

Abstract
Direct solar-powered production of value-added chemicals from CO2 and H2O, a process that mimics natural photosynthesis, is of fundamental and practical interest. In natural photosynthesis, CO2 is first reduced to common biochemical building blocks using solar energy, which are subsequently used for the synthesis of the complex mixture of molecular products that form biomass. Here we report an artificial photosynthetic scheme that functions via a similar two-step process by developing a biocompatible light-capturing nanowire array that enables a direct interface with microbial systems. As a proof of principle, we demonstrate that a hybrid semiconductor nanowire-bacteria system can reduce CO2 at neutral pH to a wide array of chemical targets, such as fuels, polymers, and complex pharmaceutical precursors, using only solar energy input. The high surface-area silicon nanowire array harvests light energy to provide reducing equivalents to the anaerobic bacterium, Sporomusa ovata, for the photoelectrochemical production of acetic acid under aerobic conditions (21% O2) with low overpotential (η < 200 mV), high Faradic efficiency (up to 90%), and long-term stability (up to 200 hours). The resulting acetate (~ 6 g/L) can be activated to acetyl coenzyme A (acetyl-CoA) by genetically engineered Escherichia coli and used as a building block for a variety of value-added chemicals, such as n-butanol, polyhydroxybutyrate (PHB) polymer, and three different isoprenoid natural products. As such, interfacing biocompatible solid-state nanodevices with living systems provides a starting point for developing a programmable system of chemical synthesis entirely powered by sunlight.
Natural photosynthesis, which harvests 130 terawatts of solar energy to generate up to 115 billion metric tons of biomass annually from the reduction of CO2, provides motivation for the development of artificial systems that can capture the energy of the sun to convert CO2 and H2O to value-added chemicals of societal benefit1–9. However, such an approach has not been fully realized owing to a host of unmet basic scientific challenges10. For example, enzymes isolated from microorganisms and plants can selectively catalyze CO2 reduction with low energy barriers11–13; however, they do not self-repair outside their native cellular context and are often intolerant to oxygen. Consequently, bio-derived CO2-reducing catalytic systems are not directly applicable to oxygen-containing CO2 sources such as flue gas. Another challenge for artificial photosynthesis is the selective synthesis of complex organic molecules13–16. Nature transforms CO2 into a variety of complex molecules using a limited number of biosynthetic intermediates as building blocks17. However, in the case of artificial photosynthesis, the selection of such an intermediate is difficult3,13,17: ideally, mass transport requires it to be water-soluble and it should also be easily incorporated into many biosynthetic pathways18.
We sought to develop a strategy for artificial photosynthesis, where biocatalysts in their native cellular environments are interfaced directly with semiconductor light-absorbers for unassisted solar CO2 reduction. Specifically, we envisioned a two-step strategy that mimics natural photosynthesis, where light capture by a biocompatible nanowire array can interface and directly provide reducing equivalents to living organisms for the targeted synthesis of value-added chemical products from CO2 fixation (Fig. 1). Such an integration between materials science and biology separates the demanding dual requirements for light-capture efficiency and catalytic activity, respectively, and provides a route to bridge efficient solar conversion in robust solid-state devices with the broad synthetic capabilities of living cells10. This artificial photosynthesis strategy is distinct from the active area of microbial electrosynthesis,19 in that the nanomaterials carry out both light-harvesting and delivery of reducing equivalents. Here, as a first step, we demonstrate a stand-alone, solar-powered system6,20–24 comprised of silicon (Si) and titanium dioxide (TiO2) nanowire arrays as the light-capturing units22 to mimic the “Z-scheme”2,25,26 and S. ovata as the cellular catalyst27,28, which can effectively reduce CO2 under mild conditions (e.g. aerobic atmosphere, neutral pH, and temperatures under 30 °C) and produce acetate for up to 200 hours under simulated sunlight, with an energy-conversion efficiency of up to 0.38%. Such an system, where CO2-reducing bacteria are in direct interface with a photo-active semiconductor, to the best of our knowledge, represents the first example of microbial photoelectrosynthesis, which is different from the conventional microbial electrosynthesis that microbes do not directly interact with light-absorbing devices.19,28 The nanowire-bacteria hybrids possess high reaction rate of CO2 reduction; and the presence of nanowire array creases a local anaerobic environment that allows strict anaerobes to continue CO2 reduction aerobically (21% O2), important for practical application. Finally, the acetate intermediate represents a biosynthetic precursor to a wide variety of potential fine and commodity chemicals via acetyl coenzyme A (acetyl-CoA), including functionalized aliphatics and aromatics, lipids, alkanes, as well as complex natural products. By simply selecting specific genetically engineered E. coli strains18,29–31, our strategy of artificial photosynthesis can be programmed to produce a variety of products with minimal modification, providing a versatile and amenable platform for solar-driven CO2 reduction to value-added fuels, chemicals, and materials.
