

Abstract
Activating autoantibodies (AAb) to β-adrenergic receptors (βAR) are associated with atrial fibrillation in patients with Graves’ disease. In the present study, we examined the interaction of thyroid hormone with β1/2AR-AAb in inducing atrial tachyarrhythmias in the rabbit. Immunization of rabbits with a β1AR or β2AR second extracellular loop peptide produced high titers of β1AR-AAb or β2AR-AAb. Thyroid hormone in combination with β1AR-AAb or β2AR-AAb induced a significant number of sustained sinus tachycardia and atrial tachycardia, respectively. Both combinations resulted in significantly increased inductions of sustained arrhythmias compared to AAb alone. Thyroid hormone alone induced sustained sinus and junctional tachycardia. Sera from immunized rabbits specifically bound to and activated β1AR or β2AR in transfected cells in vitro. This study demonstrates thyroid hormone qualitatively accentuates the specific arrhythmogenic action of these AAb and quantitatively enhances their rate. Our data support a dual role of AAb and thyroid hormone in Graves’-associated tachyarrhythmias.
Keywords: Atrial tachyarrhythmias, Activating autoantibodies, β-adrenergic receptors, Thyroid hormone, Rabbit
Introduction
Hyperthyroidism is associated with atrial tachyarrhythmias and an increased incidence of atrial fibrillation (AF) [1–3]. The prevalence of AF in hyperthyroidism increases in a stepwise fashion to reach greater than 20 % in patients older than 70 years. The causation of AF in hyperthyroidism is complex and incompletely understood. One proposed mechanism is shortening of the action potential duration in the atrial myocardium from excess thyroid hormone facilitating formation of multiple circuit reentry [4, 5]. Graves’ disease, the most common cause of hyperthyroidism, is an autoimmune disorder caused by thyroid-stimulating autoantibodies [6, 7].
There is now emerging evidence supporting a pathophysiological role for receptor-activating autoantibodies in cardiac arrhythmias [8, 9]. Recent studies from our group have shown an association between the presence of activating autoantibodies (AAb) for the β-adrenergic and M2 cholinergic receptors in patients with Graves’ disease and concurrent AF. We found that in hyperthyroid patients, the frequency of these AAb differed significantly between those with AF and sinus rhythm, and the co-presence of these AAb was the strongest predictor of AF [10]. To determine the electrophysiological effects of individual AAb in an animal model, we immunized rabbits with a receptor-specific peptide to induce expression of AAb to the β2-adrenergic receptor (β2AR) [11]. Using each rabbit as its own control, after markedly raising the titer for β2AR-AAb, there was a significant increase in the induction of sustained atrial tachycardia compared to the pre-immune control state. On the other hand, using the same approach for expressing AAb to the β1-adrenergic receptor (β1AR) in another group of rabbits, the predominant atrial tachyarrhythmia induced was sustained sinus tachycardia [12]. These data support the concept of receptor specificity for the site of origin of certain types of atrial tachyarrhythmias associated with AAb.
In the present study, we used this same format in those rabbits previously immunized with peptides for expressing either β1AR-AAb or β2AR-AAb whose treatment was then combined with increased thyroxine (T4) treatment to determine their combined impact on inducibility for cardiac arrhythmia. In a separate group of rabbits, the arrhythmogenic effects of T4 alone were tested.
Methods
Animal Immunization and Thyroxine Treatment
Nine New Zealand white rabbits (average weight 3 kg) were immunized with 1 mg of the highly conserved second extra-cellular loop (ECL2 ) peptide for β 1AR (HWWRAESDEARRCYNDPKCCDFVTNR) (n=5) or β2AR (HWYRATHQEAINCYANETCCDFFTNQ) (n=4) in 0.5 ml of complete Freund’s adjuvant. The animals were boosted with the same peptide plus incomplete Freund’s adjuvant (1 mg/0.5 ml) at 2 and 4 weeks. These animals were a subgroup taken from our previous studies [11, 12] for examination of the effect of superimposed thyrotoxicosis. At 6 weeks, the rabbits were injected with T4 (50 μg/kg/day) for 2 weeks. This dosage had previously been demonstrated to produce overt hyperthyroidism in this rabbit model with suppression of thyroid-stimulating hormone (TSH) and elevation of free triiodothyronine (T3) and T4. Four additional rabbits were treated with T4 alone. Pre- and post-immune sera were obtained from all animals for ELISA and activity assays of the expected antibodies generated during immunization.
