

Abstract
Rice yield and quality are adversely affected by increasing global surface temperature, and are strongly attributed to high night temperature (HNT) than high daytime temperature. However, the molecular mechanism underlying the heat‐tolerant characteristics of rice remains unclear. In the present study, we compared the proteomes of heat‐tolerant and ‐sensitive lines of rice at early milky stage using an iTRAQ method. We have identified 38 differentially expressed proteins between the two lines, of which 32 proteins have been functionally annotated in NCBI and/or the UniProt database. These proteins were then classified into seven functional subgroups, which include signal transduction, transcript regulation, oxidation, defense response, transport, energy metabolism, and biosynthesis. Further analysis indicated that HNT stress could disrupt the redox equilibrium of plant cells, which in turn triggers the calcium‐dependent protein kinase and COP9 signalosome, thereby regulating downstream genes/proteins that are involved in the HNT response. The candidate proteins may provide genetic resources for the improvement of heat‐tolerant characteristics in rice, and the proposed model for signal transduction and transcriptional regulation may facilitate in the elucidation of the molecular mechanism underlying the response to HNT stress in rice.
Keywords: High night temperature, iTRAQ, Plant proteomics, Rice
Abbreviations
- bHLH
basic helix‐loop‐helix
- BP
biological process
- CC
cellular component
- DEP
differentially expressed protein
- FC
fold‐change
- HNT
high night‐time temperature
- HSL
heat‐sensitive line
- HTL
heat‐tolerant line
- RFC
relative fold‐change
- TCA
tricarboxylic acid
- ZFP
zinc finger protein
1. Introduction
In recent years, global climate change has caused serious abiotic stress including high temperature stress, thereby hindering crop production 1, 2. The grain‐filling stage in the rice (Oryza sativa L.) life cycle is an important period for rice yield establishment, and this stage is highly susceptible to high temperature stress 3, 4. High temperature stress increases the grain growth rate during the early ripening period, which reduces the duration of grain growth 5. However, this increased rate in grain growth fails to compensate for the reduction in the duration of grain growth, which ultimately results in a decrease in the final grain weight 6, 7. High night temperatures (HNTs) are thought to cause carbohydrate deficiency in leaves and culms due to increased respiratory loss, thus adversely affecting grain weight in rice 8, 9. After high temperature stress, rice grain quality, and weight cannot be restored, even under optimal temperature conditions 10, 11. Rice is a staple food for more than 50% of the global population, and in order to meet the growing food demand of the increasing population worldwide, rice production must increase by more than 1% annually 1. Unfortunately, the global surface temperature is steadily rising, and a more rapid increase in nighttime temperature compared to the maximum during the day presents an even greater challenge toward achieving higher crop yields 12, 13. Therefore, it is essential to elucidate the molecular mechanisms underlying the response of rice to HNT stress and breed new cultivars with high temperature‐tolerant characteristics 12.
Significance of the study
The study focused on high nighttime temperature stress and analyzed the differentially expressed proteins between paired, genetically similar heat‐tolerant and heat‐sensitive rice lines. The differentially expressed proteins, especially the proteins involved in signal transduction and transcript regulation, may facilitate future studies of the molecular mechanisms underlying high nighttime temperature response in plants. Additionally, the differentially expressed proteins may provide genetic resources for the improvement of heat‐tolerant characteristics in rice.
The typical symptoms caused by high temperature stress during rice grain filling include increased growth rate 11, 14, low amylose content, increased chalkiness, and poor milling quality 15, 16. High‐temperature stress suppresses the expression of the amylose synthesis gene Waxy and the granule‐bound starch synthase gene GBSS, thereby resulting in lower amylose content, as well as the expression of starch branching enzymes. It additionally leads to alterations in the fine structure of amylopectin 16, 17, 18. High‐temperature environments induce the expression of α‐amylase genes Amy1A, Amy1C, Amy3A, Amy3D, and Amy3E as well as enhance α‐amylase activity, resulting in chalky grains 16, 17. Recently, several novel molecules that are produced in response to high temperature stress have been identified at the metabolomic, proteomic, and transcriptomic levels 19, 20, 21. At the metabolomic level, oxaloacetate/aspartate, α‐ketoglutarate, 3‐phosphoglycerate, and pyruvate families, which are involved in the tricarboxylic acid (TCA) cycle, are upregulated in heat‐sensitive rice cultivars at the vegetable stage under HNT conditions 20. At the proteomic level, the activity of ADPase, which cleaves sucrose and accelerates starch accumulation, is suppressed, and UDP‐glucose pyrophosphorylase, which serves as a glycosyl donor for glutelin synthesis, decreases during rice grain development at early milky stage under high‐temperature conditions 19, 22, 23. In our previous transcriptomics study, the expression of transcripts that were related to steroid hormones, namely, glycosyl, glutamine, and aspartic peptidase synthetases in early milky rice was also affected by HNT stress 21. While previous studies facilitate in better understanding the molecular mechanisms and gene regulatory networks of this important crop in response to global warming, the genes and proteins contributing to high temperature tolerance and the molecular mechanisms underlying the response of rice to high temperature (especially HNT) stress, are poorly understood 12.
In our previous investigation, we showed that HNT largely influences the expression of genes that are involved in electron transport from the inner membrane to the outer membrane of mitochondria in rice grains at early milky stage. The disruption of electron transport further affects the expression of genes involved in the TCA cycle, thereby leading to lower carbohydrate accumulation and low grain weight in rice 21. Protein expression is the key regulator of biochemical reactions and plays an important role in response to environmental stresses. The expression patterns of mRNA do not necessarily match that of their respective proteins because mRNA splicing, altered protein turnover, protein degradation, or a combination of these may mediate protein translation from mRNA. Posttranslational modifications and regulations may also play a role in the differential expression patterns between mRNAs and proteins 24. ITRAQ technique is a non‐gel‐based protein identification and quantification method for the absolute and relative quantification of proteins via isotope labeling that has a greater depth of proteome coverage. It has become one of the major quantification tools used in proteomics research 25, 26.
The molecular mechanism underlying the response of rice to high temperature stress is a relatively complex process and thus difficult to elucidate; however, it is possible to identify differentially expressed genes or proteins between heat‐tolerant and ‐sensitive varieties under high temperature stress. The potential candidate genes or proteins may then be utilized in elucidating the molecular mechanism underlying the response of rice to high temperature stress, as well as provide genetic resources for the improvement of heat‐tolerant characteristics in rice. For this, we employed the iTRAQ technique to screen for global proteomic changes in a rice heat‐tolerant line (HTL) XN0437T and a heat‐sensitive line (HSL) XN0437S under HNT stress at early milky stage, to identify proteins that contribute to HNT stress tolerance.
2. Materials and methods
2.1. Plant materials
Two rice lines, HTL XN0437T and HSL XN0437S, which were derived from RIL populations that were produced by crossing N22/Xieqingzao B//Xieqingzao B in our previous study, were used as plant materials 21. When the XN0437T and XN0437S lines were planted in the early season of double cropping rice in Nanchang City (Lat: 28°68N, Long: 115°85′E), Jiangxi Province, China, the respective average plant height was 81.3 cm and 81.1 cm, average fertile spikelet was 11.2 spikes and 11.1 spikes per plant, average ear length was 18.4 cm and 18.1 cm, total number of grains per panicle was 94.3 grains and 95 grains on average, 1000‐grain weight was 28.3 g and 27.9 g, and growth period was all 106 days. These lines had a genomic polymorphism rate of only 1.8% when 887 simple sequence repeat markers were used in screening the genomic DNA of both lines 27. However, when the HTL and HSL were exposed to HNT stress (38 ± 0.5°C for two dark periods), their grain plumpness during the grain ripening period were 93.1 and 73.9%, respectively, which indicated significant differences 21, 27.