Figure 1. Schematics of a general artificial photosynthetic approach.
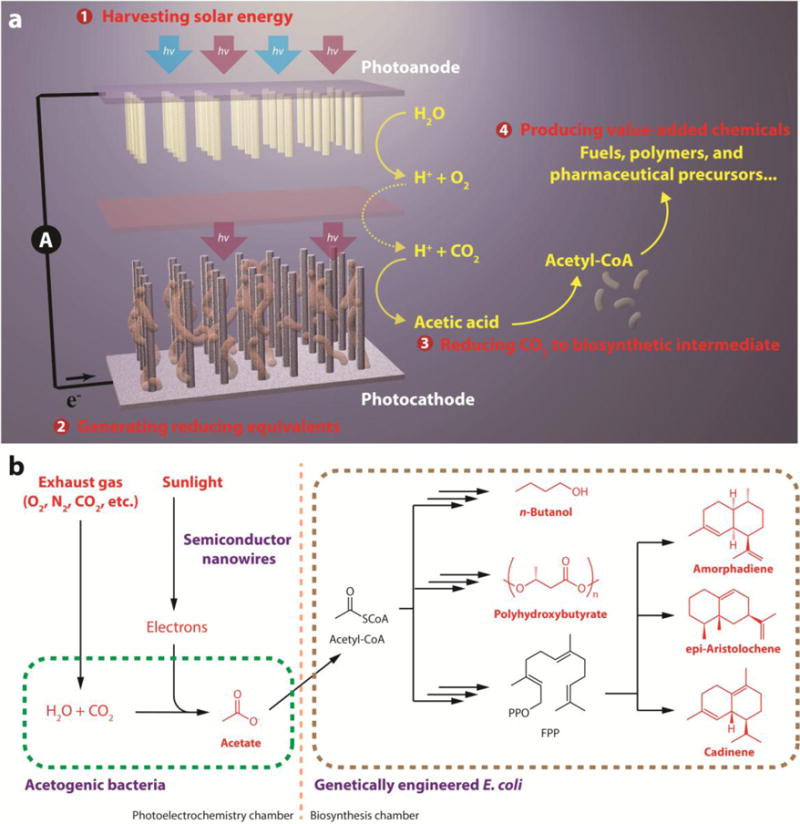
a, The proposed approach for solar-powered CO2 fixation includes four general components: 1) harvesting solar energy; 2) generating reducing equivalents; 3) reducing CO2 to biosynthetic intermediates; 4) producing value-added chemicals. An integration of materials science and biology, such an approach combines the advantages of solid-state devices with living organisms. b, As a proof of concept, we demonstrate that under mild condition sunlight can provide the energy to directly treat exhaust gas and generate acetate as the biosynthetic intermediate, which is upgraded into liquid fuels, biopolymers, and pharmaceutical precursors. For improved process yield, S. ovata and E. coli are placed in two separate containers. FPP, farnesyl pyrophosphate.
