

Abstract
Mesenchymal stromal cells (MSCs) play a pivotal role in modern therapeutic approaches in bone‐healing disorders. Although bone marrow‐derived MSCs are most frequently used, the knowledge that many other adult tissues represent promising sources for potent MSCs has gained acceptance. In the present study, the osteogenic differentiation potential of porcine skin fibroblasts (FBs), as well as bone marrow‐ (BMSCs), adipose tissue‐ (ASCs) and dental pulp‐derived stromal cells (DSCs) were evaluated. However, additional application of BMP‐2 significantly elevated the delayed osteogenic differentiation capacity of ASC and FB cultures, and in DSC cultures the supplementation of platelet‐rich plasma increased osteogenic differentiation potential to a comparable level of the good differentiable BMSCs. Furthermore, microarray gene expression performed in an exemplary manner for ASCs and BMSCs revealed that ASCs and BMSCs use different gene expression patterns for osteogenic differentiation under standard media conditions, as diverse MSCs are imprinted dependent from their tissue niche. However, after increasing the differentiation potential of ASCs to a comparable level as shown in BMSCs, a small subset of identical key molecules was used to differentiate in the osteogenic lineage. Until now, the importance of identified genes seems to be underestimated for osteogenic differentiation. Apparently, the regulation of transmembrane protein 229A, interleukin‐33 and the fibroblast growth factor receptor‐2 in the early phase of osteogenic differentiation is needed for optimum results. Based on these results, bone regeneration strategies of MSCs have to be adjusted, and in vivo studies on the osteogenic capacities of the different types of MCSs are warranted. Copyright © 2016 The Authors Tissue Engineering and Regenerative Medicine published by John Wiley & Sons, Ltd.
Keywords: osteogenesis, osteogenic differentiation potential, mesenchymal stromal cells, BMP‐2, platelet‐rich plasma
1. Introduction
Every year more than two million bone grafts are used worldwide in therapeutic approaches to musculoskeletal injuries (Giannoudis et al., 2005). At present, the autologous cancellous bone graft represents the therapeutic gold standard in the treatment of long bone defects (Fillingham and Jacobs, 2016). It supports tissue regeneration by its osteoinductive properties mediated by growth factors and osteoprogenitor cells. Due to its osteoconductivity it also serves as a scaffold (Fillingham and Jacobs, 2016). Significant drawbacks of autologous bone grafting include donor‐side morbidity (Fillingham and Jacobs, 2016). But besides limited quantity, the unpredictable quality of the cancellous graft exhibits strong intra‐individual variations in regenerative capacity (Fillingham and Jacobs, 2016).
Furthermore, cell therapeutics such as mesenchymal stromal cells (MSCs) in combination with osteoconductive bone substitutes are being used at an increasing rate today (Wittig et al., 2016). Indeed, the therapeutic application of MSCs represents a very promising approach for bone regeneration, due to their ability to differentiate in the osteogenic lineage (Keating, 2012). And as MSCs are able to secrete growth factors, they have additional osteoinductive properties (Keating, 2012). MSCs can be well expanded, giving hope to heal even large bone defects (Caplan and Bruder, 2001). Furthermore, MSCs have some feasible features – they have low immunogenic and high immunosuppressive properties (Keating, 2012).
Although bone marrow was initially considered to be the best source for therapeutically relevant MSCs, in the past 7–8 years many other adult tissues, i.e. dental pulp, umbilical cord blood, muscle biopsy, menstrual blood, adenoid tissue and particularly adipose tissue, have gained acceptance as promising sources for potent MSCs (Sousa et al., 2014).
The role of the growth factors in MSC‐based therapeutic approaches, their specific characteristics and application are discussed controversially (Keating, 2012). The knowledge about the osteoinductive characteristics of BMPs (Kuhn et al., 2013) and growth factor‐containing platelet‐rich plasma (PRP; Lee et al., 2011) as well as the corresponding receptors for these growth factors on MSCs allows the combined use of pro‐osteogenic humoral factors and MSCs as a therapeutic alternative for bone regeneration.
But, prior to secure and standardized clinical application of the optimal autologous MSC type, the molecular mechanisms of the osteogenic differentiation potential need to be analysed in versatile and larger numbers of preclinical models. The mini‐pig is the most suitable model for evaluating bone tissue regeneration capacity of autologous MSCs, as bone regeneration capacity and physiology of the mini‐pig most resembles the human conditions (Pearce et al., 2007).
Furthermore, the osteogenic differentiation potential of MSCs strongly varies, depending on multiple factors, i.e. donor age (Hempel et al., 2016), growth factors (Kuhn et al., 2013) and anatomic location of the donor site (Volk et al., 2012). Moreover, osteogenic differentiation is strongly cell type specific, but may be increased by individually matched growth factor supplementation to reach a comparable differentiation level for various MSCs. Up till now, the genetic expression pattern causing these cell type‐specific variations of the differentiation potential has not been analysed in detail. Which key molecules have to be induced or inhibited for an optimal osteogenic differentiation, apart from the already known factors as RUNX‐2 (Baroni et al., 2005) and Osterix (Sinha and Zhou, 2013), still remains uncertain. Consequently, the present study evaluates and compares the osteogenic differentiation potential of porcine bone marrow stromal cells (BMSCs), adipose tissue stromal cells (ASCs), dental pulp‐derived stromal cells (DSCs) and fibroblasts (FBs) in vitro with several growth factor supplementations. In addition, detailed gene microarray analyses of BMSCs and ASCs as examples for MSCs with high and low osteogenic differentiation potential are provided.
2. Materials and methods
2.1. Materials and experiment design
If not indicated otherwise, all chemicals were obtained from Sigma (Deisenhofen, Germany).
2.2. Animal study design
Six female Goettingen mini‐pigs obtained from the same breed (aged 18–30 months, weight 25–35 kg) were used in this study. Animal selection, management and surgery protocol were approved by the local Animal Care and Use Committee of the Heinrich Heine University and Bezirksregierung Duesseldorf (protocol number: 84‐02.04.2013.A080).
2.3. Isolation of BMSCs
In brief, the extracted bone marrow aspirates were diluted in heparin/EDTA or citrate‐phosphate‐dextrose‐solution to prevent coagulation. The mononuclear cells were separated by centrifugation over Percoll gradient (density 1.090). The centrifugation was carried out at 840 g for 25 min without break. The interphase containing the mononuclear cells was separated and washed with ammonium chloride solution (pH 7.4) to lyse extant erythrocytes. After a further step of washing, the pellet was resuspended in standard cultivation medium (see Section 2.9). Non‐adherent cells were removed after 24 h by medium change.
2.4. Isolation of ASCs
Adipose tissue‐derived stromal cells were isolated from the abdominal fat tissue as described previously (Zuk et al., 2002). Briefly, the fat tissue was washed with phosphate‐buffered saline (PBS) containing 250 U/ml penicillin and 50 μg/ml streptomycin. Adipose tissue was cut into small pieces and incubated overnight with 0.2% dispase II‐solution. In the next step, the samples were incubated in the presence of 0.2% collagenase (type: CLS 255 U/mg) buffer (1 mm CaCl, 5 mm glucose, 0.1 m HEPES, 0.12 m NaCl, 50 mm KCl in aqua dest.) for 2 h at 37 °C in a shaking water bath to release the cells from the tissue matrix. After digestion, the suspension was passed through a 100‐μm filter, washed with PBS and centrifuged at 800 g for 5 min. The cell pellet was resuspended in the respective culture medium. After 24 h, non‐adherent cells were removed by medium change.
2.5. Isolation of FBs
Fibroblasts were isolated from skin tissue as described previously (Junker et al., 2010). Briefly, skin samples were cut into small pieces and incubated overnight with 0.2% dispase II‐solution. The samples were then incubated with 0.2% collagenase (type: CLS 255 U/mg) buffer (1 mm CaCl, 5 mm glucose, 0.1 m HEPES, 0.12 m NaCl, 50 mm KCl in aqua dest.) for 2 h at 37 °C in a shaking water bath to release the cells from the tissue matrix. Following the digestion step, the suspension was passed through a 100‐μm filter, washed with PBS and centrifuged at 800 g for 5 min at room temperature (RT). The cell pellet was resuspended in the standard culture medium.
2.6. Isolation of DSCs
Dental pulp‐derived stromal cells were isolated from the pulp of the sawed third molars as described by Lee et al. (2011). The pulp was cut into small pieces and incubated in 5 ml of 0.2% collagenase buffer (type: CLS 255 U/mg) for 2 h at 37 °C in a preheated water bath. Later the suspension was passed through a 100‐μm filter, washed with PBS and centrifuged at 800 g for 5 min at RT, cell pellets were resolved in the respective culture medium.
2.7. Preparation of autologous PRP
Autologous PRP was prepared as described previously by Hakimi et al. (2010) by obtaining blood from the jugular vein of each individual mini‐pig. PRP was obtained from whole blood by using the GPS® III Platelet Separation System (Biomet Biologics, Warsaw, IN, USA). To 6 ml of citrate anticoagulant, 54 ml of autologous whole blood was added and centrifuged at 600 g for 15 min. The platelet‐poor plasma was separated, resulting in 20 ml of PRP. For the activation of the platelets, autologous thrombin was produced from 7 ml of whole blood by centrifugation, and used in combination with 2 ml of 10% calcium chloride. In previous experiments of our study group using the same animal species and PRP preparation method, the thrombocyte concentrations in whole blood and the resulting PRP were analysed in an automatic counter using veterinary software (ADVIAs 120, Bayer Diagnostics GmbH, Leverkusen, Germany). The concentration factor of the PRP was calculated from the quotient of the thrombocyte count in whole blood and PRP. By this method, a significant concentration increase of the platelets by a factor of 4.7 on average in PRP as compared with native blood could be achieved (Jungbluth et al., 2014). Moreover, the concentrations of PDGF‐bb were quantified in serum and PRP, whereas the concentrations of transforming growth factor beta 1 (TGF‐β1) were quantified in plasma and in PRP using a commercially available ELISA (Quantikine® ELISA‐Kits, R&D Systems, MN, USA) as recommended by the manufacturer in previous investigations of our study group. Here, a significant increase of the PDGF‐bb concentration in PRP compared with serum of native blood by 103.2 times on average could be determined, while the concentration of TGF‐β1 in PRP compared with plasma of native blood increased significantly by 6.8 times on average (Jungbluth et al., 2014).
