

Abstract
Samarium diiodide in the presence of water and THF (SmI2(H2O)n) has in recent years become a versatile and useful reagent, mainly for reducing carbonyl-type substrates. This work reports the reduction of several enamines by SmI2(H2O)n. Mechanistic experiments implicate a concerted proton-coupled electron transfer (PCET) pathway, based on various evidence against initial outer-sphere electron transfer, proton transfer or substrate coordination. A thermochemical analysis indicates that the C-H bond formed in the rate determining step has a bond dissociation free energy (BDFE) of ~32 kcal mol−1. The O–H BDFE of the samarium aquo ion is estimated to be 26 kcal mol−1, which is among the weakest known X–H bonds of stable reagents. Thus SmI2(H2O)n should be able to form very weak C-H bonds. The reduction of these highly electron rich substrates by SmI2(H2O)n shows that this reagent is a very strong hydrogen atom donor as well as an outer sphere reductant.
Graphical abstract
Introduction
Samarium diiodide (SmI2) is a ubiquitous reagent in organic synthesis, typically used as a single electron transfer (SET) agent to reduce alkyl halides and carbonyl functionalities.1 Additives are commonly used to increase the effectiveness and versatility of SmI2. These are Lewis bases which make SmI2 more reducing and often provide protons, such as methanol and water. Water has been the most unique and useful additive, allowing reduction of the most challenging substrates.2 Solutions of SmI2 and water in THF likely contain an equilibrium mixture of species,3 which are denoted here as SmI2(H2O)n.
Kinetic investigations of SmI2 reductions with proton donors have been reported by Hoz and coworkers.4 The broad scope of SmI2(H2O)n has been explored by Procter and coworkers with different carbonyl type functionalities and polycyclic aromatic compounds.2,5 They defined an “effective redox potential” of SmI2(H2O)n as the boundary between the outersphere (pure electron) reduction potentials of reactive and non-reactive aromatic substrates.5b Flowers et al. showed that H2O makes SmI2 more reducing by cyclic voltammetry (CV),6, and they showed how the coordination strength of proton donors affects SmI2 reductions.7 A mechanistic study by this group later indicated that SmI2(H2O)n reduces anthracene through a concerted proton coupled electron transfer (PCET) mechanism.8 Most recently, they proposed that the electrostatic interaction between carbonyl substrates and SmI2 can tune the PCET character of the reaction coordinate.9 Overall, Flowers et al. have shown that SmI2(H2O)n reductions of widely used substrates can involve PCET reactivity of SmI2(H2O)n.
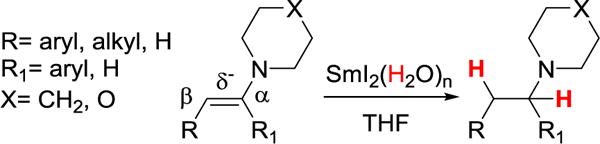
Reported here are SmI2(H2O)n reductions of a new substrate class, enamines (Scheme 1), as well as evidence that these reactions proceed through a concerted PCET pathway. A thermochemical analysis shows that the related aqueous Sm2+ ion has an extraordinarily weak O–H bond. These studies thus show that the PCET perspective significantly broadens the range of substrates that are likely to be reduced by SmI2(H2O)n.
Scheme 1.
SmI2(H2O)n Reduction of Enamines
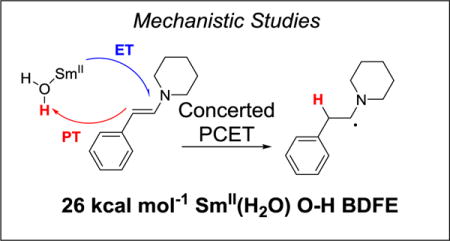
Enamines have very electron rich carbon-carbon double bonds due to the resonance donation of the nitrogen lone pair into the C=C π-bond. They are very resistant to the addition of an electron,10 but they can add a hydrogen atom (e− + H+) to form a neutral, stabilized α-amino radical (Figure 1). Therefore, enamines were chosen as an attractive substrate class to explore the ability of SmI2(H2O)n to react by concerted PCET. This mechanism involves transfer of H+ and e− together in a single kinetic step, which for enamines bypasses the high energy radical anion. From this perspective, SmI2(H2O)n should be able to reduce enamines, which are resistant to accepting an electron but have a moderate ability to accept a hydrogen atom. Additionally, this reactivity can be rationalized through analysis of the bond dissociation energies (BDEs) of the broken O–H bond in SmI2(H2O)n and the formed C–H bond in the neutral intermediate.
Figure 1.

