keeffe M, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012;12:367–382. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti J, Blander JM. Cell-autonomous stress responses in innate immunity. J Leukoc Biol. 2017;101:77–86. doi: 10.1189/jlb.2MR0416-201R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, Szabo G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A. 2013;110:16544–16549. doi: 10.1073/pnas.1308331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. 2013;340:701–706. doi: 10.1126/science.1233028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao C, Benhabib H, Ensminger AW. Phylogenetic reconstruction of the Legionella pneumophila Philadelphia-1 laboratory strains through comparative genomics. PLoS One. 2013;8:e64129. doi: 10.1371/journal.pone.0064129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. 2016;138:28–36. doi: 10.1016/j.jaci.2016.05.009. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S, Gallagher CM, Walter P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci. 2014;127:4078–4088. doi: 10.1242/jcs.154716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100–1104. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- Song S, Tan J, Miao Y, Zhang Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: involvement of UPR and the core autophagy machinery. J Cell Physiol. 2017 doi: 10.1002/jcp.26137. [DOI] [PubMed] [Google Scholar]
- Stifter SA, Feng CG. Interfering with immunity: detrimental role of type I IFNs during infection. J Immunol. 2015;194:2455–2465. doi: 10.4049/jimmunol.1402794. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6:10–21. doi: 10.1016/j.chom.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe. 2015;17:799–810. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe. 2015;17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley AT, Pollock AJ, Portnoy DA. The PAMP c-di-AMP Is Essential for Listeria monocytogenes Growth in Rich but Not Minimal Media due to a Toxic Increase in (p)ppGpp. [corrected] Cell Host Microbe. 2015;17:788–798. doi: 10.1016/j.chom.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte CE, Whiteley AT, Burke TP, Sauer JD, Portnoy DA, Woodward JJ. Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. MBio. 2013;4:e00282–00213. doi: 10.1128/mBio.00282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A–D, and F) WB on C57BL/6J wild-type (A–C) and Ulk−/+ or Ulk−/− (F) BMDM at 6 hr (A, C, F) and at different time points (B) post indicated stimuli. (C) BMDM pre-treatment with 50 μM Leupeptin and 1 μM Bafilomycin A1.
(E)Confocal micrographs on BMDM 3 hr post indicated stimuli visualized for GFP-LC3 puncta. Scale bar = 10 μm.
WB probed for proteins indicated on left of panels. LC3-II/LC3-I densitometric ratios indicated numerically in A, C, D, and F, and as bar graphs in B.
(A)WB on concentrated culture supernatants (conc. sup.) and WCE from BMDM of indicated genotypes at 20 and 6 hr, respectively, post indicated stimuli. WB probed for proteins indicated on left of panels. Bar graphs show cytokine levels measured by ELISA 20 hr post indicated stimuli.
(B)IFN-I levels in supernatants of BMDM 20 hr post indicated stimuli. BMDM pre-treatment with dimethyl sulfoxide (DMSO) vehicle, 10 μM Wortmannin or 250nM Rapamycin.
Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments. # = not detected.
(A–C) WB on BMDM at different time points (A), at 6 hr (B, C) post indicated stimuli. WB quantifications in A shown in Figure 4E. In B, BMDM pre-treatment with 4 μg/mL Tunicamycin or 1 μg/mL Brefeldin A, and bar graphs show quantification of indicated WB signals. All BMDM wild-type except of indicated genotypes in C.
(D)WB 4 hr and cytokine production 20 hr post indicated stimuli of PerkFL⁄FL or Perk-deleted PerkFL⁄FL BMDC expressing Cre after lentiviral BM progenitor transduction.
WB probed for proteins indicated on left of panels. Error bars, mean ± s.e.m. ***P≤0.001, Student’s t-test, combining results from at least two independent experiments.
(A–D) WB on BMDM of indicated genotypes 6 hr (A–C) and 4 hr (D) post indicated stimuli.
WB probed for proteins indicated on left of panels. Bar graphs show quantification of indicated signals. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, NS = non-significant, Student’s t-test, combining results from 3 independent experiments.
