

Abstract
Nonalcoholic fatty liver disease is growing in prevalence worldwide. It is started by the presence of macrosteatosis on liver histology but is often clinically asymptomatic. However, it can progress into nonalcoholic steatohepatitis which is a more severe form of liver disease characterized by inflammation and fibrosis. Further progression leads to cirrhosis, which predisposes patients to hepatocellular carcinoma or liver failure. The mechanism by which simple steatosis progresses to steatohepatitis is not entirely clear. However, multiple pathways have been proposed. A common link amongst many of these pathways is disruption of the homeostasis of bile acids. Other than aiding in the absorption of lipids and lipid-soluble vitamins, bile acids act as ligands. For example, they bind to farnesoid X receptor, which is critically involved in many of the pathways responsible for maintaining bile acid, glucose, and lipid homeostasis. Alterations to these pathways can lead to deregulation in energy balance and increased inflammation and fibrosis. Repeated insults over time may be the key to development of steatohepatitis. For this reason, current drugs target aspects of these pathways to try to reduce and halt inflammation and fibrosis. This review will focus on the role of bile acids in these various pathways and how changes in these pathways may result in steatohepatitis. While there is no approved pharmaceutical treatment for either hepatic steatosis or steatohepatitis, this review will also touch upon the multitude of potential therapies.
Keywords: Bile acids, Nonalcoholic fatty liver disease (NAFLD), Nonalcoholic steatohepatitis (NASH), Farnesoid X receptor (FXR), Gut-liver crosstalk, Enterohepatic circulation
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is growing in prevalence worldwide. Currently, it is reported to affect about 30% of the population in the United States (Younossi et al., 2016). Yet, prevalence is likely higher than reported since NAFLD is asymptomatic and requires a tissue biopsy for diagnosis. Prevalence is also increasing in adolescents and children, with approximately 10–20% of this population affected (Temple et al., 2016). NAFLD is a spectrum of diseases ranging from simple steatosis, nonalcoholic steatohepatitis (NASH), and fibrosis. Simple steatosis can progress into NASH, which is a more severe form of liver disease marked by the presence of hepatocyte ballooning and inflammation. The prevalence of NASH in the developed world is at least 2–3% (Satapathy and Sanyal, 2015). About 30% of patients with NAFLD are estimated to develop NASH (Younossi et al., 2016). The mechanism by which this occurs is not well known. NASH can subsequently progress into liver cirrhosis and hepatocellular carcinoma (HCC). Currently, the second most common indication for liver transplants in the United States is HCC secondary to NASH but this is expected to become the number one indication in the near future (Wong et al., 2014).
Due to the growing prevalence of NAFLD, there will be also an inevitable increase in the prevalence of NASH, liver cirrhosis, and HCC. Except for life style modification, no therapies exist to halt or reverse NAFLD or NASH. However, there is a lot of interest in discovering such treatments, since curing patients of NAFLD would not only prevent NASH-associated HCC, but it would also improve the multitude of comorbidities often associated with NAFLD. NAFLD and NASH often occur in conjunction with obesity, hypertension, dyslipidemia, and insulin resistance. Patients with NAFLD or NASH are also at increased risk of cardiovascular disease. Thus, understanding the mechanism behind the progression of NAFLD to NASH is crucial as this would offer some insights into potential targets for the development of drug therapies that can successfully reverse or halt NAFLD progression.
Bile acids (BAs) are well known for their role in fat absorption (Hofmann, 1963). However, they also act as signaling molecules involved in a variety of pathways that regulate BA, glucose, and lipid homeostasis (Patti et al., 2009; Qi et al., 2015; Watanabe et al., 2011). For this reason, pathways linked to BAs have been implicated as targets for NAFLD and NASH drug therapies. While several drugs have been developed, their success has been variable and further research still needs to be done to develop better, more reliable therapies.
2. NAFLD and NASH
Obesity predisposes individuals to the development of a fatty liver. With obesity on the rise, it is not surprising that NAFLD is becoming more prevalent. NAFLD, however, can develop in patients with normal or lean body weight. NAFLD is defined by the presence of macrovesicular fat accumulation in more than 5% of hepatocytes in patients who consume less than 20 grams of alcohol per day (Yuan and Bambha, 2015). Alterations in lipid metabolism that ultimately lead to increased fat accumulation by hepatocytes lead to steatosis.
NASH is marked by the addition of lobular inflammation and hepatic ballooning to macrovesicular fat accumulation (Ludwig et al., 1980). NASH can progress to liver fibrosis and cirrhosis. Briefly, liver fibrosis develops secondary to an imbalance in extracellular matrix (ECM) synthesis and degradation (Ebrahimi et al., 2016). Hepatic stellate cells have been implicated in this process since they are the major contributors of the deposition of ECM in the liver (Ebrahimi et al., 2016). With increasing fibrosis, the liver becomes grossly smaller, develops nodules, and liver function is compromised. Cirrhosis is characterized by irreversible hepatic scarring and is a risk factor for the development of HCC. The only potentially curative treatment for advanced, non-metastatic HCC is liver transplantation.
The exact mechanism of progression of simple steatosis to NASH is still unclear but is sure to involve a complex interplay of multiple factors and pathways among adipose tissue, the liver, and the gastrointestinal system. Multiple mechanisms have been proposed (Ebrahimi et al., 2016; Magee et al., 2016; Serviddio et al., 2016) but it is likely that progression of NAFLD is dependent on repeated hepatic insults via several different pathways (Buzzetti et al., 2016; Day and James, 1998). For example, steatosis may start with insulin resistance and obesity, which predispose to higher hepatic lipid levels. Increased lipids in the hepatocytes results in increased oxidative stress and cellular damage. Adipose tissue releases cytokines to increase inflammation and fibrosis. Gut microbiota is also believed to play a role in the development of NASH via alterations in the BA pool level and composition (Jiang et al., 2015; Liu et al., 2016b; Mei et al., 2015; Wang et al., 2016). Recently, vitamin D deficiency has been proposed as another possible factor as mice fed a high fat diet (HFD) exhibited increased steatosis and hepatic ballooning when coupled with a vitamin D deficiency (Kong et al., 2014).
There are undoubtedly numerous more pathways yet to be discovered that could help explain the link between simple steatosis and NASH, but many of these proposed pathways involve BAs. Total fasting and post-prandial serum BAs are increased in patients with NASH compared to patients with healthy livers (Ferslew et al., 2015). In fact, it has been proposed that patients with steatohepatitis have a shift in BA composition such that there is an increase in taurine- and glycine-conjugated BAs and increased secondary BAs (Ferslew et al., 2015; Lake et al., 2013). Another study demonstrated altered BA composition in rats fed a HFD (Suzuki et al., 2013). Secondary BAs can have harmful effects (Bartram et al., 1998; Ridlon et al., 2013) and for this reason, an increase in them may very well contribute to repeated insults of inflammation that ultimately contributes to the progression to NASH.
While NAFLD itself is asymptomatic and does not often affect liver function, it can progress into end-stage liver disease. For this reason, much research is being devoted towards finding a drug that can successfully stop NAFLD progression and, ideally, reverse its progress. To do this, a good grasp on the mechanism behind NASH development is crucial. A common factor involved with several of the pathways believed to contribute to NAFLD progression is BAs. BAs are well known for their role in lipid absorption, but they also act as ligands of various receptors to regulate BA, glucose, and lipid homeostasis. For this reason, BAs and the pathways in which they are involved are important targets for NAFLD treatments.
3. BA Synthesis
BAs are amphipathic molecules synthesized from cholesterol in the liver and are a component of bile. Bile is stored in the gallbladder and upon food consumption, released into the duodenum in response to cholecystokinin (CCK). BAs aid in lipid emulsification and absorption of fat and fat-soluble vitamins.
BAs are synthesized via one of two pathways: the classical pathway (or neutral pathway) and the alternative pathway (or acidic pathway) (Zhu et al., 2016). The majority (75%) of BAs are synthesized under the classical pathway in hepatocytes (Asgharpour et al., 2015). The first step in this pathway, which is the rate-limiting step, is catalyzed by the enzyme cholesterol 7α-hydroxylase (CYP7A1) to produce 7α-hydroxycholesterol (Russell and Setchell, 1992). This enzyme is found exclusively in the liver. The BA produced via this pathway in humans is cholic acid (CA). The enzyme sterol 27-hydroxylase (CYP27A1) initiates the first step in the alternative pathway. CYP27A1 is a mitochondrial enzyme more widely distributed and found in macrophages and various tissues. The end products of this pathway include 25-hydroxycholesterol and 27-hydroxycholesterol. Chenodeoxycholic acid (CDCA) is synthesized via the alternative pathway. CA and CDCA are termed primary BAs. In mice, CDCA is further converted to muricholic acid (MCA) by Cyp2c70 and therefore the murine primary BAs are CA and MCA (Takahashi et al., 2016). The primary BAs are conjugated often with either glycine or taurine in humans or taurine in rodents, via the enzyme bile acid-CoA:amino acid N-acyltransferase (BAAT) (Johnson et al., 1991; Killenberg and Jordan, 1978). Glycine conjugation predominates over taurine conjugation in humans (Johnson et al., 1991).
4. BA Transport
After BAs are synthesized, they are exported into the gallbladder and secreted into the duodenum. Most are recirculated back to the liver from the terminal ileum. The remainder enter the colon where some are reabsorbed back into the liver while others are excreted. This enterohepatic circulation of BAs involves multiple transporters (Figure 1). These transporters are important to note since they are potential targets for therapeutic interventions. Briefly, BAs are actively transported from hepatocytes into the bile duct by canalicular BA transporters. Bile salt export pump (BSEP) is the main BA efflux transporter. Multidrug resistance-associated protein 2 (MRP2; ABCC2) belongs to the ATP binding cassette (ABC) superfamily of transporter proteins. It transports organic anions from hepatocytes into the bile duct to become a part of bile (Konig et al., 1999). As BAs are shuttled towards the gallbladder, they are combined with various substances that are excreted from the liver, including cholesterol, phospholipids, and water, to form bile, which is stored in the gallbladder. Bile is excreted into the duodenum after ingestion of a meal rich in lipids.
