

Key Points
TCRαβ+/CD19+-depleted haplo-HCT is associated with a reduced incidence of graft failure in patients with hemoglobinopathies.
The major challenges of haplo-HCT in hemoglobinopathies are delayed immune reconstitution and associated morbidity and mortality.
Abstract
We examined outcomes of haploidentical hematopoietic cell transplantation (haplo-HCT) using T-cell receptor αβ+ (TCRαβ+)/CD19+-depleted grafts (TCR group, 14 patients) in children with hemoglobinopathies. Patients received a preparative regimen consisting of busulfan, thiotepa, cyclophosphamide, and antithymocyte globulin preceded by fludarabine, hydroxyurea, and azathioprine. The median follow-up among surviving patients was 3.9 years. The 5-year probabilities of overall survival (OS) and disease-free survival (DFS) were 84% and 69%, respectively. The incidence of graft failure was 14%. We compared outcomes to a historical group of 40 patients with hemoglobinopathies who received CD34+-selected grafts (CD34 group). The median follow-up of surviving patients for the CD34 group was 7.5 years. The 5-year probabilities of OS and DFS were 78% and 39%, respectively. The CD34 group had a significantly higher incidence of graft failure (45%) than the TCR group (14%) (P = .048). The incidences of grades 2 to 4 acute graft-versus-host disease (GVHD) in the TCR and CD34 groups were 28% and 29%, respectively, and 21% and 10% (P = .1), respectively, for extensive chronic GVHD. Viral reactivation was common in both groups. The overall incidence of posttransplant lymphoproliferative disorders for the entire group was 16%. Among all patients, 5 developed autoimmune hemolytic anemia or thrombocytopenia, with the overall cumulative incidence of 11%. The 2 groups showed suboptimal CD4+ recovery within the first 6 months of transplantation with no significant difference between groups. These data demonstrate that TCRαβ+/CD19+-depleted grafts are associated with a reduced incidence of graft failure, but delayed immune reconstitution and associated morbidity and mortality remain a significant challenge.
Visual Abstract
Introduction
Hematopoietic cell transplantation (HCT) from an HLA-matched related or unrelated donor remains the only curative therapy for hemoglobinopathies.1,2 However, only 25% of patients have an HLA-matched sibling donor, and in a recent study from the US National Marrow Donor Program, <52% of non-Europeans found a matched unrelated donor.3 Furthermore, the lack of donor registries and cost of recruiting unrelated donors makes this approach unaffordable for developing countries, where many patients with hemoglobinopathies reside.
A haploidentical related donor is often available and represents an alternate source of stem cells.4,5 Recent advances in graft engineering with effective ex vivo T-cell depletion through positive selection of CD34+ cells, depletion of CD3+/CD19+ cells, or T-cell receptor αβ+ (TCRαβ+)/CD19+ depletion have significantly improved the outcome of haploidentical HCT (haplo-HCT).4,6-8 In our preliminary report of haplo-HCT for thalassemia using CD34+-selected and/or CD3+/CD19+-depleted grafts, we had reported a high graft failure rate,9 which was not reduced in a subsequent cohort of transplanted patients with hemoglobinopathies. Extensive T-cell depletion can significantly reduce the incidence of graft-versus-host disease (GVHD) but is associated with delayed immune recovery and an increased risk of graft rejection, especially in nonmalignant diseases.10 These results have prompted us to change the graft manipulation technique for haplo-HCT for hemoglobinopathies. We hypothesized that using a new graft manipulation method that removes αβ+ T lymphocytes while retaining γδ+ lymphocytes, natural killer (NK) cells, and other accessory cells11,12 would reduce the graft failure rate and improve outcomes. Since June 2012, we have been using this new graft manipulation technique with selective depletion of TCRαβ+ and CD19+ lymphocytes. Here, we report outcomes of patients with hemoglobinopathies treated with TCRαβ+/CD19+-depleted grafts and compare them with a historical similar cohort of patients who received CD34+-selected grafts. We also evaluated long-term outcomes for patients treated with CD34+-selected grafts.
