

Abstract
Istradefylline, a selective adenosine A2A inhibitor, is under development for the treatment of Parkinson's disease. The effect of oral steady‐state rifampin 600 mg/day, a potent cytochrome P450 (CYP) 3A4 inducer, on the disposition of a single oral dose of istradefylline 40 mg was determined in a crossover study in 20 healthy subjects by measuring plasma concentrations of istradefylline and its M1 and M8 metabolites and their derived pharmacokinetic parameters. Based on the geometric mean ratio of log‐transformed data, rifampin reduced istradefylline exposure: Cmax, 0.55 (90%CI, 0.49–0.62); AUClast, 0.21 (90%CI, 0.19–0.22); and AUCinf, 0.19 (90%CI, 0.18–0.20), indicating nonequivalence. These changes were primarily because of the effect of rifampin on the elimination parameters of istradefylline; mean CL/F was increased from 4.0 to 20.6 L/h, and mean t1/2 was reduced from 94.8 to 31.5 hours. The effect of rifampin coadministration on the disposition of the istradefylline M1 and M8 metabolites was inconsistent and variable. Furthermore, as exposure of the istradefylline M1 and M8 metabolites in plasma was generally <9% of total drug exposure, it would be expected to have a negligible impact on the pharmacodynamic effect of istradefylline. Caution should be exercised when istradefylline is administered concurrently with strong CYP3A4 inducers and dose adjustment considered.
Keywords: istradefylline, rifampin, pharmacokinetics, drug interaction
Istradefylline (Nouriast, KW‐6002; Kyowa Kirin Pharmaceutical Development, Inc., Princeton, New Jersey) is currently under development as a treatment for the signs and symptoms of Parkinson's disease (PD) and has been approved for the adjunctive treatment of PD in Japan.1 It is a selective adenosine A2A inhibitor, showing higher in vitro affinity toward the A2A receptor than for A1, A2B, and A3 receptors among the adenosine receptor subtypes located within the human central nervous system.2 Istradefylline is active in numerous pharmacology studies using animal PD models.3, 4, 5, 6, 7, 8, 9, 10, 11, 12 The clinical efficacy of istradefylline as monotherapy or adjunctive therapy for PD has been demonstrated in phase 2 and 3 clinical studies.13, 14, 15, 16, 17, 18, 19, 20, 21, 22 The recommended dose of istradefylline in Japan is usually 20 mg once daily orally, with a maximum dose of 40 mg once daily orally, which is the highest intended dose currently being investigated.
The pharmacokinetics of istradefylline are best described by a 2‐compartment model with first‐order absorption.23 The absolute oral bioavailability of istradefylline cannot be determined because of its poor aqueous solubility, which precludes intravenous administration (data on file; Kyowa Kirin Pharmaceutical Development, Inc.). Exposure based on observed maximum plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) is directly proportional to istradefylline dose (up to 80 mg) in healthy subjects and PD patients.24, 25 Istradefylline is highly bound (∼98%) to plasma protein but is still extensively distributed in the body, which suggests that high lipophilicity determines its distribution (data on file; Kyowa Kirin Pharmaceutical Development, Inc.). Terminal elimination half‐life (t1/2; 67–95 hours), apparent clearance (CL/F; 4.1–6.0 L/h), and apparent volume of distribution (Vz/F; 448–557 L) are independent of dose in healthy subjects.25 It is not known whether istradefylline undergoes first‐pass metabolism (data on file; Kyowa Kirin Pharmaceutical Development, Inc.). A mass balance study showed that ∼80% of total radioactivity was detected as unchanged drug in plasma 2 hours after oral administration, and because unchanged drug was not detected within the limits of the assay in urine, istradefylline was estimated to be primarily eliminated by oxidation metabolism, with the main metabolites being the 4′‐O‐monodesmethylated derivative of istradefylline (M1) in urine and 1‐β‐hydroxylated istradefylline (M8) in plasma (data on file; Kyowa Kirin Pharmaceutical Development, Inc.). The in vitro inhibitory activity of the M1 metabolite toward the adenosine A2A receptor is similar to that of istradefylline, whereas data for the M8 metabolite are unknown (data on file; Kyowa Kirin Pharmaceutical Development, Inc.). The main cytochrome P450 (CYP) isoenzymes involved in istradefylline metabolism are CYP1A1, 3A4, and 3A5, whereas CYP1A2, 2B6, 2C8, 2C9, 2C18, and 2D6*1 are partly involved: all these CYP isoforms affect metabolism to M1 and CYP3A affects metabolism to M8; M1 and M8 are further metabolized by CYP enzymes to various oxidative metabolites (data on file; Kyowa Kirin Pharmaceutical Development, Inc.). A population pharmacokinetic analysis in 1449 healthy subjects and PD patients estimated that the AUC during the dosing interval at steady state (AUCss) was increased by 38% in those who received various CYP3A4 inhibitors.23 Coadministration of ketoconazole, a potent CYP3A4 inhibitor, has been shown to increase istradefylline AUC from time zero to infinity (AUCinf) 2.5‐fold in healthy subjects.26
Because of the importance of CYP3A4 in the disposition of istradefylline, the effect of an enzyme inducer on istradefylline pharmacokinetics was investigated. The effect of rifampin, a strong inducer of CYP3A4 (among other isozymes including CYP2B6, 2C8, 2C9, 2C19, 3A5, and 3A7) has been used for drug–drug interaction studies.27, 28 This study was planned to investigate the effect of multiple doses of rifampin on the single‐dose pharmacokinetic parameters of istradefylline in healthy subjects and to determine whether istradefylline dose adjustment is required for patients who receive concomitant administration of both istradefylline and a CYP3A4 inducer.