Figure 1. Enterohepatic circulation of BAs.
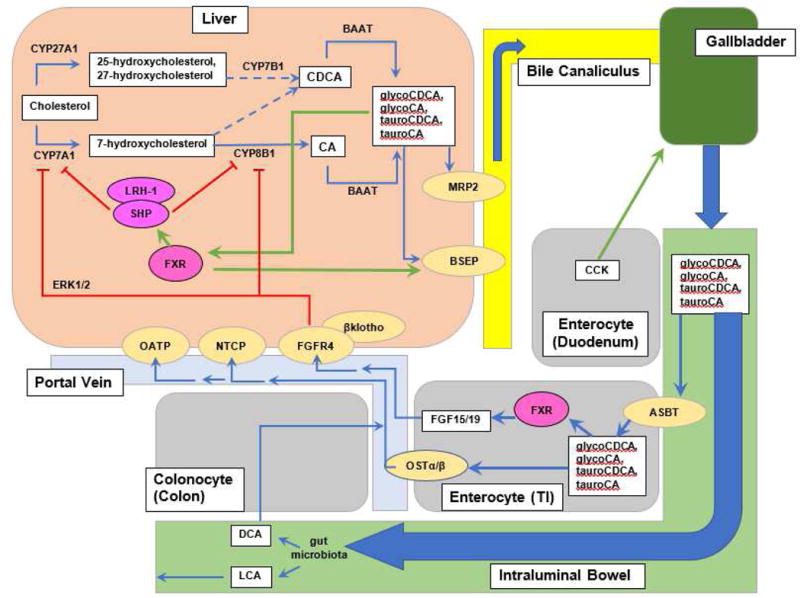
BAs are produced from cholesterol, enter bile canaliculi via BSEP and MRP2, empty into the duodenum upon stimulation of the gallbladder with CCK, get mostly reabsorbed in the terminal ileum via ASBT, then shuttled into the portal vein via OSTα/β where they are recycled back to the hepatocyte. Gut microbiota deconjugate BAs into LCA and DCA. LCA is insoluble and excreted in feces but DCA can be reabsorbed into the portal vein and recycled. BAs act on FXR in the liver to induce SHP to downregulate gene expression of CYP7A1/Cyp7a1 and CYP8A1/Cyp8b1. BAs also downregulate their own synthesis via a SHP-independent pathway involving FGF15 (FGF19 in humans) produced in the ileum. FGF15 leads to phosphorylation of FGFR4, which then activates ERK1/2 pathways to downregulate gene expression of CYP7A1/Cyp7a1 and CYP8B1/Cyp8b1. FGFR4 requires a cofactor, βklotho, to function. Green arrows indicate upregulation or stimulation. Red lines indicate downregulation. Broken arrows indicate that multiple steps are involved but not depicted.
Once BAs reach the terminal ileum, the majority (95%) are actively reabsorbed into enterocytes via apical sodium-dependent bile acid transporter (ASBT) (Shneider et al., 1995; Wong et al., 1994) and then exported by organic solute transporters (OSTα and OSTβ) to return to the liver via portal circulation (Dawson et al., 2005). Once in the portal vein, BAs are actively transported into hepatocytes via sodium taurocholate cotransporting polypeptide (NTCP) (Hagenbuch and Meier, 1994) and organic anion-transporting polypeptide (OATP) (Stieger and Hagenbuch, 2016). The remaining BAs enter the colon where bacteria deconjugate and dehydroxylate them to form the main secondary BAs, deoxycholic acid (DCA) and lithocholic acid (LCA). LCA is insoluble and lost in stool. In contrast, DCA can be passively reabsorbed from the colon and recycled (Dowling, 1973). Some BAs can be taken up into the portal circulation but not recycled into hepatocytes; these are excreted in urine.
Under normal conditions, BA pool size remains in a steady state. This is achieved via multiple “checkpoints” throughout the enterohepatic circulation. Upregulation and downregulation of the various above-listed transporter genes can increase BA recycling or decrease the BA pool via excretion. The balance is important, as BAs are not just molecules that aid in lipid absorption but play crucial roles in regulating lipid and glucose homeostasis.
5. BAs as Ligands
5.1 Nuclear Hormone Receptors
BA regulation of other pathways occurs through BA binding and mainly activation of nuclear hormone receptors (NHR), such as farnesoid X receptor (FXR), vitamin D receptor (VDR), and pregnane X receptor (PXR) (Guo et al., 2003; Makishima et al., 2002; Makishima et al., 1999). Activation of these NHRs as transcription factors requires that they interact with retinoid X receptor (RXR) as a heterodimer (Lu et al., 2000). They then act on various regulatory regions of genes to up- and down-regulate transcription by binding to hormone response elements (HRE), which may be direct, inverted, or everted repeats of the sequence ~AGGTCA separated by a variable number of nucleotides (Edwards et al., 2002). FXR, for example, normally binds to an inverted repeat separated by 1 nucleotide (IR-1), but can also bind to an everted repeat separated by 2 nucleotides (ER2) (Thomas et al., 2010). Most BAs and the secondary BA, LCA, are known to bind to FXR and PXR, respectively (Edwards et al., 2002).
5.2 FXR and TGR5
FXR is a NHR known to play a crucial role in BA, glucose, and lipid regulation. It is most highly expressed in the liver, ileum, kidneys, and adrenal glands and is most strongly activated by CDCA, followed by DCA, CA, and LCA (Makishima et al., 1999). While LCA is a weak activator of FXR, it interestingly strongly downregulates FXR activation in the presence of the FXR agonist GW4064 (Yu et al., 2002) and thus may be termed an FXR partial agonist. A study using tissue-specific intestinal and liver FXR knockout (KO) mice evaluated their specific roles in BA metabolism and showed that the tissue-specific location of FXR alters the mechanistic role they play in maintaining BA homeostasis (Kim et al., 2007). Intestinal-specific FXR but not liver-specific FXR is required for suppression of the Cyp7a1 gene expression via induction of Fgf15 transcription, whereas liver-specific FXR plays a similarly important role in the repression of the expression of the Cyp8b1 gene (Kim et al., 2007).
BAs, acting as ligands on FXR, clearly help regulate the BA pool and lipid metabolism. FXR KO mice exhibit increased serum BAs, an increased BA pool, and increased serum lipid profile compared to wild-type (WT) mice (Sinal et al., 2000). FXR also regulates the expression of various BA transporters, including NTCP, BSEP, and ileal BA transporters, especially OSTβ (Kast et al., 2002; Sinal et al., 2000). Furthermore, activation of FXR leads to increased transcription of ileal bile acid binding protein (I-BABP) (Hwang et al., 2002).
BAs are well known to suppress their own synthesis by various mechanisms. One important mechanism is via activation of FXR. An early report showed that in the liver, activation of FXR by BAs leads to upregulation of the short heterodimer partner (SHP) encoded by the NR0B2 gene (Goodwin et al., 2000). SHP then interacts with liver receptor homolog-1 (LRH-1) to repress CYP7A1 gene transcription. LRH-1 is an orphan nuclear receptor that acts as a transcription factor and is critical for the gene expression of CYP7A1 and CYP8B1 (Goodwin et al., 2000; Lu et al., 2000; Xu et al., 2002).
Recently, it is apparent that an intestinal pathway is critical in regulating BA synthesis in the liver. Fibroblast growth factor receptor 4 (FGFR4) is a transmembrane tyrosine kinase receptor activated by fibroblast growth factor 15 (FGF15; FGF19-human homolog) and this activation plays a critical role in BA, lipid, and glucose metabolism (Chen et al., 2011; Huang et al., 2007; Inagaki et al., 2005). FGF15/19 are produced by ileal enterocytes in response to BAs and FXR highly induces the transcription of FGF15/19 (Inagaki et al., 2005). FGF15/19 enters the liver via the portal circulation and leads to phosphorylation of FGFR4. This results in activation of the extracellular signal-regulated kinases (ERK1/2) to repress Cyp7a1/CYP7A1 gene expression (Inagaki et al., 2005; Kong et al., 2012; Song et al., 2009). Under physiological condition, this intestine-initiated pathway appears to be the major pathway to suppress Cyp7a1 gene expression after FXR activation (Kong et al., 2012). FGFR4 is believed to play a role in NAFLD progression, as FGFR4 KO mice are resistant to the development of hepatic steatosis (Huang et al., 2007). This is interesting and is opposite to a study showing that FGF19 increases fatty acid oxidation via repression of acetyl-CoA carboxylase 2 (ACC2) (Schreuder et al., 2010), as ACC2 normally decreases mitochondrial fatty acid oxidation (Schreuder et al., 2010).
TGR5 is a G-protein coupled membrane receptor that BAs activate and will only be briefly discussed here. Among the main primary and secondary BAs, TGR5 is most strongly activated by LCA (Li et al., 2013). TGR5 is widely expressed and found in the gallbladder, ileum, colon, liver, brown adipose tissue (BAT), nervous system, and muscle (Li et al., 2013). Like FXR, TGR5 is involved in BA, glucose, and lipid homeostasis but it also plays a role in increasing energy expenditure via browning of white adipose tissue (WAT) and inducing BAT gene expression in thermogenesis (Scheja and Heeren, 2016). TGR5 KO mice did not significantly change Cyp7a1 gene expression but had altered BA composition (Donepudi et al., 2016). Another study in which vertical sleeve gastrectomy was performed on TGR5 KO mice demonstrates that TGR5 is important in improved glucose control, decreased hepatic steatosis, and increased energy expenditure post-surgery (Ding et al., 2016). TGR5 also has anti-inflammatory properties as TGR5 inhibits activation of NF-kβ (Wang et al., 2011).