The system starts by interfacing light-absorbing Si nanowire arrays with an acetogenic organism, S. ovata27. Si nanowire arrays capture light for efficient solar energy conversion and provide high surface areas to interface with catalysts22,26. The strictly anaerobic homoacetogen S. ovata metabolizes CO2 via the energy-efficient Wood-Ljungdahl pathway, and has been reported to accept electrons from graphite electrodes to reduce CO2 into acetic acid.28 The integration was realized by directly culturing S. ovata within a Si nanowire array passivated by a 30 nm TiO2 protection layer, using buffered brackish water medium with trace vitamins as the only organic component (see Methods, Supplementary Fig. 1a). After an initial incubation period, a steady-state nanowire-bacteria hybrid structure was formed. In such a structure, the bacteria formed an interconnected network among the nanowires (Fig. 2a, Supplementary Fig. 2). Careful characterization with scanning electron microscopy (SEM) indicates that bacteria populate the array quite uniformly without apparent mass transport issues (Fig. 2b,c). The cell loading of S. ovata within the nanowire array is 4.4 ± 1.0 times of that observed on a planar Si electrode (1.4 ± 0.1 vs. 0.32 ± 0.07 cells per geometric μm2, n = 4) (Supplementary Fig. 2d), revealing increased contact interfaces between bacteria and electrodes in this high surface-area platform. The proposed half-reaction of CO2 reduction is:
From a classic electrochemical analysis without solar illumination (Fig. 2d), the nanowire-bacteria hybrids were capable of reducing CO2 to acetate under continuous sparging with 20% CO2/80% N2 with an overpotential η less than 200 mV at 0 V vs. reversible hydrogen electrode (RHE) (see Methods), similar as reported in literature.28 Additionally the Tafel slope of bacterial catalyzed CO2 reduction is distinctly different from that of abiotic proton reduction, implying different reaction mechanisms (n = 2). On average, each cell could produce (1.1 ± 0.3) × 106 molecules of acetate every second or ca. 1012 molecules of acetate over the course of about five days at −0.2 V vs. RHE (Supplementary Fig. 2c,d), comparable with its intrinsic rate of acetogenic metabolism (Supplementary Note). Such nano-bio hybrids, which operate at ambient temperature, possess a volumetric reaction rate of ca. 2 , comparable to the rates in conventional gas phase catalysts (0.1 ~ 10 ) that require much higher temperatures (higher than 100 °C)32. It also corresponds to ca. 8 electrons per second per nm2 across the semiconductor/electrolyte interface (at −0.2 V vs. RHE), suitable to couple with efficient solar devices at 10 mA/cm2 when integrated into a high surface-area electrode1,6,24,26.
Figure 2. Unassisted solar-powered acetate production from the nanowire-bacteria hybrid device.
