

New drugs, particularly those belonging to novel drug classes, can dramatically improve clinical outcomes but may also lead to serious toxicities. Given an emphasis on disease outcomes, drug-related toxicities may go unrecognized for years1-6 or be subject to biased reporting,7 and rarer toxicities may emerge during postmarketing surveillance.8,9 Delayed toxicity recognition in rapidly adopted drugs can impede clinicians’ ability to weigh potential drug benefits against harms and lead to unintended patient harm.10 A 2016 National Cancer Policy Forum workshop on drug development emphasized balancing the benefits and harms of new drugs, noting that poor knowledge of adverse events limits risk-benefit modeling.11 Optimizing accuracy and transparency of toxicity reporting in clinical trials and facilitating pooling of these data across studies would enable earlier understanding of toxicities, protect patients, and facilitate risk-benefit modeling.8 Ongoing efforts to more rapidly identify toxic drug effects have not leveraged data available on ClinicalTrials.gov.
The ClinicalTrials.gov Web site was created to improve transparency, patient access to clinical trial information, and evidence quality.12,13 The US Food and Drug Administration (FDA) Amendments Act established the legal mandate, and the 2016 Final Rule clarified reporting requirements, including provision of information about drug toxicities in the form of adverse events.13,14 Although primarily designed to improve transparency regarding outcomes, ClinicalTrials.gov also has the potential to facilitate understanding of toxicities of new drugs. However, the FDA mandates no specific reporting terminology, and lack of uniformity coupled with poor-quality adverse event data15 could hinder pooling of these data across trials.