BAs act as ligands of FXR and TGR5 to regulate crucial pathways involved in BA, glucose, and lipid homeostasis. Many of these pathways are also believed to play a role in the development of NAFLD and subsequent progression to NASH. For this reason, BAs and their associated pathways are desirable targets for drug therapies.
6. BAs and Glucose, Cholesterol and Lipid Metabolism Pathways
6.1 BAs and Glucose Metabolism
BAs help regulate glucose metabolism via FXR and TGR5, but the mechanism by which this occurs is still unclear, as studies have shown dissimilar results. BA activation of TGR5 results in increased secretion of GLP-1 and decreased insulin resistance in obese mice (Thomas et al., 2009). FXR decreases hepatic gluconeogenesis, glycolysis, and increases glycogen synthesis (Jiao et al., 2015). Type II diabetes is associated with increased gluconeogenesis and, therefore, BAs, via FXR, may play a role in the development of medications to combat type II diabetes. Phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) are the first and last enzymes, respectively, in the gluconeogenesis pathway (Zhang et al., 2006a). FXR mediates glucose metabolism via modulation of PEPCK and G6P expression in addition to upregulation of the expression of genes involved with glycogen synthesis (Zarrinpar and Loomba, 2012). The FXR-SHP pathway is believed to be responsible for decreased PEPCK and G6P activity (Jiao et al., 2015). In contrast, another study showed that BAs increase hepatic expression of PEPCK via FXR (Stayrook et al., 2005). Likewise, some studies show that FXR KO mice have increased insulin resistance (Cariou et al., 2006; Ma et al., 2006) whereas other studies suggest the opposite (Prawitt et al., 2011; Stayrook et al., 2005). Another study demonstrates reduced glucose tolerance in FXR KO mice following sleeve gastrectomy compared with WT mice (Ryan et al., 2014). These contrasting results suggest that FXR is likely involved in many pathways of the regulation of gluconeogenesis and development of type II diabetes.
BA regulation of insulin sensitivity involves WAT cross-talk. Adiponectin is an adipokine produced in adipocytes that has anti-inflammatory and anti-fibrotic properties. Adiponectin stimulates uptake of glucose in multiple tissues, which then decreases gluconeogenesis in the liver and inhibits production of pro-inflammatory cytokines like IL-6 (Silva et al., 2014). Adiponectin is believed to be negatively regulated by BAs, as patients with NASH had high levels of BAs but low levels of adiponectin (Bechmann et al., 2013). One study found increased levels of adiponectin in patients with cirrhosis possibly indicating that adiponectin levels begin to increase again after NASH progression into fibrosis and cirrhosis (Balmer et al., 2010). Adiponectin also activates ceramidase, which has recently been implicated in the progression of NAFLD to NASH (Dasarathy et al., 2011; Holland et al., 2013; Silva et al., 2014). Interplay between BAs and adipokines needs to be further determined.
6.2 BAs and Cholesterol Metabolism
Since BAs are produced from cholesterol, it is crucial to understand the mechanisms behind cholesterol homeostasis, as the availability of cholesterol can affect the BA pool. Cholesterol can be synthesized de novo from acetate and/or taken up into the liver by low-density lipoprotein (LDL) (Russell and Setchell, 1992). One important mechanism of cholesterol degradation is via conversion to BAs. In fact, cholesterol stimulates Cyp7a1 transcription in rodents (Wang et al., 1999). BA binding resins also reduce cholesterol levels and increase Cyp7a1/CYP7A1 expression in both rodents and humans.
Cholesterol can also enter the liver in the form of high-density lipoprotein (HDL), a process termed reverse cholesterol transport. ABCA1 is the transporter responsible for the first step in reverse cholesterol transport; it transports cholesterol from the peripheral tissues to apolipoproteins (Oram and Lawn, 2001). Scavenger receptor class B type I (SR-B1) mediates the uptake of cholesteryl esters from HDL into the liver (von Eckardstein et al., 2001). FXR is also known to reduce cholesterol levels via inducing SR-B1 expression to enhance HDL removal from the blood into the liver (Li et al., 2012).
Liver X receptor (LXR) and FXR are transcriptionally activated by oxysterols and BAs, respectively. Administration of RXR agonists in mice has been shown to decrease cholesterol absorption in a dose-dependent manner despite feeding the mice a high-cholesterol diet (Repa et al., 2000). The two mechanisms found to be involved with this process were increased reverse cholesterol transport via LXR-RXR and decreased BA synthesis via FXR-RXR (Repa et al., 2000). Increased expression of LXR also leads to increased expression and activity of ABCG5/G8, a pair of plasma membrane transporters that transports cholesterol out of enterocytes into the intestinal lumen (Wang et al., 1999). To maintain cholesterol homeostasis, the expression of genes involved in cholesterol synthesis were found to be upregulated (Wang et al., 1999).
6.3 BAs and Lipid Metabolism
BAs are known to facilitate lipid absorption from the intestines and activation of NHRs and other signaling pathways by BAs affects lipid homeostasis. FXR induction of SHP leads to decreased transcription of sterol regulatory element binding protein 1c (SREBP1c), a gene involved in FA, triglyceride (TG), and very low density lipoprotein (VLDL) synthesis (Watanabe et al., 2004). Recently, BAs can regulate lipid metabolism via activation of sphingosine-1 phosphate receptor 2 (S1PR2). S1PR2 KO mice fed a HFD were found to have increased hepatic lipid accumulation (Studer et al., 2012). Compared to intestinal FXR, hepatic FXR is more crucial in the prevention of hepatic lipid accumulation (Schmitt et al., 2015).
Taurine-conjugated CA (TCA) activates the S1PR2-ERK1/2 pathway and protein kinase B (AKT) pathways, which have been shown to be involved in glucose and lipid metabolism (Kwong et al., 2015; Studer et al., 2012). Treatment of human hepatocytes with CDCA showed CDCA-dependent changes in expression of genes that regulate lipid homeostasis, such as LDLR, APOL3, FABP3, and SLC27A2 (Krattinger et al., 2016). Similarly, CDCA, in addition to LCA and DCA, increases LDL receptor gene expression via a MAP kinase pathway (Nakahara et al., 2002). Changes in the expression of several microRNAs (miRNAs) involved in lipid homeostasis were also identified, which could partially account for the effects of CDCA (Krattinger et al., 2016).
HDL is known to help prevent the development of heart disease. FXR has been linked to changes in HDL, but the relationship is controversial. A protective role of FXR in atherosclerosis formation has been reported in male mice (Guo et al., 2006; Zhang et al., 2006b). In a study showing SR-B1 induction by FXR, FXR KO mice exhibit increased total and serum HDL cholesterol suggesting that reverse cholesterol transport is disrupted without FXR (Li et al., 2012). In contrast, some studies have suggested the opposite. A study using FXR KO mice demonstrated increased HDL and phospholipids but decreased ApoA-1 levels (Sinal et al., 2000). ApoA-1 is a component of HDL that promotes reverse cholesterol transport, thereby decreasing serum cholesterol levels. Another study showed that BAs downregulate ApoA-1 expression via FXR to decrease serum HDL (Claudel et al., 2002). Results of this study suggests that FXR antagonists can increase serum HDL levels and be cardioprotective, although the exact mechanism by which this would occur likely still involves other pathways not yet discovered.
7. BAs and NAFLD/NASH
BAs have emerged as a therapeutic target for NAFLD prevention and/or treatment. Alterations in both total BA levels and composition have been noted in both rodents and humans with NAFLD/NASH (Aranha et al., 2008; Lake et al., 2013; Tanaka et al., 2012). Livers from rats fed a HFD were found to have higher glycine-conjugated BAs rather than taurine-conjugated BAs (Jia et al., 2014). Further, the total glycine-conjugated BA level positively correlated with macrovesicular steatosis score (Jia et al., 2014). Normally, rodents have higher taurine-conjugated BAs so this suggests that a shift in BA conjugation exists in NAFLD pathogenesis. A change in BA composition is also observed in humans. In comparing liver samples from humans with NAFLD, NASH, and healthy livers, mRNA levels of CYP8B1 were decreased while mRNA levels of CYP7B1 were increased in NASH livers suggesting that there may be a shift towards the alternative pathway as patients progress towards NASH (Lake et al., 2013). In contrast, metabolomic analysis has revealed significantly increased serum levels of glycocholate, taurocholate, and glycochenodeoxycholate in patients with NASH compared with healthy patients (Kalhan et al., 2011).
BAs may be protective against NAFLD progression through the activation of FXR. There is increased NAFLD and NASH development in FXR KO mice in a HFD-induced NAFLD/NASH model (Kong et al., 2009). Administration of CA and ursodeoxycholic acid (UDCA) to ob/ob mice led to marked improvement in hepatic steatosis (Quintero et al., 2014). Similarly, administration of FA-BA conjugates to mice and rats with diet-induced NAFLD showed decreased hepatic fat (Leikin-Frenkel et al., 2008).
A recent study in humans found decreased protein levels of FXR, SHP, and NTCP in patients with NASH compared to patients with NAFLD suggesting that FXR plays a protective role in the progression of NAFLD to NASH (Aguilar-Olivos et al., 2015). Studies using mice models have also demonstrated this protection (Bjursell et al., 2013; Zhang et al., 2006a). In comparison to WT mice, FXR KO mice were leaner, more resistant to weight gain, and have better glucose tolerance but also have higher liver weights, serum total BAs, ALT, and increased hepatic steatosis and inflammation (Bjursell et al., 2013). Thus, while FXR seems to mediate weight gain, it also exhibits hepatoprotective effects and FXR agonists may play a role in preventing progression of NAFLD. However, a study evaluating the effects of GW4064, a synthetic FXR agonist, showed increased weight gain and insulin resistance in mice fed a HFD but reversion of these effects when diets were supplemented with CA (Watanabe et al., 2011). This suggests that FXR agonists may not in fact be protective and that, as mentioned above, increased BAs instead play a protective role against NASH development.