Patients and methods
This is a retrospective, single-center study investigating outcomes of haplo-HCT in 14 consecutive children with hemoglobinopathies given TCRαβ+/CD19+-depleted grafts between June 2012 and March 2017 (TCR group). Patients were eligible for haploidentical transplantation if HLA-matched unrelated bone marrow (BM) donors were not available within at least 6 months of search initiation. The characteristics of the patients are summarized in Table 1. Patients were <17 years old and received a haplo-HCT (≥2 HLA-mismatched antigens) for thalassemia (n = 11) or sickle cell disease (SCD; n = 3). We also report outcomes of a historical group of patients who received halpo-HCT for hemoglobinopathies using CD34+-selected peripheral blood stem cell (PBSC) and BM grafts (n = 32) or CD34+-selected PBSC and CD3+/CD19+-depleted BM grafts (n = 8) between December 2005 and December 2011 (CD34 group) at our institution (Table 1). Seven patients with thalassemia who received CD34+-selected PBSC and BM grafts and reported in preliminary report9 were not included in the study because they had undergone transplantation at other institution and were lost to follow-up. Before transplantation, most patients were heavily transfused and had moderately severe iron overload, as evidenced by high median serum ferritin. The 2 groups showed similar demographic characteristics. More patients in CD34 group received rituximab prophylaxis (P = .01). The treatment protocol was approved by the Mediterranean Institute of Hematology Institutional Review Board, and the parents of all patients provided written informed consent in accordance with the Declaration of Helsinki.
Table 1.
Patient and transplant characteristics
| Variables | TCR group | CD34 group | P |
|---|---|---|---|
| Number of patients | 14 | 40 | |
| Age, median (range), y | 7 (3-15.2) | 7 (2.4-17) | .63 |
| Male sex, n | 12 | 23 | .1 |
| Thalassemia, n | 11 | 34 | .68 |
| Sickle cell anemia, n | 3 | 4 | — |
| HbS-β thalassemia, n | — | 2 | — |
| Risk class | .08 | ||
| Class 1/2/3, n | 0/7/4 | 10/16/10 | — |
| Not applicable | 3 | 4 | — |
| Indications for transplantation, n | |||
| Chronic blood transfusion | 11 | 36 | .35 |
| Stroke | 1 | — | — |
| Recurrent vaso-occlusive crisis | 3 | 4 | — |
| Splenectomy, n | 2 | 11 | .47 |
| Median packed RBC units received pretransplant (range) | 55 (0-300) | 55 (7-310) | .53 |
| Median serum ferritin (range), ng/mL | 1391 (32-6649) | 1633 (125-8696) | .54 |
| Donor, n | .35 | ||
| Mother | 12 | 32 | — |
| Father | 2 | 4 | — |
| Brother | — | 4 | — |
| Graft type, n | — | ||
| CD34+ selection of PBSCs and BM | — | 32 | — |
| CD34+ selection of PBSCs and CD3+/CD19+ depletion of BM | — | 8 | — |
| TCRαβ+/CD19+ depletion of PBSCs | 14 | — | — |
| Donor–recipient sex, n | .06 | ||
| Female to male | 11 | 19 | — |
| Other combinations | 3 | 21 | — |
| Donor–recipient CMV status, n | |||
| Both positive | 10 | 24 | .53 |
| Any positive | 2 | 14 | .18 |
| Both negative | 2 | 2 | .27 |
| Number of mismatches, n | |||
| 5 | 10 | 20 | .21 |
| 4 | 3 | 12 | .73 |
| 3 | 1 | 6 | .66 |
| 2 | 0 | 2 | 1 |
| Conditioning regimen, n | |||
| BuTT10Cy200ATG preceded by HuAzFlu (day −59 to −12)* | 14 | 37 | — |
| BuTT10Cy200ATG preceded by Flu (day −17 to −12)* | — | 3 | — |
| Rituximab prophylaxis (yes/no) | 2/12 | 23/17 | .01 |
| GVHD prophylaxis (from day −2 to day +60), n | |||
| CSA + methylprednisolone | 12 | 40 | — |
| CSA + mycophenolate mofetil | 2 | — | — |
| Graft content, median (range) | |||
| CD34+ × 106/kg | 15.7 (8.1-39.2) | 16 (4.3-28.1) | .43 |
| CD3+ × 105/kg | — | 2.8 (0.06-31.6) | — |
| CD19+ × 106/kg | — | 0.2 (0.01-2.11) | — |
| TCRαβ+ CD3+ × 104/kg | 4 (1-10.0) | — | — |
| TCRγδ+ CD3+ × 106/kg | 9 (2.8-40.2) | — | — |
| CD3−CD16+CD56+ × 106/kg | 28.7 (9.8-194.3) | — | — |
| CD20+ × 106/kg | 0.06 (0.01-1.7) | — | — |
Az, azathioprine; ATG, anti-thymocyte globulin; Flu, fludarabine; Bu, busulfan; CD34 group, CD34+-selected and/or CD3+/CD19+-depleted grafts; CSA, cyclosporine; Cy, cyclophosphamide; HbS, hemoglobin S; Hu, hydroxyurea; RBC, red blood cell; TCR group, TCRαβ+/CD19+-depleted grafts; TT, thiotepa.