2.2. Rice cultivation, temperature treatments, and sampling
Rice cultivation, temperature treatments, and sampling were conducted using a previously described method 21. Briefly, rice were cultured using a tub‐planting method and rice ears were labeled on the same heading and flowering date to ensure that only uniformly developed samples were used for iTRAQ analysis. On the 8th day after flowering of the labeled florets, both the HTL and HSL with the same date labels were transferred to chambers and maintained at a temperature of 38.0 ± 0.5°C (treatment) or 25.0 ± 0.5°C (control) for the dark period (10 h), and 26.0 ± 0.5°C (both treatment and control) for the light period (14 h). CO2 concentrations ranged from 367 ppm to 403 ppm, and the relative humidity of the chambers was set to 70 ± 5% and 80 ± 5% for daytime and nighttime, respectively. The temperature treatments were performed in duplicate. After two dark and two light treatment periods (48 h total), the plants were taken out of the chambers and placed in normal growth conditions until maturity. Meanwhile, florets with labels from the middle to the bottom region of the labeled ears were harvested, packed in aluminum foil, flash‐frozen in liquid nitrogen, and stored at –80°C until use. In total, we harvested eight samples of rice grains, which were designated as TC1 and TC2, and TT1 and TT2, which served as the two control and two treatment samples of the HTL, respectively, whereas SC1 and SC2, and ST1 and ST2 represented the two experimental groups of the HSL. After rice maturity, six plants from each treatment group and control were harvested to investigate the grain weight per plant and genotypic variation of granules.
2.3. Protein isolation
Approximately 1 g of each sample was ground in liquid nitrogen and resuspended in 5 mL of 10% w/v TCA in acetone with 0.07% w/v β‐mercaptoethanol at −20°C for 1 h, followed by centrifugation for 15 min at 35 000 g. To remove the sugars and pigments in the pellets, the pellets were resuspended in acetone with 0.07% w/v β‐mercaptoethanol and incubated at −20°C for 1 h, and then centrifuged at 35 000 g for 15 min at 4°C. The purification step was repeated three times, after which the pellets were lyophilized. The crude protein powder was dissolved in lysis buffer (8 M urea, 2 M thiourea, 4% CHAPS, 0.5% pH 3–10 ampholine, 50 mM DTT, and 1 mM PMSF) for 1 h at room temperature, followed by centrifugation at 15 000 g for 15 min. The supernatant was collected in a 1.5‐mL tube, and a 30‐μL aliquot was collected to determine the protein concentration using a Bradford assay with BSA as standard (Bio‐Rad Laboratory, Hercules, CA, USA). The total proteins of the samples were detected by SDS‐PAGE and CBB staining according to the previous method 28.
2.4. Protein digestion and iTRAQ labeling
The protein extract was digested according to the filter‐aided sample preparation procedure 29, and the resulting peptide mixture was labeled using the 8‐plex iTRAQ reagent (AB Sciex, Foster City, CA, USA) according to the manufacturer's instructions. Briefly, 200 μg of protein from each sample was mixed with 30 μL of STD buffer (4% SDS, 100 mM DTT, 150 mM Tris‐HCl pH 8.0). The detergent DTT and other low‐molecular‐weight components were removed using UA buffer (8 M Urea, 150 mM Tris‐HCl pH 8.0) by repeat ultrafiltration (Microcon units, 30 kD). Then, 100 μL of 0.05 M iodoacetamide in UA buffer was added to block the reduced cysteine residues, and the samples were incubated for 20 min in the dark. The filters were washed three times with 100 μL of UA buffer, and then twice with 100 μL of DS buffer (50 mM triethylammonium bicarbonate at pH 8.5). Finally, the protein suspensions were digested with 2 μg of trypsin (Promega, Madison, WI, USA) in 40 μL of DS buffer overnight at 37°C, and the resulting peptides were collected as filtrate. The peptides were then labeled using the iTRAQ reagents 8‐plex kit (AB Sciex, Foster City, CA, USA) according to the manufacturer's instructions, and the peptides from samples TC1, TC2, TT1, TT2, SC1, SC2, ST1, and ST2 were labeled with 113, 114, 115, 116, 117, 118, 119, and 121 tags, respectively. All labeled samples were multiplexed and vacuum dried.
2.5. SCX fractionation
The iTRAQ‐labeled peptides were fractionated by SCX chromatography using an AKTA Purifier system (GE Healthcare, London, UK). Fractionation was performed according to the manufacturer's instructions. Briefly, the dried peptide mixture was reconstituted and acidified with 2 mL of buffer A (10 mM KH2PO4 in 25% ACN, pH 2.7) and loaded onto a polysulfoethyl (5 μm, 200 Å, 4.6 × 100 mm) column (PolyLC Inc., Colombia, MD, USA). The peptides were eluted at a 1 mL/min flow rate with buffer B (500 mM KCl, 10 mM KH2PO4 in 25% ACN, pH 2.7) using a gradient of 0–10% for 2 min, 10–20% for 25 min, 20–45% for 5 min, and 50–100% for 5 min. To collect almost equivalent amounts of peptides from each pool, the elution was monitored based on A at a wavelength of 214 nm, and the fractions were collected in 1‐min intervals. The collected fractions were finally combined into 20 pools that comprised almost equivalent amounts of fractions according to the peak area at an A at a 214 nm wavelength and desalted on C18 cartridges (standard density).
2.6. ITRAQ nanoflow LC‐MS/MS analysis
From each fraction, 10 μg of the peptides was trapped on a precolumn (200 μm x 0.5 mm) and then eluted onto an analytical column (75 μm × 15 cm) for separation. Both columns were packed with Reprosil‐Pur (RP) C18‐AQ 3 μm 120 Å phase (Eksigent, Dublin, CA, USA). The reversed‐phase mobile phase A was 98% water, 2% ACN and 0.1% formic acid), whereas mobile phase B was 98% ACN, 2% H2O, and 0.1% formic acid. The peptides were separated over 90 min using a linear gradient of 12–30% of RP mobile B at a flow rate of 300 nL/min. MS analysis was performed using a 5600 TripleTOF analyzer (AB SCIEX, Boston, MA, USA) in information‐dependent mode. Precursor ions were selected across a mass range of 350–1250 m/z using a 250‐ms accumulation time per spectrum. A maximum of 30 precursors per cycle from each MS spectrum were selected for MS/MS analyses with 100 ms minimum accumulation time for each precursor and dynamic exclusion for 25 s. The MS/MS spectra were recorded in high sensitivity mode using rolling collision energy with the iTRAQ reagent collision energy adjustment on.
2.7. Protein identification and quantification
All MS/MS spectra were searched in the NCBInr Oryza sativa sequence databases and UniProtKB/Swiss‐Prot database using the MASCOT software (Matrix Science, UK; version 2.3.02). The search parameters were as follows: (1) allowed one missed cleavage in the protein trypsin digests; (2) fixed modifications of carbamidomethylation at cysteine, variable modifications of oxidation at methionine and iTRAQ 8‐plex at tyrosine; (3) peptide tolerance was set at ± 0.02 Da and MS/MS tolerance was set at ± 0.05 Da. The peptide charge was set as Mr, and the monoisotopic mass was chosen. The iTRAQ 8‐plex was chosen for quantification during the search. The search results were filtered before data exportation. The filters were used for protein identification with the following options: significance threshold of p < 0.05 (with 95% confidence) and ion score or expected cutoff of < 0.05 (with 95% confidence). For protein quantification, the filters were set as follows: (1) “median” was chosen for the protein ratio type; (2) the minimum precursor charge was set to 2, minimum peptides were set to 2, and only unique peptides were used for quantitation; and (3) normalization by median intensities, and outliers were removed automatically.