Increased BAs due to cholestasis can lead to an upregulation of FXR and activation of lipogenic genes. For example, livers from ob/ob mice revealed development of hepatic steatosis without fibrosis, decreased expression of Oatp1a1, increased cholestasis, and upregulation of FXR protein levels (Martin et al., 2010). Similarly, rat hepatocytes that were treated with oleic acid to develop steatosis were more susceptible to apoptosis when exposed to glycoCDCA (Pusl et al., 2008).
Alterations in fatty acid oxidation have been implicated as a potential pathway involved in the development of NASH. A downstream target of FXR is peroxisome proliferator-activated receptor-α (PPARα). PPARα increases hepatic fatty acid oxidation via many known pathways and a novel one is through induction of FGF21. FGF21 serum levels are higher in patients with NASH (Dasarathy et al., 2011). Intralipid is an intravenous lipid emulsion that is used for patients requiring extended parenteral nutrition. When humans are infused with intralipid, the higher plasma concentration of fatty acids results in a greater uptake of fatty acids by the liver, which leads to higher hepatic fatty acid oxidation. There is also an increase in FGF21 and reactive oxygen products that lead to hepatic injury, but only in patients with NASH (Dasarathy et al., 2011). Along with this, it has been shown that with increased hepatic oxidation, there is an increase in the plasma level of ceramides (Dasarathy et al., 2011). Ceramides mediate apoptotic pathways (Kuzmenko and Klimentyeva, 2016) and, therefore, can increase hepatocyte apoptosis.
Diabetes and obesity are major risk factors for NAFLD. Increased insulin levels may alter BA composition via suppression of CYP8B1 activity (Ishida et al., 2000). Lipid and glucose metabolism have been shown to be altered in rats fed a HFD (Pozzo et al., 2016). These rats exhibited hepatic steatosis resembling NAFLD, had increased inflammatory markers (TNFα, TNFβ), decreased glycogen stores, increased expression of the genes encoding SREBP-1c, LXRα, PPARα, Cyp7a1, and Cyp8b1, and decreased expression of SREBP2C, LDLR, ACC1, BSEP, NTCP, and SHP (Pozzo et al., 2016). Thus, diabetes and regular consumption of food rich in fats may be the inciting factors towards development of NAFLD. BAs and FXR no doubt play a crucial role in the development of NAFLD and NASH. However, despite multiple studies conducted to date, the exact roles that they play remain undetermined. Developing drugs to treat NAFLD and NASH without a clear understanding of the mechanisms of NAFLD progression is difficult. Hence, several drugs have been proposed and studied with variable results.
8. Current Therapies
To date, no medications can reverse NAFLD or NASH. Again, understanding the mechanism behind NAFLD progression is an important step towards development of medications. BAs regulate multiple pathways that likely play important roles in NAFLD progression. Thus, there are various targets for drugs (Figure 2). However, as one can imagine, there is an intricate interplay among the different pathways to maintain BA, lipid, and glucose homeostasis and a disruption in one pathway may lead to unexpected and undesired impacts on other pathways. For instance, while an FGFR4 deficiency prevents hepatic steatosis, it also leads to increased systemic lipids and insulin resistance (Huang et al., 2007). Here, the various drugs currently being studied are reviewed.
Figure 2. Potential pathways of NAFLD and NASH development and therapeutic targets for NAFLD/NASH treatment.
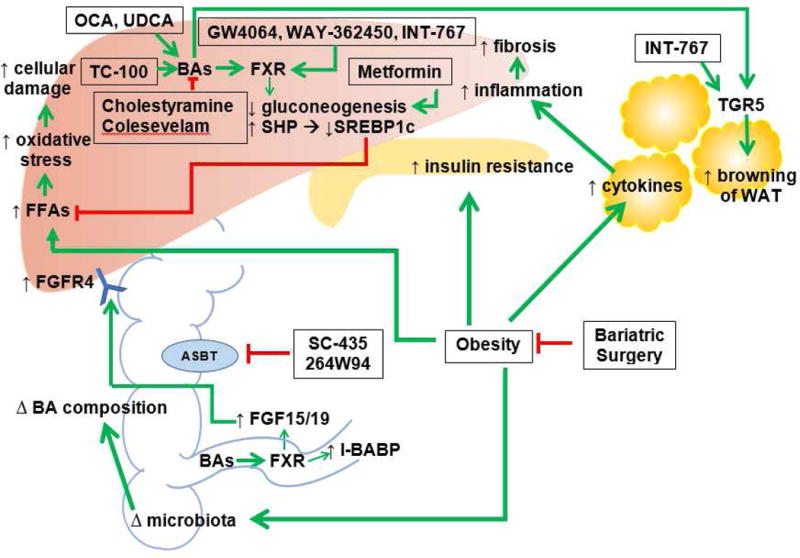
The exact mechanism for NAFLD development and progression to NASH is unknown. However, obesity and insulin resistance/diabetes are believed to be two significant contributing factors. Obesity leads to increased hepatic lipids, insulin resistance, cytokines in WAT, and changes in intestinal flora. Increased hepatic lipids results in increased oxidative stress and apoptosis. Insulin resistance can result in diabetes. Cytokines result in increased hepatic inflammation and with repeated insults, can lead to fibrosis. Changes in microbiota can cause changes in BA composition that can disrupt many pathways, since BAs are important regulators of lipid and glucose metabolism. BA conjugates and derivatives (such as OCA, UDCA, and TC-100) decrease hepatic gluconeogenesis and hepatic lipids. BA conjugates and derivatives can increase hepatic and intestinal FXR. Increased intestinal FXR can lead to increased FGF15/19 and increased FGFR4. BA binding resins (such as cholestyramine and colesevelam), are medications being used by patients with diabetes and despite having an opposite effect on BAs compared to BA conjugates, have been shown to help lower systemic lipids. However, they do not currently have a role in improving hepatic steatosis. Metformin, also used in patients with diabetes, decreases gluconeogenesis. FXR and TGR5 agonists have shown promising results with improvement in glucose tolerance and decrease in hepatic inflammation. ASBT inhibitors (such as SC-435 and 264W94) also show promising results, with decreased expression of SREBP1c and increased FXR (not depicted). Hepatic steatosis and glucose intolerance were demonstrated to be improved in rodent studies using ASBT inhibitors. Finally, bariatric surgery is emerging as another potential therapy for NAFLD and NASH. Bariatric surgery not only leads to weight loss, but has been shown to affect microbiota and improve insulin resistance independent of weight loss (not depicted).
8.1 BA Binding Resins and Other Drugs Approved for Other Diseases
Medications that may influence the total BA pool or alter glucose or lipid metabolism are expected to play a role in halting or reversing NAFLD and NASH. BA binding resins, such as cholestyramine and colesevelam, are useful medications for patients with type II diabetes due to their lipid-lowering effects. Cholestyramine improved glucose tolerance, but leptin-deficient mice were also found to have increased ALT and no changes in steatosis or inflammation on histology (Solis et al., 2013). In contrast, use of sevelamer, a phosphate binder, in mice with NAFLD decreased hepatic steatosis and pro-inflammatory cytokines (McGettigan et al., 2016). Metformin, a medication that decreases hepatic glucose production, has been shown to improve glucose tolerance in patients with liver disease, but it does not stop the progression of NAFLD, perhaps because it only protects hepatocytes against BA-induced apoptosis but no other pathways of apoptosis (Woudenberg-Vrenken et al., 2013).
A small study involving non-obese patients with NAFLD found improved ALT levels with the use of the cholesterol inhibitor, ezetimibe, but no decrease in hepatic steatosis after twelve months (as evaluated via ultrasonography) (Enjoji et al., 2010). N-3 polyunsaturated FAs (PUFAs) may be protective against HFD-induced fatty liver disease as fat-1 mice, which produce N-3 PUFAs, have improved hyperlipidemia, decreased hepatic steatosis and fibrosis, and decreased serum cholesterol and TGs (Kim et al., 2014).
8.2 FXR and TGR5 Agonists
As already discussed, FXR and TGR5 are involved in BA, lipid, and glucose homeostasis. Moderate increases in BA have been shown in studies to be hepatoprotective, decreasing hepatic inflammation and fibrosis, and therefore may play a role in the treatment of NAFLD (Liu et al., 2016a). To this end, FXR and TGR5 agonists have been developed. GW4064 is an FXR specific agonist (Maloney et al., 2000) that has been shown to lower serum glucose and triglyceride levels in mice (Zhang et al., 2006a). WAY-362450 is an FXR agonist that decreases hepatic inflammation in mice, as evidenced by a decrease in AST, ALT, expression of inflammatory genes, and hepatic collagen (Zhang et al., 2009). INT-767 is a dual FXR and TGR5 agonist with lipid-lowering effects (Rizzo et al., 2010), and its use in leptin deficient mice improved hepatic steatosis and inflammation perhaps via increased intrahepatic monocytes and production of IL-10 (McMahan et al., 2013).
8.3 Obeticholic Acid
Obeticholic acid (OCA, INT-747) is a CDCA derivative that selectively and potently activates FXR. In a multicenter prospective randomized trial, patients with NASH received either OCA or a placebo and those who received OCA had improved NAFLD activity scores by at least 2 points compared with the placebo group (Neuschwander-Tetri et al., 2015). However, lipid profiles of patients receiving OCA were worse, with increased total cholesterol and LDL but decreased HDL (Neuschwander-Tetri et al., 2015). In a phase II trial comparing placebo with OCA in patients with NAFLD and type II diabetes, OCA was associated with decreased ALT and GGT, improved insulin sensitivity, and decreased markers of liver fibrosis via an FXR-dependent pathway involving FGF19 (Mudaliar et al., 2013). While OCA shows promise in reverting NASH and increasing insulin sensitivity, it is also associated with a worsening lipid profile. Phase III clinical trials evaluating the effect of OCA on NASH have begun.