From July 2004 to June 2006, patients received 14 mg/kg total dose of oral busulfan and weight-based intravenous Bu thereafter.
Patient–donor pairs were analyzed by 4-digit sequence-based typing or sequence-specific polymerase chain reaction (PCR) at the HLA-A, HLA-B, HLA-C, HLA-DRB, and HLA-DQB1 loci. Four-digit HLA-DPB1 typing was prospectively performed by sequence-specific PCR and PCR sequence-specific oligonucleotide probing. Anti-HLA antibodies were determined retrospectively using Luminex single antigen beads (One Lambda, Canoga Park, CA).
Patients were prepared according to the established regimen (Table 1), as described previously.9 Briefly, during the preconditioning phase, patients received hydroxyurea and azathioprine from day −59 pretransplant and fludarabine from day −17 through day −12. The conditioning regimen consisted of untargeted oral busulfan (n = 5) or weight-based targeted intravenous busulfan (n = 49), thiotepa, and cyclophosphamide. Anti-T-cell globulin (Thymoglobulin [Genzyme, Lyon, France] at a dose of 12.5 mg/kg over 4 days [n = 6 patients] or ATG-Fresenius [Neovii Biotech GmbH, Gräfelfing, Germany] at a dose of 50 to 25 mg/kg over 5 days [n = 48 patients]) was administered for prevention of rejection and GVHD. Twenty-five patients received rituximab at a dose of 375 mg/m2 on day −1 to reduce the risk of Epstein-Barr virus (EBV)–related lymphoproliferative disorders (PTLD). Patients received GVHD prophylaxis with cyclosporine and methylprednisolone (n = 52) or cyclosporine and mycophenolate mofetil (n = 2) until day +60. Acute GVHD (aGVHD) and chronic GVHD were diagnosed, and severity was scored following published criteria.13,14
Chimerism analysis was first performed on whole BM samples 20 days posttransplantation. Percentages of donor–recipient DNA were determined by PCR-based analysis of short tandem repeats (Profiler Plus Applera, Foster City, CA). Myeloid and lymphoid chimerism was determined at 60, 90, 180, and 365 days posttransplantation.
Autologous BM was cryopreserved in case of graft failure for all patients. In the TCR group, donor PBSCs were mobilized with granulocyte colony-stimulating factor (12 μg/kg per day) from day −5 until collection on day 0. In 3 donors with CD34+ cell counts <0.04 × 109/L on day −1, plerixafor (240 μg/kg) was administered. Ex vivo TCRαβ+/CD19+ depletion was performed using an immunomagnetic method (CliniMACS, Miltenyi Biotech, Bergisch Gladbach, Germany).15 Donors in CD34 group underwent PBSC collection 3 to 4 months before BM harvest. CD34+ cells were isolated using the CliniMACS device (Miltenyi Biotech) and were cryopreserved. On day 0, patients received thawed CD34+ cells and CD34+-selected and/or CD3+/CD19+-depleted cells from freshly harvested BM.