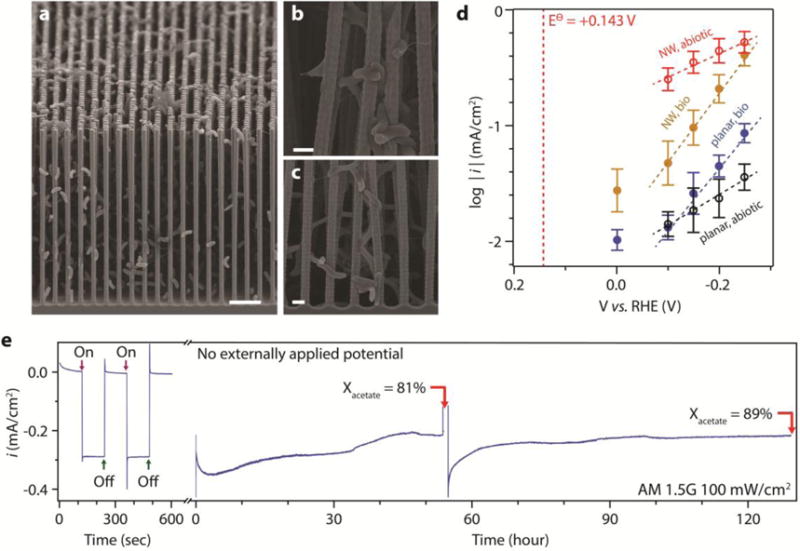
a, Cross-sectional SEM image of the three-dimensional network in the nanowire-bacteria hybrid. b & c, Magnified images at different depths of the nanowire array. d, Tafel plots, the logarithmic current density versus applied electrochemical voltage, are plotted for different electrode configurations (n = 2). Detailed data is summarized in Supplementary Fig. 2d. Filled blue circle (“planar, bio”), planar electrode loaded with bacteria; filled yellow circle (“NW, bio”), nanowire electrode loaded with bacteria; open black circle (“planar, abiotic”), bare planar electrode; open red circle (“NW, abiotic”), bare nanowire electrode. e, Measurement of unassisted solar-powered CO2 reduction for more than five days, n = 6. During the experiment the system was purged with 20% CO2/80% N2. In the plot Xacetate is the product selectivity (Faradic efficiency) of acetic acid generation. The scale bars are 5 μm (a) and 1 μm (b & c).
The high reaction rate of the nanowire-bacteria hybrids allows us to construct a solar-powered CO2-reduction device for the production of acetate as a common biosynthetic feedstock (Fig. 1a). Analogous to the two photosystems found in nature2,25,26, Si and TiO2 nanowires were applied as two robust semiconductor light-absorbers to provide the thermodynamic driving force for CO2 reduction22. Specifically, an ion-conductive membrane was placed in between the two electrodes to separate reaction products, and the water-oxidizing TiO2 nanowire electrode22 was placed in front of the nanowire-bacteria composite to absorb UV light and prevent possible bacterial photodamage (Fig. 1a, Supplementary Fig. 1b). Without any additional energy input, non-zero photocurrent was observed (light chopping experiment in Fig. 2e), and CO2 reduction to acetate was confirmed (see Methods). The overall system produced about 0.3 mA/cm2 photocurrent under simulated sunlight (AM 1.5G, 100 mW/cm2), and was stable for more than 120 hours (Fig. 2e, and the photocathode in Supplementary Fig. 3). Starting from an electrolyte free of organic compounds, acetic acid was steadily produced with a product selectivity (Faradic efficiency) of 86 ± 9% (n = 6) (Fig. 2e). The peak photocurrent reached 0.35 mA/cm2, which corresponds to an energy conversion efficiency of 0.38% for acetic acid production (requires 1.08 V thermodynamically, see Methods). The acetic acid titers were ca. 1.2 g/L (20 mM) within 5 days and could reach over 6 g/L (ca. 100 mM) in M9-MOPS minimal medium (see Methods and Supplementary Note). In separate control experiments, no acetic acid was detected without incorporation of S. ovata, and an isotope-labeling experiment proves that the acetate is produced from CO2 (Supplementary Fig. 4). These results highlight our ability to upgrade CO2 to chemicals beyond one-carbon targets. In next-generation designs, we are targeting even higher efficiencies through further improvement on peripheral limitations such as CO2 mass transport in the electrolyte and the large band gap of the photoanode26. Nevertheless, it represents a unique materials-biological hybrid for artificial photosynthesis, which demonstrates unassisted light-driven CO2 fixation to acetic acid.