Subjects and Methods
The study was conducted in accordance with US Code of Regulations and the International Conference on Harmonization of Good Clinical Practice guidelines, adhered to the ethical principles of the Declaration of Helsinki, and was registered at ClinicalTrials.gov (NCT02174250). The protocol was approved by the institutional review board at the participating clinical study center (Celerion, Inc., Tempe, Arizona). All subjects provided written informed consent prior to participation in the study.
Study Subjects
Nonsmoking, healthy adults 18–65 years old of either sex with a body mass index of 18–35 mg/m2 were eligible for study entry. Medical history had to show no clinically significant or ongoing pathology, which would preclude a subject's participation in or influence the outcome of the study. Screening routine hematology and chemistry laboratory tests of blood and urine had to be within the normal range or show no clinically meaningful deviations. Women had to be of nonchildbearing potential, defined as surgically sterile (hysterectomy, bilateral oophorectomy, or bilateral tubular ligation) or postmenopausal (amenorrhea ≥24 consecutive months and serum follicle‐stimulating hormone ≥30 IU/L in the absence of hormone replacement therapy). Men with procreative potential had to use medically acceptable, double‐barrier methods of birth control when engaging in sexual relations with a female partner of childbearing potential. Exclusion criteria were: women who were lactating or breastfeeding; the presence of gastrointestinal disease or history of malabsorption in the previous year; known history of psychiatric disorders in the previous 2 years that required hospitalization or medication; any condition or disease detected during medical interview/physical examination that would render the subject unsuitable for the study, place the subject at undue risk, or interfere with the ability of the subject to complete the study; use of pharmacologic agents known to significantly induce or inhibit drug‐metabolizing enzymes (especially inducers and inhibitors of CYP3A4 or CYP1A) within at least 4 weeks prior to dosing; administration of an investigational drug within 30 days or 5 elimination half‐lives of such investigational drug, whichever is longer, prior to study drug administration, or planned administration of another investigational product or procedure during the subject's participation in this study; known history of treatment for drug or alcohol addiction in the previous year; average alcohol intake of more than 2 units per day or 14 units per week; donation or loss of >500 mL of blood in the 3 months prior to the first dose; clinically relevant abnormalities on screening electrocardiogram (ECG); positive human immunodeficiency virus, hepatitis B surface antigen, or hepatitis C antibody; positive test results for drugs of abuse at screening; inability or unwillingness to tolerate multiple venipunctures; difficulty fasting or eating standard meals; and use of tobacco or nicotine‐containing products within the 3 months prior to study start to follow‐up visit (confirmed by urine cotinine test). Food or drink/beverage containing alcohol, grapefruit, or grapefruit juice, apple or orange juice, vegetables from the mustard green family (eg, kale, broccoli, watercress, collard greens, kohlrabi, Brussels sprouts, mustard), and charbroiled meats were not allowed from 7 days before the istradefylline dose until after the follow‐up visit.
Study Design
The primary objective of this US registration study was to investigate the effect of multiple doses of rifampin, a strong CYP3A4 enzyme inducer, on the single‐dose pharmacokinetics of istradefylline in healthy subjects. The secondary objective was to assess the safety and tolerability of istradefylline when taken alone and in combination with rifampin.
This was a single‐center, open‐label, 1‐sequence, 2‐period crossover study in 20 healthy subjects. Following the 4‐week screening period, eligible subjects were confined to the clinical research unit (CRU) for 5 days on day‐1 of period 1 until completion of day 5 assessments. Baseline assessments were undertaken on day‐1. After overnight fasting ≥8 hours, a single oral tablet of istradefylline 40 mg was administered in the morning of day 1. Subjects returned to the CRU on days 6, 7, 8, 10, 12, and 14. Following a washout period of at least 21 days, subjects were confined to the CRU on days‐1 to 2 of period 2. Baseline assessments were undertaken on day‐1. Rifampin 600 mg (2 × 300‐mg commercial capsules) was administered once daily throughout period 2 on days 1–20. Subjects were discharged on days 3–6 but returned to the CRU for rifampin administration and assessments. Subjects were then confined for 5 days from days 7–12. After overnight fasting ≥8 hours, a single oral tablet of istradefylline 40 mg was administered on the morning of day 8, with the rifampin dose administered 2 hours later. Subjects were discharged on day 12 but returned to the CRU on days 13, 14, 15, 17, 19, and 21. A final follow‐up visit was undertaken on days 28–31. All doses of istradefylline and rifampin were taken with 240 mL of water in the morning. Treatment compliance was ensured by oral cavity and hand inspection in the CRU at the time of drug administration, with the exception of days 16, 18, and 20 of period 2, when no pharmacokinetic sampling was scheduled.
Subjects could voluntarily withdraw from the study at any time for any reason. In addition, they could be withdrawn by the investigator in the event of any clinical adverse event (AE), laboratory abnormality, intercurrent illness, or other medical condition that the investigator would consider that continued participation would not be in the best interest of the subject, requirement for a concomitant medication prohibited in the study, noncompliance, administrative reasons, or pregnancy.
Safety and tolerability were determined by AEs, physical examination findings, ECG readings, vital sign measurements, and clinical laboratory test results (serum chemistry, hematology, coagulation, and urinalysis). AEs were recorded following observation by the investigator in response to nonleading questioning or spontaneous reporting by the subject. Treatment‐related AEs were those classified as possibly, probably, or definitely related to istradefylline by the investigator. The safety analysis population included all subjects who received at least 1 dose of study medication. Serious AEs (SAEs) were reported in an expedited manner.