Study end points and outcome definitions
Study end points were graft failure/rejection, disease-free survival (DFS), overall survival (OS), GVHD, and transplant-related mortality. DFS was defined as survival without graft rejection or death and OS as the time from transplant to death from any cause. Primary graft failure was defined as failure to achieve absolute neutrophil count >0.5 × 109/L by day 28 and the presence of <10% donor cells according to the BM chimerism assay. Secondary graft failure was defined as donor engraftment followed by graft loss, as evidenced by a persistent decrease in the absolute neutrophil count of <0.5 × 109/L and loss of donor chimerism in BM/peripheral blood. The day of neutrophil engraftment was defined as the first of 3 consecutive days during which the absolute neutrophil count was ≥0.5 × 109/L. Platelet engraftment was defined as the first of 7 consecutive days during which platelet counts exceeded 20 × 109/L without transfusion.
Supportive care
For infectious disease prophylaxis, the patients received systemic antibacterial and antifungal drugs until the neutrophil level was >1.0 × 109/L. They also received acyclovir for herpes virus prophylaxis and trimethoprim/sulfamethoxazole for Pneumocystis jirovecii prophylaxis. From preconditioning until at least 180 days posttransplantation, patients were monitored weekly for EBV and cytomegalovirus (CMV) in the blood and for adenovirus and BK polyomavirus (BKV) in blood and urine using sensitive reverse-transcription PCR.
Statistical analysis
Patient-, disease-, and transplant-related variables were compared between the 2 groups using χ2 statistics for categorical variables and the Mann-Whitney U test for continuous variables. Probabilities of OS and DFS were estimated using the Kaplan-Meier method and compared between groups using a log-rank test.16 The cumulative incidence of graft failure/rejection and GVHD was estimated, accounting for competing risks for each outcome.17 Death with sustained engraftment was considered a competing risk for rejection and rejection and death before GVHD as competing risks for GVHD. Between-group differences were assessed using Gray’s test. All P values were 2 tailed and considered significant if <.05. Statistical analyses were performed using IBM SPSS Statistics 21.0 statistical software and the EZR statistical program.18
Results
TCRαβ+/CD19+-depleted haplo-HCT
Engraftment.
Thirteen out of 14 patients (92%) received >10 × 106 CD34+ cells/kg. The median αβ+ T-cell dose was 4 × 104/kg, and the median γδ+ T cell dose was 9 × 106/kg (Table 1). Engraftment of neutrophils and platelets occurred in 93% and 86% of patients, respectively. Recovery of neutrophils and platelets occurred after a median of 13 days (range, 10-15 days) and 15 days (range, 10-21 days), respectively.
Graft failure/rejection.
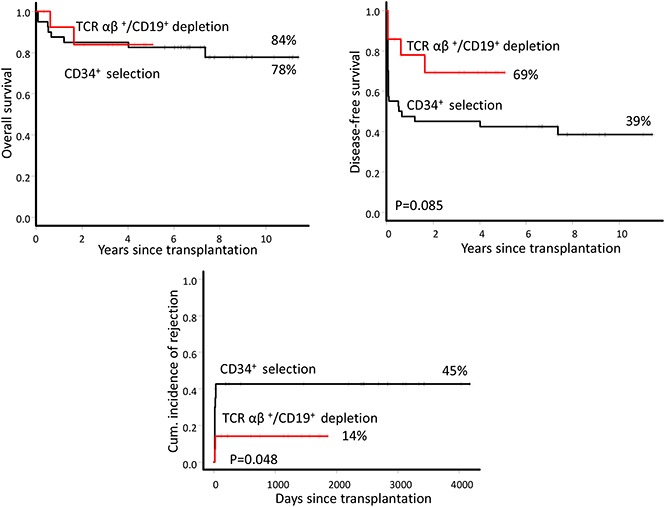
All but 2 patients had sustained engraftment. Two patients with thalassemia had primary graft failure or secondary graft failure. Both patients were given an autologous backup stem cell infusion as soon as graft failure was confirmed and showed complete autologous hematopoietic recovery. Despite primary graft failure, one of these patients had an unusually high level of fetal hemoglobin (99%) after transplantation and became transfusion independent in >4 years with hemoglobin >10 to 11 g/dL. No graft failure episode occurred beyond 1 month. The cumulative incidences of graft failure was 14% (95% confidence interval [CI], 0% to 30%) (Figure 1A). In a retrospective analysis of 5 patients whose pretransplant serum samples were available, we investigated anti-HLA antibodies.19 Among these patients, 1 who had graft failure was positive for anti-HLA antibodies, but not for donor-specific antibodies (DSAs) (Table 2). All patients with sustained engraftment had complete donor chimerism.