Checkpoint inhibitors are important new drugs, the toxicities of which are not fully understood.16 They can dramatically improve clinical outcomes across cancers but may lead to serious immune-mediated toxicities affecting any organ.17-21 Delayed recognition of toxicities could lead to patient harms.10 Although published clinical trial reports include descriptions of common and severe toxicities, published toxicity data tend to be incomplete and focus on expected complications; reliable data sources are needed to enable timely understanding of previously unrecognized toxic effects. We explored the consistency of adverse event terminology reported on ClinicalTrials.gov in clinical trials of checkpoint inhibitors, a paradigm of a new potentially revolutionary drug class, to evaluate opportunities for data pooling.
We extracted adverse event information from ClinicalTrials.gov and found wide-ranging terminology, posing challenges to the pooling of toxicity data and understanding of the potential harms of this new drug class. In this editorial, we draw attention to how the current system of adverse event reporting on ClinicalTrials.gov represents a missed opportunity to enhance early understanding of the potential harms of new classes of cancer therapy.
Variability in Adverse Event Reporting
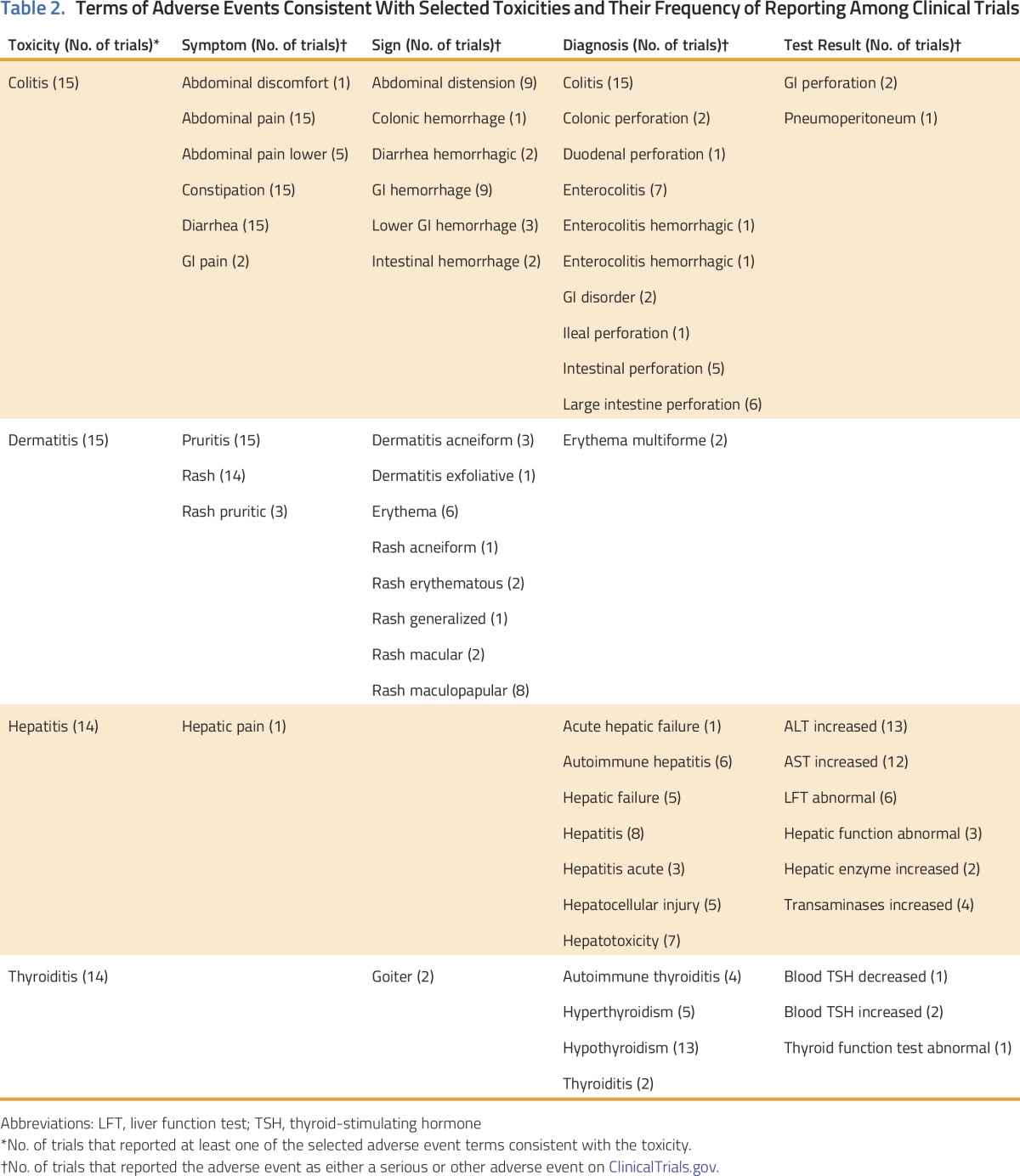
We present the scope of adverse events reported by randomized clinical trials (RCTs) of checkpoint inhibitors that reflect four immune-mediated toxicities: colitis, dermatitis, hepatitis, and thyroiditis.17-19,22 We systematically searched PubMed to identify published phase II/III RCTs of checkpoint inhibitors with full trial results posted on ClinicalTrials.gov by December 9, 2016 (Table 1). We generated a list of descriptive adverse event terms from ClinicalTrials.gov that might reflect each toxicity, categorizing them as symptoms (patient-reported clinical problems), signs (clinician-detected abnormalities), abnormal test results, or diagnoses (defined clinical entities; Table 2). For each toxicity, we determined the number of trials reporting each term. We included any term reported as either a serious or other adverse event on ClinicalTrials.gov.
Table 1.