2.8. Characterization of the cell population doubling time
The cell population doubling time of MSCs was evaluated with cells from Passages 3–25 under normal cell culture conditions. MSCs of Passage 3 were seeded in a density of 3.75 × 105 cells/75‐cm2 culture flask and were allowed to grow in a humidified atmosphere containing 95% air/5% CO2. After reaching confluence (approximately 80%), cells were detached by trypsinization and the amount of viable cells was determined by trypan blue (0.2%) exclusion with a Neubauer haemocytometer. For the next cycle (Passage 4), these cells were seeded again in a density of 3.75 × 105 cells/75‐cm2 culture flask under identical conditions. This procedure was repeated till Passage 25. Cumulative cell‐doubling values of the respective MSCs were calculated in accordance with the equation published by Cristofalo et al. (1998): N H/N I = 2X or [log10(N H) – log 10(N I)]/log10(2) = X, where N I = inoculum number, N H = cell harvest number, X = population doublings.
2.9. Standard cultivation of MSCs
Standard cultivation medium comprising DMEM (4.5 g/l glucose), 2 mm α‐glutamine with 10% fetal bovine serum (FBS; Atlas Biological, CO, USA), 100 U/ml penicillin and 100 μg/ml streptomycin was used. The cells were cultured in 75‐cm2 culture flasks, six‐ or 24‐well‐plates, and incubated at 37 °C in a humidified atmosphere containing 5% CO2.
2.10. Osteogenic differentiation medium (OM)
Osteogenic differentiation medium consisted of standard cultivation medium supplemented with dexamethasone (100 nm), α‐ascorbin‐2‐phosphat (100 μm) and β‐glycerophosphate (10 mm; Pittenger et al., 1999). Additionally, the OM was supplemented with 450 ng/ml BMP‐2 (PeproTech, Hamburg, Germany) or 1% PRP.
2.11. Adipogenic differentiation media
Adipogenic differentiation was induced with standard cultivation medium supplemented with isobutylmethylxanthine (0.5 mm), insulin (66 nm), dexamethasone (1 μm), trijodthyronin (10 μg/ml) and pioglitazone (0.1 μg/ml; Pittenger et al., 1999). After 7 days, the cells were incubated with maturation medium for an additional 7 days. Maturation medium was prepared with DMEM (4.5 g/l glucose), 2 mm α‐glutamine with 10% FBS (Atlas Biological, CO, USA), 100 U/ml penicillin and 100 μg/ml streptomycin supplemented with 66 nm insulin, 1 μm dexamethasone, 1 nm trijodthyronin, transferrin 10 μg/ml and pioglitazone 0.1 μg/ml.
2.12. Chondrogenic differentiation medium
For chondrogenic differentiation, the BMSCs, ASCs, DSCs and FBs were maintained for up to 21 days in the standard cultivation medium supplemented with insulin‐transferrin‐selenium 1%, TGF‐β (10 ng/ml) and α‐ascorbin‐2‐phosphat (1 μg/ml; Pittenger et al., 1999).
2.13. CellTiter‐Blue assay
Following the manufacturing protocol, cell viability was determined by incubation for 1 h with CellTiter‐Blue (Promega, Mannheim, Germany) used in a 1:20 dilution in medium. The obtained fluorescence signals were proportional to the number of viable cells, and were analysed with the Elmer Victor 2 plate reader at 560Ex/590EM.
2.14. Determination of alkaline phosphatase (ALP) activity
Cells were washed with PBS and incubated for 10 min with 10 mm 4‐nitrophenol solution. The resultant changes in solution absorbance were quantified by a photo‐spectrometer at 450 nm (Perkin Elmer Victor 2 plate reader). The absorbance values obtained were normalized to the respective sample using CellTiter‐Blue assay.
2.15. Alizarin red S staining
In order to determine the presence of extracellular calcium deposits secreted, after osteogenic differentiation, the cell cultures were washed with PBS, fixed with 4% (v/v) paraformaldehyde for 15 min, rinsed twice with PBS, incubated for 20 min at 37 °C with 1 ml alizarin red S (0.5% in aqua dest., pH 4.1), and finally washed twice with dH2O (Dahl, 1952). The stained cells were documented by phase contrast microscopy (Zeiss Axiovert 200 microscope). In order to quantify the amount of alizarin red S incorporated in the calcificated matrix, the stained samples were incubated with cetylpyridinium chloride (10% in 10 mm sodium phosphate, pH 7.0), and the extracted alizarin S dye was quantified by determining the absorbance at 600 nm (Gregory et al., 2004).
2.16. Oil red O staining
After 14 days of adipogenic differentiation, cells were washed with PBS, fixed with paraformaldehyde (10%), and intracellular lipid accumulation was detected by oil red O solution [0.5% oil red, O‐isopropyl alcohol/H2O (3:2, v/v)] as described previously (Zuk et al., 2002). Cells were then washed with aqua dest., and the staining was observed and documented using the Zeiss Axiovert 200 microscope.
2.17. Alcian blue staining
After chondrogenic differentiation, the MSCs were washed with PBS, fixed with paraformaldehyde (4%), washed and incubated for 10 min with 0.1 m HCl. Then samples were incubated overnight with alcian blue staining solution (Merck, Darmstadt, Germany), then washed with aqua dest. and analysed using a Zeiss Axiovert 200 microscope.
2.18. Antigen phenotype characterization by flow cytometer analysis
The antigen phenotype of three representatives in each case of ASCs, BMSCs, DSCs and FBs was characterized with a flow cytometer (FACSCalibur analyzer and Cell Quest Software, Becton Dickinson Biosciences, Heidelberg, Germany), using conjugated antibodies against pig CD45, HLA‐DR, CD29, CD79alpha, CD14, CD31 (AbDSerotec), CD105, CD26 (Novus), CD73 (R&D Systems), CD90 (BD), CD34 (abcam), and against mouse CD44 (BD) [was used because of cross‐reactivity (Boxall and Jones, 2012)]. An appropriate isotype‐matched control antibody was used as control in all analyses. FACS‐analysis was performed with cells of Passage 1.
For flow cytometer analysis, the respective cells were detached with 0.5% trypsin/0.02% EDTA, washed, centrifuged at 200 g for 5 min, and reestablished on ice for 15 min. The cells were then centrifuged at 200 g for 5 min again and resuspended in CellWash® (Becton Dickinson Company) containing 3% FBS, and stained for 30 min with the fluorophore‐conjugated antibodies. After a further washing step with CellWash/FCS, the samples were analysed.
2.19. Quantification of BMP‐2 protein expression of MSCs
In order to characterize BMP‐2 protein expression, the respective cell cultures were quantified using specific enzyme‐linked immunosorbent assay (ELISA) according to the manufacturer's recommendations (R&D Systems, Wiesbaden, Germany). The Perkin Elmer Victor 2 plate reader was used for this analysis. The values were normalized to the protein content of the respective sample (DC Protein Assay, Bio‐Rad Laboratories GmbH, München, Germany). ASCs and BMSCs were incubated with OM for 24 h, 48 h and 72 h. In the following, cells were treated with standard cultivation media (see above) for 24 h, 48 h and 72 h, and then supernatants were analysed.
2.20. Microarray gene expression analyses of ASCs and BMSCs
RNA of donor‐matched ASC and BMSC samples from two individual pigs were isolated with miRNeasy Mini Kit from Qiagen (Hilden, Germany) following the manufacturer's protocol, before and after 3 days of osteogenic differentiation. Total RNA preparations were checked for RNA‐integrity by Agilent 2100 Bioanalyzer quality control. All samples in this study revealed high‐quality RNA‐integrity numbers (RIN; at least 9.8). RNA was further analysed by photometric Nanodrop measurement and quantified by fluorometric qubit RNA assays (Life Technologies, Darmstadt, Germany).
Synthesis of biotin‐labelled cDNA was performed according to the manufacturer's protocol (WT Plus Reagent Kit; Affymetrix, Santa Clara, USA). Briefly, 100 ng of total RNA was converted to cDNA. After amplification by in vitro transcription and second cycle synthesis, cDNA was fragmented and biotin‐labelled by terminal transferase. Finally, the end‐labelled cDNA was hybridized to Affymetrix Porcine Gene 1.0 ST Gene Expression Microarrays for 16 h at 45 °C, stained by streptavidin/phycoerythrin conjugate and scanned as described in the manufacturer's protocol. Data analyses on Affymetrix CEL files were conducted with GeneSpring GX software (Vers. 12.5; Agilent Technologies). Probes within each probe set were summarized by GeneSprings’ExonRMA16 algorithm after quantile normalization of probe level signal intensities across all samples to reduce inter‐array variability (Bolstad et al., 2003). Input data pre‐processing was concluded by baseline transformation to the median of all samples. After grouping of the replicated samples according to their respective experimental conditions, a given probe set had to be expressed above the background (i.e. fluorescence signal of a probe set was detected within the 20th and 100th percentiles of the raw signal distribution of a given array) in both replicates, and at least one of the two was further analysed in pairwise comparisons. Differential gene expression was statistically determined by moderated t‐tests. The significance threshold was set to P = 0.01. Hierarchical cluster analysis on differentially expressed genes was performed by using Euclidian similarity measures and Ward's linkage. Functional gene ontology enrichment analysis of the differentially expressed genes was performed by using GeneSpring GX (enrichment P < 0.1).