In Vivo Catheter Electrophysiological Study
Each animal was anesthetized with ketamine/xylazine (35 mg/5 mg/kg) and subjected to a catheter-based electrophysiological study. Standard electrocardiograms (leads 1-aVF) were continuously monitored. After shaving the neck area and application of Betadine antiseptic, the right jugular vein was dissected and cannulated with a 4-French multi-electrode catheter. Under electrographic control, the catheter was passed into the right atrium to record atrial potentials in conjunction with the standard 6-lead ECG. Atrial tachyarrhythmia susceptibility was tested by bursts of stimuli (3–5 s duration) at a high frequency (20 Hz) and voltages that were at least twice the diastolic pacing threshold before and after the infusion of acetylcholine (ACh) in three incremental concentrations (10 μM, 100 μM, and 1 mM) at a rate of 1 ml/min. Non-sustained (<10 s) and sustained (≥10 s) arrhythmia occurrence was determined in response to burst pacing at 2× diastolic threshold, at baseline, and then with each of the three concentrations of ACh infusion for 2 min before initiating burst pacing. The number of burst pacings ranged from 3 to 10. In the pre-immune state, the number of bursts were most likely to be closer to 10 because it was more difficult to induce any non-sustained or sustained arrhythmia with or without ACh infusions, whereas after immunization, particularly with ACh infusion, 2–3 burst pacing events readily induced either non-sustained or sustained arrhythmias. When this study was completed, the wound was closed and antibiotic treatment was instituted. A second and third electrophysiological study was performed after the 6-week immunization and 2-week T4 treatment intervals. The various types of arrhythmias induced in the rabbit heart have been detailed previously [11, 12] and are defined as follows:
Non-sustained arrhythmia: any arrhythmia lasting <10 s.
Sustained arrhythmia: any arrhythmia lasting ≥10 s.
Sinus tachycardia: a regular, rapid heart rate ≥250 beats/min showing 1:1 atrioventricular (AV) conduction arising from the sinus node (upright P waves in leads II, III, and aVF) with the earliest atrial electrogram occurring ≥ 10 ms prior to the onset of the P wave.
Atrial tachycardia: a regular, rapid heart rate ≥250 beats/min showing 1:1 or 2:1 AV conduction with P wave morphology different from the sinus P wave with electrograms occurring during the P wave.
Junctional tachycardia: a regular, rapid heart rate ≥250 beats/min showing 1:1 AV conduction in which case there was a reversal of the sequence of the recorded atrial electrograms from the ones closest to the ventricles occurring first and those at the high right atrium occurring last.
Atrial fibrillation: rapid, irregular, fractionated atrial electrograms with a rapid but irregular ventricular response as noted on the ECG.
Ventricular tachycardia: three or more beats arising from the ventricles at a rate of ≥200 beats/min.
ELISA
Antibodies produced in the sera were detected by ELISA. Briefly, microtiter plates were coated with β1AR ECL2 or β 2AR ECL2 peptide at 10 μg/ml in coating buffer. To determine antibody titer, sera were diluted 1:10,000 in 1 % BSA in PBS and thereafter serially diluted twofold. Goat anti-rabbit IgG conjugated with alkaline phosphatase (Sigma) and its substrate paranitrophenyl-phosphate 104 were used to detect antibody binding. Titers were determined as the highest dilution with an optical density (OD) value of 0.10 at 60 min.
Immunofluorescence Staining
Chinese hamster ovary (CHO) cells expressing human β1AR or β2AR were cultured on glass cover slips in 6-well plates for 24 h. The cells were fixed with 4 % paraformaldehyde, blocked with 5 % normal goat sera, and incubated with pre-immune and post-immune rabbit anti-β1AR or anti-β2AR sera (1:100) for 1 h, followed by incubation with fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG (Jackson ImmunoResearch). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescence images were obtained using a fluorescence microscope (Olympus).
β 1AR/β2AR Activation of cAMP Production
Rabbit sera were tested for β1AR/β2AR activation using the cAMP Hunter eXpress GPCR Assay kit (DiscoveRx, Fremont, CA). Briefly, 30,000 CHO cells expressing human β1AR or β2AR were dispensed into each well of 96-well culture plate and incubated overnight. The medium was removed, and assay buffer containing the cAMP antibody and rabbit sera (1:100) in the presence and absence of βAR blocker propranolol (1 μM) were sequentially added and incubated for 30 min. Preincubation of sera with a 10-fold excess of β1AR/β2AR ECL2 peptide was also tested for neutralization studies. cAMP standard, negative (buffer) and positive (isoproterenol 100 nM) controls were included in each assay. Samples were tested in triplicate. Following sample treatment, cAMP detection reagent and solution were added, and luminescent signal was read on a TD-20/20 Luminometer (Turner BioSystems). The cAMP values are expressed as a percentage of buffer baseline to normalize the individual data.
Statistical Analysis
Data are expressed as mean±SD. Chi-square analysis (2×2 contingency table) followed by a two-tailed Fisher’s exact test was used to determine the difference in occurrence of arrhythmias. Differences in cAMP production were assessed by a paired or unpaired Student’s t test as appropriate. A p value of <0.05 was considered statistically significant.
Results
Electrophysiological Studies
Arrhythmias were induced by burst pacing at baseline and at each of the three incremental concentrations of infused ACh. If no arrhythmia could be elicited, no response was registered. From a pathophysiological standpoint, we compared the induction of various sustained arrhythmias in each rabbit before and after antibody expression and the additional 2-week T4 treatment. Each rabbit served as its own control. Table 1 summarizes the electrophysiological effects of T4 treatment alone in the rabbits. There was only 1 sustained arrhythmia, viz., sinus tachycardia in the pretreatment control state out of 19 induction attempts. After T4 treatment, 8 out of 15 inductions resulted in sustained arrhythmias (53 vs. 5 % pretreatment, p=0.004) including 5 junctional tachycardias (33 vs. 0 % pretreatment, p=0.01) and 3 sinus tachycardias. In addition, the starting heart rate after T4 treatment was significantly faster than in the control state (278±135 vs. 165±24 beats/min, p=0.01).