Figure 1.

Outcomes of TCRαβ+/CD19+-depleted haploidentical HCT. Cumulative incidence of graft failure (A) and Kaplan-Meier probability of OS and DFS (B).
Table 2.
Anti-HLA antibodies in patients who underwent haplo-HCT
| Number | Graft type | Anti-HLA antibodies | Donor-specific anti-HLA antibodies | Outcome | ||
|---|---|---|---|---|---|---|
| Class I | Class II | Class I | Class II | |||
| 1 | CD34+ selected* | No | No | No | No | Stable |
| 2 | CD34+ selected | No | No | No | No | Rejection |
| 3 | CD34+ selected | No | No | No | No | Rejection |
| 4 | CD34+ selected | No | No | No | No | Stable |
| 5 | CD34+ selected | No | Yes | No | No | Rejection |
| 6 | CD34+ selected | No | Yes | No | No | Stable |
| 7 | CD34+ selected | No | No | No | No | Stable |
| 8 | CD34+ selected | Yes | Yes | A24; MFI = 21 520 | No | Rejection |
| 9 | CD34+ selected | Yes | No | B57; MFI = 777 | No | Rejection |
| 10 | CD34+ selected | No | No | No | No | Stable |
| 11 | CD34+ selected | No | No | No | No | Rejection |
| 12 | CD34+ selected | No | No | No | No | Stable |
| 13 | CD34+ selected | Yes | Yes | A23; MFI=16891 | DQ2; MFI = 2358 | Rejection |
| B45; MFI = 21 993 | ||||||
| 14 | TCRαβ depleted† | No | No | No | No | Stable |
| 15 | TCRαβ depleted | Yes | Yes | No | No | Rejection |
| 16 | TCRαβ depleted | No | No | No | No | Stable |
| 17 | TCRαβ depleted | No | No | No | No | Stable |
| 18 | TCRαβ depleted | No | No | No | No | Stable |
“No” indicates that antibodies were not found.
MFI, mean fluorescence intensity.
CD34+-selected and/or CD3+/19+-depleted grafts.
TCRαβ+/CD19+-depleted grafts.
GVHD.
Four patients developed grade 2 to 3 aGVHD; grade 3 aGVHD occurred in 1 patient. All but 1 patient developed skin aGVHD. The incidence of grade 2 to 3 aGVHD was 28% (95% CI, 1% to 49%). Three patients developed extensive chronic GVHD with the incidence of 21% (95% CI, 0% to 40%). One patient had generalized steroid-dependent skin involvement, a second patient had liver and lung involvement, and the third patient had skin and gastrointestinal involvement. Chronic GVHD contributed to death of 1 patient. The remaining 2 patients are off immunosuppression.
Survival.
The median follow-up among surviving patients was 3.9 years (range, 0.5-5.2 years). The 5-year probabilities of OS and DFS were 84% (95% CI, 49% to 96%) and 69% (95% CI, 37% to 87%), respectively (Figure 1B). Two patients died of gastrointestinal bleeding (day +222) or diffuse large B-cell lymphoma (+1.7 years).
Immune reconstitution.
The kinetics of immune reconstitution is shown in Table 3. All patients with sustained engraftment had the absolute numbers of CD4+ cells less then <200/μL within 6 months of transplantation. At 6 months, the absolute numbers of CD3+, CD4+, CD8+, CD19+, and NK cells were 707 ± 179, 190 ± 38, 272 ± 33, 270 ± 91, and 274 ± 76, respectively; at 12 months, they were 832 ± 250, 295 ± 74, 415 ± 140, 307 ± 103, and 182 ± 49, respectively.