2.8. Criterion for differentially expressed proteins
To identify significant differentially expressed proteins (DEPs), we first determined the DEPs between the treatments and controls from the HTL and HSL, respectively (Fig. 1). The proteins with a fold‐change (FC) of > 1.5 (p < 0.05) or < 0.67 (p < 0.05) were considered upregulated and those with FC < 0.67 (p < 0.05) as downregulated between the treatments versus controls. Second, the relative fold change (RFC) for the upregulated and downregulated proteins between the HTL and HSL were further compared, and the proteins with an RFC > 2.0 or < 0.50 were respectively considered as significant upregulated or downregulated.
Figure 1.
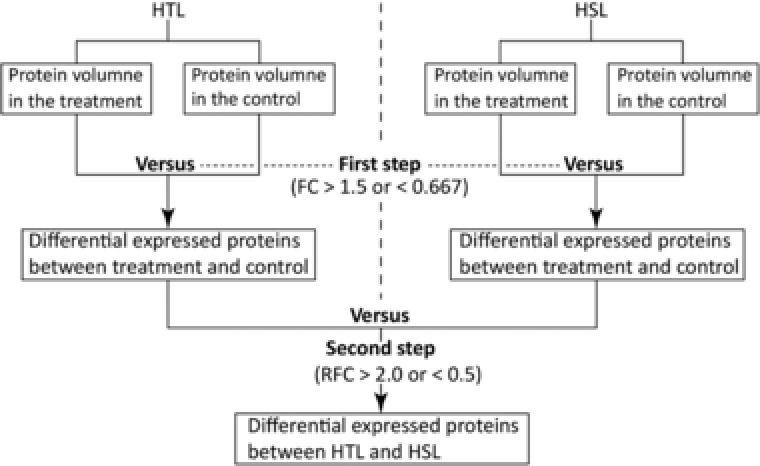
Workflow for the differential expressed proteins analysis. HTL indicated the heat‐tolerant line; HSL indicated the heat‐sensitive line. FC indicated the fold change of differential expressed proteins between the treatment and control; RFC indicated the relative fold change of the differential expressed proteins between the HTL and HSL.
2.9. Protein hierarchical cluster analysis
To assess the reproducibility of the MS data, cluster analysis on quantification of the identified proteins from the biological replicates was conducted using Ward's method, which is a hierarchical technique. After normalization of the quantification to log2 ratios of the identified proteins, a function heatmap from the R package g‐plots was used to in constructing a dendrogram 30. Pearson's correlation coefficient and scatter plots were used to estimate the relationships among the eight samples. Lowess was used to smooth the scatter plots using locally weighted regression. All the statistical analyses were performed in the R environment, using various CRAN packages
2.10. GO enrichment analysis
To compare differences in response between the HTL and HSL under HNT stress, GO enrichment (http://geneontology.org/) analysis was performed to annotate the DEPs under the categories of biological process (BP), molecular function (MF), and cellular component (CC) 31.
2.11. Validation by Western blotting
To validate the reliability of the iTRAQ data, we monitored the expression patterns of each sample by Western blotting. The BEPITOPE software 32 was used to predict unique antigenic fragments in the rice genome, and the PrimerCE software 33 was used to design the primers based on the coding sequence downloaded from the TIGR database (http://rice.plantbiology.msu.edu/). The polyclonal antibodies were generated by immunizing healthy rabbits with purified fusion proteins or synthesized peptides as antigens. Protein conjugations, immunizations, and antiserum purifications were performed by Beijing Protein Innovation Co., Ltd., Beijing, China. SDS‐PAGE and Western blotting were conducted as previously described 34. Briefly, equal amounts of protein samples isolated as described in Section 2.3 were separated using SDS‐PAGE and electrotransferred onto a PVDF membrane (Millipore Corporation, Bedford, MA, USA) at 100 V for 65 min. The membrane was immersed in 5% nonfat milk in a TTBS solution (0.2 M Tris‐HCl (pH 7.6), 1.37 M NaCl, 0.1% Tween‐20) at room temperature for 1 h. The proteins were incubated with the primary antibodies in 5% nonfat milk in TTBS at room temperature for 3 h and subjected to three successive 5‐min rinses in TTBS. The membrane was then incubated with a horseradish peroxidase‐conjugated goat anti‐rabbit antibody (Zhongshan Goldenbridge Biotechnology Co., Ltd, Beijing, China) for 1 h at room temperature, and subjected to three successive 5‐min rinses in TTBS. The blot was developed with a SuperECL Plus kit (Applygen, Beijing, China) according to the manufacturer's instructions, and the signal was exposed to an X‐ray film. The images were scanned and the intensity of each band was captured using ImageMaster 2D Platinum version 5.0 (Amersham Pharmacia Biotech, Uppsala, Sweden).
3. Results
3.1. Grain‐filling ratio, grain weight, and SDS‐PAGE
After application of short‐term HNT stress at early milky stage, the HSL brown rice showed significant shrinkage, whereas that of the HTL showed almost no morphological changes (Fig. 2A). The grain weight per plant of the HSL exhibited a significant decrease compared to that of HTL (Fig. 2B). These findings indicated that short‐term HNT stress imparted different effects on the HSL and HTL. Although we could not distinguish the differences between the total proteins of treatment and control groups based on SDS‐PAGE (Fig. 2C), we have determined that our total protein isolation was successful.
Figure 2.

Variation in granule genotype, grain weight, and total proteins SDS‐PAGE. CK and T indicate the control and treatment, respectively. In panel C, each lane of SDS‐PAGE was loaded 150 μg total proteins of sample.
3.2. Protein identification and quantification
After iTRAQ and LC‐MS/MS analysis, a total of 588 063 spectra were detected, and 352 916 spectra were identified, of which 14 497 spectra could be matched to peptides in the database and 5447 were unique peptides (Fig. 3A). In total, 3124 proteins were identified using the Oryza sativa UniProt database with a FDR < 0.01 as stated in the methods.
Figure 3.
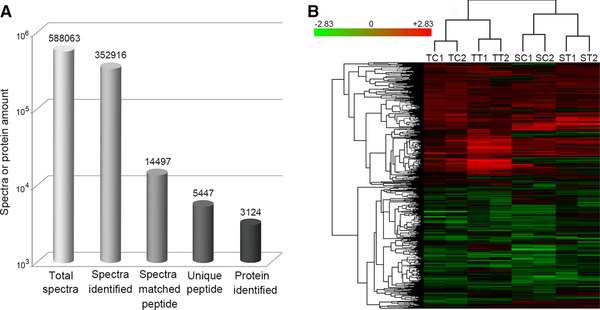
Results of LC‐MS/MS (A) and cluster analysis for the expressed proteins in the HSL and HTL (B). SC1 and SC2 indicate the duplicates of the controls; ST1 and ST2 indicate the duplicates of the treatments, in the HSL. TC1 and TC2 indicate the duplicates of the controls; TT1 and TT2 indicate the duplicates of the treatments, in the HTL.
To evaluate the reliability of the data generated from iTRAQ analysis, the reproducibility of the protein volume of the two biological replicates were compared by using Cluster 3.0, which indicated that the two samples could be clustered into the same class (Fig. 3B).
3.3. DEPs in the HTL and HSL
Proteins with an FC >1.5 (p < 0.05) between the treatment and control groups were considered upregulated, whereas those with an FC of < 0.67 (p < 0.05) were downregulated; these DEPs were hence considered as HNT responsive proteins. Based on these criteria, 136 and 42 proteins in the HTL (Fig. 4) and 104 and 45 in the HSL were respectively up‐ and downregulated. The two lines had 38 upregulated and 11 downregulated proteins in common.