Table 1.
Arrhythmia induction in thyroxine (T4) alone-treated rabbits (n=4)
| EP response | Pretreatment | T4 alone |
|---|---|---|
| Heart rate (beats/min) | 165±24 | 278±135* |
| Sustained arrhythmias (n)/induction attempts (n) | 1/19 | 8/15* |
| Sustained JT | 0/19 | 5/15* |
| Sustained ST | 1/19 | 3/15 |
JT junctional tachycardia, ST sinus tachycardia
p<0.05 vs. pretreatment
Table 2 summarizes the effects of β1AR-AAb and combination of β1AR-AAb and T4 on arrhythmia induction. In the pre-immune state, 4 episodes of sustained arrhythmias including 2 sinus tachycardias, 1 AF and 1 junctional tachycardia were induced out of 23 induction attempts. In the post-immune state, 15 episodes of sustained arrhythmias (65 vs. 17 % pre-immune, p=0.002) including 11 sinus tachycardias (48 vs. 9 % pre-immune, p=0.007), 1 AF, 2 junctional tachycardias, and 1 atrial tachycardia were induced out of 23 induction attempts. In the post-immune + T4 state, there were 27 episodes of sustained arrhythmias (90 vs. 17 % pre-immune, p<0.0001) including 15 sinus tachycardias (50 vs. 9 % pre-immune, p=0.002), 6 AF (20 vs. 4 % pre-immune, p=0.12), 2 atrial tachycardias, 1 junctional tachycardia, and 3 ventricular tachycardias out of 30 induction attempts. The induced sustained arrhythmias also significantly increased from the post-immune state to the post-immune + T4 state (65 vs. 90 %, p=0.041). All animals were in sinus rhythm in the pre-immune, post-immune, and post-immune + T4 states before burst pacing and ACh infusions, but the starting heart rates in the post-immune and post-immune + T4 states were faster than in the pre-immune state (202±36 and 207±38 vs. 171±32 beats/min, p=0.003 and p=0.036, respectively).
Table 2.
Arrhythmia induction in β1AR-immunized and thyroxine-treated rabbits (n=5)
| EP response | Pre-immune | Post-immune | Post-immune + T4 |
|---|---|---|---|
| Heart rate (beats/min) | 171±32 | 202±36* | 207±38* |
| Sustained arrhythmias (n)/induction attempts (n) | 4/23 | 15/23* | 27/30**,# |
| Sustained ST | 2/23 | 11/23* | 15/30* |
| Sustained AF | 1/23 | 1/23 | 6/30 |
| Sustained VT | 0/23 | 0/23 | 3/30 |
| Sustained JT | 1/23 | 2/23 | 1/30 |
| Sustained AT | 0/23 | 1/23 | 2/30 |
AF atrial fibrillation, AT atrial tachycardia, JT junctional tachycardia, ST sinus tachycardia, VT ventricular tachycardia
p<0.05,
p<0.001 vs. pre-immune;
p<0.05 vs. post-immune
Table 3 summarizes the effects of β2AR-AAb and combination of β2AR-AAb and T4 on arrhythmia induction in the rabbits. In the pre-immune state, only 1 out of 17 induction attempts produced a sustained arrhythmia, viz., sinus tachycardia. In the post-immune state, 6 episodes of sustained arrhythmias (38 vs. 6 % pre-immune, p=0.039) were observed including 5 atrial tachycardias (31 vs. 0 % pre-immune, p=0.018) and 1 AF during 16 induction attempts. In the post-immune + T4 state, 13 episodes of sustained arrhythmias (76 vs. 6 % pre-immune, p<0.0001) including 10 atrial tachycardias (59 vs. 0 % pre-immune, p=0.0003), 2 AF, and 1 ventricular tachycardia were induced out of 17 induction attempts. The starting heart rate was significantly faster in the post-immune + T4 state than in the pre-immune state (263±21 vs. 176±27 beats/min, p<0.0001) and post-immune state (263±21 vs. 154±10 beats/min, p<0.0001). In addition, there was a significant increase in sustained arrhythmia induction in the post-immune + T4 state compared to the post-immune state (76 vs. 38 %, p=0.037).
Table 3.
Arrhythmia induction in β2AR-immunized and thyroxine-treated rabbits (n=4)
| EP response | Pre-immune | Post-immune | Post-immune + T4 |
|---|---|---|---|
| Heart rate (beats/min) | 176±27 | 154±10 | 263±21**,# |
| Sustained arrhythmias (n)/induction attempts (n) | 1/17 | 6/16* | 13/17**,# |
| Sustained AT | 0/17 | 5/16* | 10/17** |
| Sustained AF | 0/17 | 1/16 | 2/17 |
| Sustained VT | 0/17 | 0/16 | 1/17 |
| Sustained ST | 1/17 | 0/16 | 0/17 |
AF atrial fibrillation, AT atrial tachycardia, ST sinus tachycardia, VT ventricular tachycardia
p<0.05,
p<0.001 vs. pre-immune;
p<0.05 vs. post-immune
Examples of atrial burst pacing-induced sinus tachycardia, atrial tachycardia, and junctional tachycardia are shown in Fig. 1.