2.21. Statistical analysis
Significant differences were evaluated by using the unpaired two‐way ANOVA with post hoc Bonferroni, P < 0.05 was considered as significant.
3. Results
3.1. Phenotypic and functional characterization of porcine MSCs and skin FBs
The porcine BMSCs, ASCs, DSCs as well as FBs showed plastic‐adherence and a fibroblastoid‐like cell morphology (Figure 1A). Furthermore, a MSC‐like antigenic phenotype (Figure 1C) was determined (Keating, 2012). Consequently, MSCs were detected positive for CD90+, CD14−, CD31−, CD45−, CD34−, CD79alpha−, HLA‐DR−, CD29+ and CD44+, but only to some extent positive for CD105 and CD73. BMSC and ASC cultures exhibited the capacity to differentiate into the osteogenic, chondrogenic as well as adipogenic lineages. However, DSCs and FBs showed osteogenic differentiation potential but were unable to demonstrate chondrogenic and adipogenic differentiation potential (Figure 1D; Table 1). Besides these differences, the MSCs also displayed a divergent performance in their cumulative doubling values in the descending order: BMSC > ASC > DSC > FB (Figure 1B).
Figure 1.
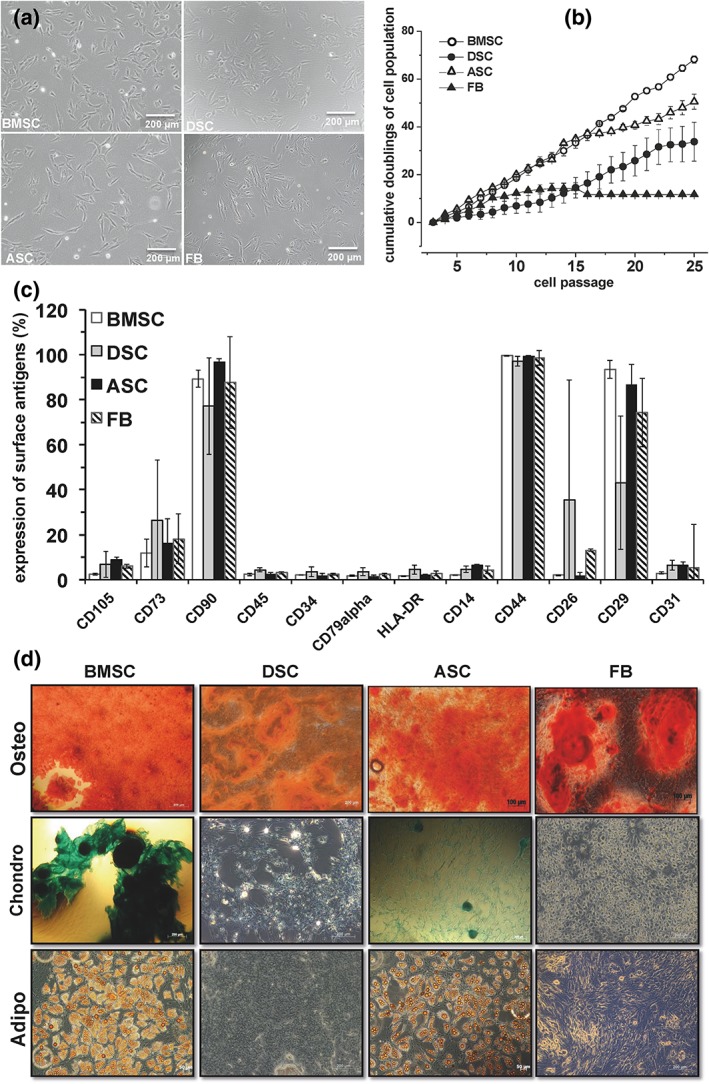
Phenotypic and functional characterization of porcine mesenchymal stromal cells (MSCs) obtained from bone marrow (BMSCs), dental pulp (DSCs), adipose tissue (ASCs) and skin fibroblasts (FBs). (A) Plastic adherence and cell morphology of BMSCs, DSCs, ASCs and FB cultures. A representative illustration of three individual experiments with identical results is illustrated. Scale bar: 100 μm. (B) Cumulative doublings of the cell populations. Values represent mean ± SD of three individual experiments. (C) Surface antigen phenotype expression. Values represent mean ± SD of three–six individual experiments. (D) Osteogenic (alizarin red S staining), chondrogenic (Alcian blue staining) and adipogenic (oil red O staining) differentiation potential of porcine MSCs and FBs under standard differentiation conditions. One representative illustration of three individual experiments with identical results is demonstrated. Scale bar: 200 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
Significant changes (down‐ or upregulated) in expression of osteogenesis‐relevant genes of ASCs incubated with osteogenic standard medium (ASCOM)
| Fold | Regulation | Gene description/gene symbol |
|---|---|---|
| 1,69 | down | ADAM metallopeptidase with thrombospondin type‐1 motif‐20/ADAMTS20 |
| 4,87 | down | ADAM metallopeptidase with thrombospondin type‐1 motif‐6/ADAMTS6 |
| 2,35 | down | ADAM TS‐like protein 5/LOC100512586 |
| 3,48 | down | BMP‐binding endothelial regulator/BMPER |
| 2,34 | down | Catenin (cadherin‐associated protein) alpha‐1/CTNNAL1 |
| 3,06 | down | Chemokine (C‐X‐C motif) ligand 2/CXCL2 |
| 1,64 | down | Chemokine (C‐X‐C motif) receptor 7/CXCR7 |
| 4,69 | down | Cyclic AMP‐dependent transcription factor ATF‐3/LOC100738612 |
| 1,40 | down | Cyclin‐dependent kinase1/CDK1 |
| 1,37 | down | Cyclin‐dependent kinase 6/CDK6 |
| 3,40 | down | Cyclin‐dependent kinase inhibitor 3/CDKN3 |
| 1,55 | down | Fibroblast growth factor 19/LOC100518950 |
| 1,82 | down | Inositolpolyphosphate‐5‐phosphatase/INPP5B |
| 2,37 | down | Insulin‐like growth factor binding protein 4/IGFBP4 |
| 3,69 | down | Insulin‐like growth factor binding protein 6/IGFBP6 |
| 9,60 | down | Interleukin‐33/LOC100518643 |
| 23,72 | down | Interleukin‐6/IL6 |
| 1,99 | down | Mitogen‐activated protein kinase 8/LOC100622217 |
| 9,67 | down | Protein kinase (cAMP‐dependent, catalytic) inhibitor alpha/PKIA |
| 2,36 | down | Sestrin‐2/LOC100620966 |
| 3,21 | down | Toll‐like receptor 3/TLR3 |
| 1,57 | down | Wntless homologue/WLS |
| 1,48 | up | ADAM metallopeptidase domain 10/ADAM10 |
| 2,51 | up | ADAM metallopeptidase domain 19/ADAM19 |
| 2,97 | up | ADAM metallopeptidase with thrombospondin type‐1 motif‐1/ADAMTS1 |
| 2,14 | up | ADAM metallopeptidase with thrombospondin type‐1 motif‐12/ADAMTS12 |
| 1,43 | up | Bone morphogenetic protein 1/BMP1 |
| 1,33 | up | Bone morphogenetic protein 2/BMP2 |
| 1,52 | up | cAMP‐responsive element binding protein 3‐like‐4/CREB3L4 |
| 2,60 | up | Catalase/CAT |
| 1,93 | up | Cathepsin F/CTSF |
| 1,52 | up | Cathepsin Z/CTSZ |
| 1,82 | up | CCAAT/enhancer‐binding protein delta/LOC100153946 |
| 1,44 | up | CCAAT/enhancer‐binding protein delta/LOC100621023 |
| 1,57 | up | CCAAT/enhancer‐binding protein(C/EBP) beta|/CEBPB |
| 2,62 | up | Collagen, type IV, alpha 2/COL4A2 |
| 1,58 | up | Collagen, type IV, alpha 5/LOC100737091 |
| 1,51 | up | Collagen, type V, alpha 3/COL5A3 |
| 2,61 | up | Collagen and calcium‐binding EGF domain‐containing protein1/LOC100510995 |
| 7,13 | up | Complement component 7/C7 |
| 14,66 | up | Complement component C9/LOC100037951 |
| 1,53 | up | CREB‐binding protein/LOC100738967 |
| 2,57 | up | Cyclin‐dependent kinase‐5/CDKL5 |
| 1,55 | up | Cyclin‐dependent kinase 19/CDK19 |
| 2,46 | up | Cyclin‐dependent kinase 9/CDK9 |
| 1,51 | up | F‐box and leucine‐rich repeat protein 5/FBXL5 |
| 1,64 | up | F‐box protein 21/FBXO21 |
| 2,05 | up | Forkhead box O1/FOXO1 |
| 3,02 | up | Forkhead box O3/FOXO3 |
| 1,86 | up | Homeobox A5/HOXA5 |
| 2,79 | up | Homeobox protein cut‐like2/LOC100153673 |
| 1,60 | up | Homeobox protein Hox‐A6/LOC100519284 |
| 1,98 | up | Homeodomain interacting protein kinase 3/LOC100738110 |
| 9,39 | up | Insulin‐like growth factor binding protein 3/IGFBP3 |
| 2,12 | up | Insulin receptor/INSR |
| 1,56 | up | Interleukin‐18 receptor alpha chain/IL‐18RA |
| 1,65 | up | Interleukin‐1 receptor‐associated kinase‐2/LOC100523467 |
| 2,45 | up | Interleukin‐1 receptor type 1/LOC100626904 |
| 2,42 | up | Interleukin‐1 receptor type 2/IL1R2 |
| 2,02 | up | Interleukin‐6 receptor/IL6R |
| 1,73 | up | Janus kinase 1/JAK1 |
| 1,47 | up | MAP kinase‐activated protein kinase 2/MAPKAPK2 |
| 1,85 | up | Mitogen‐activated protein kinase 3/MAP3K3 |
| 3,22 | up | SH2 domain‐containing protein 4A/LOC100518097 |
| 1,64 | up | SH3 and PX domains 2A/SH3PXD2A |
| 2,20 | up | Transforming growth factor beta‐1/TGFBI |
| 1,44 | up | Transforming growth factor beta‐1 receptor 1/TGFBR1 |
| 1,87 | up | WNT1 inducible signalling pathway protein 1/WISP1 |
| 3,88 | up | Wnt‐16/LOC100511484 |
3.2. Comparison of the osteogenic differentiation potentials of porcine BMSCs, ASCs, DSCs and FBs
All porcine MSCs as well as the FBs had the ability for osteogenic differentiation even though the potency for differentiation strongly varied within the respective cell types (Figure 2). On days 7, 14 and 21, the BMSC and DSC cultures showed nearly identical elevated osteogenic differentiation but, on days 28 and 35, the expression of the deposited calcificated matrix in the BMSC cultures increased over‐proportionally and was almost twofold higher compared with DSC cultures. In contrast, ASCs and FBs did not exhibit any significant osteogenic differentiation on days 7, 14 and 21. Not until days 28 and 35 was a significant formation of calcium deposition in the extracellular matrix in the ASC and FB cultures determined, significantly lower compromised compared with BMSC or DSC cultures.