SmI2(H2O)n reductions of enamines via ET and concerted PCET Pathways
Results
I. Preparative Results
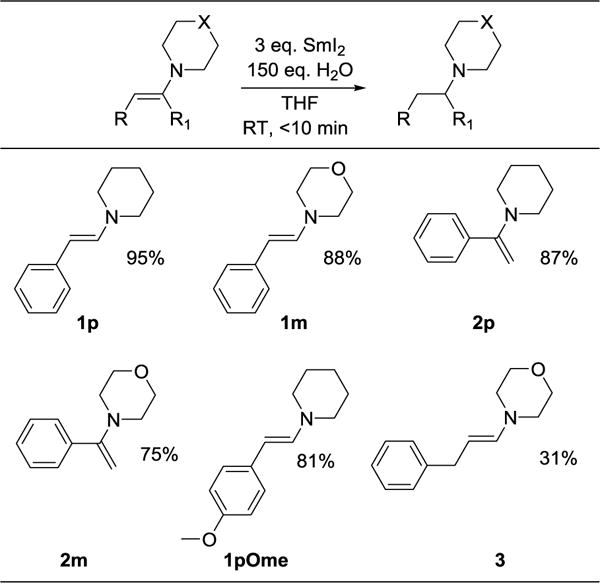
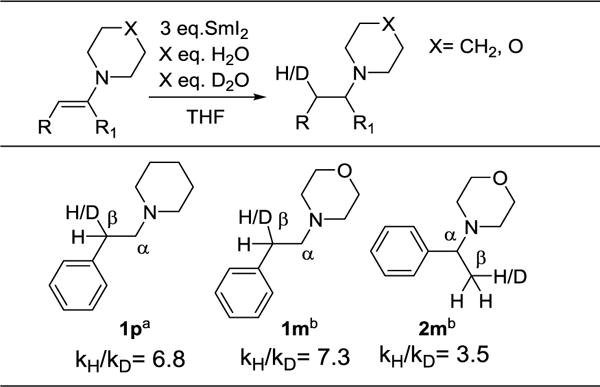
Enamines are rapidly reduced to amines by SmI2(H2O)n (Scheme 1). The addition of enamines to air-free SmI2(H2O)n solutions caused bleaching of the red color and formation of a white Sm3+ precipitate within 10 min at ambient temperatures. The piperidine and morpholine substrates in Table 1 are generally reduced in good isolated yields. Simple methods of product isolation, such as filtration of the crude reaction mixtures, were unsuccessful because the amine product was bound to the insoluble metal salts. This also prevented straightforward 1H NMR analysis of products. Acid-base workup conditions to isolate the amine product were similarly unsuccessful, so basic alumina or reverse phase column chromatography was used. Substrates include compounds bearing conjugated phenyl groups either β (1p, 1m and 1pOMe) or α (2p and 2m) to the nitrogen. An enamine with a benzyl group β to the nitrogen (3) is also reduced, albeit in lower yield. The substrates were chosen with varying electronic character, pKas, and conjugation in order to answer specific mechanistic questions (see below). Nitrogen containing aromatic compounds such as imidazole, indole, pyrrole, and their N-methyl analogues did not react under these conditions, despite formally containing enamine motifs.
Table 1.
Enamine Reduction Substratesa
|
Non-optimized conditions: 0.024 M substrate, 0.072 M SmI2, 3.6 M H2O, 4.9 mL THF. Isolated yields of products. p = piperidinyl; m = morpholinyl.
II. Mechanistic Results
Four mechanistic possibilities have been considered for the enamine reductions: (i) initial outer-sphere electron transfer followed by proton transfer (ET/PT); (ii) initial proton transfer followed by electron transfer (PT/ET); (iii) concerted PCET with transfer of a proton and an electron in a single kinetic step; and (iv) initial substrate coordination. The first three cases are illustrated in Scheme 2 and would all be considered PCET mechanisms within the current wider PCET framework,11 though we note that within the organic chemistry literature “PCET” still typically refers only to the concerted process. Mechanistic experiments probing each of these are discussed in turn below.
Scheme 2.
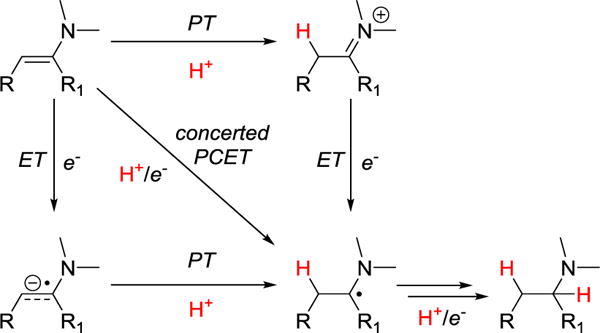
Square Scheme showing PCET Mechanisms for Enamine Reduction by SmI2(H2O)n
(a) Initial electron transfer
Initial ET is very unlikely because of the electron rich character of the enamine substrates. This is evident from the cyclic voltammetry (CV) of 1p in THF, which displayed no reduction wave within the solvent window, to −3.5 V vs. Fc0/+ (Supporting Information (SI) Section 4.1).