Fig. 1.

Atrial burst pacing-induced sinus tachycardia, atrial tachycardia, and junctional tachycardia. ECG leads I through aVF and the sequence of bipolar electrograms from the superior vena cava (SVC) just above the right atrial entrance, high right atrium (HRA), mid-right atrium (MID RA), and the area of the AV junction (AVJ) are shown. a Induced ventricular premature contractions followed by a sinus tachycardia. b Induced atrial tachycardia with a constant A-A interval (118 ms) and 2:1 atrioventricular block. c Induced junctional tachycardia. The sequence of atrial activation starts at the AVJ area and proceeds toward the HRA (↑). Note that the atrial electrogram at the SVC is coincident with the ventricular activation until at the end of the trace. At this point, the SVC electrograms appear before the ventricular activation (↓). Reproduced with permission from Li et al. [12]
β1AR/β2AR Antibody Binding and Activity
All immunized rabbits developed high antibody titers to β1AR and to β2AR ranging from 1:320,000 to 1:1.28 million. To analyze antibody binding to β1AR and to β2AR, immunofluorescence was performed with CHO cells expressing human β1AR or β2AR. A representative stain is shown in Fig. 2. Rabbit anti-β1AR and anti-β2AR sera strongly reacted with β1AR and β2AR, respectively, in CHO cells, while pre-immune sera did not show any significant reactivity. Preincubation of rabbit anti-β1AR and anti-β2AR sera with an excess of the β1AR and β2AR ECL2 peptide, respectively, diminished fluorescence signal in CHO cells (data not shown), confirming the specific reactivity to β1AR and β2AR.
Fig. 2.

Immunofluorescence staining of β1-adrenergic receptor (β1AR) and β2-adrenergic receptor (β2AR) in transfected Chinese hamster ovary (CHO) cells. CHO cells expressing human β1AR (a, b) or β2AR (c, d) were stained with rabbit β1AR pre-immune sera (a), rabbit anti-β1AR sera (b), rabbit β2AR pre-immune sera (c), rabbit anti-β2AR sera (d), and FITC (green)-labeled secondary antibody. Nuclei were counterstained with DAPI (blue). The anti-β1AR and anti-β2AR sera demonstrated strong reactivity, while the pre-immune sera did not show any significant binding. All images were obtained at 40× magnification
Rabbit anti-β1AR sera were able to stimulate cAMP production in β1AR-transfected CHO cells in vitro (Fig. 3a). Sera-induced β1AR activation was abolished by the non-selective βAR blocker propranolol and by preincubation of the sera with the β1AR ECL2 peptide. Rabbit anti-β2AR sera stimulated cAMP production in β2AR-transfected CHO cells in a similar fashion, which was also inhibited by propranolol and by preincubation with the β2AR ECL2 peptide (Fig. 3b). No significant increase in cAMP production was found with the pre-immune sera compared to buffer baseline.
Fig. 3.

Rabbit sera-induced cAMP production in transfected CHO cells. Compared to the pre-immune sera, the anti-β1AR sera (a) and anti-β2AR sera (b) significantly increased cAMP production (*p<0.01, n=4), while the β-blocker propranolol or preincubation with the second extracellular loop (ECL2) peptide for β1AR and β2AR both effectively blocked the sera-induced β1AR and β2AR activation of cAMP production in β1AR and β2AR transfected CHO cells, respectively (#p<0.01, n=4)
To check the specificity of rabbit antisera, rabbit anti-β1AR and anti-β2AR sera were examined for cross-reactivity with β2AR and β1AR, respectively, by the ELISA and cAMP assay. As shown in Fig. 4a, the anti-β1AR sera reacted specifically with the β1AR and not the β2AR ECL2 peptide in ELISA. Similarly, the anti-β2AR sera reacted specifically with the β2AR but not the β1AR ECL2 peptide. The anti-β1AR sera also stimulated significant cAMP production in β1AR-transfected CHO cells, while no significant cAMP stimulation was observed in β2AR-transfected CHO cells (Fig. 4b). The anti-β2AR sera demonstrated similar receptor-specific stimulation of cAMP production in β2AR but not β1AR-transfected CHO cells.
Fig. 4.