List of Included Trials and Corresponding Studies
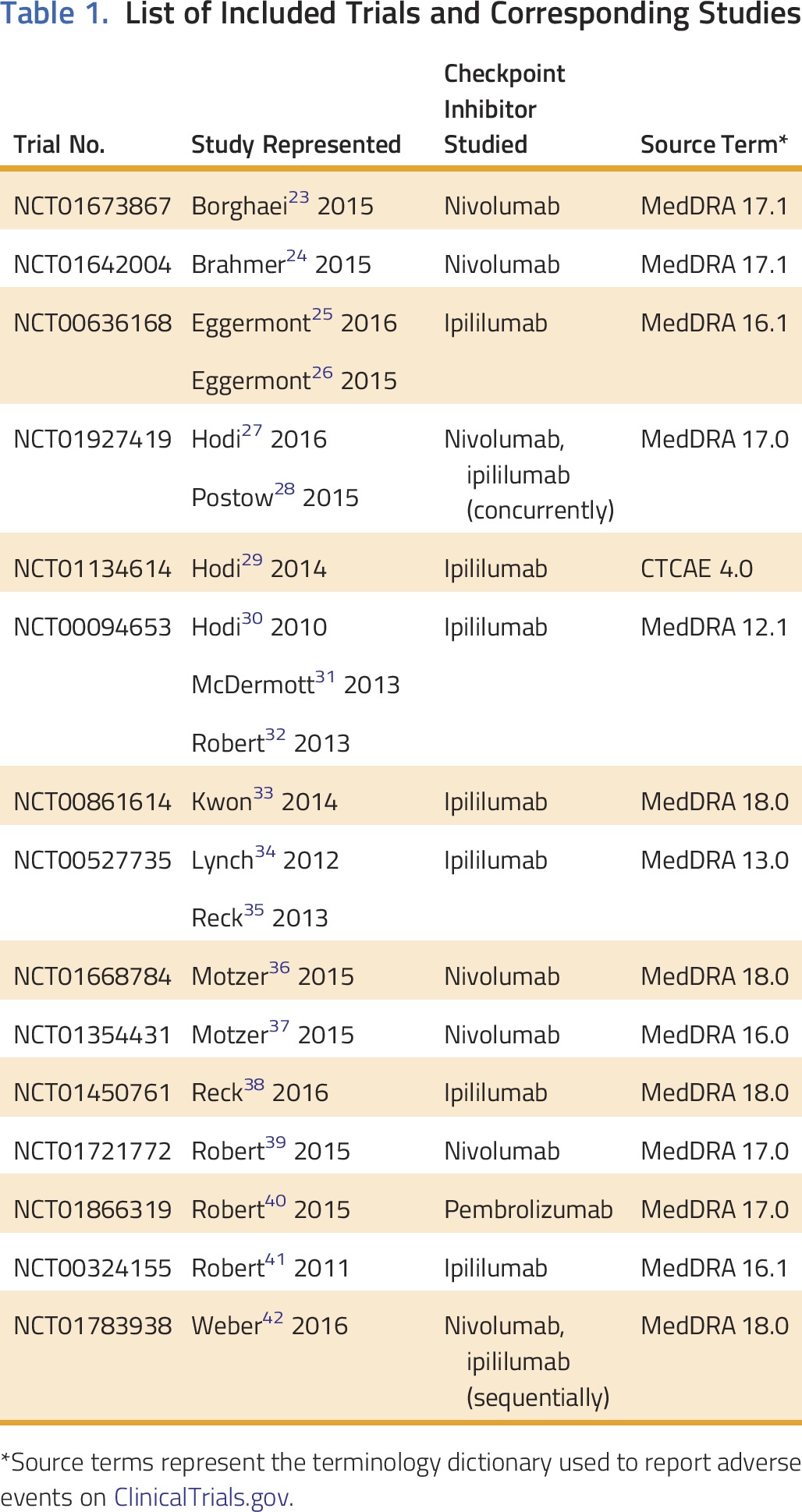
Table 2.
Terms of Adverse Events Consistent With Selected Toxicities and Their Frequency of Reporting Among Clinical Trials
Our search identified 325 studies; we extracted data from 20 studies representing 15 RCTs. Most trials (n = 15) reported adverse events using MedDRA (Table 1); all trials collected data using Common Terminology Criteria for Adverse Events. Study drugs included ipilimumab, nivolumab, nivolumab and ipilimumab, and pembrolizumab (Table 1).
The number of descriptive terms ranged from eight for thyroiditis to 24 for colitis (Table 2). For all four toxicities, terms from at least three of the four categories were reported by at least one trial, and few terms were included across all trials. For colitis, all trials used the terms abdominal pain, constipation, and diarrhea; other terms varied. For dermatitis, all trials reported pruritis (n = 15); most reported rash (n = 14). For hepatitis and thyroiditis, 14 trials reported at least one adverse event term, most often a diagnosis or test result.
Limitations of Current Adverse Event Reporting
Checkpoint inhibitors are being rapidly adopted into clinical practice; reliable data sources are needed to capture and understand their toxicities. Given researcher comfort with traditional chemotherapy, and because toxicities are often immediate and dose related, potential chemotherapy toxicities are easily recognized during clinical trials. However, new drug classes may differ, so mechanisms for enhancing recognition of toxicities are needed to protect patient safety. In the case of checkpoint inhibitors, we found that ClinicalTrials.gov included multiple terms potentially representing select toxicities, sometimes including symptoms, signs, diagnoses, and laboratory results with inconsistency across trials. These inconsistencies limit our ability to understand the prevalence of toxicities and pool data across trials, hindering the power of ClinicalTrials.gov to meaningfully enhance the safety of patients with cancer.