Several studies have shown that istradefylline doses of 20 and 40 mg/day provide statistically significant improvement in OFF time and are well tolerated in PD patients,14, 21 with the 40 mg/day dose the highest dose currently being evaluated in clinical efficacy studies. As istradefylline exhibits dose‐linear pharmacokinetics,24, 25 the 40‐mg dose was selected to maximize the possibility of demonstrating drug–drug interaction. Rifampin 600 mg/day for 7 days was selected, as this generally achieves full steady‐state induction of CYP3A4.29 As CYP3A4 induction may last >1 week after cessation of rifampin treatment,30, 31, 32 rifampin 600 mg/day was continued for 20 days during the istradefylline pharmacokinetic sampling period to ensure that maximal CYP3A4 induction continued to cover 4–5 half‐lives for istradefylline.
Pharmacokinetic Measurements
Blood samples (4‐mL aliquots) were taken by direct venipuncture at the following times relative to the istradefylline dose during periods 1 and 2 (without and with rifampin coadministration, respectively): 0 (predose), 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 60, 72, 96, 120, 144, 168, 216, 264, and 312 hours. All blood samples were collected into sodium heparin tubes and mixed. Whole blood was processed as soon as possible and within 30 minutes of collection at ∼1500g–2000g for 10 minutes at 4°C and kept on ice and protected from light until frozen at −20°C ± 10°C or lower as soon as possible and within 45 minutes. Frozen plasma samples were stored until shipment on an adequate supply of dry ice (≥3 days) to the bioanalytical laboratory (Celerion, Inc., Lincoln, Nebraska). Blood and plasma samples were always protected from light by covering with foil or processing under yellow or red lighting or in a dark room throughout to prevent light‐induced degradation of istradefylline.
Validated liquid chromatography–tandem mass spectrometry (LC‐MS/MS) methods were used to assay for istradefylline and for the M1 and M8 metabolites. Plasma aliquots (100 μL) plus internal standard (5 μL of 13Cd3‐istradefylline, 13Cd3‐istradefylline‐M1, or 13Cd3‐istradefylline‐M8) were mixed with 400 μL of 50 mM acetonitrile and 2 mL methyl tert‐butyl ether and centrifuged. The aqueous layer was collected, dried, and reconstituted with 400 μL of 10:90 acetonitrile:water before injection into a 50 × 3.0‐mm (5‐μm particle size) chromatography column (ACE Ca8, Advanced Chromatography Technologies, Aberdeen, UK). The mobile phase used acetonitrile:water:formic acid (60:40:1 for istradefylline and 35:65:1 for the M1 and M8 metabolites) with elution at a flow rate of 1.2 mL/min at 50°C. MS used an API 4000 detector (Applied Biosystems Sciex, Foster City, California) at the following transitions: 385.1→314.2 m/z for istradefylline and 389.1→218.2 m/z for its standard; 371.2→343.1 m/z for the M1 metabolite and 375.1→347.1 m/z for its standard; and 401.2→355.1 m/z for the M8 metabolite and 405.2→359.3 m/z for its standard. For istradefylline, the calibration range was 1–500 ng/mL, lower limit of quantification was 1 ng/mL, assay precision was 2.1%–11.9%, and accuracy (relative error) was −2.7% to 0.5%. For the M1 and M8 metabolites, the calibration range was 1–50 ng/mL, and the lower limit of quantitation was 1 ng/mL. For the M1 and M8 metabolites, assay precision was 3.2%–4.0% and 4.7%–6.1%, respectively, and accuracy was −1.9% to 3.3% and 0.3%–4.6%, respectively.
Pharmacokinetic parameters were determined using noncompartmental methods with Phoenix WinNonlin version 6.3 and included: AUC from time zero to the last quantifiable concentration (AUClast), AUCinf, Cmax, time to reach Cmax (Tmax), and t1/2 for istradefylline and its M1 and M8 metabolites; CL/F and Vz/F for istradefylline; and metabolite‐to‐parent ratio for pharmacokinetic exposure (M/P ratio) of Cmax, AUClast, and AUCinf for the M1 and M8 metabolites.
Statistical Analysis
Statistical analyses were performed with SAS version 9.3. Sample size for this study was based on previous pharmacokinetic data estimated for istradefylline (data on file; Kyowa Kirin Pharmaceutical Development, Inc., Princeton, New Jersey), which showed that intersubject coefficient of variation for AUCinf and Cmax following a single dose of istradefylline is approximately 40% and 28%, respectively. A sample size of 16 would produce a 2‐sided 90% confidence interval (CI) of the geometric mean ratios (GMRs) comparing administration with and without rifampin within the interval of 80%–125% with a probability of at least 80% for both AUCinf and Cmax when the coefficient of variation is as high as 40%. Target recruitment was 20 subjects to take into account potential discontinuations.
The data of the primary pharmacokinetic parameters (AUClast, AUCinf, and Cmax) were logarithmically transformed and subjected to analysis of variance (ANOVA) with factors for both treatment and subject. Based on the residual variation of the ANOVA, 90%CIs for the GMRs were calculated with an 0.80–1.25 ratio being indicative of no significant drug–drug interaction (equivalence). Primary analysis comparison with and without rifampin was performed where paired data were available. Descriptive statistics were used to summarize all pharmacokinetic parameters.
Data from subjects who experienced emesis during the pharmacokinetic sampling period time course of the study for istradefylline were excluded from the summary statistics for the given treatment and from the statistical comparison of pharmacokinetic parameters if vomiting occurred at or before twice the median Tmax for that treatment.
Results
Study Population
All 20 subjects completed the study, and their baseline characteristics are summarized in Table 1. Most were male (70%, n = 14), all were white, and mean age was 46 years.
Table 1.