Figure 2.
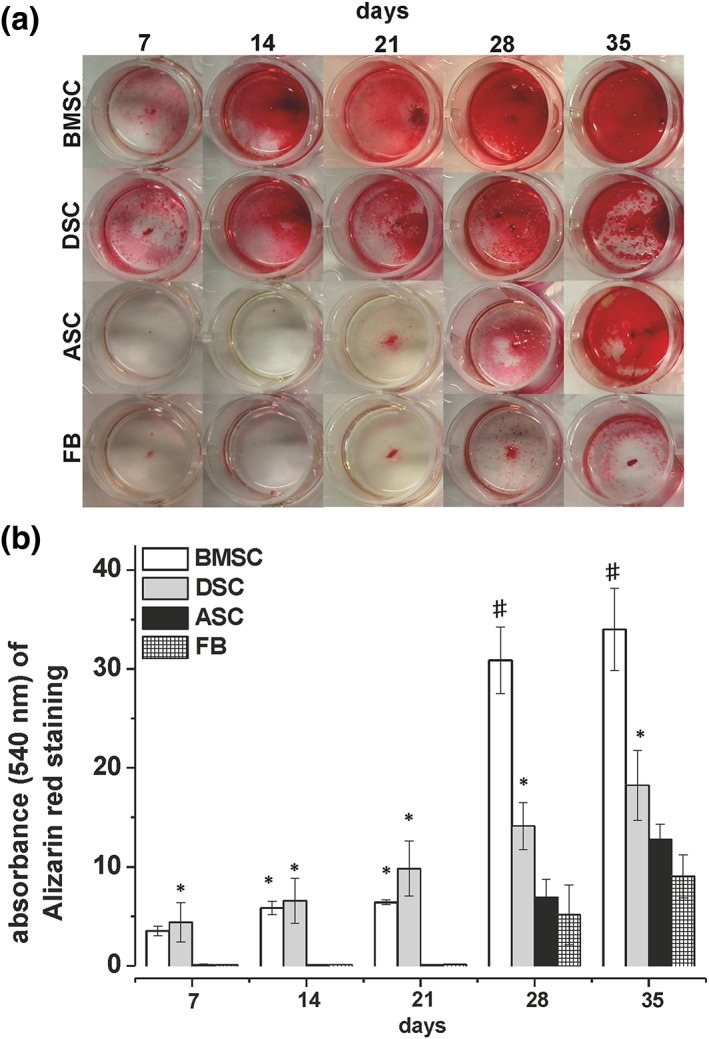
Comparison of the osteogenic differentiation potentials of porcine mesenchymal stromal cells (MSCs) obtained from bone marrow (BMSCs), dental pulp (DSCs), adipose tissue (ASCs) and fibroblasts (FBs). (A) Qualitative evaluation of osteogenic differentiation by alizarin red S staining of deposited calcificated matrix. Shown is one representative illustration of six individual experiments with identical results. (B) Quantification of osteogenic differentiation of BMSCs (white bars), DSCs (grey bars), ASCs (black bars) and FB cultures (squared bars) by alizarin red S extraction with cetylpyridiniumchlorid and photospectral analysis at 540 nm. Values represent mean ± SD of four–six individual experiments. * P < 0.05 compared with ASC and FB cultures. # P < 0.05 compared with the respective DSC cultures. [Colour figure can be viewed at wileyonlinelibrary.com]
3.3. Molecular basis of osteogenic‐deficiency of MSCs
Under osteogenic culture conditions, BMSC but not ASC cultures revealed an apparent ALP activity (Figure 3A). When comparing BMSC and ASC cultures, here examined as the representatives for osteogenic high‐ and low‐responder, respectively, a high ALP activity observed in BMSC cultures was paralleled by a significantly higher BMP‐2 protein expression detected in ASC cultures (Figure 3B).
Figure 3.
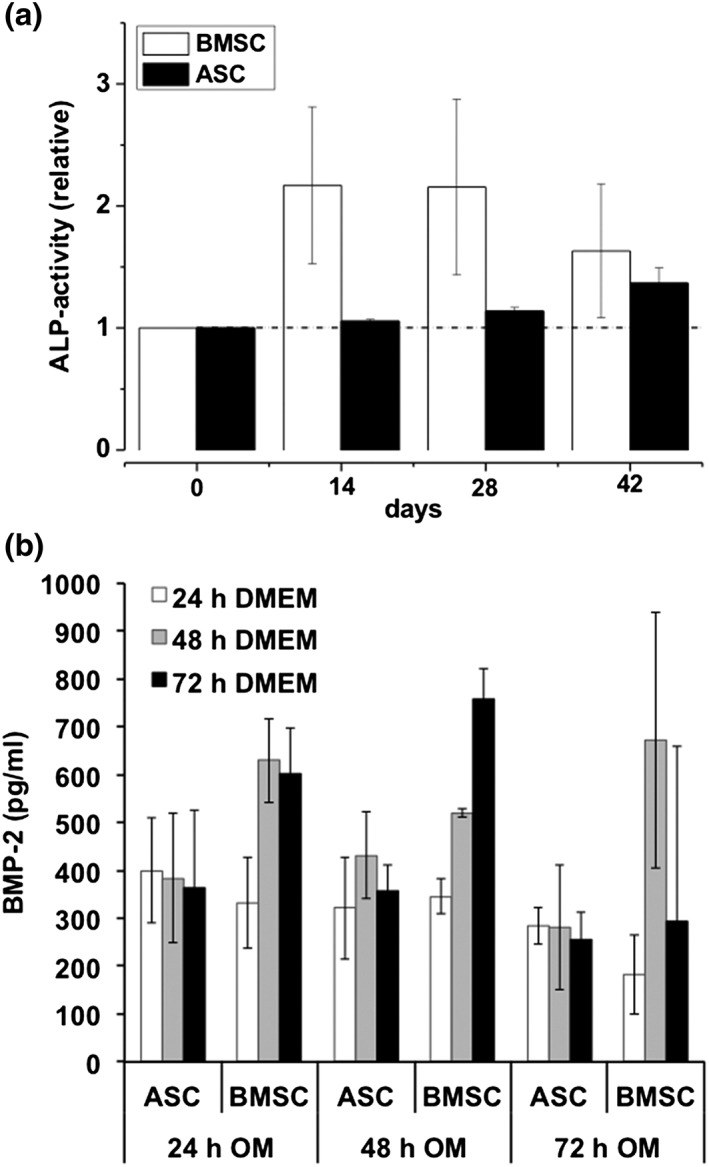
Quantification of alkaline phosphatase (ALP) activity and BMP‐2 in resting and osteogenic‐differentiated porcine mesenchymal stromal cells (MSCs) obtained from bone marrow (BMSCs) and adipose tissue (ASCs). (A) ALP activity in osteogenic differentiated BMSC (white bars) and ASC (black bars) cultures. ALP activity was normalized to cell viability of the respective sample. Values represent mean ± SD of five–six individual experiments. * P < 0.05 compared with values indicated. (B) BMP‐2 protein expression in osteogenic‐differentiated BMSC (white bars) and ASC (black bars) cultures as quantified by a specific ELISA. Values represent mean ± SD of four–five individual experiments. * P < 0.05
3.4. Impact of BMP‐2 and PRP on osteogenic differentiation of MSCs and FBs
The data shown above indicate an impaired BMP‐2 expression as cellular basis for a deficient osteogenesis in ASC and FB cultures. To prove this assumption, the standard OM was supplemented with BMP‐2 or PRP. The results shown in Figures 4 and 5 demonstrate that medium supplementation with BMP‐2 significantly enhanced osteogenic differentiation of ASC and FB cultures, and led to differentiation stages that were comparable to those seen in BMSC and DSC cultures. However, the osteogenic potency of BMSCs and DSCs could not be significantly altered by a BMP‐2 addition.
Figure 4.