(A) WB on sucrose density gradient fractions F1 to F12 from BMDM of indicated genotypes 3 hr post stimulation with live L. innocua. No genotype inherent differences in fractionation of ER proteins Calreticulin and ERP72, Golgi proteins GM130 and Syntaxin 6, or the mitochondrial protein VDAC.
(B) WB on 20 μg PNS proteins and 2 μg F5 proteins from BMDM of indicated genotypes 3 hr post stimulation with L. monocytogenes.
(C and D) WB on BMDM (C) or BMDC (D) of C57BL/6J wild-type (WT) and Fam134b−/− genotypes 6 hr post indicated stimuli. Bar graphs show IFN-I production 20 hr post indicated stimuli.
(E–H) 3 independent replicates of the Becn1Fl/Fl (WT#1-3) or Becn1Fl/Fl LysM-Cre (KO#1-3) F5 fractions analyzed by label free quantitation (LFQ) mass spectrometry. (E) The average numbers of total or exclusive proteins identified in either WT, KO or both WT and KO samples. Of 3467 proteins identified, 2840 were common to WT and KO F5 showing a global qualitative similarity. (F) The percentage of proteins from Mus musculus genome or Listeria innocua genome in each sample. (G) Detailed information on the ER proteins shown in Figure 6H that are more abundant in WT versus KO F5. (H) Heat-maps showing hierarchical clustering of proteins from Mitochondria (135 proteins, Z-scores generated based on the Log2 transformed LFQ intensity values from samples WT#1-3 and KO#1-3). Scatter plot show the distribution of 135 proteins from Mitochondria, based on the Log2 LFQ average intensity for each selected protein. Line shows the equal distribution between WT and KO samples.
(A–G, and I) WB on BMDM of indicated genotypes 6 hr post indicated stimuli. F, BMDM pre-treatment with DMSO vehicle or 50 μM Piceatannol to inhibit Syk-dependent PRRs such as Dectin 1 or Fc receptors. G and I, IFN-I production 20 hr post indicated stimuli. G, addition of 10 μg/mL total RNA (RNAtot) purified from L. innocua failed to augment IFN-I production in response to HK L. innocua. I, c-di-AMP at 10 and 30 μM added to BMDM concomitantly with bacteria.
(H) IL-1β production 20 hr post stimulation of BMDM with L or HK E. coli. Addition of 10 μg/mL L. innocua RNAtot enabled IL-1β secretion in response to dead E. coli showing no inherent immune stimulatory differences between RNA from Gram-positive or Gram-negative bacteria.
WB probed for proteins indicated on left of panels. In C, E, G, LC3-II/LC3-I densitometric ratios are indicated to quantify LC3 lipidation. In D and I, bar graphs show densitometric quantification of indicated WB signals. Error bars, mean ± s.e.m. *P≤0.05, **P≤0.01, NS = non-significant, Student’s t-test, combining results from independent experiments.
Examination of the innate immune response to live or dead gram-positive bacteria revealed a heightened IFN-I response induced specifically by live bacteria. The path to this IFN-I response involves unique and pre-existing cell autonomous responses mobilized quickly within 30 minutes and only in response to live bacteria. After phagocytosis of live bacteria, the second messenger, cyclic di-adenosine monophosphate, gains access to the cytosol and activates the ER-resident transmembrane pattern recognition receptor STING. Detection of c-di-AMP by STING, best known for its induction of an IFN-I response, leads to the initiation of an integrated ER stress response characterized by the phosphorylation of ER proteins PERK and IRE1α. The metabolic sensor mTORC1 detects these cellular perturbations releasing its suppression of autophagy. Upon induction of autophagy, autophagosomes sequester membranes from the stressed ER. This ER-phagy response to live bacteria serves to resolve ER stress and relocate STING from ER membranes to autophagosomal membranes, an event critical for the augmented IFN-I response. Notably, this elaborate cell-autonomous response is mobilized specifically by c-di-AMP, the second member of vita-PAMPs, and not a classic PAMP like peptidoglycan for example shared by either live or dead gram-positive bacteria. The model illustrates PRR-dependent mobilization of preformed constitutive cell-autonomous responses upstream of IFN-I-inducible immunity.