To further test this mechanistic possibility, a competition experiment was done with 1p and the more electron rich 1pOMe.
A reaction with a 1 : 1 mixture of 1p and 1pOMe with limited SmI2(H2O)n gave a 1.2 : 1 mixture of reduced products (Table 2). This is inconsistent with initial rate-limiting ET because the p-OMe substituted 1pOMe would form a much less stable radical anion intermediate than the unsubstituted 1p, and consequently would be reduced with a much slower rate. This result could in principle be consistent with pre-equilibrium ET followed by rate limiting PT. However, this seems very unlikely because the difference in the rate constants for protonation of the radical anions would have to precisely balance the difference in ET equilibrium constants. For the related oxidation of these styrenes, the difference in the CV oxidation peak in MeCN is 0.56 V;12 if the difference in reduction potentials are similar, this would imply a ~109 difference in Keq for ET.
Table 2.

|
Non-optimized conditions: 0.025 M substrate 1, 0.025 M substrate 2, 0.020 M SmI2, 1.0 M H2O, 8.0 mL THF.
Under these conditions, less than 20% total substrate conversion was achieved. S = stilbene, A = anthracene.
(b) Initial proton transfer
The possibility of initial PT was similarly probed with a competition experiment between 1p and 1m. The β-carbon is the most basic site on an enamine, and the pKa’s of piperidine-containing enamines in acetonitrile are ca. 2.5 units more basic than the corresponding morpholine-containing enamines.13 Reduction of a 1 : 1 mixture of 1p and 1m gave a 4.0 : 1 ratio of products (Table 2). A larger rate difference in protonation rates would have been expected for a difference of 2.5 pKa units.
To further explore this mechanistic scenario, reductions were investigated with an added bulk solvent acid, trifluoroethanol (TFE), 1:1 with the added H2O. TFE is known to be non-coordinating with SmI2.4a TFE has an aqueous pKa of ca. 12.4,14 which is likely more acidic than aqueous Sm2+.15 Monitoring reactions by optical spectroscopy with and without substantial TFE showed that the time courses increased, but by less than a factor of 1.3 in all cases (SI Section 5.1). Thus, both in changing the enamine and in adding an acid, significant changes in basicity or acidity caused only small changes in rate, arguing against rate-limiting PT.
The occurrence of pre-equilibrium proton transfer was explored via isotope-exchange experiments.16 Enamine reductions were done with D2O and with half of the stoichiometric requirement of SmI2(H2O)n, and the unreacted starting material was examined by 1H NMR. For both 1p and 1m, no exchange of the β-H for deuterium was observed (eq 1 and SI Sections 3.6 and 7.4). Pre-equilibrium PT is also very unlikely because deprotonation of the C–H bond of the cationic imine in the slightly acidic solution would have to be faster than electron transfer from the highly reducing SmI2(H2O)n. This path is also unlikely based on the large product isotope effects reported in the next section.
 |
(1) |
(c) Product isotope effects
Enamine reductions were performed using SmI2 in THF with H2O/D2O mixtures. The relative ratios of protonated and deuterated products were determined by integrating the relevant CH peaks in the 1H NMR spectra of the products. Large H/D product isotope effects (PIEs) were found in the reductions of substrates 1p, 1m, and 2m (Table 3). Substrates 1m and 2m were reduced using a 1:1 ratio of D2O:H2O, while a 4.5:1 ratio was used for 1p to obtain more reliable integrations. Effects at the β-carbons range from 3.5 - 7.3, indicating that H/D transfer to the β-carbon is involved in the selectivity-determining step of reduction. This is consistent with a concerted PCET mechanism or an ET/PT pathway, but would be difficult to rationalize with pre-equilibrium PT. Effects at the α-carbons are much closer to unity, but accurate determinations were precluded by overlap of 1H NMR resonances for these protons and piperidinyl or morpholinyl α protons.
Table 3.
Deuterium product isotope effects
|
Non-optimized conditions: 0.024 M substrate, 0.072 M SmI2, 0.65 M (27 eq.) H2O, 2.9 M (123 eq.) D2O, 4.9 mL THF.
Same as a except 1.8 M (75 eq.) H2O and 1.8 M (75 eq.) D2O.