Baseline Demographic Characteristics (n = 20)
| Characteristic | |
|---|---|
| Age (years), mean (SD) | 45.9 (9.7) |
| Sex, n (%) | |
| Male | 14 (70) |
| Female | 6 (30) |
| Race, n (%) | |
| White | 20 (100) |
| Ethnicity, n (%) | |
| Hispanic or Latino | 16 (80) |
| Not Hispanic or Latino | 4 (40) |
| Weight (kg), mean (SD) | 74.5 (12.1) |
| Height (cm), mean (SD) | 165 (8.5) |
| BMI (kg/cm2), mean (SD) | 27.1 (3.3) |
BMI, body mass index; SD, standard deviation
Istradefylline Pharmacokinetics
Mean plasma istradefylline concentrations over time following the administration of a single oral dose of istradefylline 40 mg alone or in combination with oral steady‐state rifampin 600 mg/day are shown in Figure 1. Nontransformed istradefylline pharmacokinetic parameters after administration of istradefylline alone and in combination with steady‐state rifampin are summarized in Table 2. During coadministration of istradefylline and rifampin, istradefylline Tmax was unchanged, and exposure (Cmax, AUClast, and AUCinf) was substantially increased. At the same time, t1/2 was decreased, Vz/F was increased, and CL/F was considerably increased.
Figure 1.
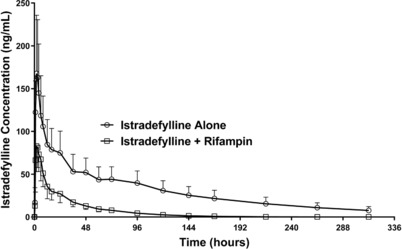
Mean ± SD plasma concentration–time profiles of istradefylline following a single oral dose of istradefylline 40 mg alone or in combination with steady‐state rifampin 600 mg/day in healthy subjects.
Table 2.
Nontransformed Pharmacokinetic Parameters for Istradefylline and its M1 and M8 Metabolites Following Oral Administration of a Single Dose of Istradefylline 40 mg Alone or in Combination With Steady‐State Rifampin 600 mg/day in Healthy Subjects
| Pharmacokinetic Parametera | Istradefylline (n = 20) | Istradefylline + Rifampin (n = 20) |
|---|---|---|
| Istradefylline | ||
| Cmax, ng/mL | 181.1 (68.6) | 97.8 (24.4) |
| Tmax, h | 2.0 (1.0, 12.0) | 1.9 (1.0, 6.0) |
| AUClast, ng·h/mL | 9854 (2853) | 2037 (513) |
| AUCinf, ng·h/mL | 11 100 (3388) | 2096 (514) |
| t1/2, h | 94.8 (36.5) | 31.5 (8.6) |
| Vz/F, L | 515 (188) | 925 (336) |
| CL/F, L/h | 4.0 (1.5) | 20.6 (6.7) |
| M1 metabolite | ||
| Cmax, ng/mL | 4.34 (2.49) | 7.25 (2.27) |
| Tmax, h | 3.5 (2.0, 12.0) | 3.0 (1.0, 6.0) |
| AUClast, ng·h/mL | 123 (77.6) | 118 (44.3) |
| AUCinf, ng·h/mL | na | 159 (47.6)b |
| t1/2, h | na | 17.6 (4.95)b |
| M/P ratio (Cmax) | 0.0241 (0.0045) | 0.0778 (0.0161) |
| M/P ratio (AUClast) | 0.0134 (0.0078) | 0.0613 (0.0195) |
| M/P ratio (AUCinf) | na | 0.0821 (0.0138)b |
| M8 metabolite | ||
| Cmax, ng/mL | 12.6 (5.13) | 14.1 (4.27) |
| Tmax, h | 3.0 (2.0, 24.0) | 3.0 (1.0, 6.0) |
| AUClast, ng·h/mL | 484 (202) | 146 (36.9) |
| AUCinf, ng·h/mL | 610 (272)c | 170 (34.8)d |
| t1/2, h | 62.0 (33.0)c | 10.8 (4.66)d |
| M/p ratio (Cmax) | 0.0674 (0.0131) | 0.1377 (0.0199) |
| M/P ratio (AUClast) | 0.0461 (0.0104) | 0.0705 (0.0145) |
| M/P ratio (AUCinf) | 0.0513 (0.0093)c | 0.0785 (0.0134)d |
AUCinf, area under the plasma concentration–time curve (AUC) from time zero to infinity; AUClast, AUC from time zero to the last quantifiable concentration; CL/F, apparent clearance; Cmax, observed maximum plasma concentration; M/P ratio, metabolite‐to‐parent ratio for pharmacokinetic exposure; na, not applicable; Tmax time to reach Cmax; t1/2, terminal elimination half‐life; Vz/F, apparent volume of distribution.
Data are shown as arithmetic mean (standard deviation) except for Tmax values, which are presented as median (min, max).
n = 13.
n = 14.
n = 19.
Analysis of the effect of rifampin coadministration on the primary log (ln)‐transformed pharmacokinetic parameters (Cmax and AUC values) showed decreases in istradefylline exposure during coadministration with rifampin, that is, nonequivalence (Table 3). The GMR of istradefylline Cmax, AUClast, and AUCinf was decreased to 0.55 (90%CI, 0.49–0.62), 0.21 (90%CI, 0.19–0.22), and 0.19 (90%CI, 0.18–0.20), respectively.
Table 3.