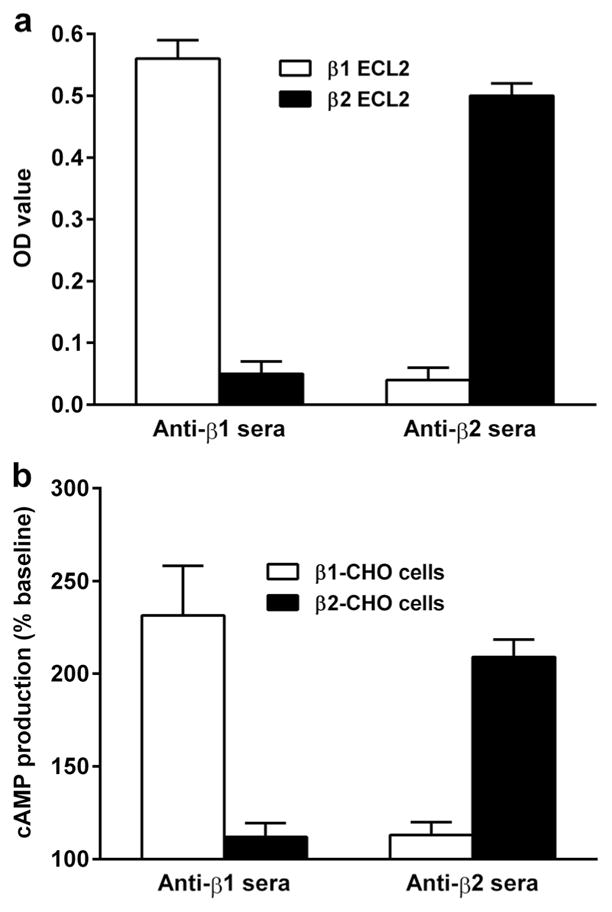
Binding and agonist specificity of rabbit anti-β1AR and anti-β2AR sera. Rabbit anti-β1AR and anti-β2AR sera were tested for reactivity with β1AR vs. β2AR ECL2 peptide in ELISA (a) and activation of cAMP production in β1AR-CHO vs. β2AR-CHO cells in cAMP assay (b). The anti-β1AR and anti-β2AR sera demonstrated subtype-specific reactivity and activity. No subtype cross-reactivity was observed
Discussion
In the present study, we used ACh and burst atrial pacing for induction of arrhythmias in rabbits with T4-induced hyperthyroidism alone and in immunized rabbits with β1AR or β2AR AAb expression and subsequently with concurrent T4 treatment. The rationale for this combination was discussed previously [11, 12]. T4 alone induced both sustained junctional and sinus tachycardias. Notably, the combination of β1AR-AAb and T4 was associated with a significant increase in the inducibility of sustained sinus tachycardia compared to that of the pre-immune control state measured in the same animals. In contrast, the combination of β2AR-AAb and T4 induced a significantly greater number of only sustained atrial tachycardia.
We have previously reported the electrophysiological effect of β2AR-AAb in rabbits immunized with a specific peptide to induce expression of β2AR-AAb [11]. Using each rabbit as its own control, after markedly raising the titer for β2AR-AAb, there was a significant increase in the induction of sustained atrial tachycardia compared to the control state. On the other hand, using the same approach for expressing β1AR-AAb in another group of rabbits, the predominant atrial tachyarrhythmia that was induced was sustained sinus tachycardia [12]. In the present experiments, the addition of T4 to those rabbits with high titers of β1AR-AAb and β2AR-AAb induced a significant increase in the number of sinus tachycardia and atrial tachycardia, respectively. Indeed, in the latter group, 9 of 16 of the starting heart rhythm were atrial tachycardia. By contrast, in the group with β1AR-AAb and T4, the starting heart rhythm was consistently sinus in the control and post-immune state. When T4 was given concurrently in the rabbits with β1AR-AAb, there was a significant increase in induced arrhythmia events over those observed with the β1AR-AAb alone, as when T4 was added to animals harboring β2AR-AAb.
There have been many studies dealing with the arrhythmogenic effects of thyroid hormone. Most of these have reported that frequent complications of documented hyperthyroidism are sinus tachycardia, premature atrial beats, and AF [13–15]. In a clinical case study of five women treated with levothyroxine, reentrant junctional tachycardias were documented in three of the five cases [16]. In our rabbit study, T4 alone in conjunction with burst pacing and/or ACh infusions elicited 5 inductions of junctional tachycardia and 3 inductions of sinus tachycardia out of a total of 15 events.
Experimental studies have investigated some of the underlying mechanisms that might be responsible for the arrhythmogenic effects of thyroid hormone. In vitro experiments have reported that thyroid hormone increased the I(f) current in rabbit sinus node myocytes and was a possible mechanism for the increased sinus rate associated with thyroid hormone [17]. Wang et al. investigated the effect of thyroid hormone on cat atrial myocytes and found that thyroid hormone-stimulated Na+/Ca2+ exchanger activity could increase Ca2+-mediated atrial tachycardia [18]. We have no direct evidence from the present study to support specific ionic channel changes from the 2-week exposure to increased thyroid hormone as causative. There are several thyroid hormonal alterations in channels which have a net effect of shortening the atrial action potential duration and refractory period [1, 19]. We believe these changes would be additive to the expected impact of β1/2AR-AAb activity on atrial cell repolarization and sensitivity to serve as a trigger for atrial tachyarrhythmias (Fig. 5).
Fig. 5.