Table 3.
Immune reconstitution of patients after haplo-HCT
| 1 mo | 3 mo | 6 mo | 12 mo | 24 mo | |
|---|---|---|---|---|---|
| TCRαβ+/CD19+-depleted HCT* | |||||
| CD3+ T cells/µL | 265 ± 110 | 366 ± 229 | 707 ± 179 | 832 ± 250 | 1640 ± 414 |
| CD3+CD4+ T cells/µL | 33 ± 17 | 58 ± 23 | 190 ± 38 | 295 ± 74 | 608 ± 106 |
| CD3+CD8+ T cells/µL | 175 ± 73 | 97 ± 17 | 272 ± 33 | 415 ± 140 | 868 ± 347 |
| CD19+ B cells/µL | 4 ± 2 | 287 ± 109 | 270 ± 91 | 307 ± 103 | 205 ± 60 |
| CD3−CD56+ NK cells/µL | 199 ± 49 | 174 ± 53 | 274 ± 76 | 182 ± 49 | 429 ± 141 |
| CD34+* selected- and/or CD3+/CD19+-depleted HCT | |||||
| CD3+ T cells/µL | 140 ± 88 | 626 ± 252 | 736 ± 216 | 1014 ± 238 | 2390 ± 559 |
| CD3+CD4+ T cells/µL | 17 ± 11 | 78 ± 29 | 148 ± 43 | 423 ± 97 | 934 ± 203 |
| CD3+CD8+ T cells/µL | 120 ± 77 | 531 ± 218 | 528 ± 147 | 559 ± 147 | 1314 ± 311 |
| CD19+ B cells/µL | 9 ± 2 | 320 ± 94 | 151 ± 54 | 600 ± 241 | 539 ± 169 |
| CD3−CD56+ NK cells/µL | 255 ± 51 | 152 ± 39 | 127 ± 27 | 413 ± 151 | 481 ± 246 |
Data are expressed as means ± standard error of the mean.
There was no statistically significant difference between the 2 groups with respect to each cell subset determined at different time points.
Infectious complications.
Viral reactivation was common. The incidences of CMV, EBV, adenovirus, and BKV reactivation were 64% (95% CI, 28% to 82%), 28% (95% CI, 0% to 49%), 7% (95% CI, 0% to 19%), and 23% (95% CI, 1% to 64%), respectively. None of the patients developed CMV or adenovirus disease. The incidence of BKV-associated late-onset hemorrhagic cystitis was 35% (95% CI, 5% to 57%). Three patients developed EBV-related PTLD, with a cumulative incidence of 23% (95% CI, 0% to 43%). None of these patients received rituximab prophylaxis. Bacterial infections were also frequent; 5 (38%) patients developed gram-negative or gram-positive sepsis and 7 (50%) patients pneumonia. Invasive fungal infections occurred in 2 (14%) patients.
Autoimmune disorders.
Four patients developed autoimmune disorders: 3 patients had autoimmune hemolytic anemia at 183, 415, and 875 days, and 1 patient had autoimmune thrombocytopenia at 478 days after transplantation. The incidence of autoimmune disorders was 37% (95% CI, 0% to 60%). None of these patients received rituximab prophylaxsis. The first-line treatment of all patients included methylprednisolone and immunoglobulins. Three patients achieved complete remission after rituximab was administered as second-line treatment.
Outcomes of TCR group patients were compared with a historical group of 40 patients with hemoglobinopathies who received CD34+-selected and/or CD3+/CD19+-depleted grafts (CD34 group) in our institution. The median follow-up was 7.5 years (range, 5.7-11.6 years) for the CD34 group. The cumulative incidence of graft failure was significantly higher in the CD34 group (45%; 95% CI, 27% to 58%) than the TCR group (14%; 95% CI, 0% to 31%) (P = .048). There was no association between graft failure and patient-, disease-, and treatment-related variables (age, sex, serum ferritin, number of blood transfusion, risk classes, splenectomy, and busulfan concentration), donor type, HLA mismatches, type of disease, and CD34+ cell dose (data not shown). In a retrospective analysis of 13 CD34 group patients (6 patients with sustained engraftment and 7 patients with graft failure), we investigated anti-HLA antibodies.19 Anti-HLA antibodies were identified in 5 (38%) patients. Three of these patients also had DSAs (Table 2). Four of 5 patients (80%) with anti-HLA antibodies had rejection compared with 3 of 8 patients (38%) who were negative for anti-HLA antibodies (P = .26, Fisher’s exact test). All 3 patients with DSAs had rejection vs 4 of 10 patients (40%) without DSAs (P = .19).