An interesting benefit of the nanowire array arises from its selective control of mass transport within the wire assembly26,33. Specifically, the design of nanowire-bacteria hybrids allow for the continuation of CO2 reduction, a reaction catalyzed by strict anaerobe S. ovata, under a headspace containing 21% oxygen when an oxygen reduction reaction electrocatalyst (in current case, Pt, see Methods) was loaded. The similar Tafel slopes of CO2-reduction for planar and nanowire electrodes (Fig. 2d) inform us that mass transport of protons and CO2 was not a limiting factor within the nanowire array. However, with its limited solubility in water, oxygen can be depleted within the nanowire array logarithmically, distinctly different from a planar counterpart (Supplementary Note, Supplementary Fig. 5a,b). This arrangement effectively creates a local anaerobic environment at the bottom of the nanowire arrays, as supported by numerical simulation (Fig. 3a, Supplementary Fig. 5c). Experimentally, after S. ovata had colonized the electrode anaerobically, we switched to an aerobic gas environment with 21% oxygen partial pressure (21% O2/10% CO2/69% N2) (see Methods). Only the nanowire array loaded with Pt maintained its ability to reduce CO2 and consistently produce acetic acid with a Faradic efficiency of about 70% (t = 85 hour, Fig. 3b). Compared to the data in anaerobic condition (20% CO2/80% N2), the loss of Faradic efficiency (~15%) is related to the oxygen reduction reaction (Supplementary Fig. 6), which can be greatly minimized with improved design of the nanowire-bacteria hybrids (Supplementary Note). In general, our observation implies that 1) in our system, most, if not all, of the acetate is produced from bacteria interfacing directly with nanowires; 2) combining nanowire arrays with CO2-reducing microorganisms can allow anaerobes to be used in a wider range of applications, such as CO2 scrubbing from exhaust gas or even open air operation.
Figure 3. Enhanced oxygen tolerance for nanowire-bacteria hybrids.
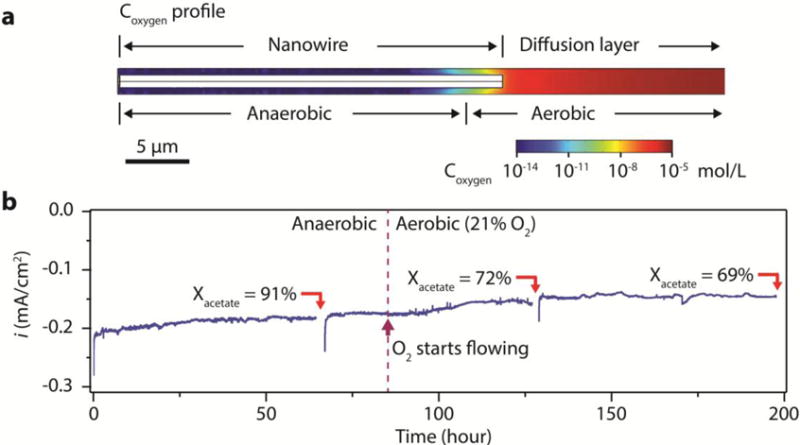
a, A numerical simulation illustrates that integrating bacteria into a nanowire array allows for the survival of strict anaerobes in an aerobic environment. The oxygen concentration in the electrolyte decreases logarithmically from the nanowire array’s entrance, creating a local anaerobic environment. This is in contrast to the linear decrease of oxygen concentration for a planar electrode (Supplementary Note and Supplementary Fig. 5). b, Experimental demonstration of aerobic CO2 reduction by S. ovata when Pt was additionally loaded onto the nanowire electrode, n = 3. Constant electrochemical bias (−0.2 V vs. RHE) was applied to the Si nanowire electrode and the current was plotted against time. As highlighted in the plot, the sparging gas of the setup was switched from anaerobic (20% CO2/80% N2) to aerobic (21% O2/10% CO2/69% N2) at t = 85 hour.