Figure 4.
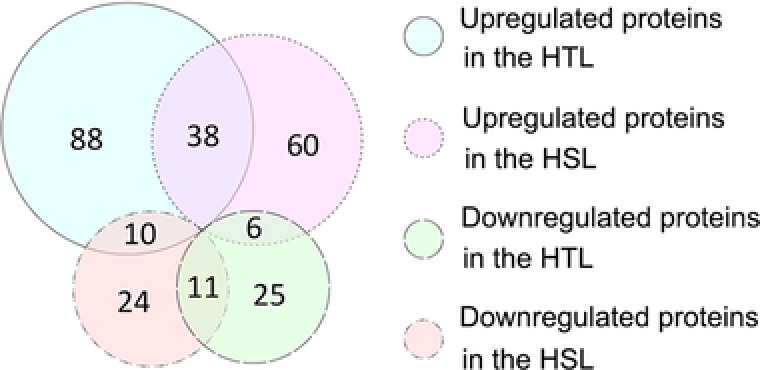
Venn diagram the up‐ and downregulated proteins between the HSL and HTL. HSL and HTL indicate the heat‐sensitive line and heat‐tolerant line, respectively.
3.4. Gene functional description and GO analysis of HNT responsive proteins
To annotate the function of the HNT response proteins, protein IDs were searched against the NCBI database (http://www.ncbi.nlm.nih.gov/) and/or the Uniprot database (http://www.uniprot.org/). Among the 178 HTL‐responsive proteins, 104 upregulated proteins and 40 downregulated proteins were annotated functions, and 32 upregulated proteins and two downregulated proteins were of unknown function. The summaries and expression patterns of the proteins are listed in Supporting Information Table 1. Among the 149 proteins in the HSL responding to HNT stress, 82 upregulated proteins and 36 downregulated proteins showed functional annotation, and 22 upregulated proteins and nine downregulated proteins were of unknown. The summaries and expression patterns of these proteins are presented in Supporting Information Table 2.
To determine the MF, BP, and CC categories of GO for the HNT response protein, we searched the GO database using their protein IDs 31. GO analysis showed that the HNT‐related DEPs in both rice lines were involved in ten subgroups of MF, eight subgroups of BP, and four subgroups of CC (Fig. 5). Compared to the HSL, the HTL proteins were involved in protein‐binding transcript factor activity, receptor activity, antioxidant activity, and structural molecular activity of MF, whereas the HSL proteins that were involved in apoptotic and development processes of BP were significantly affected by HNT.
Figure 5.

Gene Ontology enrichment analysis of the DEPs between the HSL and HTL. HSL and HTL indicate the heat‐sensitive line and heat‐tolerant line, respectively; DEPs indicate the differential expressed proteins.
3.5. DEPs
To identify the DEPs between the HTL and HSL, the HNT responsive proteins with an FC > 1.5 (p < 0.05) or < 0.67 (p < 0.05) between the treatment and control groups were selected for further calculation of the RFC between the two lines. RFC values of > 2.0‐fold or < 0.5‐fold (1/2.0) were acknowledged as DEPs between the HSL and HTL, as previously described in the Materials and methods section (Fig. 1). According to our criteria, a total of 38 proteins were differentially expressed between the HTL and HSL. Comparing to the HSL, 26 proteins were upregulated and 12 proteins were downregulated in the HTL. After searching against the NCBI and UniProt databases, 32 proteins were functional annotated, whereas six proteins had unknown functions. According to their functional description, DEPs were further classified into subgroups (Table 1, Fig. 6). Of the 38 DEPs, three proteins were involved in oxidation (7.89%), two proteins in signal transduction (5.26%), three proteins in defense response (7.89%), four proteins in transcript regulation (10.53%), seven proteins in transport (18.42%), one protein in energy metabolism (2.63%), 12 proteins in biosynthesis (31.58%), and another six were of unknown function (15.79%).
Table 1.
Functional description and FC for the HNT‐responsive DEPs between rice HTL and HSL
| Protein accession | Gene symbol | Functional description | FC | ||
|---|---|---|---|---|---|
| HTL | HSL | HTL/HSL | |||
| Signal transduction | |||||
| Q6ZJ47 | LOC4344535 | COP9 signalosome complex subunit 3 | 4.594 | 0.662 | 6.945 |
| Q6I587 | LOC4339751 | Calcium‐dependent protein kinase 3 | 3.007 | 1.057 | 2.845 |
| Transcript regulation | |||||
| Q67TK9 | B1012G11.2 | Zinc knuckle containing protein‐like | 3.559 | 1.095 | 3.252 |
| Q0JD64 | LOC4335858 | Transcription factor bHLH95 | 1.84 | 0.644 | 2.857 |
| Q5QMN3 | LOC9268869 | DEAD‐box ATP‐dependent RNA helicase 20 | 1.737 | 0.658 | 2.639 |
| Q2QYK4 | LOC4351322 | Elongation factor P | 1.396 | 0.659 | 2.119 |
| Oxidation | |||||
| Q6ZCP8 | LOC4344752 | Protein chlororespiratory reduction 6, chloroplastic | 3.049 | 0.661 | 4.612 |
| Q8GTK2 | LOC4344152 | Serine carboxypeptidase II‐3 | 2.746 | 1.097 | 2.502 |
| Q0E225 | LOC4329011 | Ent‐cassadiene C2‐hydroxylase‐like | 0.326 | 0.665 | 0.491 |
| Defense response | |||||
| Q9AUU6 | OSJNBa0040E01.6 | Putative uncharacterized protein | 1.623 | 0.662 | 2.452 |
| Q0DPX0 | LOC4333610 | Dehydrin HIRD11 | 1.689 | 0.65 | 2.6 |
| Q84Q72 | LOC4332361 | 18.1 kDa class I heat shock protein | 1.579 | 3.366 | 0.469 |
| Transport | |||||
| Q2R237 | LOC4350785 | Sec‐independent protein translocase protein TATB, chloroplastic | 5.125 | 1.199 | 4.275 |
| C7JA46 | LOC9266530 | Sucrose transport protein SUT2 | 2.573 | 0.651 | 3.954 |
| Q69XN2 | P0012B02.41 | Putative yip1 interacting factor | 1.495 | 0.445 | 3.359 |
| Q6ZCC9 | LOC4344489 | Uncharacterized acetyltransferase At3g50280 | 0.907 | 0.425 | 2.131 |
| Q2R1N0 | LOC9266321 | UDP‐glycosyltransferase 72B3 | 0.402 | 1.064 | 0.377 |
| Q5N9C8 | LOC4327685 | Trafficking protein particle complex subunit 5 | 0.439 | 1.567 | 0.280 |
| Q7XIM1 | LOC4342455 | Outer envelope protein 61 | 0.329 | 1.713 | 0.192 |
| Energy metabolism | |||||
| Q336M3 | LOC4349518 | ADP‐ribosylation factor | 0.664 | 1.528 | 0.435 |
| Biosynthesis | |||||
| Q7XQ85 | LOC4336750 | 1‐aminocyclopropane‐1‐carboxylate synthase | 1.042 | 0.326 | 3.197 |
| Q10KT9 | LOC4332935 | H/ACA ribonucleoprotein complex subunit 4 | 1.026 | 0.328 | 3.129 |
| Q0DW47 | LOC4331259 | Callose synthase 3 | 1.469 | 0.648 | 2.266 |
| Q7Y1N5 | OSJNBa0053G10.26 | Putative polyprotein | 1.375 | 0.653 | 2.106 |
| Q7Y007 | LOC4333736 | GDSL esterase/lipase At5g03820 | 2.767 | 1.367 | 2.025 |
| Q0E3F2 | LOC4328488 | FGGY carbohydrate kinase domain‐containing protein | 2.229 | 1.11 | 2.008 |
| Q6EPN6 | LOC4330320 | 2‐C‐methyl‐D‐erythritol 2,4‐cyclodiphosphate synthase, chloroplastic | 0.579 | 1.202 | 0.482 |
| Q94GQ6 | LOC4334098 | Late embryogenesis abundant protein D‐34 | 1.07 | 2.27 | 0.472 |
| C7IZW5 | LOC9272175 | Putative dihydroflavonol‐4‐reductase | 0.665 | 1.533 | 0.434 |
| Q688M5 | LOC4338719 | Chitinase 9 | 0.646 | 1.554 | 0.416 |
| Q9FVZ0 | OSJNBb0073N24.10 | Putative folylpolyglutamate synthetase | 0.427 | 1.096 | 0.39 |
| Q658I1 | LOC4339986 | 39S ribosomal protein L47, mitochondrial | 0.664 | 2.029 | 0.327 |
| Functional unknown protein | |||||
| Q53M16 | LOC_Os11g13690 | Putative uncharacterized protein | 2.272 | 0.524 | 4.337 |
| Q6Z7I3 | LOC4329203 | Os02g0329800 protein | 1.788 | 0.604 | 2.96 |
| Q851I5 | LOC107278586 | Putative uncharacterized protein | 1.852 | 0.648 | 2.859 |
| Q69MT6 | OSJNBb0034B12.21 | Putative uncharacterized protein | 2.948 | 1.158 | 2.546 |
| Q9LWS6 | LOC4339919 | Os06g0115100 protein | 2.349 | 0.997 | 2.356 |
| Q0JQP1 | Os01g0149200 | Os01g0149200 protein | 1.463 | 0.661 | 2.212 |
FC indicate fold change; DEPs indicate differentially expressed proteins; HTL indicate heat‐tolerant line; HSL indicate heat‐sensitive line; HNT indicate high night temperature.