(d) Substrate coordination
It is also possible that the substrates can coordinate to SmI2 during the reaction, increasing their electrophilicity and allowing initial ET to occur. No coordination of 1p was observed by optical spectroscopy when monitoring the reactions, as the initial spectra are unchanged upon addition of substrate (as the reaction starts). The visible absorption spectrum of SmI2 in THF is known to be sensitive to changes in coordination.6,17 However, this does not exclude a small equilibrium amount of coordinated substrate.
The similar reactivity of 1p and 1m also indicates the unimportance of coordination. These substrates should have different affinities for the metal center due to their different basicities.13b This should result in significantly different rates of reduction if binding through the alkene or the nitrogen moiety occurred along the reaction pathway.
The reactivity of 1p was also compared with substrates incapable of coordination to the metal center (Table 2). 1p reacted 4.3 times faster than trans-stilbene (S), and 2.0 times slower than anthracene (A), both of which have no coordinating group. This is not a simple comparison because the driving forces for the reactions are different.18 However, the results suggest that the presence of a potentially coordinating nitrogen atom does not substantially increase the rate of reduction. In sum, these experiments are not definitive, but they suggest that coordination does not play a major role in the enamine reductions.
Discussion
I. Mechanism
All the experimental results above are consistent with a concerted PCET mechanism for the first step of enamine reduction by SmI2(H2O)n. We note that this is not an ideal system for mechanistic study, as the speciation of the samarium reactant is not well defined, the samarium product is insoluble, the acidity of the solutions is not easily controlled over the course of the reactions, and the remaining organic reactant is unstable to the workup conditions. Still, the body of experiments point to the concerted transfer of e− and H+ from SmI2(H2O)n to the enamines. This is consistent with recent studies by Flowers for the reduction of anthracene.
The concerted PCET mechanism can be described as transfer of a proton from a H2O coordinated to Sm(II) to the β-carbon of the enamine concerted with transfer of an electron from Sm(II) to the α-carbon of the enamine (Scheme 3, top left). This same step could equivalently be described as the transfer of a hydrogen atom from SmI2(H2O)n to the enamine β-carbon. Subsequent conversion of the radical to the saturated amine product occurs by transfers of an additional electron and proton, which could be stepwise or concerted.
Scheme 3.

Two Descriptions of the Proposed Mechanism for SmI2(H2O)n Reduction of Enamines
II. Thermochemistry
Describing these reductions as the transfer of a hydrogen atom implies that the free energy of the concerted PCET step can be quantified as the difference between the bond dissociation energies (BDFEs) of the O–H bond being cleaved and the C–H bond being formed. While the use of bond dissociation enthalpies (BDEs) is common in organic free radical chemistry, free energies are preferred because they connect with equilibrium constants and a Marcus theory approach.19
(a) Strength of the C–H bond formed
While no thermochemical data is available for the specific enamines used in this study, C–H bond strengths can be well estimated for 3 based on values for related compounds. We take the heat of heat of hydrogenation of 3 to be the same as that for closely related compound N-(1-propenyl)piperidine (4), −23.6 kcal mol−1.20 The resulting hydrogenated compound 3H2 is taken to have a BDE of its α-C–H bond that is that same as that of triethylamine. 90.7 kcal mol−1.21 Using these values, the thermochemical cycle in Scheme 4 gives the BDE of the C–H bond formed upon reduction of 3, labelled (i) in the Scheme. Steps (i)-(iii) in the Scheme separate the overall hydrogenation (step iv) into the two BDEs. This analysis gives the BDE of the initially formed C–H bond to be ~37 kcal mol−1 using eqs 2 and 3.
Scheme 4.
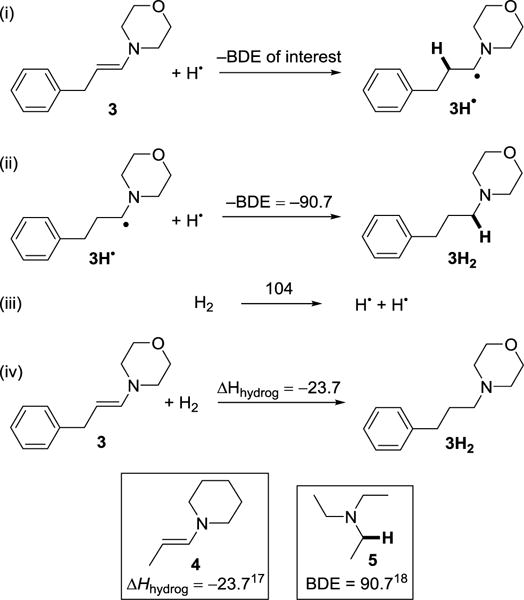
Estimation of the initially formed C–H BDE in the reduction of 3.a
a All values in kcal mol–1. Step (ii) is assumed to be equivalent to that of 5, and step (iv) is assumed to be equal to that of 4.