Currently, adverse event reporting on ClinicalTrials.gov does not enable determination of the total number of patients experiencing a given toxicity, because reporting of a single toxic event might use multiple terms across domains (eg, including both symptoms and diagnoses), obscuring the number of individuals represented. At the same time, many descriptive terms are nonspecific. For example, colitis might be reported under diarrhea, abdominal pain, and/or colitis; however, diarrhea or abdominal pain could be unrelated to colitis. This overlap and poor specificity may relate to the inclusion of multiple terms that capture a single clinical problem in clinical trial case report forms, limiting transparency and accurate estimates of the prevalence of toxicities.
Policy Implications
There are other potential methods for aggregating toxicity information, including systematic reviews of published toxicity data and FDA pharmacovigilance databases43; however, the public nature of ClinicalTrials.gov and its use during initial clinical trials enable early and complete compiling of information. Changes to ClinicalTrials.gov policy could help optimize its usefulness for understanding toxicities. The FDA Amendments Act and the 2016 Final Rule recognized the importance of capturing all-cause mortality, requiring trials to report all-cause deaths without mandating reporting of other specific outcomes.44 The FDA could establish standards for reporting of the number of patients experiencing specific toxicities, perhaps those that are most severe or most common, for use across all trials of a particular class of drugs, such as checkpoint inhibitors, and/or facilitate public access to narrative descriptions of these toxicities. Alternatively, the oncology research community could describe specific toxicities of interest for public reporting of clinical trials of particular drugs, which would be easy to quickly implement and would avoid political challenges while still leveraging the potential of ClinicalTrials.gov. Either approach would improve reporting clarity and facilitate pooling across trials to elucidate the true prevalence of toxic drug effects, complementing calls for better mechanisms of sharing clinical trial data45 and efforts to optimize ClinicalTrials.gov.46
Comparability of adverse event reporting across trials requires shared language and definitions. Currently, trials reporting on ClinicalTrials.gov can use different dictionaries with vague or variable definitions of specific terms, although primary data are collected using Common Terminology Criteria for Adverse Events. The FDA rule opted against implementing a standard vocabulary, recognizing the potential burden on researchers.44 However, the lack of standard terminology is a barrier to aggregation and data compilation. Thus, despite potential burden, a standard lexicon for adverse event reporting would benefit researchers, clinicians, and the public and would help ensure patient safety. This lexicon could be developed for specific drug classes to ensure relevance, with toxicity classification using syndrome-specific checklists. In addition, redundancy of reporting could be addressed by requiring reporting of a toxicity in a single patient in only one domain, facilitated by electronic reporting of adverse events. The electronic system could recognize groups of symptoms to define syndromes of interest, prompting the investigator to consider a unifying diagnosis that would then be reported. Such a system might also pull from standard electronic medical records, reducing reporting burden. Alternatively, programming could capture the evolution of adverse events. For example, for a patient who has diarrhea eventually confirmed as colitis, investigators could be prompted to relabel the diarrhea as early colitis if appropriate rather than reporting both as adverse events.
In conclusion, novel drug classes have the potential to dramatically improve outcomes in patients with cancer, but rapid understanding of their toxicities is critical. Although ClinicalTrials.gov has the potential to facilitate more complete understanding of toxicities, the wide-ranging terminology in current use impedes transparency and possibly patient safety. A standard vocabulary, required reporting for select adverse events, and electronic systems to identify syndromes and optimize reporting would clarify data, allow better estimates of toxicity rates, and facilitate pooling of toxicity data across trials. In this way, investigators and clinicians could better understand potential patient harms and optimize safety for patients receiving potentially lifesaving, but also potentially toxic, therapies.
ACKNOWLEDGMENT
Supported in part by a Cancer Center Support Grant No. P30 CA008748 from the National Cancer Institute to Memorial Sloan Kettering Cancer Center (S.B., D.K.).