Taking advantage of the power of synthetic biology18, a wide spectrum of complex organic molecules was synthesized by directly using the solar-derived acetate from oxygen-containing CO2 feedstock. Under aerobic or microaerobic conditions, genetically engineered E. coli can activate acetate into the common biochemical intermediate acetyl-CoA, which will be then used for the biosynthesis of a variety of complex molecules (Fig. 4a). In principle, the acetate-consuming E. coli and the solar-powered CO2-reducing nano-bio hybrids can be positioned in a single aerobic reactor. However for optimized yield these two processes are conducted in separate containers (Supplementary Note). Here, as a proof of concept, the production of n-butanol29, PHB biopolymer31, and three isoprenoid compounds30 is demonstrated with H2O and CO2 as the starting materials, and sunlight as the energy source (Fig. 1b, Supplementary Fig. 7,8, see Methods). After the solar-powered acetate-production step, the accumulation of the target molecules is correlated with the consumption of acetate (n = 3, Supplementary Fig. 9a), implying the conversion of acetate into the desired products. The yield of target molecules was as high as 26% for n-butanol, 25% for one of the isoprenoid compounds (amorphadiene), and up to 52% for PHB biopolymer (Fig. 4b, Supplementary Fig. 9b), comparable with literature values29–31. Combining with the 0.38% efficiency from CO2 to acetic acid, a solar energy-conversion efficiency of 0.20% is achieved from CO2 to PHB biopolymer, a renewable and biodegradable plastic. Overall, the production of different organic products with vastly different synthetic pathways (Supplementary Fig. 7) proves the versatility of the integrated approach starting from one common biochemical building block, analogous to natural photosynthesis.
Figure 4. Biocatalytic production of diverse organic compounds using genetically engineered E. coli.
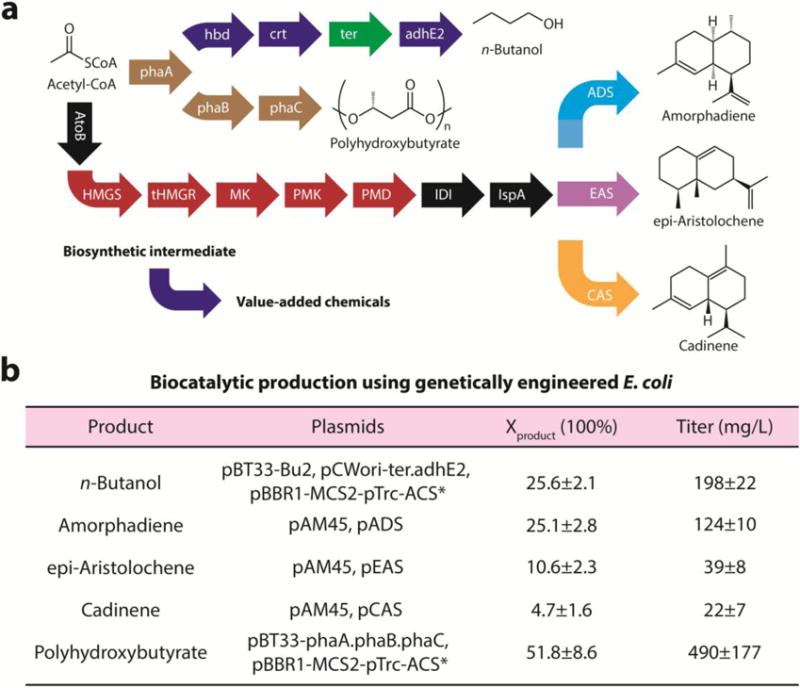
a, Synthetic pathways for the production of a variety of value-added chemicals. Here the names of proteins are listed, and the colors differentiate their genetic origins. In addition to these described pathways, some of the acetyl-CoA are expected to be diverted into the TCA cycle for redox balancing. b, Solar-derived acetic acid from nanowire-bacteria hybrids was used as the feedstock to yield a variety of chemicals in M9-MOPS medium (t = 5 days). No organic substrates were provided except the solar-derived acetic acid, and the acetate-containing medium solution was yielded at aerobic conditions (21% O2/10% CO2/69% N2) under simulated sunlight. Here Xproduct is acetate-to-product conversion efficiency. n = 3 for all reported values. Blue, Clostridium acetobutylicum; green, Treponema denticola; brown, Ralstonia eutropha; black, E. coli; red, Saccharomyces cerevisiae; light blue, Artemisia annua; purple, Nicotiana tabacum; yellow, Gossypium arboreum. phaA, acetoacetyl-CoA thiolase/synthase; hbd, phaB, 3-hydroxybutyryl-CoA dehydrogenase; crt, crotonase; ter, trans-enoyl-CoA reductase; adhE2, bifunctional butyraldehyde and butanol dehydrogenase; phaC, PHA synthase; AtoB, acetyl-CoA acetyltransferase; HMGS, hydroxymethylglutaryl-CoA synthase; tHMGR, truncated hydroxymethylglutary-CoA reductase; MK, mevalonate kinase; PMK, phosphomevalonate kinase; PMD, phosphomevalonate decarboxylase; IDI, isopentenyl diphosphate-isomerase; IspA, farnesyl diphosphoate synthase; ADS, amorphadiene synthase; EAS, epi-aristolochene cyclase; CAS, cadinene synthase.