Figure 6.

Subgroups and percentage of the differential expressed proteins (DEPs).
Among the two DEPs involved in signal transduction (Table 1), the proteins of COP9 signalosome complex subunit 3 (Q6ZJ47) and calcium‐dependent protein kinase 3 (Q6I587) were upregulated by more than threefold in the HTL, whereas none of these proteins were altered in the HSL. For the transcript regulation related‐proteins, zinc knuckle containing protein‐like (Q67TK9), transcription factor bHLH95 (Q0JD64), DEAD‐box ATP‐dependent RNA helicase 20 (Q5QMN3), and elongation factor P (Q2QYK4) were upregulated more than 2.1‐fold in the HTL compared to that in the HSL. Interestingly, the six proteins of unknown functions were upregulated almost 1.5‐fold in the HTL, whereas downregulated 1.5‐fold (including Q53M16, Q6Z7I3, Q851I5, and Q0JQP1) or not altered (including Q69MT6 and Q9LWS6) in the HSL.
3.6. Western blotting validation
To monitor the expression pattern of the proteins identified by iTRAQ technology, six representative proteins, including Ent‐cassadiene C2‐hydroxylase‐like protein (Q0E225, LOC4329011), glutelin‐type‐B 1 (P14323, LOC4328884), UDP‐glycosyltransferase 72B3 (Q2R1N0, LOC9266321), COP9 signalosome complex subunit 3 (Q6ZJ47, LOC4344535), transcription factor bHLH95 (Q0JD64, LOC4335858), and 18.1‐kDa class I HSP (Q84Q72, LOC4332361) were selected for Western blotting analysis. The generated specific antigens for the six representative proteins were showed in Supporting Information Table 3. Compared to the expression patterns of the six target proteins analyzed by iTRAQ and Western blotting technologies, the proteins identified by iTRAQ technology and Western blotting analyses showed almost the same expression patterns (Fig. 7), further verifying the reliability of the iTRAQ technology.
Figure 7.
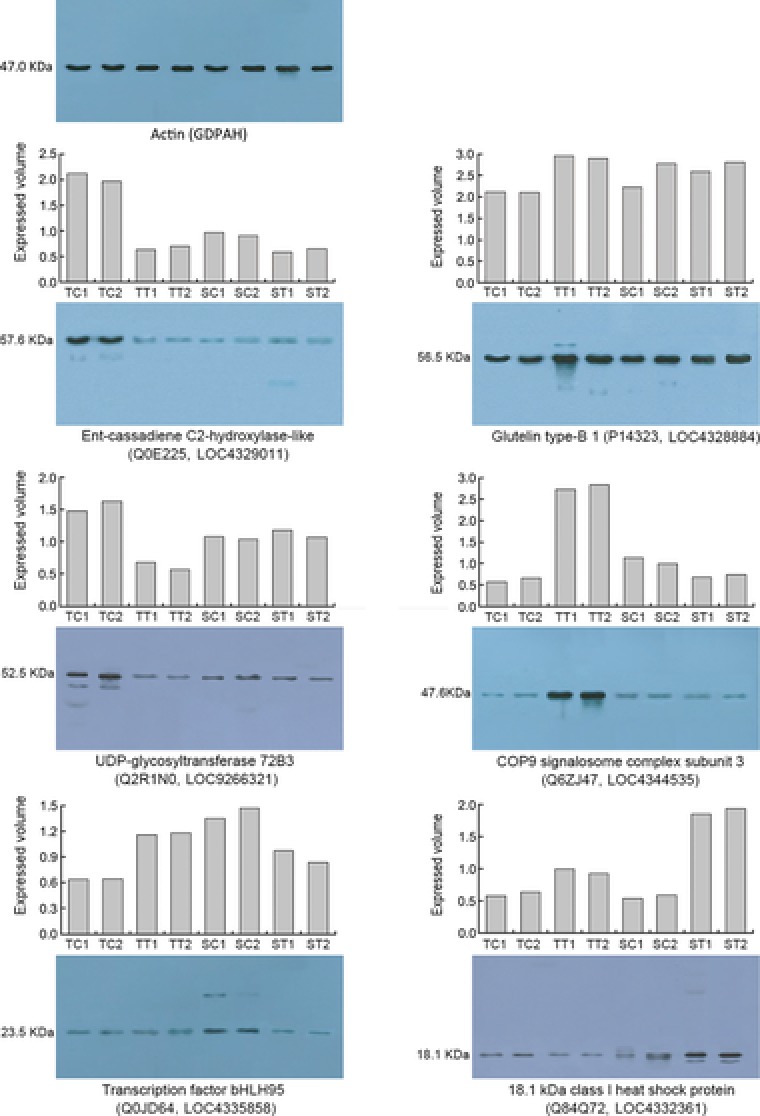
Expression patterns of six selected proteins based on iTRAQ analysis (histogram) and Western blotting analyses (gel map). SC1 and SC2 indicate the duplicates of the controls; ST1 and ST2 indicate the duplicates of the treatments, in the HSL. TC1 and TC2 indicate the duplicates of the controls; TT1 and TT2 indicate the duplicates of the treatments, in the HTL.
4. Discussion
4.1. Summary
The iTRAQ technology in combination with LC‐MS/MS is an effective method for investigating altered protein levels in plant cells during environmental stress 25. In the present study, the iTRAQ technique with 8‐plex DiLeu isobaric labels was used in the quantitative proteomics analysis of both lines in response to HNT stress and to identify DEPs between the HTL and HSL. By comparing the relative expression pattern (treatment versus control) of the proteins in the HSL, a total of 38 DEPs were identified. Of these 38 DEPs, 26 proteins were upregulated and 12 proteins were downregulated. Further bioinformatics analysis showed that only 32 DEPs could be functionally annotated, and these were further classified into seven subgroups, including oxidation, signal transduction, defense response, transcript regulation, transport, energy metabolism, and biosynthesis.