OS was similar between the 2 groups (78%; 95% CI, 23% to 54% versus 84%; 95% CI, 49% to 96%) (P = .9). DFS was higher in the TCR group (69%; 95% CI, 37% to 87%) than the CD34 group (39%; 95% CI, 23% to 54%), although the difference was not statistically significant (P = .08), likely because of the small number of patients in the TCR group. In the entire group, patients aged ≤10 years had significantly higher OS than patients >10 years old (94%; 95% CI, 80% to 99% vs 47%; 95% CI, 19% to 71%; P=.0004). Respective DFS was 57% (95% CI, 39% to 71%) and 27% (95% CI, 8% to 50%) (P = .25). Of note, for the whole cohort, there was a significantly higher incidence of PTLD in patients who did not receive rituximab prophylaxis (26%; 95% CI, 7% to 41%) compared with patients who did (4%; 95% CI, 0% to 10%) (P = .032). Five patients (4 in TCR group and 1 in CD34 group) developed autoimmune hemolytic anemia or thrombocytopenia with the overall cumulative incidence of 11% (95% CI, 1% to 19%). The TCR group had a significantly higher incidence of autoimmune disorders (37%) than the CD34 group (3%) (P = .002). It is noteworthy that none of these patients received rituximab prophylaxis. In fact, the incidence of autoimmune disorders was significantly higher in patients who did not receive rituximab (19%; 95% CI, 2% to 33%) compared to those who did (0%) (P = .041).
All 18 patients in the CD34 group with graft failure were given an autologous backup stem cell infusion as soon as graft failure was confirmed. Despite initial recovery of autologous hematopoiesis, 2 of these patients died of infectious complications. The remaining 16 patients are alive with complete recovery of autologous hematopoiesis.
In the CD34 group, 8 patients died of pneumonia (day +29), perianal abscess (day +33), CMV pneumonia (day +190), diffuse large B-cell lymphoma (day +199), disseminated aspergillosis (day +243), toxic megacolon (+1.2 years), acute heart failure (+4 years), and overwhelming postsplenectomy sepsis (+7.4 years). There were no statistically significant differences between the 2 groups with respect to incidences of vial reactivation, acute and chronic GVHD, infectious complications, immune reconstitution, and PTLD (data not shown).
Discussion
The role of haploidentical HCT in hemoglobinopathies and the long-term outcomes are unclear. Our present results showed that TCRαβ+/CD19+-depleted grafts were associated with a significantly reduced incidence of graft failure compared with CD34+-selected grafts in patients with hemoglobinopathies. The major reason for transplant failure in our patients given CD34+-selected grafts was graft failure. This finding is corroborated by the most recent observation that the incidence of graft failure was 50% in 8 patients with SCD who received CD34+-selected and CD3+-depleted grafts from haploidentical parents.20 Extensive T-cell depletion through CD34+ selection and infusion of megadose CD34+-cell (>10 × 106 CD34+ cells/kg)–containing grafts resulted in a high engraftment rate and low GVHD after haplo-HCT in patients with hematologic malignancies.4 It has been suggested that high numbers of CD34+ cells can overcome the HLA barrier to engraftment. Extensive T-cell depletion can significantly reduce the incidence of GVHD but is associated with delayed immune recovery and an increased risk of graft rejection, especially in nonmalignant diseases.10,21 In the present study, the incidence of graft failure in the CD34 group was unacceptably high despite a megadose of CD34+ cells and an intensive myeloablative and immunosuppressive conditioning regimen, indicating that cells removed because of extensive T-cell depletion are important for promoting engraftment in immunocompetent patients with hemoglobinopathies. In fact, donor T cells play an important role in promoting engraftment following transplantation.22,23 Recently, Gilman et al reported on 10 SCD patients who underwent CD34+-selected, T-cell–depleted PBSC transplantation from mismatched family or unrelated donors.24 Seven of these patients received transplantation from related donors with >2 mismatches. Patients