8.4 BA Conjugates
Administration of BAs has also been studied. A study evaluating chenodeoxycholyl-arginine ethyl ester conjugate (CDCArg) showed promising results in the treatment of NASH as it was found to reduce hepatic steatosis and lobular inflammation and to suppress SREBP1 activity in HFD fed mice (Voloshin et al., 2014). Recently, 3α, 7α,11β-trihydroxy-6α-ethyl-5β-cholan-24-oic acid (TC-100), which is a hydrophilic semi-synthetic BA derivative that specifically binds to FXR, has been synthesized and studied for its in vivo effects. It showed greater binding ability to FXR compared to OCA and similarly regulated FXR target genes, such as Cyp7a1, Shp, Bsep, and Ostα (Pellicciari et al., 2016). It has the potential to decrease BA toxicity via increasing BA efflux from the liver as it was noted to cause higher expression of Bsep than OCA and CDCA (Pellicciari et al., 2016).
A study from Israel examined the role of a fatty acid-BA conjugate, Aramchol (3β-arachidyl-amido, 7α–12α-dihydroxy, 5β-cholan-24-oic acid), in patients with NAFLD (Safadi et al., 2014). Patients receiving a high dose of Aramchol had significantly lower liver fat content and increased adiponectin levels (Safadi et al., 2014). However, no histological end points were assessed.
8.5 Ursodeoxycholic Acid
Due to the hepatoprotective effects seen in cholestasis, UDCA was thought to play a role in NASH. However, multiple trials have shown a lack of improvement in NAFLD or NASH and it is not currently recommended for use in patients with NAFLD or NASH (Dufour et al., 2006; Haedrich and Dufour, 2011; Leuschner et al., 2010; Lindor et al., 2004; Parikh et al., 2016). C57BL/6 mice fed a HFD given UDCA in conjunction with omega-3 had decreased liver fibrosis and inflammation compared with a group of mice given only omega-3 (Kim et al., 2014). This study also demonstrates decreased mRNA expression of SREBP1c in mice given UDCA and omega-3. Taurine-conjugated UDCA (TUDCA) administered to ob/ob mice resulted in improved hepatic steatosis, decreased serum ALT and AST, and decreased expression of FA synthesis genes, but no improvement in glucose intolerance (Yang et al., 2010). Since NASH is believed to be a progression of NAFLD secondary to increased fibrosis, inflammation, and hepatic lipid deposition, these studies support the notion that UDCA may play a role in reversing NASH.
The use of UDCA alone did not result in any significant improvements on body weights, liver function profile, or liver histology on rats with NASH (Fan et al., 2005). In a randomized controlled trial conducted by Parikh et al., patients with NAFLD were given either vitamin E or UDCA (Parikh et al., 2016). UDCA was not superior to vitamin E when comparing improvements in ALT and NAFLD fibrosis score. Similarly, other randomized controlled clinical trials showed no improvements in histology compared with placebo or vitamin E in patients with NASH given UDCA (Dufour et al., 2006; Leuschner et al., 2010; Lindor et al., 2004). While a randomized controlled study conducted by Ratziu et al. showed significant improvements in ALT and serum markers of fibrosis, this study did not include histological end points (Ratziu et al., 2011). In a more recent study, morbidly obese patients receiving UDCA had decreased serum free fatty acids (FFAs), total and LDL cholesterol, and increased triglycerides and BAs compared with the placebo group (Mueller et al., 2015). BA synthesis was thought to be increased by decreased FGF19 and decreased FXR activation. Further, cholesterol synthesis was increased due to increased SREBP2 and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) (Mueller et al., 2015).
UDCA in conjunction with losartan, an angiotensin receptor blocker (ARB), decreased NAFLD activity score and have had a synergistic role in decreasing hepatic fibrosis in rats via decreased expression of pro-inflammatory cytokines, TGFβ, toll-like receptor 4 (TLR4), and hepatic stellate cells (Namisaki et al., 2016).
8.6 ASBT Inhibitors
Inhibition of the ASBT receptor is another potential mechanism to improve NAFLD and NASH. Administration of an ASBT inhibitor in mice fed a HFD led to increased clearance of BAs in feces, increased hepatic BA synthesis, decreased FGF15, increased I-BABP, and decreased hepatic steatosis (Rao et al., 2016). Furthermore, the hepatic BA pool shifted towards FXR agonistic BAs and SREBP1c expression decreased, but the hepatic steatosis score was not significantly improved in diet-induced obese mice given a short course of an ASBT inhibitor (Rao et al., 2016). Another study using a different ASBT inhibitor, 264W94, demonstrated improved glucose tolerance and upregulation of hepatic FXR in Zucker Diabetic Fatty rats (Chen et al., 2012). Like other therapeutic interventions, ASBT inhibitors appear to show promising results. However, interrupting the enterohepatic circulation of BAs could have undesired effects and, therefore, more studies need to be performed.
8.7 Bariatric Surgery
Although bariatric surgery, which most commonly refers to vertical sleeve gastrectomy and Roux-en-Y gastric bypass, is not currently approved treatment of NAFLD and NASH, it is emerging as a new potential treatment for NAFLD and NASH due to post-surgical improvements in glucose and lipid homeostasis. Its role will only be briefly mentioned in this article. Bariatric surgery increases BA levels, changes BA composition, decreases hepatic steatosis, and improves insulin resistance (Bhutta et al., 2015; Dutia et al., 2015; Myronovych et al., 2014; Noel et al., 2016). SHP is also believed to play a role in preventing liver inflammation after sleeve gastrectomies (Myronovych et al., 2014). However, more human studies need to be done to show histological improvements after bariatric surgery. Only then will NAFLD and NASH be more likely to be approved indications for bariatric surgery.
9. BAs and Microbiota
Bacteria in the distal small bowel and colon break BAs down into secondary BAs, the main ones being DCA and LCA. Thus, gut microbiota can alter BA composition and affect BA metabolism. Results to date have been extensively reviewed (Bashiardes et al., 2016; He et al., 2016; Mouzaki et al., 2013; Mouzaki et al., 2016) and will only be briefly discussed here. One study looked at the role of intestinal FXR on NAFLD and found an increased level of tauro-β-muricholic acid (T-β-MCA) in the ileum in antibiotic- and tempol-treated mice on a HFD (Jiang et al., 2015). T-β-MCA is a known FXR antagonist. The activity of bile salt hydrolase (BSH), an enzyme responsible for breaking down taurine-conjugated BAs, was significantly decreased in antibiotic-treated mice (Jiang et al., 2015). It was also found that levels of certain ceramides in the ileum and serum were decreased, which leads to decreased activity of SREBP1c and subsequently decreased accumulation of hepatic lipids (Jiang et al., 2015). Comparison of fecal BAs in humans with healthy livers, NAFLD, and NASH revealed higher total fecal BAs, higher glycine- and taurine-conjugated LCA in patients, and decreased levels of Bacteriodetes in patients with NASH (Mouzaki et al., 2013; Mouzaki et al., 2016). In contrast, another study showed increased levels of Bacteriodetes and decreased levels of Firmicutes in patients with NASH compared with patients with healthy livers (Zhu et al., 2013). C57BL/6 mice fed a HFD exhibited markedly increased Lactobacillus in fecal samples (Zeng et al., 2013). Many studies on gut microbiota and NAFLD look to quantify and qualify changes in gut microbiota. However, there is still a lack of data regarding a mechanism by which gut microbiota may lead to NAFLD progression and more studies need to be pursued.