The results reported here outline a solar-energy conversion process that combines the strengths of semiconductor nanodevices and bacterium-based biocatalysts (Fig. 1a). Key advantages of the nanowire-based device are the enhanced oxygen tolerance that allows exhaust gas to be directly fed into the system, thereby enabling use of strict anaerobes with aerobes, as well as the high measured CO2 fixation activity of the nanowire-bacteria hybrid. Moreover, this modular platform simplifies the overall system design by allowing for the production of a variety of molecular targets, without any setup change in the components for light capture and CO2 reduction into acetate, by varying only the downstream microorganisms.
Supplementary Material
Acknowledgments
We would like to thank Dr. Miao Wen for use of the ACS and PHB plasmids. We also would like to acknowledge Yude Su, Dr. Jongwoo Lim, and Dr. Sarah F. Brittman for helpful discussion. P. Y. thanks support from DOE/LBNL DE-AC02-05CH11231 (PChem, P. Y.). C. J. C. and M. C. Y. C. thank support from DOE/LBNL DE-AC02-05CH11231, FWP no. CH030201 (C. J. C. and M. C. Y. C.). C. J. C. is an Investigator with the Howard Hughes Medical Institute. J. J. G., K. K. S. and E. M. N. would like to acknowledge the NSF-GRFP for funding. J. J. G. also thanks NIH Training Grant 1 T32 GMO66698 for support.
Footnotes
Supplementary Information is available free of charge via the Internet at http://pubs.acs.org.
Author contributions C. L. and K. K. S. cultured the S. ovata; C. L. prepared the semiconductor nanowires and conducted the solar-powered acetate production; J. J. G. prepared the genetically engineered E. coli strains and conducted production experiment; J. J. G., C. L., and E. M. N. analyzed the composition of upgraded organic compounds; C. L., C. J. C., M. C. Y. C., and P. Y. designed the study; C. L. analyzed the data and wrote the paper. C. L. and J. J. G. contribute equally to the study. All authors discussed the results and commented on the manuscript.
Author Information The authors declare no competing financial interests.