| (2) |
| (3) |
A C–H bond dissociation enthalpy, such as that derived in Scheme 4 and eq 3, is related to its bond dissociation free energy by the differences in entropy between the reaction components, RH, R• and H•. For the cleavage of C–H bonds in organic solvents, the entropies of RH and R• are typically very similar,11b so the BDFE and BDE are related by TS°(H•)solv, which is ~4.6 kcal mol−1 in polar organic solvents (eq 4).11b Thus, the BDFE of the C–H bond formed upon addition of H• to 3 is ~32 kcal mol–1.
| (4) |
(b) SmIIO–H bond strength
In order for the hydrogen atom transfer reaction in Scheme 3 to occur at a reasonable rate, the O-H BDFE in SmI2(H2O)n cannot be much stronger than 32 kcal mol−1. Because the O–H bond in water is incredibly strong (BDFE(HO–H) = 111 kcal mol−1 in the gas phase),11b this means the O–H BDFE has decreased dramatically upon coordination to SmI2, by ca. 79 kcal mol−1. Such O–H bond weakening upon coordination to a reducing metal center is well precedented,22 and Flowers and coworkers have estimated a 73 kcal mol−1 bond weakening for SmI2(H2O)n based on its successful reduction of anthracene.8 The same group also demonstrated that coordination of SmI2 to the carbonyl of 2-pyrrolidinone weakens the N-H bond by approximately 70 kcal mol−1.23
To obtain a more direct estimate of the O–H bond strength for a water molecule bound to Sm2+, we determine here the O-H BDFE of the related Sm2+ aquo ion, Sm(H2O)n2+ (aq). This can be determined directly11b from the aqueous Sm3+/2+ standard reduction potential of –1.55 V24 and the pKa of Sm(H2O)n3+ (aq) as shown in Scheme 5. This determination of X–H bond strengths from the separate thermochemistry of removal of the electron (E°) and proton (pKa) has a long history and was developed extensively by Bordwell; in some cases when there is separation of the e− and H+, what is determined might better be called an ‘effective BDFE.’11b The pKa of Sm(H2O)n3+ (aq) has not been reported but is taken to be the same as that of Eu(H2O)n3+ (aq), 3.3,25 because radii and pKa’s are fairly constant across the lanthanide row.26 The analysis in Scheme 5 gives an O-H BDFE of 26 kcal mol−1 for Sm(H2O)n2+ (aq) [BDE = ~25 kcal mol−1].11b
Scheme 5.
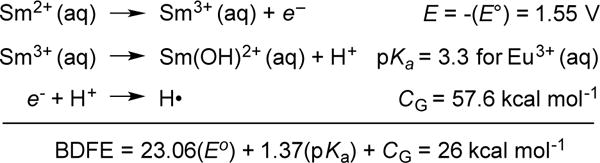
The O-H bond dissociation enthalpy in Sm(H2O)n2+(aq)
A similar analysis was very recently reported by Szostak and coworkers for SmI2 in a THF/water mixture.27 They obtained BDFE = 37.4 kcal mol−1 but this calculation unfortunately is in error because it used values from different solvents and it used an estimated pKa for Sm2+ instead of the appropriate Sm3+ value (Scheme 5).
To our knowledge,11b the O-H BDFE of the Sm2+ aquo ion, Sm(H2O)n2+ (aq), and the N–H bond in the related pyrrolidinone complex23 are the weakest X–H bonds for any species stable enough to be used as a reagent under ambient conditions.
We believe that the value for SmI2(H2O)n in THF is likely similar to the aqueous value derived above. While many thermochemical quantities such as reduction potentials and pKa values are very solvent-dependent, bond strengths typically vary only small amounts between solvents. This is why, for instance, gas-phase BDEs are typically given in chemistry textbooks to rationalize various solution reactivity. As we have shown in a recent review,11b the origin of limited solvent dependence can be illustrated in Scheme 6, which shows that solution and gas-phase BDFEs are related by the free energies of solution. For most large molecular systems, the solvation of XH and X• are similar, except perhaps due to a difference in the number of hydrogen bonds.11b For instance, Sm(H2O)n2+ and Sm(H2O)n-1(OH)2+ are the same size and charge, so they are likely solvated similarly in different solvents.
Scheme 6.