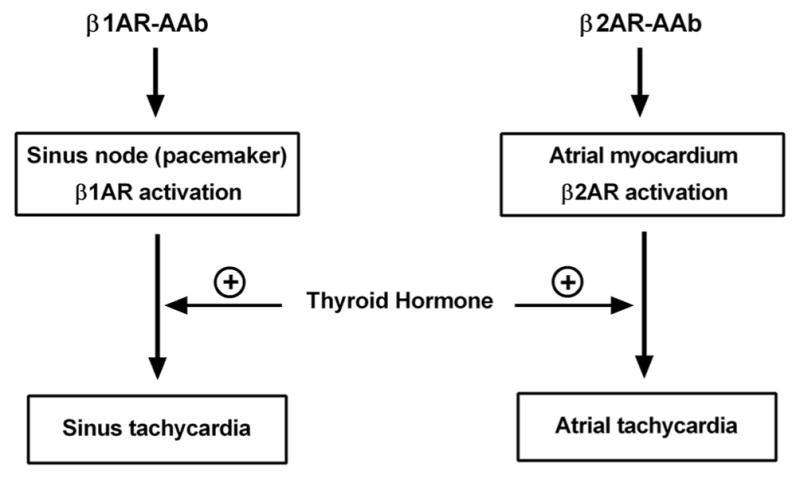
Proposed mechanisms of activating autoantibodies (AAb) and thyroid hormone-induced atrial tachyarrhythmias in the rabbit. Activation of β1AR in the sinus node by β1AR-AAb leads to increased induction of sustained sinus tachycardia, while activation of β2AR in the atrial myocardium by β2AR-AAb results in increased induction of sustained atrial tachycardia. The co-presence of excess thyroid hormone provides an additive effect on arrhythmia induction by genomic regulation of specific cardiac gene transcription and non-genomic modulation of cardiac membrane ion channels
It is important in interpreting these data to recognize that the relative densities of β1AR and β2AR in the atrium and their physiological roles in humans are different from those in the rabbit. In human atria, although β1AR is the predominant subtype, β2AR density is significantly higher in the sinoatrial node than that in the working atrial myocardium [20], which is consistent with physiologic studies indicating the involvement of β2AR in the regulation of cardiac chronotropism [21–23]. β1AR and β2AR also coexist in the rabbit atria [24]. However, β1AR is the predominant subtype in both density and physiological significance, while β2AR plays little role in regulating the chronotropic responses [25]. These data suggest a relatively low β2AR density in specialized tissues, i.e., sinus node and other sites of pacemaker activity, in the rabbit. The β2AR, however, may be involved in mediating the electro-physiologic responses in rabbit atrial myocardium as evidenced by studies using canine atrial myocytes [26]. These findings and our data support the concept that β1AR-AAb activation of β1AR in the sinus node and β2AR-AAb activation of β2AR in the atrial myocardium predispose the rabbit heart to increased induction of sustained sinus tachycardia and atrial tachycardia, respectively (Fig. 5).
It should be noted that in the present study, the combination of T4 and β1AR-AAb significantly increased the number of occurrences of sustained sinus tachycardia and increased the number of instances of sustained AF, but the occurrence of the latter did not achieve significance. However, based on our previous report associating M2 cholinergic autoantibodies with AF [10], the probability remains that the further addition of M2 cholinergic autoantibodies to this combination may allow the induction of a significant number of AF events in our rabbit model.
The effect of thyroid hormone on atrial tachyarrhythmias in susceptible older subjects is best illustrated in Graves’ hyper-thyroidism [6]. This association, although present, is less pronounced than observed in other forms of non-autoimmune hyperthyroidism such as toxic multinodular goiters. The unique autoimmune status of subjects with Graves’ is well documented by the causative association in most cases with production of thyroid-stimulating autoantibodies which activate the thyrotropin receptor. Our recent documentation that a very high proportion of subjects with Graves’ and AF also produce β1AR and M2 cholinergic autoantibodies [10] demonstrates the need to mechanistically determine if there is an autoimmune component participating in the pathophysiology of the multiple forms of atrial tachyarrhythmias. Witebsky’s postulates for proof of an autoimmune-related disease include demonstration of the events in an animal model [27]. The present study is a logical progression for development of such a model in the rabbit, an animal which is convenient for induction of autoantibodies in contrast to larger animals such as the dog or pig. A disadvantage of the rabbit, however, is the relative resistance to sustaining tachyarrhythmias owing to the myocardial relative refractory period extinguishing a wavefront that more rapidly approaches in the smaller heart. We have incorporated the addition of graded concentrations of ACh along with burst pacing to shorten the myocardial relative refractory period to allow us to use this otherwise convenient model for modeling of the interaction of adrenergic autoantibodies and thyroid hormone. We have examined the role of both β1AR and β2AR autoantibodies in the absence and presence of excess thyroid hormone in this animal model, and their unique receptor specificity provides support for this association. It will now be logical to extend these studies to include the co-presence of the M2 cholinergic autoantibodies since activation of this cardiac receptor also would be expected to shorten the refractory period and lead to hyperpolarization. These autoantibodies may contribute to the resistance for conversion of AF to a normal rhythm after correction of the hyperthyroidism. Recognition of the contribution of these receptor-activating autoantibodies in Graves’-associated AF will be followed by specific autoantibody blockade rather than that by receptor blockade using new decoy peptides now in development in our laboratory.
Clinical Implications
The importance of the present study is to provide a model for demonstrating the synergistic arrhythmogenic effect of βAR-AAb and thyroid hormone. It also provides direct evidence linking specific forms of supraventricular arrhythmias to receptor-specific AAb as opposed to the indirect or observed associations described clinically. It is likely that future therapies targeting these pathological autoantibodies may improve or prevent the persistence of tachyarrhythmias in hyperthyroid patients who harbor these autoantibodies.