Geometric Mean Ln‐Transformed Pharmacokinetic Parameters for Istradefylline and its M1 and M8 Metabolites Following Oral Administration of a Single Dose of Istradefylline 40 mg Alone or in Combination With Steady‐State Rifampin 600 mg/day in Healthy Subjects
| Parametera | Istradefylline (n = 20) | Istradefylline + Rifampin (n = 20) | Geometric Mean Ratio (90%CI) |
|---|---|---|---|
| Istradefylline | |||
| Cmax (ng/mL) | 170.8 (35.2) | 94.8 (26.6) | 0.55 (0.49–0.62) |
| AUClast (ng·h/mL) | 9457 (30.6) | 1965 (29.5) | 0.21 (0.19–0.22) |
| AUCinf (ng·h/mL) | 10 575 (33.9) | 2026 (28.7) | 0.19 (0.18–0.20) |
| M1 metabolite | |||
| Cmax (ng/mL) | 3.90 (46.3) | 7.0 (29.0) | 1.79 (1.59–2.01) |
| AUClast (ng·h/mL) | 105 (61.4) | 110 (41.8) | 1.04 (0.90–1.21) |
| M8 metabolite | |||
| Cmax (ng/mL) | 11.8 (39.9) | 13.5 (32.2) | 1.14 (1.03–1.27) |
| AUClast (ng·h/mL) | 443 (46.5) | 142 (26.0) | 0.32 (0.28–0.36) |
| AUCinf (ng·h/mL) | 552 (53.5) | 163 (20.7) | 0.30 (0.25–0.34) |
AUCinf, area under the plasma concentration–time curve (AUC) from time zero to infinity; AUClast, AUC from time zero to the last quantifiable concentration; CI, confidence interval; Cmax, observed maximum plasma concentration; CV, coefficient of variation.
Geometric mean (CV%) data are presented.
Istradefylline Metabolite Pharmacokinetics
Mean plasma istradefylline metabolite (M1 and M8) concentrations over time following the administration of a single oral dose of istradefylline 40 mg alone or in combination with oral steady‐state rifampin 600 mg/day are shown in Figure 2. Nontransformed pharmacokinetic parameters for the M1 and M8 metabolites after administration of istradefylline alone and in combination with steady‐state rifampin are summarized in Table 2. GMR analysis of the effect of rifampin coadministration on the ln‐transformed pharmacokinetic parameters (Cmax and AUC values) for the M1 and M8 metabolites are shown in Table 3.
Figure 2.
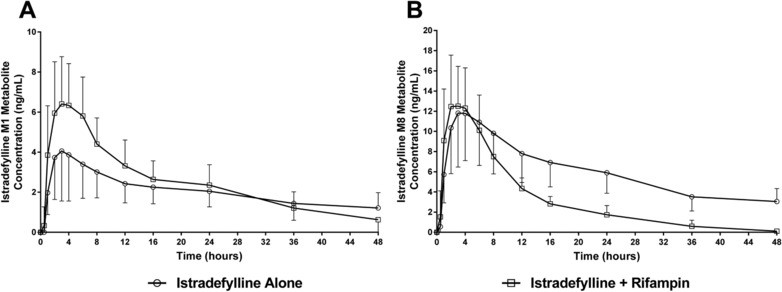
Mean ± SD plasma concentration–time profiles of istradefylline M1 (A) and istradefylline M8 (B) metabolites following a single oral dose of istradefylline 40 mg alone or in combination with steady‐state rifampin 600 mg/day in healthy subjects.
For the istradefylline M1 metabolite, nontransformed pharmacokinetic parameters showed no change in median Tmax and mean AUClast and increased Cmax during coadministration of istradefylline and rifampin. Certain istradefylline M1 metabolite parameters (t1/2 and AUCinf) could not be estimated because of the lack of an apparent terminal phase during administration of istradefylline alone and therefore were not available for comparison during coadministration of istradefylline and rifampin. M/P ratios for the M1 metabolite based on Cmax and AUCinf were increased following coadministration of istradefylline and rifampin compared with istradefylline alone. All M/P ratios for the M1 metabolite were <9% following administration of istradefylline alone or with rifampin. The GMR of istradefylline M1 Cmax was increased (1.79; 90%CI, 1.59–2.01) with no change in AUClast (1.04; 90%CI, 0.90–1.21) following coadministration.
For the istradefylline M8 metabolite, nontransformed pharmacokinetic parameters showed no change in median Tmax or mean Cmax and decreased mean AUC values (AUClast and AUCinf) and mean t1/2 during coadministration of istradefylline and rifampin. M/P ratios for the M8 metabolite based on Cmax, AUClast, and AUCinf were increased following coadministration of istradefylline and rifampin compared with istradefylline alone. All M/P ratios for the M8 metabolite were <8% following administration of istradefylline alone or with rifampin with the exception of the M/P ratio for Cmax after coadministration of istradefylline and rifampin (∼14%). The GMR of istradefylline M8 AUC values decreased (AUClast, 0.32; 90%CI, 0.28–0.36; AUCinf, 0.30; 90%CI, 0.25–0.34) with minimal change in Cmax (1.14; 90%CI, 1.03–1.27) following coadministration.
Safety
Most treatment‐emergent AEs were transient and mild in intensity. Similar numbers of subjects experienced AEs during administration of istradefylline alone (n = 4, 20%) and istradefylline + rifampin (n = 5, 25%), but more during administration of rifampin alone (n = 17, 85%). The most frequent AE for subjects receiving rifampin alone was chromaturia (n = 14, 70%). All other AEs were reported by no more than 1 subject each. Treatment‐related AEs (considered related to istradefylline) occurred in a single subject (while receiving istradefylline alone). This subject experienced mild, transient disturbance in attention, asthenia, and intermittent dizziness, which was considered possibly related to istradefylline. No subjects died, experienced SAEs, or were discontinued from the study because of AEs. There were no clinically significant changes from baseline in any clinical laboratory parameters, vital signs, ECG recordings, or physical examinations.