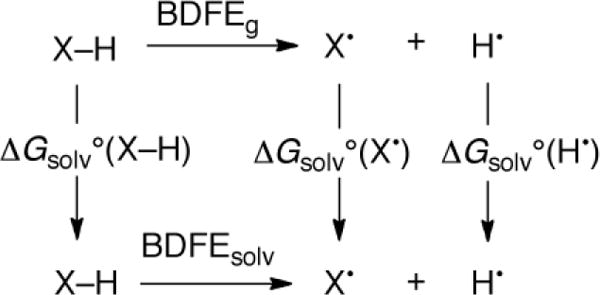
Relationship between gas-phase and solution bond dissociation free energies (from11b).
The samarium(II) system discussed here, however, could be an exception to the general rule of low solvent dependence of BDEs and BDFEs, due to differences in the speciation of the cation in different solvents. The analysis in Scheme 5 involves Sm(H2O)n2+ [equivalently, Sm2+ (aq)], while the speciation of SmI2 in THF/H2O mixtures is likely complicated. Some iodide anion coordination to the 2+ cation would be expected given the low dielectric constant of THF with 6.5% water (ε≅ 10)17c,28 and the iodide-coordinated crystal structures obtained from SmI2/THF solutions.29 Given the uncertainty in speciation and the insolubility of the SmIII product, rigorous thermodynamic measurements on this system would be very challenging. We conclude that the aqueous BDFE of 26 kcal mol−1 for Sm(H2O)n2+ is the best available value and is likely to be a good approximation.
III. Reactivity
Using the BDFE values from the last section, transfer of a hydrogen atom from SmI2(H2O)n to 3 is exoergic by ΔG° = −6 kcal mol−1. This thermochemical analysis thus supports the mechanistic conclusion of a concerted PCET/hydrogen atom transfer mechanism (Scheme 3).
The uniqueness of Sm2+ PCET chemistry is not just the weakness of the O–H but also that these solutions do not rapidly evolve H2.30 Based on the BDFE above, the formation of H2 gas (eq 5) should be ~45 kcal mol−1 exoergic. The traditional ‘rule of thumb’ is that H2 will be produced by reducing agents that are ca. 0.5 V (~25 kcal mol−1) below the reversible H2 potential (RHE).31 Still, Sm2+ has some stability in water despite its −1.55 V standard potential, and solutions of SmI2(H2O)n in THF/H2O are stable for hours under our reaction conditions. There is evidently no facile kinetic pathway for Sm2+ to make H2, despite the large driving force.
| (5) |
Kinetic factors also must be responsible for the lack of hydrogenation of simple alkenes by SmI2(H2O)n in THF/H2O. The transfer of H. to cyclohexene is ~ 4 kcal mol−1 less favorable than for enamine 3 (S.I. Section 6.1.1),32 yet we observed no reduction over an hour at ambient temperatures.33 This energetic difference should translate into a rate difference of roughly 30 based on the Marcus cross relation [Δ(ΔG‡) ≅ ½Δ(ΔG°), other things being equal].19 Given the rapidity of the reduction of enamines (typically seconds by preliminary kinetic studies, SI section 5), reduction of cyclohexene should have been observed. Similarly puzzling is that 1,1-diphenylethylene is not reduced while trans-stilbene is readily hydrogenated. The origin of these kinetic factors is not known, but we note that reaction conditions more forcing than a few hours at 25 C are not possible. Perhaps the faster reactions of enamines are due to a polar effect, well known in organic systems, that H-atom transfers from an acidic X–H bond are more facile to a reagent that has a higher HOMO.34
Conclusion
SmI2(H2O)n in THF has been shown to reduce a new substrate class, enamines. Enamines would not normally have been thought of as a possible substrate for reduction by SmI2 because of their resistance to outer-sphere electron transfer. The mechanism has been probed with competition experiments, product isotope effects, and other studies using substrates of varying electron richness, basicity, and coordinative capability. The results are all consistent with a mechanism of initial concerted transfer of e− and H+ (H atom) from SmI2(H2O)n to the enamine. There are strong arguments against stepwise PCET mechanisms of initial electron transfer or proton transfer, and there is no indication of a mechanism involving coordination of the enamine substrate. A thermochemical analysis indicates that the O–H bond in Sm(H2O)n2+ (aq), and by implication SmI2(H2O)n, is incredibly weak. This O–H bond has a bond dissociation free energy of only 26 kcal mol−1, being the weakest O–H bond for any stable material. This BDE analysis therefore shows why SmI2(H2O)n can reduce such recalcitrant substrates, but also raises questions about the origin of the larger kinetic barriers for other substrates. This study thus provides a more detailed understanding of the important and varied chemistry of SmI2(H2O)n and opens the door to new applications of this reagent as a hydrogen atom donor.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by U.S. NIH grant 5R01GM050422.