Conclusions
In summary, we have developed a small animal model demonstrating T4 in combination with β1AR-AAb significantly manifested sustained sinus tachycardia. T4 in conjunction with β2AR-AAb induced sustained atrial tachycardia, whereas T4 alone induced both sustained sinus tachycardia and junctional tachycardia. In previous studies, using the same formats, we found that β1AR-AAb alone induced a significant number of sustained sinus tachycardia while β2AR-AAb alone induced significant numbers of sustained atrial tachycardia. T4 acted to potentiate the specific arrhythmogenic action of these two β-adrenergic autoantibodies. These data cumulatively support the concept that the highly specific receptor activity inherent with these β-adrenergic AAb may increase site-specific vulnerability of atrial cells as triggers and/or to demonstrate an enhanced substrate susceptibility to generate and/or sustain various atrial tachyarrhythmias. Alternatively, it may be presumed that the presence of these autoantibodies potentiates the effect of excess thyroid hormone on atrial arrhythmogenesis.
Acknowledgments
Funding This work was supported in part by grants from a Veterans Affairs Merit Review Award (to D.C.K. and X.Y.), National Heart, Lung, and Blood Institute HL56267 (to M.W.C. and D.C.K.), American Heart Association Postdoctoral Fellowship (to H.L.), and the Helen and Will Webster Arrhythmia Research Fund of the University of Oklahoma Foundation (to B.J.S. and D.C.K.).
Footnotes
Animal Subjects This study protocol was approved by the Institutional Animal Care and Use Committee of the Oklahoma City Veterans Affairs Medical Center and Oklahoma University Health Sciences Center, and conforms to international standards for animal safety and comfort.
Human Subjects No human studies were carried out by the authors for this article.
Conflict of Interest The authors have no conflict of interest to disclose.
Contributor Information
Hongliang Li, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA. Endocrinology and the Harold Hamm Diabetes Center, Veterans Affairs Medical Center and University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA.
Benjamin J. Scherlag, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
David C. Kem, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA. Endocrinology and the Harold Hamm Diabetes Center, Veterans Affairs Medical Center and University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
Alexandria Benbrook, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA.
Ling Zhang, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA.
Bing Huang, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA.
Madeleine W. Cunningham, Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
Ralph Lazzara, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA.
Xichun Yu, Heart Rhythm Institute, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA. Endocrinology and the Harold Hamm Diabetes Center, Veterans Affairs Medical Center and University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA. Heart Rhythm Institute and Endocrinology, University of Oklahoma, Health Sciences Center, TCH 6E103, 1200 Everett Drive, Oklahoma, City, OK 73104, USA.
References
- 1.Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. New England Journal of Medicine. 2001;344(7):501–509. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- 2.Klein I, Danzi S. Thyroid disease and the heart. Circulation. 2007;116(15):1725–1735. doi: 10.1161/CIRCULATIONAHA.106.678326. [DOI] [PubMed] [Google Scholar]
- 3.Selmer C, Olesen JB, Hansen ML, Lindhardsen J, Olsen AM, Madsen JC, et al. The spectrum of thyroid disease and risk of new onset atrial fibrillation: a large population cohort study. BMJ. 2012;345:e7895. doi: 10.1136/bmj.e7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu Y, Jones SV, Dillmann WH. Effects of hyperthyroidism on delayed rectifier K+ currents in left and right murine atria. American Journal of Physiology - Heart and Circulatory Physiology. 2005;289(4):H1448–H1455. doi: 10.1152/ajpheart.00828.2004. [DOI] [PubMed] [Google Scholar]
- 5.Chen YC, Chen SA, Chen YJ, Chang MS, Chan P, Lin CI. Effects of thyroid hormone on the arrhythmogenic activity of pulmonary vein cardiomyocytes. Journal of the American College of Cardiology. 2002;39(2):366–372. doi: 10.1016/s0735-1097(01)01731-4. [DOI] [PubMed] [Google Scholar]
- 6.Weetman AP. Graves’ disease. New England Journal of Medicine. 2000;343(17):1236–1248. doi: 10.1056/NEJM200010263431707. [DOI] [PubMed] [Google Scholar]
- 7.Davies TF, Ando T, Lin RY, Tomer Y, Latif R. Thyrotropin receptor-associated diseases: from adenomata to Graves disease. Journal of Clinical Investigation. 2005;115(8):1972–1983. doi: 10.1172/JCI26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazzerini PE, Capecchi PL, Guideri F, Acampa M, Selvi E, Bisogno S, et al. Autoantibody-mediated cardiac arrhythmias: mechanisms and clinical implications. Basic Research in Cardiology. 2008;103(1):1–11. doi: 10.1007/s00395-007-0686-8. [DOI] [PubMed] [Google Scholar]