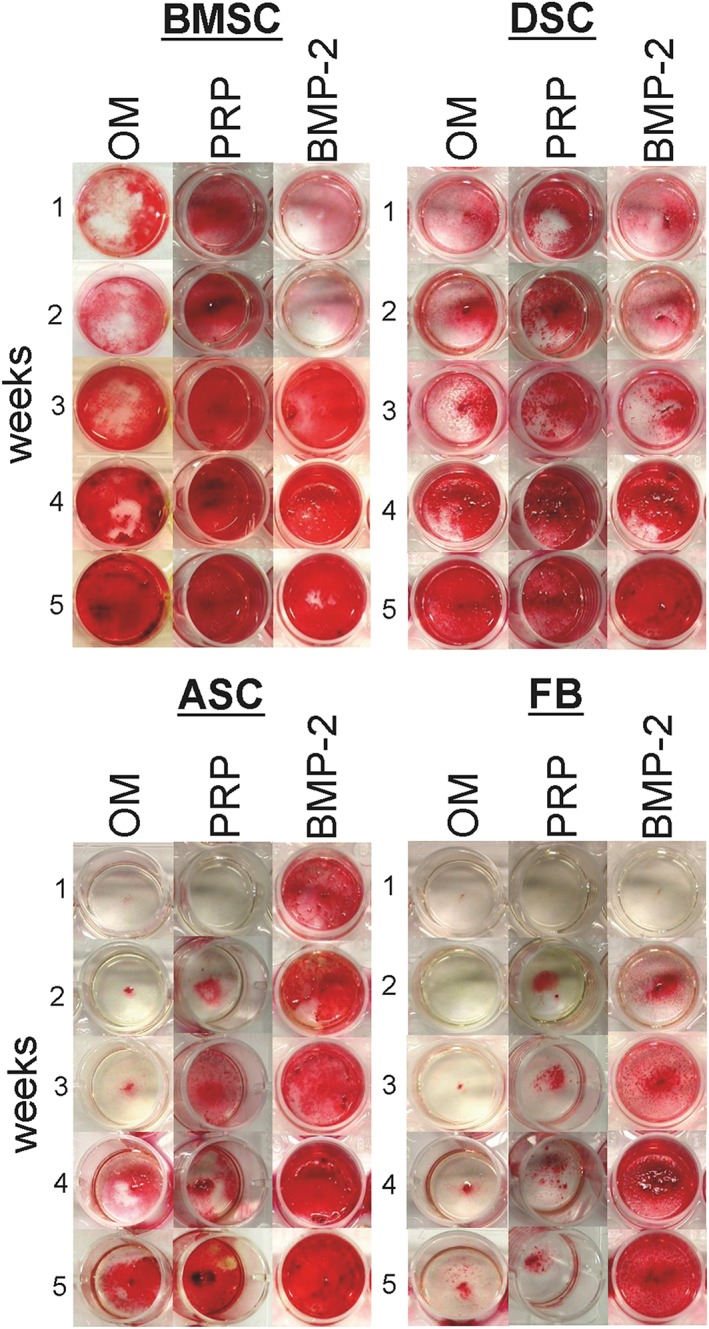
Impact of BMP‐2 and platelet‐rich plasma (PRP) on osteogenic‐differentiated mesenchymal stromal cells (MSCs) obtained from bone marrow (BMSCs), dental pulp (DSCs), adipose tissue (ASCs) and fibroblasts (FBs). BMSC, DSC, ASC and FB cultures were osteogenic differentiated using the standard osteogenic differentiation medium (OM). Additionally, OM was supplemented with BMP‐2 (450 ng/ml) or PRP (1%). At the indicated time points, the calcification of the extracellular matrix was visualized by alizarin red S staining. Shown is one representative illustration of six individual experiments with identical results. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 5.
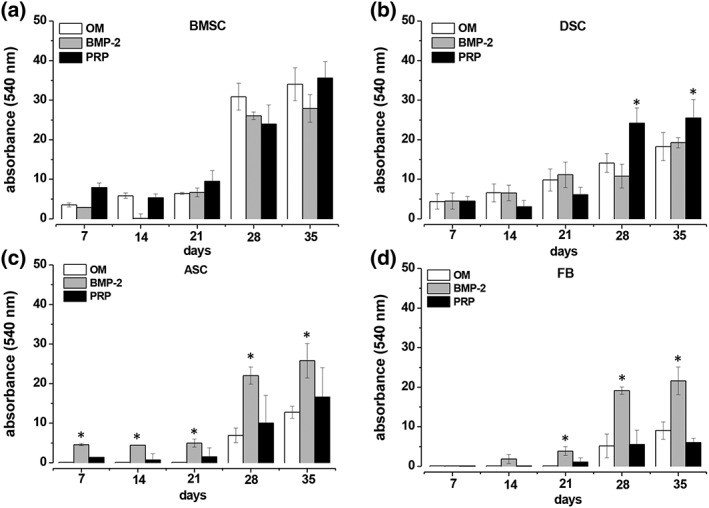
Quantitative evaluation of the impact of BMP‐2 and platelet‐rich plasma (PRP) on osteogenic‐differentiated mesenchymal stromal cells (MSCs) obtained from bone marrow (BMSCs), adipose tissue (ASCs) and fibroblasts (FBs). BMSC (A), DSC (B), ASC (C) and FB (D) cultures were osteogenic differentiated using the standard osteogenic differentiation medium (OM). Additionally, OM (white bars) was supplemented with BMP 2 (grey bars; 450 ng/ml) or with PRP (black bars; 1%). At the time points indicated, the calcification of the extracellular matrix was quantified by staining with alizarin red S, extraction of the dye by cetylpyridiniumchlorid and photospectral analysis at 540 nm. Values in (A) and (D) represent the mean ± SD of four individual experiments, and the mean ± SD of six individual experiments in (B) and (C). * P < 0.05 as compared with the other values of the respective time points
Platelet‐rich plasma supplementation of the standard osteogenic medium did not reveal any significant add‐on effects on the osteogenic differentiation potential of the BMSC, ASC and FB cultures, but significantly enhanced osteogenesis in DSC cultures on days 28 and 35 (Figures 4 and 5B).
3.5. Microarray analysis of the mRNA‐expression profile of ASCs and BMSCs
The osteogenic activation of resting ASCs with OM (ASCRES vs. ASCOM) was accompanied by significant changes in 1166 differentially expressed genes. The intersection of both data sets revealed 107 differentially expressed (down or up) genes. Seventy‐five genes of the 1166 bore osteogenetic relevance (Table 1).
In contrast, successful osteogenic differentiation of resting ASC cultures by activation with OM + BMP‐2‐medium (ASCRES vs. ASCOM+BMP‐2) was accompanied by significant changes in the expression of 827 genes. The intersection with the gene expression of the osteogenetic activated BMSC cultures (BMSCRES vs. BMSCOM) revealed 152 differentially expressed genes, which apparently represent the necessary factors for successful osteogenic differentiation in BMSC as well as ASC cultures. Of the mentioned 827 genes, 40 are known for osteogenetic relevance (Table 2), but only four differentially expressed transcripts, cyclic‐AMP‐dependent transcription factor‐3 (ATF‐3, down), interleukin‐33 (IL‐33, down), interleukin‐1 receptor (up) and TGF‐β1 (up), have been found together in OM‐ or OM + BMP‐2‐activated ASC cultures (Table 2A, marked by grey colour).
Table 2.