Footnotes
Supporting Information. Preparative procedures, substrate and product spectral data, and kinetic data and thermochemical analyses are included in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Szostak M, Fazakerley NJ, Parmar D, Procter DJ. Chem Rev. 2014;114:5959. doi: 10.1021/cr400685r. [DOI] [PubMed] [Google Scholar]; (b) Szostak M, Spain M, Procter DJ. Chem Soc Rev. 2013;42:9155. doi: 10.1039/c3cs60223k. [DOI] [PubMed] [Google Scholar]
- 2.Szostak M, Spain M, Parmar D, Procter DJ. Chem Commun. 2012;48:330. doi: 10.1039/c1cc14252f. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zhao X, Perrin L, Procter DJ, Maron L. Dalton Trans. 2016;45:3706. doi: 10.1039/c6dt00241b. [DOI] [PubMed] [Google Scholar]; (b) Kefalidis CE, Perrin L, Maron L. Eur J Inorg Chem. 2013;2013:4042. [Google Scholar]
- 4.(a) Tarnopolsky A, Hoz S. Org Biomol Chem. 2007;5:3801. doi: 10.1039/b713014g. [DOI] [PubMed] [Google Scholar]; (b) Amiel-Levy M, Hoz S. J Am Chem Soc. 2009;131:8280. doi: 10.1021/ja9013997. [DOI] [PubMed] [Google Scholar]; (c) Halder S, Hoz S. J Org Chem. 2014;79:2682. doi: 10.1021/jo500161s. [DOI] [PubMed] [Google Scholar]
- 5.(a) Szostak M, Spain M, Procter DJ. J Am Chem Soc. 2014;136:8459. doi: 10.1021/ja503494b. [DOI] [PubMed] [Google Scholar]; (b) Szostak M, Spain M, Procter DJ. J Org Chem. 2014;79:2522. doi: 10.1021/jo4028243. [DOI] [PubMed] [Google Scholar]; (c) Huang HM, Procter DJ. J Am Chem Soc. 2016;138:7770. doi: 10.1021/jacs.6b04086. [DOI] [PubMed] [Google Scholar]; (d) Huang HM, Procter DJ. J Am Chem Soc. 2017;139:1661. doi: 10.1021/jacs.6b12077. [DOI] [PubMed] [Google Scholar]; (e) Duffy LA, Matsubara H, Procter DJ. J Am Chem Soc. 2008;130:1136. doi: 10.1021/ja078137d. [DOI] [PubMed] [Google Scholar]
- 6.Prasad E, Flowers RA. J Am Chem Soc. 2005;127:18093. doi: 10.1021/ja056352t. [DOI] [PubMed] [Google Scholar]
- 7.Chciuk TV, Anderson WR, Flowers RA. Angew Chem Int Ed. 2016;55:6033. doi: 10.1002/anie.201601474. [DOI] [PubMed] [Google Scholar]
- 8.Chciuk TV, Flowers RA. J Am Chem Soc. 2015;137:11526. doi: 10.1021/jacs.5b07518. [DOI] [PubMed] [Google Scholar]
- 9.Chciuk TV, Anderson WR, Flowers RA. J Am Chem Soc. 2016;138:8738. doi: 10.1021/jacs.6b05879. [DOI] [PubMed] [Google Scholar]
- 10.Cook AG. Enamines: Synthesis, Structure and Reactions. 2nd. CRC Press: New York, New York, USA; 1987. [Google Scholar]
- 11.(a) Cukier RI, Nocera DG. Annu Rev Phys Chem. 1998;49:337. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]; (b) Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Chem Rev. 2012;112:4016. doi: 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- 12.Schepp NP, Johnston LJ. J Am Chem Soc. 1996;118:2872. [Google Scholar]
- 13.(a) Based on ΔpKa values of 3.07, and 2.48 for cyclopent-1-en-1-yl, cyclohex-1-en-1-yl, and cyclohept-1-en-1-yl derivatives; Cook AG, Absi ML, Bowden VK. J Org Chem. 1995;60:3169. [Google Scholar]
- 14.Donghi D, Beringhelli T, D’Alfonso G, Mondini M. Chem Eur J. 2006;12:1016. doi: 10.1002/chem.200500920. [DOI] [PubMed] [Google Scholar]
- 15.The aqueous pKa of Sm(H2O)n2+ is not known, but the acidity of ‘hard’ aquo ions is well known to vary as Z2/r, where Z is the charge on the ion and r is the radius. Comparing dications, Sm2+ is likely very similar in size to Sr2+ (1.32 Å) since neighboring Eu2+ has r = 1.31 Å. Sr2+ has a pKa of 13.3, almost an order of magnitude weaker than TFE. Principles and values from:; Wulfsberg G. Principles of Descriptive Inorganic Chemistry. Brooks/Cole; Belmont, CA: 1987. pp. 24–30. and inside back cover. [Google Scholar]
- 16.We thank the reviewer for suggesting a version of this experiment.