AUTHOR CONTRIBUTIONS
Conception and design: Shrujal Baxi, Deborah Korenstein
Administrative support: Annie Yang
Collection and assembly of data: Annie Yang
Data analysis and interpretation: Annie Yang, Deborah Korenstein
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
ClinicalTrials.gov for Facilitating Rapid Understanding of Potential Harms of New Drugs: The Case of Checkpoint Inhibitors
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jop/site/ifc/journal-policies.html.
Annie Yang
No relationship to disclose
Shrujal Baxi
Consulting or Advisory Role: Bristol-Myers Squibb, AstraZeneca, Merck
Research Funding: Bristol-Myers Squibb, AstraZeneca
Deborah Korenstein
No relationship to disclose
REFERENCES
- 1.Ribeiro-Vaz I, Silva AM, Costa Santos C, et al. How to promote adverse drug reaction reports using information systems: A systematic review and meta-analysis. BMC Med Inform Decis Mak. 2016;16:27. doi: 10.1186/s12911-016-0265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vera-Badillo FE, Al-Mubarak M, Templeton AJ, et al. Benefit and harms of new anti-cancer drugs. Curr Oncol Rep. 2013;15:270–275. doi: 10.1007/s11912-013-0303-y. [DOI] [PubMed] [Google Scholar]
- 3.Lasser KE, Allen PD, Woolhandler SJ, et al. Timing of new black box warnings and withdrawals for prescription medications. JAMA. 2002;287:2215–2220. doi: 10.1001/jama.287.17.2215. [DOI] [PubMed] [Google Scholar]
- 4.Frank C, Himmelstein DU, Woolhandler S, et al. Era of faster FDA drug approval has also seen increased black-box warnings and market withdrawals. Health Aff (Millwood) 2014;33:1453–1459. doi: 10.1377/hlthaff.2014.0122. [DOI] [PubMed] [Google Scholar]
- 5.Moore TJ, Bennett CL. Underreporting of hemorrhagic and thrombotic complications of pharmaceuticals to the U.S. Food and Drug Administration: Empirical findings for warfarin, clopidogrel, ticlopidine, and thalidomide from the Southern Network on Adverse Reactions (SONAR) Semin Thromb Hemost. 2012;38:905–907. doi: 10.1055/s-0032-1328890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett CL, Angelotta C, Yarnold PR, et al. Thalidomide- and lenalidomide-associated thromboembolism among patients with cancer. JAMA. 2006;296:2558–2560. doi: 10.1001/jama.296.21.2558-c. [DOI] [PubMed] [Google Scholar]
- 7.de Vries YA, Roest AM, Beijers L, et al. Bias in the reporting of harms in clinical trials of second-generation antidepressants for depression and anxiety: A meta-analysis. Eur Neuropsychopharmacol. 2016;26:1752–1759. doi: 10.1016/j.euroneuro.2016.09.370. [DOI] [PubMed] [Google Scholar]
- 8.Persaud N, Doshi P. North American regulatory agencies can and should make clinical trial data publicly available. CMAJ. 2016;188:96–97. doi: 10.1503/cmaj.150679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senior JR. Evolution of the Food and Drug Administration approach to liver safety assessment for new drugs: Current status and challenges. Drug Saf. 2014;37(suppl 1):S9–S17. doi: 10.1007/s40264-014-0182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeAngelis CD, Fontanarosa PB. Impugning the integrity of medical science: The adverse effects of industry influence. JAMA. 2008;299:1833–1835. doi: 10.1001/jama.299.15.1833. [DOI] [PubMed] [Google Scholar]
- 11.National Academies of Sciences . Engineering, and Medicine: The Drug Development Paradigm in Oncology: Proceedings of a Workshop. Washington, DC: National Academies Press; 2017. [PubMed] [Google Scholar]
- 12.Tse T, Williams RJ, Zarin DA. Reporting “basic results” in. Chest. 2009;136:295–303. doi: 10.1378/chest.08-3022. ClinicalTrials.gov [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz LM, Woloshin S, Zheng E, et al. ClinicalTrials.gov and Drugs@FDA: A comparison of results reporting for new drug approval trials. Ann Intern Med. 2016;165:421–430. doi: 10.7326/M15-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarin DA, Tse T, Williams RJ, et al. Trial reporting in ClinicalTrials.gov: The Final Rule. N Engl J Med. 2016;375:1998–2004. doi: 10.1056/NEJMsr1611785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorr DA, Burdon R, West DP, et al. Quality of reporting of serious adverse drug events to an institutional review board: A case study with the novel cancer agent, imatinib mesylate. Clin Cancer Res. 2009;15:3850–3855. doi: 10.1158/1078-0432.CCR-08-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Couzin-Frankel J. Breakthrough of the year 2013: Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 17.Marrone KA, Ying W, Naidoo J. Immune-related adverse events from immune checkpoint inhibitors. Clin Pharmacol Ther. 2016;100:242–251. doi: 10.1002/cpt.394. [DOI] [PubMed] [Google Scholar]