10. Conclusion
BAs are involved in multiple pathways to regulate BA, glucose, and lipid metabolism. As discussed, many of these pathways involve BAs acting on FXR and TGR5, as well as other signaling pathways. Changes in BA levels and composition, via alterations in BA receptors, negative and positive feedback mechanisms, and even gut microbiota, therefore can affect other systems. These pathways are intricately involved with one another such that an alteration in one changes the others to maintain a homeostasis. However, repeated insults inevitably result in hepatocyte damage. Thus, the progression of NAFLD to NASH likely involves not just one pathway or one inciting event, but rather repeated insults over time, that lead to an accumulation of hepatic lipids, insulin resistance, initiation of hepatocyte apoptosis, an increase in inflammation, and an increase in extracellular matrix that eventually results in fibrosis. There are currently no approved medications for either steatosis or steatohepatitis, perhaps secondary to the fact that there are in fact multiple pathways involved in steatosis progression. Potential drugs to date include medications already approved and in use for other medical problems, such as ezetimibe, BA derivatives, and NHR agonists. Future directions include looking at alterations in gut microbiota and undergoing bariatric surgery, both of which have roles in quantitative and qualitative changes in BAs. There are undoubtedly more pathways involved and more research needs to be done to understand the mechanism behind steatosis to steatohepatitis progression so that drugs that successfully treat NAFLD and NASH can be developed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar-Olivos NE, Carrillo-Cordova D, Oria-Hernandez J, Sanchez-Valle V, Ponciano-Rodriguez G, Ramirez-Jaramillo M, Chable-Montero F, Chavez-Tapia NC, Uribe M, Mendez-Sanchez N. The nuclear receptor FXR, but not LXR, up-regulates bile acid transporter expression in nonalcoholic fatty liver disease. Ann Hepatol. 2015;14(4):487–493. [PubMed] [Google Scholar]
- Aranha MM, Cortez-Pinto H, Costa A, da Silva IB, Camilo ME, de Moura MC, Rodrigues CM. Bile acid levels are increased in the liver of patients with steatohepatitis. Eur J Gastroenterol Hepatol. 2008;20(6):519–525. doi: 10.1097/MEG.0b013e3282f4710a. [DOI] [PubMed] [Google Scholar]
- Asgharpour A, Kumar D, Sanyal A. Bile acids: emerging role in management of liver diseases. Hepatol Int. 2015;9(4):527–533. doi: 10.1007/s12072-015-9656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Joneli J, Schoepfer A, Stickel F, Thormann W, Dufour JF. Significance of serum adiponectin levels in patients with chronic liver disease. Clin Sci (Lond) 2010;119(10):431–436. doi: 10.1042/CS20100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartram HP, Draenert R, Dusel G, Richter F, Liebscher E, Christl SU, Scheppach W, Kasper H. Effects of sodium selenite on deoxycholic acid-induced hyperproliferation of human colonic mucosa in short-term culture. Cancer Epidemiol Biomarkers Prev. 1998;7(12):1085–1089. [PubMed] [Google Scholar]
- Bashiardes S, Shapiro H, Rozin S, Shibolet O, Elinav E. Non-alcoholic fatty liver and the gut microbiota. Mol Metab. 2016;5(9):782–794. doi: 10.1016/j.molmet.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechmann LP, Kocabayoglu P, Sowa JP, Sydor S, Best J, Schlattjan M, Beilfuss A, Schmitt J, Hannivoort RA, Kilicarslan A, Rust C, Berr F, Tschopp O, Gerken G, Friedman SL, Geier A, Canbay A. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology. 2013;57(4):1394–1406. doi: 10.1002/hep.26225. [DOI] [PubMed] [Google Scholar]
- Bhutta HY, Rajpal N, White W, Freudenberg JM, Liu Y, Way J, Rajpal D, Cooper DC, Young A, Tavakkoli A, Chen L. Effect of Roux-en-Y gastric bypass surgery on bile acid metabolism in normal and obese diabetic rats. PLoS One. 2015;10(3):e0122273. doi: 10.1371/journal.pone.0122273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell M, Wedin M, Admyre T, Hermansson M, Bottcher G, Goransson M, Linden D, Bamberg K, Oscarsson J, Bohlooly YM. Ageing Fxr deficient mice develop increased energy expenditure, improved glucose control and liver damage resembling NASH. PLoS One. 2013;8(5):e64721. doi: 10.1371/journal.pone.0064721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD) Metabolism. 2016;65(8):1038–1048. doi: 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, Kuipers F, Staels B. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281(16):11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- Chen L, Yao X, Young A, McNulty J, Anderson D, Liu Y, Nystrom C, Croom D, Ross S, Collins J, Rajpal D, Hamlet K, Smith C, Gedulin B. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. American journal of physiology. Endocrinology and metabolism. 2012;302(1):E68–76. doi: 10.1152/ajpendo.00323.2011. [DOI] [PubMed] [Google Scholar]
- Chen Q, Jiang Y, An Y, Zhao N, Zhao Y, Yu C. Soluble FGFR4 extracellular domain inhibits FGF19-induced activation of FGFR4 signaling and prevents nonalcoholic fatty liver disease. Biochem Biophys Res Commun. 2011;409(4):651–656. doi: 10.1016/j.bbrc.2011.05.059. [DOI] [PubMed] [Google Scholar]
- Claudel T, Sturm E, Duez H, Torra IP, Sirvent A, Kosykh V, Fruchart JC, Dallongeville J, Hum DW, Kuipers F, Staels B. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109(7):961–971. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasarathy S, Yang Y, McCullough AJ, Marczewski S, Bennett C, Kalhan SC. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol. 2011;23(5):382–388. doi: 10.1097/MEG.0b013e328345c8c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, Ballatori N. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280(8):6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- Ding L, Sousa KM, Jin L, Dong B, Kim BW, Ramirez R, Xiao Z, Gu Y, Yang Q, Wang J, Yu D, Pigazzi A, Schones D, Yang L, Moore D, Wang Z, Huang W. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology. 2016;64(3):760–773. doi: 10.1002/hep.28689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donepudi AC, Boehme S, Li F, Chiang JY. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology. 2016 doi: 10.1002/hep.28707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RH. The enterohepatic circulation of bile acids as they relate to lipid disorders. J Clin Pathol Suppl (Assoc Clin Pathol) 1973;5:59–67. doi: 10.1136/jcp.s1-5.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour JF, Oneta CM, Gonvers JJ, Bihl F, Cerny A, Cereda JM, Zala JF, Helbling B, Steuerwald M, Zimmermann A, Swiss Association for the Study of the, L Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2006;4(12):1537–1543. doi: 10.1016/j.cgh.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Dutia R, Embrey M, O’Brien CS, Haeusler RA, Agenor KK, Homel P, McGinty J, Vincent RP, Alaghband-Zadeh J, Staels B, le Roux CW, Yu J, Laferrere B. Temporal changes in bile acid levels and 12alpha-hydroxylation after Roux-en-Y gastric bypass surgery in type 2 diabetes. Int J Obes (Lond) 2015;39(5):806–813. doi: 10.1038/ijo.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi H, Naderian M, Sohrabpour AA. New Concepts on Pathogenesis and Diagnosis of Liver Fibrosis; A Review Article. Middle East J Dig Dis. 2016;8(3):166–178. doi: 10.15171/mejdd.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards PA, Kast HR, Anisfeld AM. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res. 2002;43(1):2–12. [PubMed] [Google Scholar]
- Enjoji M, Machida K, Kohjima M, Kato M, Kotoh K, Matsunaga K, Nakashima M, Nakamuta M. NPC1L1 inhibitor ezetimibe is a reliable therapeutic agent for non-obese patients with nonalcoholic fatty liver disease. Lipids Health Dis. 2010;9:29. doi: 10.1186/1476-511X-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JG, Zhong L, Tia LY, Xu ZJ, Li MS, Wang GL. Effects of ursodeoxycholic acid and/or low-calorie diet on steatohepatitis in rats with obesity and hyperlipidemia. World J Gastroenterol. 2005;11(15):2346–2350. doi: 10.3748/wjg.v11.i15.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferslew BC, Xie G, Johnston CK, Su M, Stewart PW, Jia W, Brouwer KL, Barritt ASt. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig Dis Sci. 2015;60(11):3318–3328. doi: 10.1007/s10620-015-3776-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. 2003;278(46):45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- Guo GL, Santamarina-Fojo S, Akiyama TE, Amar MJ, Paigen BJ, Brewer B, Jr, Gonzalez FJ. Effects of FXR in foam-cell formation and atherosclerosis development. Biochim Biophys Acta. 2006;1761(12):1401–1409. doi: 10.1016/j.bbalip.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haedrich M, Dufour JF. UDCA for NASH: end of the story? J Hepatol. 2011;54(5):856–858. doi: 10.1016/j.jhep.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest. 1994;93(3):1326–1331. doi: 10.1172/JCI117091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Ji G, Jia W, Li H. Gut Microbiota and Nonalcoholic Fatty Liver Disease: Insights on Mechanism and Application of Metabolomics. Int J Mol Sci. 2016;17(3):300. doi: 10.3390/ijms17030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF. The Function of Bile Salts in Fat Absorption. The Solvent Properties of Dilute Micellar Solutions of Conjugated Bile Salts. Biochem J. 1963;89:57–68. doi: 10.1042/bj0890057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, Volk K, Kuo MS, Gordillo R, Kharitonenkov A, Scherer PE. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013;17(5):790–797. doi: 10.1016/j.cmet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Yang C, Luo Y, Jin C, Wang F, McKeehan WL. FGFR4 prevents hyperlipidemia and insulin resistance but underlies high-fat diet induced fatty liver. Diabetes. 2007;56(10):2501–2510. doi: 10.2337/db07-0648. [DOI] [PubMed] [Google Scholar]
- Hwang ST, Urizar NL, Moore DD, Henning SJ. Bile acids regulate the ontogenic expression of ileal bile acid binding protein in the rat via the farnesoid X receptor. Gastroenterology. 2002;122(5):1483–1492. doi: 10.1053/gast.2002.32982. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Ishida H, Yamashita C, Kuruta Y, Yoshida Y, Noshiro M. Insulin is a dominant suppressor of sterol 12 alpha-hydroxylase P450 (CYP8B) expression in rat liver: possible role of insulin in circadian rhythm of CYP8B. J Biochem. 2000;127(1):57–64. doi: 10.1093/oxfordjournals.jbchem.a022584. [DOI] [PubMed] [Google Scholar]