4.2. Signal transduction proteins triggered early in the HTL
The COP9 signalosome is a conserved protein complex that is involved in regulating the activity of cullin‐RING ligase families of ubiquitin E3 complexes in the ubiquitin‐proteasome system 35. The COP9 signalosome has been reported as a key player in DNA‐damage response, cell‐cycle control, and gene expression 36 and is also reported to sustain postembryonic meristem function in Arabidopsis 37. A previous study showed that HNT is mainly influenced in endosperm development including endosperm cell number and size 11, and in present study, a COP9 signalosome complex subunit 3 (Q6ZJ47) showed a 4.6‐fold upregulation in the HTL and was downregulated 1.5 (1/0.667)‐fold in the HSL after HNT stress at early milky stage.
Ca2+ ions, as a major secondary messenger, play a vital role and are triggered immediately after a plant encounters stress 38. To transmit the calcium ion flux signal, cells employ calcium‐binding proteins such as calmodulins, and CaM‐like proteins, as well as the Ca2+‐dependent protein kinases, which are characterized by an EF‐hand domain with a helix‐loop‐helix motif and are further phosphorylated and in turn, activate other proteins 39. In the present study, calcium‐dependent protein kinase 3 (Q6I587) was upregulated more than 3.0‐fold in the HTL compared to no‐fold change in the HSL, which suggests that temperature stress signals delivered by Ca2+ ions were received early by the HTL.
In summary, the HNT stress signals delivered by COP9 signalosome complex subunit 3 and calcium‐dependent protein kinase 3 were received early by the HTL, which is beneficial to rice responses to HNT stress and may sustain the normal process of endosperm cell division and growth of rice grains.
4.3. Transcript regulation proteins were upregulated in the HTL and restrained in the HSL
Zinc finger proteins (ZFPs) have a wide range of functions as cofactors of over 300 enzymes and proteins involved in DNA or RNA binding by site‐specific modification and/or regulation of chromatin to regulate transcription, RNA metabolism, protein–protein interactions, and other cellular functions that probably require specific protein contacts of the ZFP domain 40, 41, 42. ZFP families comprise different members that are involved in developmental processes and stress response. For example, a RING ZFP OsR‐ZFP34 regulates somata opening when rice is exposed to high temperature stress 43, and a C2H2‐type ZFP36 regulates the levels of superoxide dismutase and ascorbate peroxidase in plant cells when rice is exposed to abscisic acid and hydrogen peroxide stresses 42. In the present study, the zinc knuckle containing protein (Q67TK9) was upregulated more than 3.5‐fold in the HTL, whereas a no‐fold change was observed in the HSL, thereby suggesting that gene transcription in the HTL is regulated by ZFPs.
Basic helix‐loop‐helix (bHLH) transcription factors exist in large families in plants, and contain two functionally distinct regions, the bHLH domain and the leucine zipper domain. The bHLH domain contains two amphipathic alpha helices with a linking loop of variable length, and functions as a DNA‐binding motif at the N‐terminal side of the leucine zipper domain. The leucine zipper domain functions as a protein–protein interaction motif. Transcription factor bHLHs in rice have been characterized in relation to stress response 44, 45 and is also reported to regulate its own development, grain size, and grain number 46. In present study, we reported that a bHLH95 is involved in the response to HNT stress in rice grains, and under HNT stress, is upregulated over 1.8‐fold in the HTL, whereas downregulated in the HSL.
DEAD‐box proteins comprise a large protein family and are involved in almost all nucleic acid metabolic activities, including replication, repair, recombination, transcription, translation, ribosome biogenesis, and splicing 47, 48. It has been reported that the DEAD‐box proteins play important roles in plant growth and development, as well as abiotic stress response 49, 50, 51. In the present study, we reported that DEAD‐box ATP‐dependent RNA helicase 20 is involved in rice response to HNT stress, and RNA helicase 20 was upregulated by over 1.7‐fold in the HTL and downregulated by 1.5‐fold (1/0.658) in the HSL, compared to the controls, respectively.
According to the expression patterns of zinc knuckle containing protein, bHLH transcription factors, and DEAD‐box ATP‐dependent RNA helicase 20, we suggest that the genes involved in HNT response are triggered early, which in turn restores the intracellular environment (such as ROS levels) to its normal conditions, thereby minimizing loss of grain plumpness in the HTL.
4.4. Proteins involved in oxidation
Oxidative bursts induced by environmental stress trigger disturbances in the cellular redox balance, which are highly toxic and include oxidative modification of cellular macromolecules, inhibition of protein function, and promotion of cell death 52. For their survival, plants have evolved complex regulatory mechanisms to prevent cellular injury by regulating the steady‐state levels of ROS 53. In the present study, two proteins, chlororespiratory reduction 6 (Q6ZCP8) and serine carboxypeptidase II‐3 (Q8GTK2), showed more than 2.7‐fold upregulation in the HTL. This finding suggests that genes are triggered to regulate the levels of ROS in plant cells of the HTL to prevent cellular injury in the HTL.
4.5. Proposed model for signal transduction and gene transcript regulation of rice in response to HNT
Based on the functions and expression patterns of the proteins involved in signal transduction and gene transcript regulation, we propose a model that depicts how these proteins regulate heat‐tolerant characteristics in rice (Fig. 8). Initially, heat shock triggers oxidation, which leads to an imbalance in oxidation in plant cells. The unbalanced signals are transmitted to Ca2+ ions, which are then released into the cytoplasm and bind to calcium‐dependent protein kinases, thereby triggering the expression of oxidation‐responsive genes. Furthermore, the imbalance in oxidation is also detected by the COP9 signalosome, which then triggers the upregulation of the bHLH transcription factor and accelerates the protein degradation of the ubiquitin‐proteasome system, ultimately triggering the expression of target genes. The heat shock signals are also relayed and activate the ZFP, which in turn triggers the expression of its target genes. The target genes also trigger or regulate various metabolic pathways, which consequently influence the development of heat‐tolerant characteristics in rice.
Figure 8.
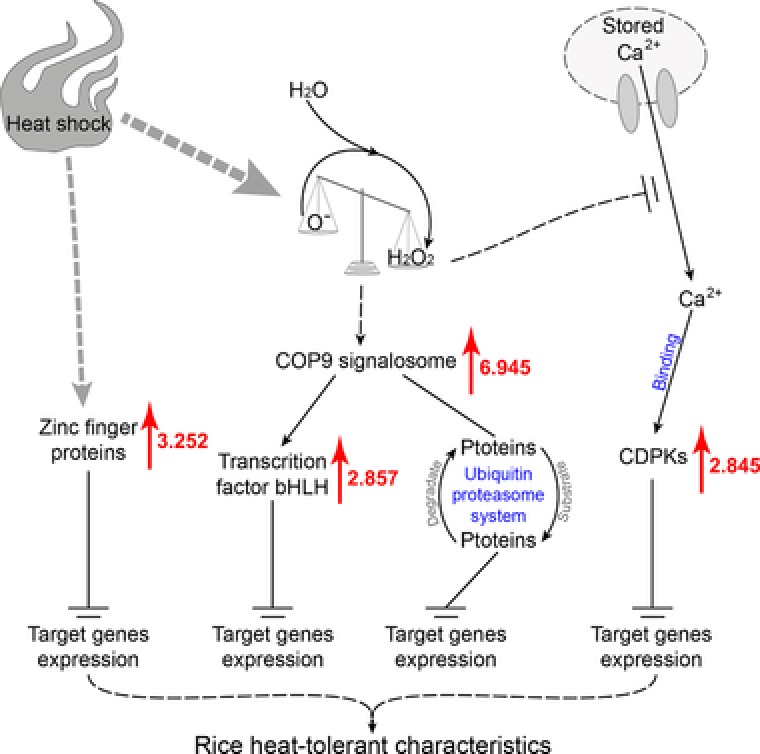
The proposed model of the DEPs involved in signal transduction and regulation of gene transcripts. DEPs indicate the differential expressed proteins; CDPKs indicate calcium‐dependent protein kinases. Red arrows and the number next to each red arrow is the fold change in upregulation.