- 18.González-Rodríguez E, Rodríguez-Abreu D. Immune checkpoint inhibitors: Review and management of endocrine adverse events. Oncologist. 2016;21:804–816. doi: 10.1634/theoncologist.2015-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Kane GM, Labbé C, Doherty MK, et al. Monitoring and management of immune-related adverse events associated with programmed cell death protein-1 axis inhibitors in lung cancer. Oncologist. 2017;22:70–80. doi: 10.1634/theoncologist.2016-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with cancer: A systematic review of case reports. PLoS One. 2016;11:e0160221. doi: 10.1371/journal.pone.0160221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–1755. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber JS, Yang JC, Atkins MB, et al. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33:2092–2099. doi: 10.1200/JCO.2014.60.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375:1845–1855. doi: 10.1056/NEJMoa1611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomized, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522–530. doi: 10.1016/S1470-2045(15)70122-1. [DOI] [PubMed] [Google Scholar]
- 27.Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomized, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558–1568. doi: 10.1016/S1470-2045(16)30366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hodi FS, Lee S, McDermott DF, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: A randomized clinical trial. JAMA. 2014;312:1744–1753. doi: 10.1001/jama.2014.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDermott D, Haanen J, Chen TT, et al. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20) Ann Oncol. 2013;24:2694–2698. doi: 10.1093/annonc/mdt291. [DOI] [PubMed] [Google Scholar]
- 32.Robert C, Schadendorf D, Messina M, et al. Efficacy and safety of retreatment with ipilimumab in patients with pretreated advanced melanoma who progressed after initially achieving disease control. Clin Cancer Res. 2013;19:2232–2239. doi: 10.1158/1078-0432.CCR-12-3080. [DOI] [PubMed] [Google Scholar]
- 33.Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomized, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–712. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non–small-cell lung cancer: Results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 35.Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24:75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 36.Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for metastatic renal cell carcinoma: Results of a randomized phase II trial. J Clin Oncol. 2015;33:1430–1437. doi: 10.1200/JCO.2014.59.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reck M, Luft A, Szczesna A, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol. 2016;34:3740–3748. doi: 10.1200/JCO.2016.67.6601. [DOI] [PubMed] [Google Scholar]
- 39.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 40.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 41.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 42.Weber JS, Gibney G, Sullivan RJ, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomized, phase 2 trial. Lancet Oncol. 2016;17:943–955. doi: 10.1016/S1470-2045(16)30126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu ZK, Kessler SJ, Schulz R, et al. Systematic approach to pharmacovigilance beyond the limits: The Southern Network on Adverse Reactions (SONAR) projects. Adv Pharmacoepidemiol Drug Saf. 2014;3:149–161. [Google Scholar]
- 44.National Institutes of Health, Department of Health and Human Services Clinical trials registration and results information submission: Final Rule. Fed Regist. 2016;81:64981–65157. [PubMed] [Google Scholar]
- 45.Taichman DB, Backus J, Baethge C, et al. Sharing clinical trial data: A proposal from the International Committee of Medical Journal Editors. JAMA. 2016;315:467–468. doi: 10.1001/jama.2015.18164. [DOI] [PubMed] [Google Scholar]
- 46. Office of the Press Secretary: FACT SHEET: Vice President Biden announces new steps to improve clinical trials essential to advancing the Cancer Moonshot. https://obamawhitehouse.archives.gov/the-press-office/2016/09/16/fact-sheet-vice-president-biden-announces-new-steps-improve-clinical.