References
- 1.Lewis NS, Nocera DG. Proc Natl Acad Sci USA. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barber J. Chem Soc Rev. 2009;38:185. doi: 10.1039/b802262n. [DOI] [PubMed] [Google Scholar]
- 3.Concepcion JJ, House RL, Papanikolas JM, Meyer TJ. Proc Natl Acad Sci USA. 2012;109:15560–15564. doi: 10.1073/pnas.1212254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magnuson A, et al. Acc Chem Res. 2009;42:1899–1909. doi: 10.1021/ar900127h. [DOI] [PubMed] [Google Scholar]
- 5.Gust D, Moore TA, Moore AL. Acc Chem Res. 2009;42:1890–1898. doi: 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]
- 6.Luo J, et al. Science. 2014;345:1593–1596. doi: 10.1126/science.1258307. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, et al. Proc Natl Acad Sci USA. 2012;109:15612–15616. doi: 10.1073/pnas.1118339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tachibana Y, Vayssieres L, Durrant JR. Nature Photon. 2012;6:511–518. [Google Scholar]
- 9.Gray HB. Nature Chem. 2009;1:7. doi: 10.1038/nchem.141. (2009) [DOI] [PubMed] [Google Scholar]
- 10.Blankenship RE, et al. Science. 2011;332:805–809. doi: 10.1126/science.1200165. [DOI] [PubMed] [Google Scholar]
- 11.Reda T, Plugge CM, Abram NJ, Hirst J. Proc Natl Acad Sci USA. 2008;105:10654–10658. doi: 10.1073/pnas.0801290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachmeier A, Hall S, Ragsdale SW, Armstrong FA. J Am Chem Soc. 2014;136:13518–13521. doi: 10.1021/ja506998b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Appel AM, et al. Chem Rev. 2013;113:6621–6658. doi: 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar B, et al. Annu Rev Phys Chem. 2012;63:541–569. doi: 10.1146/annurev-physchem-032511-143759. [DOI] [PubMed] [Google Scholar]
- 15.Barton EE, Rampulla DM, Bocarsly AB. J Am Chem Soc. 2008;130:6342–6344. doi: 10.1021/ja0776327. [DOI] [PubMed] [Google Scholar]
- 16.Li CW, Ciston J, Kanan MW. Nature. 2014;508:504–507. doi: 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Liao JC. Energy Environ Sci. 2013;6:2892–2899. [Google Scholar]
- 18.Keasling JD. Science. 2010;330:1355–1358. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]
- 19.Lovley DR, Nevin KP. Curr Opin Biotechnol. 2013;24:385–390. doi: 10.1016/j.copbio.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 20.Khaselev OA, Turner JA. Science. 1998;280:425–427. doi: 10.1126/science.280.5362.425. [DOI] [PubMed] [Google Scholar]
- 21.Brillet J, et al. Nature Photon. 2012;6:824–828. [Google Scholar]
- 22.Liu C, Tang J, Chen HM, Liu B, Yang P. Nano Lett. 2013;13:2989–2992. doi: 10.1021/nl401615t. [DOI] [PubMed] [Google Scholar]
- 23.Reece SY, et al. Science. 2011;334:645–648. doi: 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- 24.Cox CR, Lee JZ, Nocera DG, Buonassisi T. Proc Natl Acad Sci USA. 2014;111:14057–14061. doi: 10.1073/pnas.1414290111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nozik AJ. Appl Phys Lett. 1976;29:150–153. [Google Scholar]
- 26.Liu C, Dasgupta NP, Yang P. Chem Mater. 2013;26:415–422. [Google Scholar]
- 27.Möller B, Oβmer R, Howard B, Gottschalk G, Hippe H. Arch Microbiol. 1984;139:388–396. [Google Scholar]
- 28.Nevin KP, Woodard TL, Franks AE, Summers ZM, Lovley DR. mBio. 2010;1:e00103–10. doi: 10.1128/mBio.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bond-Watts BB, Bellerose RJ, Chang MCY. Nature Chem Biol. 2011;7:222–227. doi: 10.1038/nchembio.537. [DOI] [PubMed] [Google Scholar]
- 30.Chang MCY, Eachus RA, Trieu W, Ro DK, Keasling JD. Nature Chem Biol. 2007;3:274–277. doi: 10.1038/nchembio875. [DOI] [PubMed] [Google Scholar]
- 31.Sim SJ, et al. Nature Biotech. 1997;15:63–67. doi: 10.1038/nbt0197-63. [DOI] [PubMed] [Google Scholar]
- 32.Levenspiel O. Chemical Reaction Engineering. 3rd. John Wiley & Sons; 1999. p. 5. [Google Scholar]
- 33.Xiang C, Meng AC, Lewis NS. Proc Nat Acad Sci USA. 2012;109:15622–15627. doi: 10.1073/pnas.1118338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.