Discussion
This crossover study examined the effect of oral steady‐state rifampin 600 mg/day on the pharmacokinetic disposition of a single oral dose of istradefylline 40 mg in healthy subjects. Istradefylline was well tolerated when given alone or in combination with rifampin, as only a single patient experienced treatment‐related AEs (mild, transient disturbance in attention, asthenia, and intermittent dizziness, each considered possibly related to istradefylline) while receiving istradefylline alone. Sudden onset of sleep without preceding signs, sleep attacks, orthostatic hypotension, somnolence, dizziness, loss of consciousness, syncope, and so forth may occur in patients receiving istradefylline, which necessitates caution when engaging in potentially hazardous activities.33 Other AEs not considered related to istradefylline were reported in only single subjects except for rifampin‐related chromaturia, which was reported in most subjects (85%) receiving rifampin. Chromaturia is a commonly associated, benign effect of oral rifampin administration.34
Istradefylline was primarily present as unchanged drug in the healthy subjects. Rifampin coadministration had a significant effect on istradefylline exposure (AUC values and Cmax) and disposition (CL/F, Vz/F, and t1/2) with no effect on the rate of istradefylline absorption (median Tmax was unaffected). Based on GMR comparison of ln‐transformed data, rifampin reduced istradefylline Cmax, AUClast, and AUCinf to 0.55 (90%CI, 0.49–0.62), 0.21 (90%CI, 0.19–0.22), and 0.19 (90%CI, 0.18–0.20), respectively, which indicated nonequivalence. These changes would appear to be primarily from the effect of rifampin on the elimination parameters of istradefylline: mean CL/F was increased from 4.00 to 20.6 L/h, and mean t1/2 was reduced from 94.8 to 31.5 hours. Mean Vz/F was increased from 515 to 925 L.
The in vitro inhibitory activity of the M1 metabolite toward the adenosine A2A receptor is similar to that of istradefylline, whereas data for the M8 metabolite are unknown (data on file; Kyowa Kirin Pharmaceutical Development, Inc.). As the exposure of the istradefylline M1 and M8 metabolites in plasma is generally <9% of the total exposure, their impact on the pharmacodynamic effect of istradefylline would be expected to be minor, with exposure to unchanged istradefylline the relevant clinical factor. The effect of rifampin coadministration on the disposition of the 2 metabolites was inconsistent and variable. GMR comparison of ln‐transformed data for the M1 metabolite revealed an increase in Cmax (+79%) but no change in AUClast (+4%) during rifampin coadministration, whereas for the M8 metabolite, Cmax increased marginally (+14%) and AUC values decreased (AUClast, −68%; AUCinf, −70%) during rifampin coadministration. The decrease in M8 AUC values and lack of a change in M1 AUC are attributable to induction of oxidative pathways responsible for the further metabolism of M1 and M8. The effect of rifampin on the 2 metabolites was not considered clinically relevant given that the relative exposure of the metabolites to the parent drug was <9%.
Rifampicin is primarily recognized as the prototypical strong CYP3A4 inducer. It is also a P‐glycoprotein (P‐gp) inducer and able to affect the disposition of drugs that are P‐gp substrates.35, 36 However, istradefylline is not a P‐gp substrate (data on file; Kyowa Kirin Pharmaceutical Development, Inc.).
The narrow 90%CIs for GMRs might indicate that the study was overpowered for the purpose of estimating the size of the expected interaction or that the assumptions made in sample‐size calculations were inappropriate. The number of subjects recruited was increased to allow for potential dropouts and subject noncompliance given that the study design required a 21‐day washout between the treatment periods, which each lasted 14 days. Another consideration in sizing the study was the unknown variability in induction that might be observed in the study. Because there were no dropouts, all subjects were compliant and there was lower variability in the pharmacokinetic effect than anticipated, 90%CIs were narrower than anticipated.
Conclusion
The principle finding of this study was that there was a substantial decrease in istradefylline exposure by ∼80% based on AUC values after the oral administration of a single dose of istradefylline 40 mg in healthy subjects during coadministration of steady‐state oral rifampin 600 mg/day. This effect is considered clinically significant and necessitates caution when istradefylline is administered concurrently with rifampin and consequently other moderate to strong CYP3A4 inducers; dose adjustment of istradefylline may be necessary. Other CYP3A4 inducers used in clinical practice include, for example, bosentan, carbamazepine, efavirenz, etravirine, modafinil, nafcillin, and phenytoin.37 Medications commonly prescribed to treat PD are not reported to be CYP3A4 inducers and include dopamine agonists (pramipexole, ropinirole, and bromocriptine),38, 39, 40 monoamine oxidase inhibitors (selegiline and rasagiline),41, 42 and catechol O‐methyltransferase inhibitors (entacapone and tolcapone).43, 44
Acknowledgments
We thank Dr. Danielle Armas and the study team at Celerion, Inc. (Tempe, Arizona) for the clinical conduct of the study and Dr. Ginny James at Celerion, Inc. (Lincoln, Nebraska) for conducting study sample analysis and LC‐MS/MS assay validation. We thank Dr. Mark Klausner, Ms. Penny Brisco, and Dr. Indravadan H. Patel (Kyowa Kirin Pharmaceutical Development, Inc., Princeton, New Jersey) for medical monitoring, trial management, and protocol design and development, respectively, and Dr. Masuto Mizutani (Kyowa Hakko Kirin Co., Ltd, Shizuoka, Japan) for initial LC‐MS/MS assay development and validation. Medical writing assistance provided by Dr. Peter Todd of Tajut Ltd. (Kaiapoi, New Zealand) was supported financially by Kyowa Kirin Pharmaceutical Development, Inc. (Princeton, New Jersey) during the preparation of this article. We are grateful to all subjects who participated in the study.
Funding
The study was supported and funded by Kyowa Kirin Pharmaceutical Development, Inc. (Princeton, New Jersey).
Declaration of Conflicting Interests
M.M. and D.G. are employees and X.Z was an employee of Kyowa Kirin Pharmaceutical Development, Inc. (Princeton, New Jersey). T.U. is an employee of Kyowa Hakko Kirin Co. Ltd. (Tokyo, Japan). M.V. is an employee of Kyowa Kirin Inc. (Bedminster, New Jersey). M.C. is on the faculty of Robert Wood Johnson Medical School (Bedminster, New Jersey) and a consultant to Kyowa Kirin Pharmaceutical Development, Inc. (Princeton, New Jersey).