(A) Significant changes (down‐ or upregulated) in expression of osteogenesis‐relevant genes of ASCs incubated in osteogenic medium supplemented in BMP‐2 (ASCOM + BMP). Grey labelling depicts osteogenesis‐relevant genes expressed together in OM‐ (Table 1) and OM + BMP‐2‐activated MSC cultures. (B) Significant changes (down‐ or upregulated) of osteogenesis‐relevant genes expression of BMSC cultures incubated with osteogenic medium (BMSCOM). (C) Differentially expressed osteogenesis‐relevant genes expressed together in OM‐activated BMSC and OM + BMP‐2‐activated ASC cultures
| Table 2 (A) | ||
|---|---|---|
| Fold | Regulation | Gene description/gene symbol |
| 1,57 | down | ADAM metallopeptidase domain 15/ADAM15 |
| 1,63 | down | ADAM metallopeptidase domain 17/ADAM17 |
| 2,67 | down | Bone morphogenetic protein 4/BMP4 |
| 2,34 | down | Catenin (cadherin‐associated protein) alpha‐1/CTNNAL1 |
| 1,55 | down | Cathepsin K/CTSK |
| 1,57 | down | Cathepsin L1/CTSL1 |
| 1,97 | down | Cathepsin O‐like/LOC100737564 |
| 10,25 | down | Collagen typeXIV alpha 1/ COL14A1 |
| 3,87 | down | Cyclic AMP‐dependent transcription factor ATF‐3/LOC100738612 |
| 1,50 | down | Epidermal growth factor receptor pathway substrate 15/EPS15 |
| 2,01 | down | Fibroblast growth factor 9/FGF9 |
| 1,92 | down | Fibroblast growth factor receptor 1/FGFR1 |
| 1,47 | down | F‐box and WD‐repeat domain containing protein 7, E3 ubiquitin protein ligase/FBXW7 |
| 1,56 | down | Interleukin‐17/LOC100738902 |
| 9,60 | down | Interleukin‐33/LOC100518643 |
| 3,48 | down | Latent‐transforming growth factor beta‐binding protein 2/ LOC100514300 |
| 1,92 | down | microRNAmir‐143/MIR143 |
| 3,02 | down | Plasminogen activator, urokinase/PLAU |
| 1,77 | down | Platelet‐derived growth factor receptor/PDGFRB |
| 1,55 | down | Platelet/endothelial cell adhesion molecule 1/PECAM1 |
| 6,17 | up | Cartilage oligomeric matrix protein/COMP |
| 5,80 | up | Chemokine (C‐X‐C motif) receptor 4/CXCR4 |
| 1,55 | up | Collagen type III alpha 1/COL3A1 |
| 1,74 | up | Endoglin/ENG |
| 8,60 | up | Fibroblast growth factor receptor 2/FGFR2 |
| 1,51 | up | Fibroblast growth factor receptor 3/LOC100514115 |
| 3,71 | up | Homeobox protein DLX‐3/LOC100516335 |
| 2,65 | up | Homeobox protein DLX‐4/LOC100517238 |
| 3,30 | up | Interleukin‐1‐receptor‐1/IL1RL1 |
| 3,21 | up | Osteomodulin/LOC100511925 |
| 1,94 | up | Parathyroid hormone‐1 receptor/PTH1R |
| 1,47 | up | Rab‐5 GDP/GTP‐exchange factor/LOC100516668 |
| 1,77 | up | Rab‐acceptor 1/RABAC1 |
| 4,22 | up | SMAD family member 6/SMAD6 |
| 5,68 | up | SMAD family member 7/SMAD7 |
| 2,58 | up | Snailhomologue 2/SNAI2 |
| 2,58 | up | Snailhomologue 2/SNAI2 |
| 2,68 | up | Transforming growth factor beta‐1/TGFB1 |
| 1,55 | up | Transmembrane protein 229A/LOC100524656 |
| Table 2 (B) | ||
| Fold | Regulation | Gene description/gene symbol |
| 1,87 | down | Cyclic‐AMP‐dependent transcription factor ATF‐3/LOC100738612 |
| 2,47 | down | Frizzled‐related protein/FRZB |
| 1,71 | down | Inositolpolyphosphate‐1‐phosphatase/INPP1 |
| 3,76 | down | Interleukin‐33/LOC100518643 |
| 11,98 | down | Interleukin‐6/IL6 |
| 1,56 | down | microRNAmir‐23a/MIR23A |
| 1,71 | down | Plasminogen activator, urokinase/PLAU |
| 2,42 | up | Collagen type IV alpha 5/LOC100737091 |
| 1,67 | up | Collagen alpha‐6(IV)chain/LOC100620724 |
| 2,45 | up | FGF receptor 2 IIIc/FGFR2IIIC |
| 2,02 | up | Fibroblast growth factor receptor 2/FGFR2 |
| 2,82 | up | Forkhead box O1/FOXO1 |
| 3,09 | up | Forkhead box O3/FOXO3 |
| 1,73 | up |
Inositol 1,4,5‐trisphosphate receptor type 2/ LOC100517279 |
| 3,24 | up | Interleukin‐1receptor type II/IL1R2 |
| 4,87 | up | Osteomodulin/LOC100511925 |
| 2,33 | up | Transmembrane protein 229A/LOC100524656 |
| Table 2 (C) | |
| Regulation | Gene description/gene symbol |
| down | Cyclic‐AMP‐dependent transcription factor ATF‐3/ LOC100738612 |
| down | Interleukin‐33/LOC100518643 |
| down | Plasminogen activator, urokinase/PLAU |
| up | Fibroblast growth factor receptor 2/FGFR2 |
| up | Osteomodulin/LOC100511925 |
| up | Transmembrane protein 229A/LOC100524656 |
Successful osteogenic differentiation of resting BMSC cultures by osteogenic OM (BMSCRES vs. BMSCOM) was accompanied by significant changes in the expression of 335 of the 15 589 analysed genes (Figure 6A). Upon them the expression of 10 osteogenesis‐promoting factors was significantly increased and mRNA expression of seven osteogenesis‐reducing factors was significantly enhanced (Table 2B).
Figure 6.
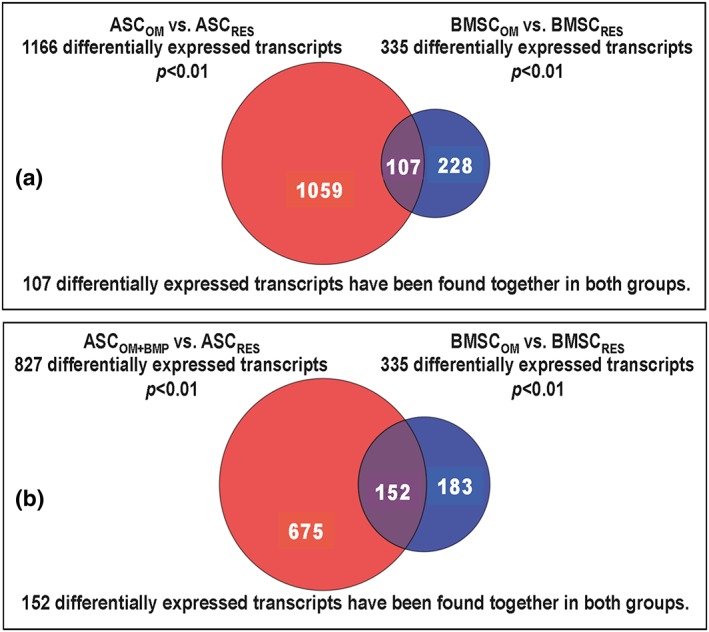
Micro‐array analysis and multi‐Venn diagram of differential expressed transcripts of adipose tissue stromal cells (ASCs) and bone marrow stromal cells (BMSC) treated with osteogenic differentiation medium (OM) compared with OM supplemented with BMP‐2. (A) Number of significantly changed mRNA expression in BMSCs (blue circle; BMSCOM) and ASCs (red circle; ASCOM) that were osteogenic activated by OM. The suffix RES indicates resting, not osteogenic activated cultures. The intersection represents 107 differentially expressed transcripts found together in both groups. (B) Number of significantly changed mRNA expression in BMSC cultures (blue circle; BMSCOM) that were osteogenic induced with OM, and ASC cultures (red circle; ASCOM + BMP) that were activated with BMP‐2‐supplemented osteogenic medium (OM + BMP). The suffix RES indicates resting, not osteogenic induced cultures. The intersection represents 152 differentially expressed transcripts found together in both groups. [Colour figure can be viewed at wileyonlinelibrary.com]
By comparing the expression of successfully osteogenetic activated BMSC and ASC cultures, only six genes that were differentially expressed together in both cell types were identified (Table 2C). The expression of genes coding for ATF‐3, IL‐33 and plasminogen activator was significantly downregulated, and the expression of genes coding for fibroblast growth factor receptor 2 (FGFR‐2), osteomodulin and transmembrane protein 229A was significantly upregulated (Table 2C).
4. Discussion
Mesenchymal stromal cells have been gaining importance as therapeutic tools in both experimental and clinical approaches for bone regeneration (Wittig et al., 2016). Although good results for osteogenic differentiation have been described with the use of BMSCs, necessary bone marrow aspiration has drawbacks, such as pain and risk of infection. Therefore, researchers as well as clinicians have begun to characterize the potential of other mesenchymal tissues to serve as possible sources for multipotent cells. In this regard, stromal cells isolated from dental pulp (Lee et al., 2011), adipose tissue (Zuk et al., 2002) as well as skin FBs (Junker et al., 2010) have proved to be suitable cell sources for bone regenerative approaches, albeit sometimes with substantial variations in their osteogenic differentiation potentials. Besides presentation of osteogenic properties in vitro, several studies have shown the influence of MSC‐based approaches on bone regeneration of bone defects in vivo (Wittig et al., 2016). Mini‐pigs are a commonly used in vivo model for the evaluation of bone regeneration (Pearce et al., 2007). The characteristics of bone metabolism closely resemble those in humans, which makes this an excellent model to compare the osteogenic potential of multipotent stromal cells (Pearce et al., 2007).
As shown for protagonists for high and low osteogenic differentiation capacities and fulfilling the mesenchymal stem cell criteria most likely, BMSC and ASC cultures were selected for microarray analysis. In this study, evaluation of the microarray analysis database of mRNA‐expression confirmed the observed humoral osteogenesis‐relevant dichotomy in MSCs, and further revealed the molecular mechanism behind a superior osteogenic differentiation potential. Osteogenic differentiation of BMSCs in OM was accompanied by significantly up‐ or downregulation of the expression of 335 genes. A significant increase was observed in the expression of genes for 11 different osteogenesis‐promoting factors accompanied by downregulation of the mRNA expression of seven osteogenesis‐reducing factors. In ASC cultures incubated in OM, significant alterations in the expression of more than 1100 genes were observed. Of those genes, 75 are of relevance in osteogenetic differentiation. However, incubation in OM led to significantly inferior osteogenic differentiation in ASCs compared with BMSCs. BMSC‐like differentiation of ASCs could only be observed with BMP‐2‐supplementation to OM (OM + BMP‐2). This was accompanied by significantly altered expression of only 827 genes, with 40 of those being of osteogenetic relevance. Remarkably, only four differentially expressed transcripts, ATF‐3 (down), IL‐33 (down), interleukin‐1‐receptor (up), and TGF‐β1 (up) have been found together in OM‐ and OM + BMP‐2‐induced ASC cultures, respectively. When comparing the expression of osteogenic activated BMSC and ASC cultures, only six genes were differentially expressed in both cell types. Those include significantly downregulated expression of genes coding for ATF‐3, IL‐33 and plasminogen activator, and a significantly upregulated expression of genes coding for FGFR‐2, osteomodulin and transmembrane protein 229A (TMEM229A), supposing that these factors are crucial for early osteogenic differentiation.