- Jia X, Suzuki Y, Naito H, Yetti H, Kitamori K, Hayashi Y, Kaneko R, Nomura M, Yamori Y, Zaitsu K, Kato M, Ishii A, Nakajima T. A possible role of chenodeoxycholic acid and glycine-conjugated bile acids in fibrotic steatohepatitis in a dietary rat model. Dig Dis Sci. 2014;59(7):1490–1501. doi: 10.1007/s10620-014-3028-3. [DOI] [PubMed] [Google Scholar]
- Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, Tanaka N, Desai D, Amin SG, Albert I, Patterson AD, Gonzalez FJ. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125(1):386–402. doi: 10.1172/JCI76738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Lu Y, Li XY. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol Sin. 2015;36(1):44–50. doi: 10.1038/aps.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MR, Barnes S, Kwakye JB, Diasio RB. Purification and characterization of bile acid-CoA:amino acid N-acyltransferase from human liver. J Biol Chem. 1991;266(16):10227–10233. [PubMed] [Google Scholar]
- Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, Milburn M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60(3):404–413. doi: 10.1016/j.metabol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, Tontonoz P, Kliewer S, Willson TM, Edwards PA. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277(4):2908–2915. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- Killenberg PG, Jordan JT. Purification and characterization of bile acid-CoA:amino acid N-acyltransferase from rat liver. J Biol Chem. 1978;253(4):1005–1010. [PubMed] [Google Scholar]
- Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48(12):2664–2672. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- Kim JK, Lee KS, Lee DK, Lee SY, Chang HY, Choi J, Lee JI. Omega-3 polyunsaturated fatty acid and ursodeoxycholic acid have an additive effect in attenuating diet-induced nonalcoholic steatohepatitis in mice. Exp Mol Med. 2014;46:e127. doi: 10.1038/emm.2014.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong B, Luyendyk JP, Tawfik O, Guo GL. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. The Journal of pharmacology and experimental therapeutics. 2009;328(1):116–122. doi: 10.1124/jpet.108.144600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56(3):1034–1043. doi: 10.1002/hep.25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong M, Zhu L, Bai L, Zhang X, Chen Y, Liu S, Zheng S, Pandol SJ, Han YP, Duan Z. Vitamin D deficiency promotes nonalcoholic steatohepatitis through impaired enterohepatic circulation in animal model. Am J Physiol Gastrointest Liver Physiol. 2014;307(9):G883–893. doi: 10.1152/ajpgi.00427.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Nies AT, Cui Y, Leier I, Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta. 1999;1461(2):377–394. doi: 10.1016/s0005-2736(99)00169-8. [DOI] [PubMed] [Google Scholar]
- Krattinger R, Bostrom A, Lee SM, Thasler WE, Schioth HB, Kullak-Ublick GA, Mwinyi J. Chenodeoxycholic acid significantly impacts the expression of miRNAs and genes involved in lipid, bile acid and drug metabolism in human hepatocytes. Life Sci. 2016;156:47–56. doi: 10.1016/j.lfs.2016.04.037. [DOI] [PubMed] [Google Scholar]
- Kuzmenko DI, Klimentyeva TK. Role of Ceramide in Apoptosis and Development of Insulin Resistance. Biochemistry (Mosc) 2016;81(9):913–927. doi: 10.1134/S0006297916090017. [DOI] [PubMed] [Google Scholar]
- Kwong E, Li Y, Hylemon PB, Zhou H. Bile acids and sphingosine-1-phosphate receptor 2 in hepatic lipid metabolism. Acta Pharm Sin B. 2015;5(2):151–157. doi: 10.1016/j.apsb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake AD, Novak P, Shipkova P, Aranibar N, Robertson D, Reily MD, Lu Z, Lehman-McKeeman LD, Cherrington NJ. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol Appl Pharmacol. 2013;268(2):132–140. doi: 10.1016/j.taap.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikin-Frenkel A, Goldiner I, Leikin-Gobbi D, Rosenberg R, Bonen H, Litvak A, Bernheim J, Konikoff FM, Gilat T. Treatment of preestablished diet-induced fatty liver by oral fatty acid-bile acid conjugates in rodents. Eur J Gastroenterol Hepatol. 2008;20(12):1205–1213. doi: 10.1097/MEG.0b013e3282fc9743. [DOI] [PubMed] [Google Scholar]
- Leuschner UF, Lindenthal B, Herrmann G, Arnold JC, Rossle M, Cordes HJ, Zeuzem S, Hein J, Berg T, Group NS. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52(2):472–479. doi: 10.1002/hep.23727. [DOI] [PubMed] [Google Scholar]
- Li G, Thomas AM, Williams JA, Kong B, Liu J, Inaba Y, Xie W, Guo GL. Farnesoid X receptor induces murine scavenger receptor Class B type I via intron binding. PLoS One. 2012;7(4):e35895. doi: 10.1371/journal.pone.0035895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol. 2013;86(11):1517–1524. doi: 10.1016/j.bcp.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor KD, Kowdley KV, Heathcote EJ, Harrison ME, Jorgensen R, Angulo P, Lymp JF, Burgart L, Colin P. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39(3):770–778. doi: 10.1002/hep.20092. [DOI] [PubMed] [Google Scholar]
- Liu H, Pathak P, Boehme S, Chiang JY. Cholesterol 7alpha-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J Lipid Res. 2016a;57(10):1831–1844. doi: 10.1194/jlr.M069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Zou WL, Chen SJ, Wei HY, Yin YN, Zou YY, Lu FG. Effects of different diets on intestinal microbiota and nonalcoholic fatty liver disease development. World J Gastroenterol. 2016b;22(32):7353–7364. doi: 10.3748/wjg.v22.i32.7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6(3):507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clinic proceedings. 1980;55(7):434–438. [PubMed] [Google Scholar]
- Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116(4):1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee N, Zou A, Zhang Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed Res Int. 2016;2016:5170402. doi: 10.1155/2016/5170402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284(5418):1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, Jones SA, Willson TM. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem. 2000;43(16):2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- Martin IV, Schmitt J, Minkenberg A, Mertens JC, Stieger B, Mullhaupt B, Geier A. Bile acid retention and activation of endogenous hepatic farnesoid-X-receptor in the pathogenesis of fatty liver disease in ob/ob-mice. Biol Chem. 2010;391(12):1441–1449. doi: 10.1515/BC.2010.141. [DOI] [PubMed] [Google Scholar]
- McGettigan BM, McMahan RH, Luo Y, Wang XX, Orlicky DJ, Porsche C, Levi M, Rosen HR. Sevelamer Improves Steatohepatitis, Inhibits Liver and Intestinal Farnesoid X Receptor (FXR), and Reverses Innate Immune Dysregulation in a Mouse Model of Non-alcoholic Fatty Liver Disease. J Biol Chem. 2016;291(44):23058–23067. doi: 10.1074/jbc.M116.731042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, Pruzanski M, Adorini L, Golden-Mason L, Levi M, Rosen HR. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288(17):11761–11770. doi: 10.1074/jbc.M112.446575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L, Tang Y, Li M, Yang P, Liu Z, Yuan J, Zheng P. Co-Administration of Cholesterol-Lowering Probiotics and Anthraquinone from Cassia obtusifolia L. Ameliorate Non-Alcoholic Fatty Liver. PLoS One. 2015;10(9):e0138078. doi: 10.1371/journal.pone.0138078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, McGilvray ID, Allard JP. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58(1):120–127. doi: 10.1002/hep.26319. [DOI] [PubMed] [Google Scholar]
- Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L, Fung S, Fischer SE, McGilvray IG, Allard JP. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS One. 2016;11(5):e0151829. doi: 10.1371/journal.pone.0151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(3):574–582 e571. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- Mueller M, Thorell A, Claudel T, Jha P, Koefeler H, Lackner C, Hoesel B, Fauler G, Stojakovic T, Einarsson C, Marschall HU, Trauner M. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol. 2015;62(6):1398–1404. doi: 10.1016/j.jhep.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myronovych A, Salazar-Gonzalez RM, Ryan KK, Miles L, Zhang W, Jha P, Wang L, Setchell KD, Seeley RJ, Kohli R. The role of small heterodimer partner in nonalcoholic fatty liver disease improvement after sleeve gastrectomy in mice. Obesity (Silver Spring) 2014;22(11):2301–2311. doi: 10.1002/oby.20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara M, Fujii H, Maloney PR, Shimizu M, Sato R. Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J Biol Chem. 2002;277(40):37229–37234. doi: 10.1074/jbc.M206749200. [DOI] [PubMed] [Google Scholar]
- Namisaki T, Noguchi R, Moriya K, Kitade M, Aihara Y, Douhara A, Nishimura N, Takeda K, Okura Y, Kawaratani H, Takaya H, Seki K, Yoshiji H. Beneficial effects of combined ursodeoxycholic acid and angiotensin-II type 1 receptor blocker on hepatic fibrogenesis in a rat model of nonalcoholic steatohepatitis. J Gastroenterol. 2016;51(2):162–172. doi: 10.1007/s00535-015-1104-x. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E, Network, N.C.R. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel OF, Still CD, Argyropoulos G, Edwards M, Gerhard GS. Bile Acids, FXR, and Metabolic Effects of Bariatric Surgery. J Obes. 2016;2016:4390254. doi: 10.1155/2016/4390254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF, Lawn RM. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. J Lipid Res. 2001;42(8):1173–1179. [PubMed] [Google Scholar]
- Parikh P, Ingle M, Patel J, Bhate P, Pandey V, Sawant P. An open-label randomized control study to compare the efficacy of vitamin e versus ursodeoxycholic acid in nondiabetic and noncirrhotic Indian NAFLD patients. Saudi journal of gastroenterology: official journal of the Saudi Gastroenterology Association. 2016;22(3):192–197. doi: 10.4103/1319-3767.182451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, Badman MK, Maratos-Flier E, Mun EC, Pihlajamaki J, Auwerx J, Goldfine AB. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 2009;17(9):1671–1677. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicciari R, Passeri D, De Franco F, Mostarda S, Filipponi P, Colliva C, Gadaleta RM, Franco P, Carotti A, Macchiarulo A, Roda A, Moschetta A, Gioiello A. Discovery of 3alpha,7alpha,11beta-Trihydroxy-6alpha-ethyl-5beta-cholan-24-oic Acid (TC-100), a Novel Bile Acid as Potent and Highly Selective FXR Agonist for Enterohepatic Disorders. J Med Chem. 2016 doi: 10.1021/acs.jmedchem.6b01126. [DOI] [PubMed] [Google Scholar]