- 9.Lee HC, Huang KT, Wang XL, Shen WK. Autoantibodies and cardiac arrhythmias. Heart Rhythm. 2011;8(11):1788–1795. doi: 10.1016/j.hrthm.2011.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stavrakis S, Yu X, Patterson E, Huang S, Hamlett SR, Chalmers L, et al. Activating autoantibodies to the beta-1 adrenergic and m2 muscarinic receptors facilitate atrial fibrillation in patients with Graves’ hyperthyroidism. Journal of the American College of Cardiology. 2009;54(14):1309–1316. doi: 10.1016/j.jacc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Scherlag BJ, Kem DC, Zillner C, Male S, Thirunavukkarasu S, et al. Atrial tachycardia provoked in the presence of activating autoantibodies to beta2-adrenergic receptor in the rabbit. Heart Rhythm. 2013;10(3):436–441. doi: 10.1016/j.hrthm.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Scherlag BJ, Kem DC, Benbrook A, Shen X, Cunningham MW, et al. Inducible cardiac arrhythmias caused by enhanced beta1-adrenergic autoantibody expression in the rabbit. American Journal of Physiology - Heart and Circulatory Physiology. 2014;306(3):H422–H428. doi: 10.1152/ajpheart.00551.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Olshausen K, Bischoff S, Kahaly G, Mohr-Kahaly S, Erbel R, Beyer J, et al. Cardiac arrhythmias and heart rate in hyperthyroidism. American Journal of Cardiology. 1989;63(13):930–933. doi: 10.1016/0002-9149(89)90142-2. [DOI] [PubMed] [Google Scholar]
- 14.Presti CF, Hart RG. Thyrotoxicosis, atrial fibrillation, and embolism, revisited. American Heart Journal. 1989;117(4):976–977. doi: 10.1016/0002-8703(89)90642-x. [DOI] [PubMed] [Google Scholar]
- 15.Forfar JC, Miller HC, Toft AD. Occult thyrotoxicosis: a correctable cause of “idiopathic” atrial fibrillation. American Journal of Cardiology. 1979;44(1):9–12. doi: 10.1016/0002-9149(79)90243-1. [DOI] [PubMed] [Google Scholar]
- 16.Biondi B, Fazio S, Coltorti F, Palmieri EA, Carella C, Lombardi G, et al. Clinical case seminar: reentrant atrio-ventricular nodal tachycardia induced by levothyroxine. Journal of Clinical Endocrinology and Metabolism. 1998;83(8):2643–2645. doi: 10.1210/jcem.83.8.5000. [DOI] [PubMed] [Google Scholar]
- 17.Renaudon B, Lenfant J, Decressac S, Bois P. Thyroid hormone increases the conductance density of f-channels in rabbit sinoatrial node cells. Receptors & Channels. 2000;7(1):1–8. [PubMed] [Google Scholar]
- 18.Wang YG, Dedkova EN, Fiening JP, Ojamaa K, Blatter LA, Lipsius SL. Acute exposure to thyroid hormone increases Na+ current and intracellular Ca2+ in cat atrial myocytes. Journal of Physiology. 2003;546(Pt 2):491–499. doi: 10.1113/jphysiol.2002.032847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocrine Reviews. 2005;26(5):704–728. doi: 10.1210/er.2003-0033. [DOI] [PubMed] [Google Scholar]
- 20.Rodefeld MD, Beau SL, Schuessler RB, Boineau JP, Saffitz JE. Beta-adrenergic and muscarinic cholinergic receptor densities in the human sinoatrial node: identification of a high beta 2-adrenergic receptor density. Journal of Cardiovascular Electrophysiology. 1996;7(11):1039–1049. doi: 10.1111/j.1540-8167.1996.tb00479.x. [DOI] [PubMed] [Google Scholar]
- 21.Arnold JM, O’Connor PC, Riddell JG, Harron DW, Shanks RG, McDevitt DG. Effects of the beta 2-adrenoceptor antagonist ICI 118,551 on exercise tachycardia and isoprenaline-induced beta-adrenoceptor responses in man. British Journal of Clinical Pharmacology. 1985;19(5):619–630. doi: 10.1111/j.1365-2125.1985.tb02689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall JA, Petch MC, Brown MJ. Intracoronary injections of salbutamol demonstrate the presence of functional beta 2-adrenoceptors in the human heart. Circulation Research. 1989;65(3):546–553. doi: 10.1161/01.res.65.3.546. [DOI] [PubMed] [Google Scholar]
- 23.Hall JA, Kaumann AJ, Brown MJ. Selective beta 1-adrenoceptor blockade enhances positive inotropic responses to endogenous catecholamines mediated through beta 2-adrenoceptors in human atrial myocardium. Circulation Research. 1990;66(6):1610–1623. doi: 10.1161/01.res.66.6.1610. [DOI] [PubMed] [Google Scholar]
- 24.Brodde OE, Leifert FJ, Krehl HJ. Coexistence of beta 1- and beta 2-adrenoceptors in the rabbit heart: quantitative analysis of the regional distribution by (−)-3H-dihydroalprenolol binding. Journal of Cardiovascular Pharmacology. 1982;4(1):34–43. doi: 10.1097/00005344-198201000-00007. [DOI] [PubMed] [Google Scholar]
- 25.Tenner TE, Jr, Young JA, Earley KJ, Yen YC. Functional characterization of beta-adrenoceptor subtypes in rabbit right atria. Life Sciences. 1989;44(10):651–660. doi: 10.1016/0024-3205(89)90469-4. [DOI] [PubMed] [Google Scholar]
- 26.Liang BT, Frame LH, Molinoff PB. Beta 2-adrenergic receptors contribute to catecholamine-stimulated shortening of action potential duration in dog atrial muscle. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(13):4521–4525. doi: 10.1073/pnas.82.13.4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited) Immunology Today. 1993;14(9):426–430. doi: 10.1016/0167-5699(93)90244-F. [DOI] [PubMed] [Google Scholar]