4.6. Conclusions
In summary, comparative proteomics analysis using iTRAQ LC‐MS/MS technologies identified 38 DEPs between HTL and HSL under HNT stress. Further bioinformatics analysis showed that the DEPs are involved in oxidation, signal transduction, defense response, transcript regulation, transport, energy metabolism, and biosynthesis. The signal transduction proteins COP9 signalosome complex subunit 3, calcium‐dependent protein kinase 3, transcript regulation proteins ZFPs, and bHLH transcription factors were upregulated in the HTL compared to that in the HSL, thereby suggesting that these have major regulation functions in the heat‐tolerant characteristics of rice. Although protein function analysis is needed to further understand the roles on symptom formation, the potential candidate proteins may provide a starting point in the elucidation of the molecular mechanism underlying HNT tolerant characteristics and also provide potential genetic resources for the improvement of heat‐tolerant characteristics in rice.
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Functional description and protein expression patterns in the HTL
Functional description and protein expression patterns in the HSL
Table S3. Proteins and its antigenic peptides validated by western blotting
Acknowledgements
The National Natural Science Foundation of China (Grant nos. 31260315 and 31471467) and the Natural Science Foundation of Jiangxi Province (Grant nos. 20133ACB21004 and 20161ACB20021) supported this study.
Zhang H.‐Y., Lei G., Zhou H.‐W., He C., Liao J.‐L., Huang Y.‐J., Proteomics 2017, 1600365.
Colour Online: See the article online to view Figs. 4–8 in colour.
Contributor Information
Jiang‐Lin Liao, Email: jlliao514815@163.com.
Ying‐Jin Huang, Email: yjhuang_cn@126.com.
5 References
- 1. Smith, P. , Gregory, P. J. , Climate change and sustainable food production. Proc. Nutr. Soc. 2013, 72, 21–28. [DOI] [PubMed] [Google Scholar]
- 2. Wheeler, T. , von Braun, J. , Climate change impacts on global food security. Science 2013, 341, 508–513. [DOI] [PubMed] [Google Scholar]
- 3. Kim, J. , Shon, J. , Lee, C. , Yang, W. et al., Relationship between grain filling duration and leaf senescence of temperate rice under high temperature. Field Crop Res. 2011, 122, 207–213. [Google Scholar]
- 4. Takai, T. , Matsuura, S. , Nishio, T. , Ohsumi, A. et al., Rice yield potential is closely related to crop growth rate during late reproductive period. Field Crop Res. 2006, 96, 328–335. [Google Scholar]
- 5. Morita, S. , Yonemaru, J. , Takanashi, J. , Grain growth and endosperm cell size under high night temperatures in rice (Oryza sativa L.). Ann. Bot. 2005, 95, 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li, H. , Chen, Z. , Hu, M. , Wang, Z. et al., Different effects of night versus day high temperature on rice quality and accumulation profiling of rice grain proteins during grain filling. Plant Cell Rep. 2011, 30, 1641–1659. [DOI] [PubMed] [Google Scholar]
- 7. Peng, S. , Huang, J. , Sheehy, J. E. , Laza, R. C. et al., Rice yields decline with higher night temperature from global warming. Proc. Natl. Acad. Sci. USA 2004, 101, 9971–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cao, Y. , Duan, H. , Yang, L. , Wang, Z. et al., Effect of high temperature during heading and early filling on grain yield and physiological characteristics in indica rice. Acta Agronomica Sinica 2009, 35, 512–521. [Google Scholar]
- 9. Fu, G. , Song, J. , Xiong, J. , Liao, X. et al., Thermal resistance of common rice maintainer and restorer lines to high temperature during flowering and early grain filling stages. Rice Sci. 2012, 19, 309–314. [Google Scholar]
- 10. Ahmed, N. , Tetlow, I. J. , Nawaz, S. , Iqbal, A. et al., Effect of high temperature on grain filling period, yield, amylose content and activity of starch biosynthesis enzymes in endosperm of basmati rice. J. Sci. Food Agric. 2015, 95, 2237–2243. [DOI] [PubMed] [Google Scholar]
- 11. Jagadish, S. V. , Craufurd, P. Q. , Wheeler, T. R. , High temperature stress and spikelet fertility in rice (Oryza sativa L.). J. Exp. Bot. 2007, 58, 1627–1635. [DOI] [PubMed] [Google Scholar]
- 12. Jagadish, S. V. , Murty, M. V. , Quick, W. P. , Rice responses to rising temperatures—challenges, perspectives and future directions. Plant Cell Environ. 2015, 38, 1686–1698. [DOI] [PubMed] [Google Scholar]
- 13. IPCC (2013) Working group I contribution to the IPCC fifth assessment report on climate change 2013: The Physical Science Basis, Summary for Policymakers.
- 14. Cao, Z. , Pan, G. , Wang, F. , Wei, K. et al., Effect of high temperature on the expressions of genes encoding starch synthesis enzymes in developing rice endosperms. J. Integr. Agr. 2015, 14, 642–659. [Google Scholar]
- 15. Jiang, H. , Dian, W. , Wu, P. , Effect of high temperature on fine structure of amylopectin in rice endosperm by reducing the activity of the starch branching enzyme. Phytochemistry 2003, 63, 53–59. [DOI] [PubMed] [Google Scholar]
- 16. Hakata, M. , Kuroda, M. , Miyashita, T. , Yamaguchi, T. et al., Suppression of alpha‐amylase genes improves quality of rice grain ripened under high temperature. Plant Biotechnol. J. 2012, 10, 1110–1117. [DOI] [PubMed] [Google Scholar]
- 17. Wei, K. , Zhang, Q. , Cheng, F. , Zhong, L. , Chen, N. , Expression profiles of rice soluble starch synthase Isoform genes in response to high temperature. Acta. Agronomica Sinica 2009, 35, 18–24. [Google Scholar]
- 18. Murata, K. , Iyama, Y. , Yamaguchi, T. , Ozaki, H. et al., Identification of a novel gene (Apq1) from the indica rice cultivar ‘Habataki’ that improves the quality of grains produced under high temperature stress. Breed Sci. 2014, 64, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liao, J. L. , Zhou, H. W. , Zhang, H. Y. , Zhong, P. A. , Huang, Y. J. , Comparative proteomic analysis of differentially expressed proteins in the early milky stage of rice grains during high temperature stress. J. Exp. Bot. 2014, 65, 655–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glaubitz, U. , Erban, A. , Kopka, J. , Hincha, D. K. , Zuther, E. , High night temperature strongly impacts TCA cycle, amino acid and polyamine biosynthetic pathways in rice in a sensitivity‐dependent manner. J. Exp. Bot. 2015, 66, 6385–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao, J. L. , Zhou, H. W. , Peng, Q. , Zhong, P. A. et al., Transcriptome changes in rice (Oryza sativa L.) in response to high night temperature stress at the early milky stage. BMC Genomics 2015, 16: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin, S. K. , Chang, M. C. , Tsai, Y. G. , Lur, H. S. , Proteomic analysis of the expression of proteins related to rice quality during caryopsis development and the effect of high temperature on expression. Proteomics 2005, 5, 2140–2156. [DOI] [PubMed] [Google Scholar]