- Pozzo L, Vornoli A, Coppola I, Croce CM, Giorgetti L, Gervasi PG, Longo V. Effect of HFD/STZ on expression of genes involved in lipid, cholesterol and glucose metabolism in rats. Life Sci. 2016;166:149–156. doi: 10.1016/j.lfs.2016.09.022. [DOI] [PubMed] [Google Scholar]
- Prawitt J, Abdelkarim M, Stroeve JH, Popescu I, Duez H, Velagapudi VR, Dumont J, Bouchaert E, van Dijk TH, Lucas A, Dorchies E, Daoudi M, Lestavel S, Gonzalez FJ, Oresic M, Cariou B, Kuipers F, Caron S, Staels B. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. 2011;60(7):1861–1871. doi: 10.2337/db11-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusl T, Wild N, Vennegeerts T, Wimmer R, Goke B, Brand S, Rust C. Free fatty acids sensitize hepatocytes to bile acid-induced apoptosis. Biochem Biophys Res Commun. 2008;371(3):441–445. doi: 10.1016/j.bbrc.2008.04.113. [DOI] [PubMed] [Google Scholar]
- Qi Y, Jiang C, Cheng J, Krausz KW, Li T, Ferrell JM, Gonzalez FJ, Chiang JY. Bile acid signaling in lipid metabolism: metabolomic and lipidomic analysis of lipid and bile acid markers linked to anti-obesity and anti-diabetes in mice. Biochim Biophys Acta. 2015;1851(1):19–29. doi: 10.1016/j.bbalip.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero P, Pizarro M, Solis N, Arab JP, Padilla O, Riquelme A, Arrese M. Bile acid supplementation improves established liver steatosis in obese mice independently of glucagon-like peptide-1 secretion. J Physiol Biochem. 2014;70(3):667–674. doi: 10.1007/s13105-014-0336-1. [DOI] [PubMed] [Google Scholar]
- Rao A, Kosters A, Mells JE, Zhang W, Setchell KD, Amanso AM, Wynn GM, Xu T, Keller BT, Yin H, Banton S, Jones DP, Wu H, Dawson PA, Karpen SJ. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Science translational medicine. 2016;8(357):357ra122. doi: 10.1126/scitranslmed.aaf4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, de Ledinghen V, Oberti F, Mathurin P, Wartelle-Bladou C, Renou C, Sogni P, Maynard M, Larrey D, Serfaty L, Bonnefont-Rousselot D, Bastard JP, Riviere M, Spenard J, Fresgun A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54(5):1011–1019. doi: 10.1016/j.jhep.2010.08.030. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289(5484):1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes. 2013;4(5):382–387. doi: 10.4161/gmic.25723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, Orlandi S, Sadeghpour B, Wang XX, Jiang T, Levi M, Pruzanski M, Adorini L. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Molecular pharmacology. 2010;78(4):617–630. doi: 10.1124/mol.110.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry. 1992;31(20):4737–4749. doi: 10.1021/bi00135a001. [DOI] [PubMed] [Google Scholar]
- Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Perez HE, Sandoval DA, Kohli R, Backhed F, Seeley RJ. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509(7499):183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, Oren R, Group F. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12(12):2085–2091 e2081. doi: 10.1016/j.cgh.2014.04.038. [DOI] [PubMed] [Google Scholar]
- Satapathy SK, Sanyal AJ. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin Liver Dis. 2015;35(3):221–235. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- Scheja L, Heeren J. Metabolic interplay between white, beige, brown adipocytes and the liver. J Hepatol. 2016;64(5):1176–1186. doi: 10.1016/j.jhep.2016.01.025. [DOI] [PubMed] [Google Scholar]
- Schmitt J, Kong B, Stieger B, Tschopp O, Schultze SM, Rau M, Weber A, Mullhaupt B, Guo GL, Geier A. Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int. 2015;35(4):1133–1144. doi: 10.1111/liv.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreuder TC, Marsman HA, Lenicek M, van Werven JR, Nederveen AJ, Jansen PL, Schaap FG. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298(3):G440–445. doi: 10.1152/ajpgi.00322.2009. [DOI] [PubMed] [Google Scholar]
- Serviddio G, Bellanti F, Villani R, Tamborra R, Zerbinati C, Blonda M, Ciacciarelli M, Poli G, Vendemiale G, Iuliano L. Effects of dietary fatty acids and cholesterol excess on liver injury: A lipidomic approach. Redox Biol. 2016;9:296–305. doi: 10.1016/j.redox.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneider BL, Dawson PA, Christie DM, Hardikar W, Wong MH, Suchy FJ. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J Clin Invest. 1995;95(2):745–754. doi: 10.1172/JCI117722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva TE, Colombo G, Schiavon LL. Adiponectin: A multitasking player in the field of liver diseases. Diabetes Metab. 2014;40(2):95–107. doi: 10.1016/j.diabet.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- Solis N, Pizarro M, Quintero P, Arab JP, Riquelme A, Padilla O, Carrasco G, Pirola CJ, Sookoian S, Arrese M. Effects of bile acid sequestration on hepatic steatosis in obese mice. Ann Hepatol. 2013;13(1):105–112. [PubMed] [Google Scholar]
- Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49(1):297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, Christe ME, Michael LF, Burris TP. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146(3):984–991. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- Stieger B, Hagenbuch B. Recent advances in understanding hepatic drug transport. F1000Res. 2016;5:2465. doi: 10.12688/f1000research.9466.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, Dent P, Spiegel S, Shi R, Xu W, Liu X, Bohdan P, Zhang L, Zhou H, Hylemon PB. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012;55(1):267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Kaneko R, Nomura M, Naito H, Kitamori K, Nakajima T, Ogawa T, Hattori H, Seno H, Ishii A. Simple and rapid quantitation of 21 bile acids in rat serum and liver by UPLC-MS-MS: effect of high fat diet on glycine conjugates of rat bile acids. Nagoya J Med Sci. 2013;75(1–2):57–71. [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, Wolf CR, Henderson CJ, Gonzalez FJ. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. 2016;57(12):2130–2137. doi: 10.1194/jlr.M071183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Matsubara T, Krausz KW, Patterson AD, Gonzalez FJ. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology. 2012;56(1):118–129. doi: 10.1002/hep.25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple JL, Cordero P, Li J, Nguyen V, Oben JA. A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence. Int J Mol Sci. 2016;17(6) doi: 10.3390/ijms17060947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology. 2010;51(4):1410–1419. doi: 10.1002/hep.23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloshin I, Hahn-Obercyger M, Anavi S, Tirosh O. L-arginine conjugates of bile acids-a possible treatment for non-alcoholic fatty liver disease. Lipids Health Dis. 2014;13:69. doi: 10.1186/1476-511X-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eckardstein A, Nofer JR, Assmann G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arteriosclerosis, thrombosis, and vascular biology. 2001;21(1):13–27. doi: 10.1161/01.atv.21.1.13. [DOI] [PubMed] [Google Scholar]
- Wang B, Jiang X, Cao M, Ge J, Bao Q, Tang L, Chen Y, Li L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-alcoholic Fatty Liver Disease. Sci Rep. 2016;6:32002. doi: 10.1038/srep32002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3(5):543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology. 2011;54(4):1421–1432. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, Mataki C, Sato H, Tanigawara Y, Schoonjans K, Itoh H, Auwerx J. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem. 2011;286(30):26913–26920. doi: 10.1074/jbc.M111.248203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MH, Oelkers P, Craddock AL, Dawson PA. Expression cloning and characterization of the hamster ileal sodium-dependent bile acid transporter. J Biol Chem. 1994;269(2):1340–1347. [PubMed] [Google Scholar]
- Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology. 2014;59(6):2188–2195. doi: 10.1002/hep.26986. [DOI] [PubMed] [Google Scholar]
- Woudenberg-Vrenken TE, Conde de la Rosa L, Buist-Homan M, Faber KN, Moshage H. Metformin protects rat hepatocytes against bile acid-induced apoptosis. PLoS One. 2013;8(8):e71773. doi: 10.1371/journal.pone.0071773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Pan LX, Erickson SK, Forman BM, Shneider BL, Ananthanarayanan M, Li X, Shefer S, Balasubramanian N, Ma L, Asaoka H, Lear SR, Nguyen LB, Dussault I, Suchy FJ, Tint GS, Salen G. Removal of the bile acid pool upregulates cholesterol 7alpha-hydroxylase by deactivating FXR in rabbits. J Lipid Res. 2002;43(1):45–50. [PubMed] [Google Scholar]
- Yang JS, Kim JT, Jeon J, Park HS, Kang GH, Park KS, Lee HK, Kim S, Cho YM. Changes in hepatic gene expression upon oral administration of taurine-conjugated ursodeoxycholic acid in ob/ob mice. PLoS One. 2010;5(11):e13858. doi: 10.1371/journal.pone.0013858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- Yu J, Lo JL, Huang L, Zhao A, Metzger E, Adams A, Meinke PT, Wright SD, Cui J. Lithocholic acid decreases expression of bile salt export pump through farnesoid X receptor antagonist activity. J Biol Chem. 2002;277(35):31441–31447. doi: 10.1074/jbc.M200474200. [DOI] [PubMed] [Google Scholar]
- Yuan L, Bambha K. Bile acid receptors and nonalcoholic fatty liver disease. World J Hepatol. 2015;7(28):2811–2818. doi: 10.4254/wjh.v7.i28.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A, Loomba R. Review article: the emerging interplay among the gastrointestinal tract, bile acids and incretins in the pathogenesis of diabetes and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;36(10):909–921. doi: 10.1111/apt.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Liu J, Jackson MI, Zhao FQ, Yan L, Combs GF., Jr Fatty liver accompanies an increase in lactobacillus species in the hind gut of C57BL/6 mice fed a high-fat diet. J Nutr. 2013;143(5):627–631. doi: 10.3945/jn.112.172460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51(2):380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, Edwards PA. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006a;103(4):1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang X, Vales C, Lee FY, Lee H, Lusis AJ, Edwards PA. FXR deficiency causes reduced atherosclerosis in Ldlr−/− mice. Arteriosclerosis, thrombosis, and vascular biology. 2006b;26(10):2316–2321. doi: 10.1161/01.ATV.0000235697.35431.05. [DOI] [PubMed] [Google Scholar]
- Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57(2):601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Liu H, Zhang M, Guo GL. Fatty liver diseases, bile acids, and FXR. Acta Pharm Sin B. 2016;6(5):409–412. doi: 10.1016/j.apsb.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]