Data from this study have been presented in part as an abstract (#419) and poster at the Annual Meeting of the American Society of Clinical Pharmacology and Therapeutics, San Diego, California, March 8–12, 2016.
References
- 1. Dungo R, Deeks ED. Istradefylline: first global approval. Drugs. 2013;73(8):875–882. [DOI] [PubMed] [Google Scholar]
- 2. Harper LK, Beckett SR, Marsden CA, McCreary AC, Alexander SPH. Effects of the A2A adenosine receptor antagonist KW6002 in the nucleus accumbens in vitro and in vivo. Pharmacol Biochem Behav. 2006;83(1):114–121. [DOI] [PubMed] [Google Scholar]
- 3. Kanda T, Jackson MJ, Smith LA, et al. Adenosine A2A antagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann Neurol. 1998;43(4):507–513. [DOI] [PubMed] [Google Scholar]
- 4. Grondin R, Bédard PJ, Hadj Tahar A, Grégoire L, Mori A, Kase H. Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP‐treated monkeys. Neurology. 1999;52(8):1673–1677. [DOI] [PubMed] [Google Scholar]
- 5. Shiozaki S, Ichikawa S, Nakamura J, Kitamura S, Yamada K, Kuwana Y. Actions of adenosine A2A receptor antagonist KW‐6002 on drug‐induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology (Berl). 1999;147(1):90–95. [DOI] [PubMed] [Google Scholar]
- 6. Aoyama S, Kase H, Borrelli E. Rescue of locomotor impairment in dopamine D2 receptor‐deficient mice by an adenosine A2A receptor antagonist. J Neurosci. 2000;20(15):5848–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kanda T, Jackson MJ, Smith LA, et al. Combined use of the adenosine A2A antagonist KW‐6002 with L‐DOPA or with selective D1 or D2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP‐treated monkeys. Exp Neurol. 2000;162(2):321–327. [DOI] [PubMed] [Google Scholar]
- 8. Koga K, Kurokawa M, Ochi M, Nakamura J, Kuwana Y. Adenosine A2A receptor antagonists KF17837 and KW‐6002 potentiate rotation induced by dopaminergic drugs in hemi‐Parkinsonian rats. Eur J Pharmacol. 2000;408(3):249–255. [DOI] [PubMed] [Google Scholar]
- 9. Bibbiani F, Oh JD, Petzer JP, et al. A2A antagonist prevents dopamine agonist‐induced motor complications in animal models of Parkinson's disease. Exp Neurol. 2003;184(1):285–294. [DOI] [PubMed] [Google Scholar]
- 10. Lundblad M, Vaudano E, Cenci MA. Cellular and behavioural effects of the adenosine A2a receptor antagonist KW‐6002 in a rat model of L‐DOPA‐induced dyskinesia. J Neurochem. 2003;84(6):1398–1410. [DOI] [PubMed] [Google Scholar]
- 11. Betz AJ, Vontell R, Valenta J, Worden L, Sink KS, Font L. Effects of the adenosine A2A antagonist KW 6002 (istradefylline) on pimozide‐induced oral tremor and striatal c‐Fos expression: comparisons with the muscarinic antagonist tropicamide. Neuroscience. 2009;163(1):97–108. [DOI] [PubMed] [Google Scholar]
- 12. Kanda T, Tahiro T. Antiparkinsonian activities of istradefylline (KW‐6002), pramipexole and entacapone in MPTP‐treated common marmosets. Mov Disord. 2009;24(suppl 1):S353–S354 [abstract no. Tu‐258]. [Google Scholar]
- 13. Bara‐Jimenez W, Sherzai A, Dimitrova T, et al. Adenosine A2A receptor antagonist treatment of Parkinson's disease. Neurology. 2003;61(3):293–296. [DOI] [PubMed] [Google Scholar]
- 14. Hauser RA, Hubble JP, Truong DD, et al. Randomized trial of the adenosine A2A receptor antagonist istradefylline in advanced PD. Neurology. 2003;61(3):297–303. [DOI] [PubMed] [Google Scholar]
- 15. Hauser RA, Shulman LM, Trugman JM, et al. Istradefylline 6002‐US‐013 Study Group. Study of istradefylline in patients with Parkinson's disease on levodopa with motor fluctuations. Mov Disord. 2008;23(15):2177–2185. [DOI] [PubMed] [Google Scholar]
- 16. LeWitt PA, Guttman M, Tetrud JW, et al. 6002‐US‐005 Study Group. Adenosine A2A receptor antagonist istradefylline (KW‐6002) reduces ‘“off’” time in Parkinson's disease: a double‐blind, randomized, multicenter clinical trial (6002‐US‐005). Ann Neurol. 2008;63(3):295–302. [DOI] [PubMed] [Google Scholar]
- 17. Stacy M, Silver D, Mendis T, et al. A 12‐week, placebo‐controlled study (6002‐US‐006) of istradefylline in Parkinson disease. Neurology. 2008;70(23):2233–2240. [DOI] [PubMed] [Google Scholar]
- 18. Factor S, Mark MH, Watts R, et al. Istradefylline 6002‐US‐007 Study Group. A long‐term study of istradefylline in subjects with fluctuating Parkinson's disease. Parkinsonism Relat Disord. 2010;16(6):423–426. [DOI] [PubMed] [Google Scholar]
- 19. Mizuno Y, Hasegawa K, Kondo T, Kuno S, Yamamoto M. Japanese Istradefylline Study Group. Clinical efficacy of istradefylline (KW‐6002) in Parkinson's disease: a randomized, controlled study. Mov Disord. 2010;25(10):1437–1443. [DOI] [PubMed] [Google Scholar]
- 20. Pourcher E, Fernandez HH, Stacy M, Mori A, Ballerini R, Chaikin P. Istradefylline for Parkinson's disease patients experiencing motor fluctuations: results of the KW‐6002‐US‐018 study. Parkinsonism Relat Disord. 2012;18(2):178–184. [DOI] [PubMed] [Google Scholar]