- 17.(a) Teprovich JA, Antharjanam PKS, Prasad E, Pesciotta EN, Flowers RA. Eur J Inorg Chem. 2008;2008:5015. [Google Scholar]; (b) Yacovan A, Hoz S, Bilkis I. J Am Chem Soc. 1996;118:261. [Google Scholar]; (c) Sadasivam DV, Teprovich JA, Procter DJ, Flowers RA. Org Lett. 2010;12:4140. doi: 10.1021/ol101722c. [DOI] [PubMed] [Google Scholar]
- 18.Assuming a rate-limiting transfer of a hydrogen atom from SmI2(H2O)n to a carbon atom of the substrate (see below), the rate constants should parallel the strengths of the C–H bonds formed. Preliminary estimates are that these fall in the order 1p < stilbene < anthracene. The dependence of rate constants on driving force in SmI2(H2O)n reactions will be analyzed in more detail in future publications.
- 19.Mayer JM. Acc Chem Res. 2011;44:36. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prochazka M, K V, Palecek M, Pecka K. Collection Czechoslov, Chem Commun. 1970;35:3813. [Google Scholar]
- 21.Luo YR. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton, Florida, USA: 2007. [Google Scholar]
- 22.(a) Bezdek MJ, Guo S, Chirik PJ. Science. 2016;354:730. doi: 10.1126/science.aag0246. [DOI] [PubMed] [Google Scholar]; (b) Spiegel DA, Wiberg KB, Schacherer LN, Medeiros MR, Wood JL. J Am Chem Soc. 2005;127:12513. doi: 10.1021/ja052185l. [DOI] [PubMed] [Google Scholar]; (c) Campaña AG, Estévez RE, Fuentes N, Robles R, Cuerva JM, Buñuel E, Cárdenas D, Oltra JE. Org Lett. 2007;9:2195. doi: 10.1021/ol070779i. [DOI] [PubMed] [Google Scholar]
- 23.Chciuk TV, Li AM, Vazquez-Lopez A, Anderson WR, Flowers RA. Org Lett. 2017;19:290. doi: 10.1021/acs.orglett.6b03664. [DOI] [PubMed] [Google Scholar]
- 24.The value of –1.55 V vs. NHE for the standard reduction potential of Sm3+/2+ appears in a number of reviews of standard electrode potentials and of the thermochemistry of lanthanide elements, including; (a) Morss LR. Chem Rev. 1976;76:827. [Google Scholar]; (b) Bratsch SG. J Phys Chem Ref Data. 1989;18:1–21. [Google Scholar]; It appears to be relatively independent of the nature of the anions present:; Timnick A, Glockler G. J Am Chem Soc. 1948;70:1347–1350. doi: 10.1021/ja01184a019. [DOI] [PubMed] [Google Scholar]
- 25.Mohapatra PK, Khopkar PK. Polyhedron. 1989;8:2071. [Google Scholar]
- 26.Shannon R. Acta Crystallographica Section A. 1976;32:751. [Google Scholar]
- 27.Shi S, Szostak R, Szostak M. Org Biomol Chem. 2016;14:9151. doi: 10.1039/c6ob01621a. [DOI] [PubMed] [Google Scholar]
- 28.Critchfield FE, Gibson JA, Hall JL. J Am Chem Soc. 1953;75:6044. [Google Scholar]
- 29.Evans WJ, Gummersheimer TS, Ziller JW. J Am Chem Soc. 1995;117:8999. [Google Scholar]
- 30.We thank a reviewer for raising this issue and encouraging us to address it.
- 31.Wulfsberg G. Principles of Descriptive Inorganic Chemistry. Brooks/Cole; Belmont, CA: 1987. pp. 146–149. [Google Scholar]
- 32.(a) Value calculated in SI Section 6.1.1. from experimental values, which matches well with the following reference;; Zhang XM. J Org Chem. 1998;63:1872. doi: 10.1021/jo981129i. [DOI] [PubMed] [Google Scholar]
- 33.Reaction done in an NMR tube with 0.024 mM cyclohexene, 0.072 mM SmI2, 3.6 M H2O, 0.6 mL THF-d8.
- 34.(a) Free Radicals. Vol. 1 Wiley; New York, N.Y., U.S.A.: 1973. [Google Scholar]; (b) Especially KU. Ingold. 1:69. chap. 2. and G. A. Russell, vol. 1, chap. 7, pp. 283-293. [Google Scholar]; (c) Tedder JM. Angew Chem Int Ed. 1982;21:401. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.