AMP‐dependent transcription factor‐3 (ATF‐3) is a member of the ATF/cAMP response binding‐protein (CREB) family of transcription factors, which suppress ALP expression and activation, and therefore regulate osteogenic differentiation. It is generally accepted that BMP‐2 reduces ATF‐3 protein expression, and overexpression of ATF‐3 negatively regulated BMP‐2‐induced ALP expression (Park et al., 2014). In line with such evidence in porcine ASCs, treated with BMP‐2, ATF‐3 is also downregulated. Unsurprisingly, in good differentiable BMSCs, demonstrated among others with elevated ALP‐activity, ATF‐3 exhibits as well a diminished expression.
A downregulation of plasminogen activator, as demonstrated here for osteogenic differentiated ASCs and BMSCs, seems to be plausible, as mice lacking plasminogen activator expression reveal increased bone mass and bone formation (Daci et al., 2003). Furthermore, it has been shown that glucocorticoids (dexamethasone was part of the used standard OM) decrease the transcription of plasminogen activator gene (Pearson et al., 1987). Moreover, plasminogen activator appears to be associated with redistribution of cytokeratins and cytoskeleton as well as changes in cell shape (Baribault et al., 1989). This phenomenon could be observed in the differentiation process: MSC phenotypes transformed from an elongated fibroblastoid character to a spherical osteoblastic shape.
Interleukin‐33 is a pleiotrophic cytokine affecting several signalling cascades and immune cells. Furthermore, IL‐33 assumes a role in bone homeostasis, influencing osteoclasts and osteoblasts. It was shown that monocytes could differentiate to osteoclasts only with the treatment of IL‐33 and macrophage colony‐stimulating factor. Moreover, IL‐33 induces the expression of some factors important for osteoclast maturation (Mun et al., 2010). In addition, IL‐33 mediates the expression of RANKL involving ERK and p38 pathways in MC3T3 cells (Mine et al., 2014) and osteoblasts, modulating the osteoclasts differentiation. Some research could detect IL‐33 in osteoblasts, but could not observe an effect on bone remodelling (Saidi et al., 2011), assuming only a role for IL‐33 in bone inflammation. Other authors demonstrated an increasing expression of IL‐33 in mice after an additional treatment with parathyroid hormone or oncostatin M (Saleh et al., 2011), and demonstrated a corresponding accelerated expression of matrix mineral deposition in osteoblasts. The latter observation could be supported by Schulze et al. (2011) in the course of 25 days in mice. On the contrary, our data indicate a decreased expression of IL‐33 in ASCs and BMSCs at the early stage of osteoblast differentiation. Comparable to our findings, Schulze et al. (2011) demonstrated a short‐lasting, decreasing effect of IL‐33 expression after 5 days of osteogenic induction.
The effect of FGF on osteogenesis is observed in multiple cases. The inhibition of FGFR signalling resulted in a complete loss of osteogenic differentiation in human MSCs (Ng et al., 2008). In in vivo studies, FGF accelerates callus remodelling and induces fracture healing. In vitro observations revealed that low doses of FGF‐2 promote differentiation in rat calvarial cells and osteoblast cell lines (Kuhn et al., 2013). Furthermore, a treatment with FGF‐2 at the beginning of the differentiation process accelerates osteogenic differentiation (Kuhn et al., 2013). The present study underlines these findings, as in good differentiable ASCs (OM + BMP‐2) and BMSCs, the expression of FGFR‐2 is strongly upregulated (Table 2). It is recognized that FGFR‐2 signalling induces the transcription of RUNX‐2, an essential factor in osteoblastogenesis (Baroni et al., 2005). In BMSCs the FGFR‐2IIIC and in ASCs the FGFR‐3 also demonstrate an increased expression (Table 2). These findings support the importance of the FGF‐signalling induction in the early phase of osteogenic differentiation.
As verified in our study, osteomodulin also demonstrated an increased expression in porcine ASCs and BMSCs. Moreover, osteomodulin is known as osteoadherin and SLRR2C, and might have a role in regulation of mineralization and as an early marker for osteoblasts producing extracellular matrix. In line with our findings, osteomodulin was consistently upregulated during initial bone formation in mice (Sugars et al., 2013).
At least, TMEM229A is a highly conserved gene in chimpanzee, rhesus monkey, dog, cow, mouse, rat, zebrafish, frog and sus scrofa with unknown function. It is assumed that TMEM229A has some transcription factor activity. Up until now, there have been no further information or publications concerning this gene. Remarkably, in our study an elevated expression of TMEM229A was confirmed in both osteogenic differentiated ASCs as well as BMSCs.
In summary, our findings indicate that MSCs from several genetic origins, with varying osteogenic differentiation potential, use different gene expression profiles for osteogenic differentiation under standard differentiation conditions. Nevertheless, we demonstrated that ASCs incubated with BMP‐2 (OM + BMP‐2) reached a comparable osteogenic differentiation to BMSCs. Under these circumstances, both BMSCs and ASCs use the same subset of above‐mentioned key molecules, which were also regulated in the same manner. Up until now, these key molecules are from subordinate interest. Here, further studies are necessary to analyse their function for osteogenesis in detail.
Furthermore, the present study analyses characteristics and the potential for osteogenic differentiation of porcine BMSCs, ASCs, DSCs and FBs.
Cells from all sources displayed fibroblastoid cell morphology, plastic‐adherence and a MSC‐like antigenic phenotype with expression of CD90+, CD45−, CD34−, CD79a−, HLA‐DR−, CD14−, CD31−, CD29+, CD44+, aside from CD105, CD73, which do not present entirely typical antigen expression. Eventually, it depends on the species of stromal cells. Furthermore, MSCs exhibited divergent performance in their cumulative doublings in the range BMSCs > ASCs > DSCs > FBs. All evaluated cell types were capable of differentiating into the osteogenic lineage, but only BMSCs and ASCs could be differentiated into adipocytes and chondrocytes. In conclusion, BMSCs and ASCs met the criteria defining multipotent MSCs (Keating, 2012). The used adipogenic and chondrogenic differentiation media established for human MSCs may have caused the porcine DSC and FB cultures to not fulfill these criteria.
The low osteogenic potential of ASCs was accompanied by an apparent lack of ALP activity as well as reduced BMP‐2 protein expression, whereas the BMSC cultures exhibited a characteristic osteogenesis‐specific pattern of ALP activity (Owen et al., 1990) and a distinctive BMP‐2 protein expression. ALP has been shown to be an essential osteogenic marker and essentially involved in matrix mineralization (Wennberg et al., 2000), whereas BMP‐2 is an important intrinsic inducer of ALP (Rawadi et al., 2003). Following BMP receptor activation, this signalling cascade further comprises BMP‐2‐induced R‐Smad activation and the expression of osteogenic transcription factors, such as Runx2, osterix (Osx), distal‐less homeobox 5 (Dlx5) and muscle segment homeobox homologue (Msx2), that finally regulate ALP expression (Ryoo et al., 2006). Our results underline the different relevance of the osteogenic capacity of BMP‐2 for osteogenic competent cells. Supplementation with BMP‐2 at the concentration of 450 ng/ml has previously been shown to be the best quantity for osteogenic differentiation in mouse BMSCs (Luong et al., 2012). In this study we could confirm this result for porcine ASCs with the help of a simple titration experiment (data not shown). Therefore, we used this concentration to increase the osteogenic differentiation capacity of ASCs and FBs to a level comparable to BMSCs or DSCs. However, BMP‐2 supplementation did not increase the osteogenic differentiation potential of DSCs. Various in vivo studies have shown osteoinductive properties of PRP in BMSCs (Hakimi et al., 2010). Consequently, we evaluated the influence of an additional application of PRP on the osteogenetic capacity of the MSC cultures and FBs. It was shown that PRP did not exhibit any significant effects on the osteogenic differentiation of BMSCs, ASCs and FBs. However, a significant increase of the osteogenic differentiation of DSCs to a level comparable to BMSCs was observed. These results are in accordance with previous findings, which showed that BMP‐2 had no positive effects on dental pulp regeneration (Ko et al., 2010), whereas PRP significantly improved the osteogenetic differentiation capacity of DSCs (Lee et al., 2011).
It is generally accepted that PRP is an excellent source for growth factors, cytokines and haemostatic factors. Besides others, it contains significant amounts of BMP‐2 (Jungbluth et al., 2014), and the overall concentration of growth factors in PRP is significantly higher compared with whole blood (Jungbluth et al., 2014). As shown recently, the mean concentration of BMP‐2 in PRP (167.8 ± 83.4 pg/ml) is below the observed osteogenesis‐promoting concentration of 450 ng/ml. Also, significant concentrations of TGF‐β1 (40.9 ± 5.5 pg/ml) and PDGF (13.9 ± 5.1 pg/ml) were measured in PRP (Jungbluth et al., 2014). The osteogenesis‐improving effects of the PRP supplementation on DSCs may not be due to BMP‐2, but caused by the growth factor mix released by PRP. Using a concentration of 1% PRP in our study appeared advisable, because a higher concentration led to coagulation of plasma proteins.
Concerning the osteogenic differentiation, significant differences were observed between the cell cultures. Using a protocol optimized for osteogenic differentiation of human cells (Pittenger et al., 1999), excellent osteogenic differentiation capacity was observed for BMSCs and DSCs, whereas ASCs and FBs showed significantly inferior osteogenic differentiation potential.
Although our results indicated that for successful osteogenic differentiation BMSCs and ASCs, due to their diversity and similarities in genetic activity, need completely different patterns of gene activation, it is possible to achieve nearly identical osteogenic differentiation yields with different MSC types or even FBs by optimizing or inducing cell type‐specific osteogenic signalling pathways. Based on these results, further investigations and in vivo animal studies on the osteogenic capacities of the different types of MSCs are warranted.
Conflict of interest
No competing financial interests exist.