- 21. Mizuno Y, Kondo T. Japanese Istradefylline Study Group. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson's disease. Mov Disord. 2013;28(8):1138–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kondo T, Mizuno Y. Japanese Istradefylline Study Group. A long‐term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin Neuropharmacol. 2015;38(2):41–46. [DOI] [PubMed] [Google Scholar]
- 23. Knebel W, Rao N, Uchimura T, et al. Population pharmacokinetic analysis of istradefylline in healthy subjects and in patients with Parkinson's disease. J Clin Pharmacol. 2011;51(1):40–52. [DOI] [PubMed] [Google Scholar]
- 24. Rao N, Uchimura T, Mori A. Evaluation of safety, tolerability, and multiple‐dose pharmacokinetics of istradefylline in Parkinson's disease patients. Clin Pharmacol Ther. 2008;83(suppl. 1):S99 [abstract no. PIII‐88]. [Google Scholar]
- 25. Rao N, Uchimura T, Mori A. Evaluation of safety, tolerability, and multiple‐dose pharmacokinetics of istradefylline in healthy subjects. Clin Pharmacol Ther. 2008;83(suppl 1):S99 [abstract no. PIII‐89]. [Google Scholar]
- 26. Rao N, Chaikin P, Dvorchik B, Mori A, Uchimura T. Evaluation of the pharmacokinetic interaction of istradefylline and ketoconazole. Parkinsonism Relat Disord. 2007;13(suppl 2):S104 [abstract no. 2.225]. [Google Scholar]
- 27. Strolin Benedetti MS, Dostert P. Induction and autoinduction properties of rifampin derivatives: a review of animal and human studies. Environ Health Perspect. 1994;102(suppl 9):101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finch CK, Christman CR, Baciewicz, Self TH . Rifampin and rifabutin drug interactions: an update. Arch Intern Med. 2002;162(9):985–992. [DOI] [PubMed] [Google Scholar]
- 29. Gorski JC, Vannaprasaht S, Hamman MA, et al. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin Pharmacol Ther. 2003;74(3):275–287. [DOI] [PubMed] [Google Scholar]
- 30. Backman JT, Kivistö KT, Olkkola KT, Neuvonen PJ. The area under the plasma concentration‐time curve for oral midazolam is 400‐fold larger during treatment with itraconazole than with rifampicin. Eur J Clin Pharmacol. 1998;54(1):53–58. [DOI] [PubMed] [Google Scholar]
- 31. Niemi M, Backman JT, Fromm MY, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42(9):819–850. [DOI] [PubMed] [Google Scholar]
- 32. Oswald S, Giessmann T, Luetjohann D, et al. Disposition and sterol‐lowering effect of ezetimibe are influenced by single‐dose coadministration of rifampin, an inhibitor of multidrug transport proteins. Clin Pharm Ther. 2006;80(5):477–485. [DOI] [PubMed] [Google Scholar]
- 33. Nouriast® tablets prescribing information. Kyowa Hakko Kirin Co., Ltd., October 2015. http://www.e-search.ne.jp/~jpr/PDF/KYOWA13.pdf. Accessed May 20, 2016.
- 34. Rifadin® (rifampin capsules USP) prescribing information. Sanofi, February 2013. http://products.sanofi.us/rifadin/Rifadin.pdf. Accessed May 20, 2016.
- 35. Guidance for Industry. Drug interaction studies – study design, data analysis, implications for dosing, and labeling recommendations. Draft guidance. FDA, February 2012. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064982.htm. Accessed May 20, 2016.
- 36. Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P‐glycoprotein and cytochrome P4503A coordinately upregulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49(2):311–318. [PubMed] [Google Scholar]
- 37. Greiner B, Eichelbaum M, Fritz P, et al. The role of intestinal P‐glycoprotein in the interaction of digoxin and rifampicin. J Clin Invest. 1999;104(2):147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mirapex® (pramipexole dihydrochloride) prescribing information. Boehringer Ingelheim. May 2007. https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020667s014s017s018lbl.pdf. Accessed May 20, 2016. [Google Scholar]
- 39. Requip® (ropinirole hydrochloride) prescribing information. GlaxoSmithKline, 2007. http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020658s018s020s021lbl.pdf. Accessed May 20, 2016. [Google Scholar]
- 40. Parlodel® (bromocriptine mesylate) prescribing information. Novartis, January 2012. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/017962s065s068lbl.pdf. Accessed May 20, 2016. [Google Scholar]
- 41. Zelapar® (selegiline hydrochloride) prescribing information. Valeant, July 2014. http://www.valeant.com/Portals/25/Pdf/PI/Zelapar-PI.pdf. Accessed May 20, 2016. [Google Scholar]
- 42. Azilect® (rasagiline mesylate) prescribing information. Teva, May 2014. https://www.azilect.com/Resources/pdf/PrescribingInformation.pdf. Accessed May 20, 2016. [Google Scholar]
- 43. Comtan® (entacapone) prescribing information. Novartis, July 2014. https://www.pharma.us.novtis.arcom/product/pi/pdf/comtan.pdf. Accessed May 20, 2016. [Google Scholar]
- 44. Tasmar® (tolcapone) prescribing information. Valeant, May 2013. http://www.accessdata.fda.argov/drugsatfda_docs/label/2013/020697s004lbl.pdf. Accessed May 20, 2016. [Google Scholar]