- 23. Timabud, T. , Yin, X. , Pongdontri, P. , Komatsu, S. , Gel‐free/label‐free proteomic analysis of developing rice grains under heat stress. J. Proteomics 2016, 133, 1–19. [DOI] [PubMed] [Google Scholar]
- 24. Pradet‐Balade, B. , Boulme, F. , Beug, H. , Mullner, E. W. , Garcia‐Sanz, J. A. , Translation control: bridging the gap between genomics and proteomics? Trends Biochem. Sci. 2001, 26, 225–229. [DOI] [PubMed] [Google Scholar]
- 25. Frost, D. C. , Greer, T. , Xiang, F. , Liang, Z. , Li, L. , Development and characterization of novel 8‐plex DiLeu isobaric labels for quantitative proteomics and peptidomics. Rapid Commun. Mass Spectrom. 2015, 29, 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neilson, K. A. , Mariani, M. , Haynes, P. A. , Quantitative proteomic analysis of cold‐responsive proteins in rice. Proteomics 2011, 11, 1696–1706. [DOI] [PubMed] [Google Scholar]
- 27. Liao, J. , Zhang, H. , Shao, X. , Zhong, P. , Huang, Y. , Identification for heat tolerance in backcross recombinant lines and screening of backcross introgression lines with heat tolerance at milky stage in rice. Rice Sci. 2011, 18, 279–286. [Google Scholar]
- 28. Liao, J. , Huang, Y. , Evaluation of protocols used in 2‐D electrophoresis for proteome analysis of young rice caryopsis. Genomics Proteomics Bioinformatics 2011, 9, 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wisniewski, J. R. , Zougman, A. , Nagaraj, N. , Mann, M. , Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- 30. Gentleman, R. C. , Carey, V. J. , Bates, D. M. , Bolstad, B. et al., Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mi, H. , Muruganujan, A. , Casagrande, J. T. , Thomas, P. D. , Large‐scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Odorico, M. , Pellequer, J. L. , BEPITOPE: predicting the location of continuous epitopes and patterns in proteins. J. Mol. Recognit. 2003, 16, 20–22. [DOI] [PubMed] [Google Scholar]
- 33. Cao, Y. , Sun, J. , Zhu, J. , Li, L. , Liu, G. , PrimerCE: designing primers for cloning and gene expression. Mol. Biotechnol. 2010, 46, 113–117. [DOI] [PubMed] [Google Scholar]
- 34. Li, X. , Bai, H. , Wang, X. , Li, L. et al., Identification and validation of rice reference proteins for Western blotting. J. Exp. Bot. 2011, 62, 4763–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wei, N. , Deng, X. W. , The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 2003, 19, 261–286. [DOI] [PubMed] [Google Scholar]
- 36. Fuzesi‐Levi, M. G. , Ben‐Nissan, G. , Bianchi, E. , Zhou, H. et al., Dynamic regulation of the COP9 signalosome in response to DNA damage. Mol. Cell Biol. 2014, 34, 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franciosini, A. , Moubayidin, L. , Du K, Matari, N. H. et al., The cop9 signalosome is required for postembryonic meristem maintenance in arabidopsis thaliana. Mol. Plant 2015, 8, 1623–1634. [DOI] [PubMed] [Google Scholar]
- 38. Raorane, M. L. , Mutte, S. K. , Varadarajan, A. R. , Pabuayon, I. M. , Kohli, A. , Protein SUMOylation and plant abiotic stress signaling: in silico case study of rice RLKs, heat‐shock and Ca(2+)‐binding proteins. Plant Cell Rep. 2013, 32, 1053–1065. [DOI] [PubMed] [Google Scholar]
- 39. DeFalco, T. A. , Bender, K. W. , Snedden, W. A. , Breaking the code: Ca2+ sensors in plant signalling. Biochem. J. 2010, 425, 27–40. [DOI] [PubMed] [Google Scholar]
- 40. Wang, W. , Liu, B. , Xu, M. , Jamil, M. , Wang, G. , ABA‐induced CCCH tandem zinc finger protein OsC3H47 decreases ABA sensitivity and promotes drought tolerance in Oryza sativa . Biochem. Biophys. Res. Commun. 2015, 464, 33–37. [DOI] [PubMed] [Google Scholar]
- 41. Zhang, Y. , Lan, H. , Shao, Q. , Wang, R. et al., An A20/AN1‐type zinc finger protein modulates gibberellins and abscisic acid contents and increases sensitivity to abiotic stress in rice (Oryza sativa). J. Exp. Bot. 2016, 67, 315–326. [DOI] [PubMed] [Google Scholar]
- 42. Zhang, H. , Liu, Y. , Wen, F. , Yao, D. et al., A novel rice C2H2‐type zinc finger protein, ZFP36, is a key player involved in abscisic acid‐induced antioxidant defence and oxidative stress tolerance in rice. J. Exp. Bot. 2014, 65, 5795–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hsu, K. H. , Liu, C. C. , Wu, S. J. , Kuo, Y. Y. et al., Expression of a gene encoding a rice RING zinc‐finger protein, OsRZFP34, enhances stomata opening. Plant Mol. Biol. 2014, 86, 125–137. [DOI] [PubMed] [Google Scholar]
- 44. Kiribuchi, K. , Jikumaru, Y. , Kaku, H. , Minami, E. et al., Involvement of the basic helix‐loop‐helix transcription factor RERJ1 in wounding and drought stress responses in rice plants. Biosci. Biotechnol. Biochem. 2005, 69, 1042–1044. [DOI] [PubMed] [Google Scholar]
- 45. Seo, J. S. , Joo, J. , Kim, M. J. , Kim, Y. K. et al., OsbHLH148, a basic helix‐loop‐helix protein, interacts with OsJAZ proteins in a jasmonate signaling pathway leading to drought tolerance in rice. Plant J. 2011, 65, 907–921. [DOI] [PubMed] [Google Scholar]
- 46. Luo, J. , Liu, H. , Zhou, T. , Gu, B. et al., An‐1 encodes a basic helix‐loop‐helix protein that regulates awn development, grain size, and grain number in rice. Plant Cell 2013, 25, 3360–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanner, N. K. , Linder, P. , DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell 2001, 8, 251–262. [DOI] [PubMed] [Google Scholar]
- 48. Tuteja, N. , Tuteja, R. , Unraveling DNA helicases. Motif, structure, mechanism and function. Eur. J. Biochem. 2004, 271, 1849–1863. [DOI] [PubMed] [Google Scholar]
- 49. Wang, D. , Qin, B. , Li, X. , Tang, D. et al., Nucleolar DEAD‐box RNA helicase TOGR1 regulates thermotolerant growth as a pre‐rRNA chaperone in rice. Plos Genet. 2016, 12, e1005844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Macovei, A. , Vaid, N. , Tula, S. , Tuteja, N. , A new DEAD‐box helicase ATP‐binding protein (OsABP) from rice is responsive to abiotic stress. Plant Signal Behav. 2012, 7, 1138–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu, J. , Liu, C. , Li, M. , Hu, J. et al., A rice DEAD‐box RNA helicase protein, OsRH17, suppresses 16S ribosomal RNA maturation in Escherichia coli . Gene 2015, 555, 318–328. [DOI] [PubMed] [Google Scholar]
- 52. Circu, M. L. , Aw, T. Y. , Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen, R. , Zhao, X. , Shao, Z. , Wei, Z. et al., Rice UDP‐glucose pyrophosphorylase1 is essential for pollen callose deposition and its cosuppression results in a new type of thermosensitive genic male sterility. Plant Cell 2007, 19, 847–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Functional description and protein expression patterns in the HTL
Functional description and protein expression patterns in the HSL
Table S3. Proteins and its antigenic peptides validated by western blotting