Sources of funding
This work was supported by the Faculty of Medicine of the Heinrich‐Heine‐University Düsseldorf. In parts this work was also supported by the German Research Foundation DFG (PAK 816 ‘Plasma and Cell Interaction in Dermatology’, SU 631/4‐1).
Declaration
All experiments conformed to the respective national guidelines.
Author disclosures
Authors state no disclosures.
Acknowledgments
The authors thank Samira Seghrouchni, Christa‐Maria Wilkens and Jutta Schneider for technical assistance.
Bayraktar, S. , Jungbluth, P. , Deenen, R. , Grassmann, J. , Schneppendahl, J. , Eschbach, D. , Scholz, A. , Windolf, J. , Suschek, C. V. , and Grotheer, V. (2018) Molecular‐ and microarray‐based analysis of diversity among resting and osteogenically induced porcine mesenchymal stromal cells of several tissue origin. J Tissue Eng Regen Med, 12: 114–128. doi: 10.1002/term.2375.
References
- Baribault H, Blouin R, Bourgon L, Marceau N. 1989; Epidermal growth factor‐induced selective phosphorylation of cultured rat hepatocyte 55‐kD cytokeratin before filament reorganization and DNA synthesis. J Cell Biol 109: 1665–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroni T, Carinci P, Lilli C et al 2005; P253R fibroblast growth factor receptor‐2 mutation induces RUNX2 transcript variants and calvarial osteoblast differentiation. J Cell Physiol 202: 524–535. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003; A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193. [DOI] [PubMed] [Google Scholar]
- Boxall SA, Jones E. 2012; Markers for characterization of bone marrow multipotential stromal cells. Stem Cells Int 2012: 975871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AI, Bruder SP. 2001; Mesenchymal stem cells: building blocks for molecular medicine in the 21st century. Trends Molec Med 7: 259–264. [DOI] [PubMed] [Google Scholar]
- Cristofalo VJ, Allen RG, Pignolo RJ, Martin BG, Beck JC. 1998; Relationship between donor age and the replicative lifespan of human cells in culture: a reevaluation. Proc Natl Acad Sci U S A 95: 10 614–10 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daci E, Everts V, Torrekens S et al 2003; Increased bone formation in mice lacking plasminogen activators. J Bone Min Res 18: 1167–1176. [DOI] [PubMed] [Google Scholar]
- Dahl LK. 1952; A simple and sensitive histochemical method for calcium. Proc Soc Exp Biol Med Soc Exp Biol Med (New York, NY) 80: 474–479. [DOI] [PubMed] [Google Scholar]
- Fillingham Y, Jacobs J. 2016; Bone grafts and their substitutes. Bone Joint J 98‐b: 6–9. [DOI] [PubMed] [Google Scholar]
- Giannoudis PV, Dinopoulos H, Tsiridis E. 2005; Bone substitutes: an update, Injury 36: S20–S27. [DOI] [PubMed] [Google Scholar]
- Gregory CA, Gunn WG, Peister A, Prockop DJ. 2004; An Alizarin red‐based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem 329: 77–84. [DOI] [PubMed] [Google Scholar]
- Hakimi M, Jungbluth P, Sager M et al 2010; Combined use of platelet‐rich plasma and autologous bone grafts in the treatment of long bone defects in mini‐pigs. Injury 41: 717–723. [DOI] [PubMed] [Google Scholar]
- Hempel U, Muller K, Preissler C et al 2016; Human bone marrow stromal cells: a reliable, challenging tool for in vitro osteogenesis and bone tissue engineering approaches. Stem Cells Int 2016: 7 842 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth P, Grassmann JP, Thelen S et al 2014; Concentration of platelets and growth factors in platelet‐rich plasma from Goettingen minipigs. GMS Interdiscipl Plastic Reconstruct Surg DGPW 3: Doc11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker JP, Sommar P, Skog M, Johnson H, Kratz G. 2010; Adipogenic, chondrogenic and osteogenic differentiation of clonally derived human dermal fibroblasts. Cells Tiss Organs 191: 105–118. [DOI] [PubMed] [Google Scholar]
- Keating A. 2012; Mesenchymal stromal cells: new directions. Cell Stem Cell 10: 709–716. [DOI] [PubMed] [Google Scholar]
- Ko H, Yang W, Park K, Kim M. 2010; Cytotoxicity of mineral trioxide aggregate (MTA) and bone morphogenetic protein 2 (BMP‐2) and response of rat pulp to MTA and BMP‐2. Oral Surg Oral Med Oral Pathol Oral Radiol Endodont 109: e103–e108. [DOI] [PubMed] [Google Scholar]
- Kuhn LT, Ou G, Charles L, Hurley MM, Rodner CM, Gronowicz G. 2013; Fibroblast growth factor‐2 and bone morphogenetic protein‐2 have a synergistic stimulatory effect on bone formation in cell cultures from elderly mouse and human bone. J Gerontol Series A Biol Sci Med Sci 68: 1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee UL, Jeon SH, Park JY, Choung PH. 2011; Effect of platelet‐rich plasma on dental stem cells derived from human impacted third molars. Regen Med 6: 67–79. [DOI] [PubMed] [Google Scholar]
- Luong LN, Ramaswamy J, Kohn DH. 2012; Effects of osteogenic growth factors on bone marrow stromal cell differentiation in a mineral‐based delivery system. Biomaterials 33: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mine Y, Makihira S, Yamaguchi Y, Tanaka H, Nikawa H. 2014; Involvement of ERK and p38 MAPK pathways on Interleukin‐33‐induced RANKL expression in osteoblastic cells. Cell Biol Int 38: 655–662. [DOI] [PubMed] [Google Scholar]
- Mun SH, Ko NY, Kim HS et al 2010; Interleukin‐33 stimulates formation of functional osteoclasts from human CD14(+) monocytes. Cell Molec Life Sci 67: 3883–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng F, Boucher S, Koh S et al 2008; PDGF, TGF‐β, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood, 112: 295–307. [DOI] [PubMed] [Google Scholar]
- Owen TA, Aronow M, Shalhoub V et al 1990; Progressive development of the rat osteoblast phenotype in vitro: reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J Cell Physiol 143: 420–430. [DOI] [PubMed] [Google Scholar]
- Park JK, Jang H, Hwang S et al 2014; ER stress‐inducible ATF3 suppresses BMP2‐induced ALP expression and activation in MC3T3‐E1 cells. Biochem Biophys Res Comm 443: 333–338. [DOI] [PubMed] [Google Scholar]
- Pearce AI, Richards RG, Milz S, Schneider E, Pearce SG. 2007; Animal models for implant biomaterial research in bone: a review. Eur Cells Mater 13: 1–10. [DOI] [PubMed] [Google Scholar]
- Pearson D, Altus MS, Horiuchi A, Nagamine Y. 1987; Dexamethasone coordinately inhibits plasminogen activator gene expression and enzyme activity in porcine kidney cells. Biochem Biophys Res Comm 143: 329–336. [DOI] [PubMed] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC et al 1999; Multilineage potential of adult human mesenchymal stem cells. Science (New York, NY) 284: 143–147. [DOI] [PubMed] [Google Scholar]
- Rawadi G, Vayssiere B, Dunn F, Baron R, Roman‐Roman S. 2003; BMP‐2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res 18: 1842–1853. [DOI] [PubMed] [Google Scholar]
- Ryoo HM, Lee MH, Kim YJ. 2006; Critical molecular switches involved in BMP‐2‐induced osteogenic differentiation of mesenchymal cells. Gene 366: 51–57. [DOI] [PubMed] [Google Scholar]
- Saidi S, Bouri F, Lencel P et al 2011; IL‐33 is expressed in human osteoblasts, but has no direct effect on bone remodeling. Cytokine 53: 347–354. [DOI] [PubMed] [Google Scholar]
- Saleh H, Eeles D, Hodge JM et al 2011; Interleukin‐33, a target of parathyroid hormone and oncostatin m, increases osteoblastic matrix mineral deposition and inhibits osteoclast formation in vitro. Endocrinology 152: 1911–1922. [DOI] [PubMed] [Google Scholar]
- Schulze J, Bickert T, Beil FT et al 2011; Interleukin‐33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J Bone Miner Res 26: 704–717. [DOI] [PubMed] [Google Scholar]
- Sinha KM, Zhou X. 2013; Genetic and molecular control of osterix in skeletal formation. J Cell Biochem 114: 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa BR, Parreira RC, Fonseca EA et al 2014; Human adult stem cells from diverse origins: an overview from multiparametric immunophenotyping to clinical applications. Cytometry Part A 85: 43–77. [DOI] [PubMed] [Google Scholar]
- Sugars RV, Olsson ML, Marchner S, Hultenby K, Wendel M. 2013; The glycosylation profile of osteoadherin alters during endochondral bone formation. Bone 53: 459–467. [DOI] [PubMed] [Google Scholar]
- Volk SW, Wang Y, Hankenson KD. 2012; Effects of donor characteristics and ex vivo expansion on canine mesenchymal stem cell properties: implications for MSC‐based therapies. Cell Transplant 21: 2189–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennberg C, Hessle L, Lundberg P et al 2000; Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. J Bone Miner Res 15: 1879–1888. [DOI] [PubMed] [Google Scholar]
- Wittig O, Romano E, Gonzalez C et al 2016; A method of treatment for nonunion after fractures using mesenchymal stromal cells loaded on collagen microspheres and incorporated into platelet‐rich plasma clots. Int Orthop 40: 1033–1038. [DOI] [PubMed] [Google Scholar]
- Zuk PA, Zhu M, Ashjian P et al 2002; Human adipose tissue is a source of multipotent stem cells. Molec Biol Cell 13: 4279–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]