were given prophylactic donor lymphocyte infusion starting 33 days after transplantation. Only one patient had graft failure. Two-year OS and DFS rates were 90% and 80%, respectively. The low incidence of graft failure in that study may in part be related to early donor lymphocyte infusion, which promoted sustained engraftment. Reduced graft failure with TCRαβ+/CD19+ grafts observed in our patients is consistent with a recent study showing a 16.2% graft failure rate following TCRαβ+/CD19+-depleted haplo-HCT for nonmalignant conditions.25 Unlike CD34+-selected grafts, TCRαβ+/CD19+-depleted grafts contain γδ lymphocytes, NK cells, and stem cells, which might promote engraftment in these patients. In fact, the γδ T cells promote engraftment of allogeneic stem cells in preclinical models.26,27 However, a higher graft failure rate (36%) has been reported in children with immunodeficiencies who underwent TCRαβ+/CD19+-depleted haplo-HSCT following a reduced-intensity conditioning regimen,28 indicating that the intensity of the conditioning regimen is an important contributor to graft failure in patients with nonmalignant conditions.
Donor-specific anti-HLA antibodies are associated with a high rate of graft rejection after haplo-HCT.29 We retrospectively analyzed pretransplant serum samples of 18 patients for the presence of DSAs. Three patients who received CD34+-selected grafts had DSAs. Although patients with DSAs had a higher rate of graft failure than patients who did not have DSAs, this was not statistically significant, likely because of the small sample size and unrecognized DSAs.19 Remarkably, in CD34 group, 4 of 10 patients (40%) who were negative for DSAs had rejection, which supports our hypothesis that graft content plays an important role in promoting engraftment in T-cell–depleted haplo-HCT in patients with hemoglobinopathies. The high sustained engraftment rate in patients treated with TCRαβ+/CD19+-depleted grafts translated into a higher DFS rate (69%) compared with patients given CD34+-selected grafts, although it was not statistically significant because of the small number of patients.
The incidence of grade 2 to 3 aGVHD (28%) observed in our patients was similar to that reported in children who received TCRαβ+/CD19+-depleted haploidentical grafts with posttransplant pharmacological GVHD prophylaxis for immunodeficiencies28 or malignant and nonmalignant diseases.30 In contrast, Bertaina et al reported a 13% acute GVHD rate in a group of 23 patients who received TCRαβ+/CD19+-depleted haplo-HCT for nonmalignant conditions and received no GVHD prophylaxis.25 It is worth noting that the median dose of αβ T cells given to our patients was similar to that reported by Bertaina et al, whereas patients in the previous 2 studies28,30 received lower doses of αβ T cells. The incidence of extensive chronic GVHD in our patients treated with TCRαβ+/CD19+-depleted grafts was higher than that reported in previously published series.25,28,30
The incidence of PTLD was relatively high (23%) in the TCR group, probably because few patients were given rituximab before transplantation. In fact, patients who received rituximab prophylaxis had a significantly lower incidence of EBV-associated PTLD than patients who did not (4% vs 26%). Therefore, we recommend rituximab prophylaxis and EBV monitoring in patients undergoing haplo-HCT. However, we cannot rule out a negative impact of anti-thymocyte globulin in the conditioning on development of PTLD.31
Remarkably, 4 patients in the TCR group and 1 patient in the CD34 group developed autoimmune disorders, with an overall cumulative incidence of 11%, which is in agreement with previously published studies.32,33 However, we found that patients who did not receive rituximab prophylaxis had significantly higher incidence of autoimmune disorders. This finding suggests the importance of rituximab prophylaxis in patients undergoing T-cell–depleted haploidentical transplantation.
A delayed immune reconstitution and associated infectious complications are common in T-cell–depleted haplo-HCT. Our finding of suboptimal CD4+ cell recovery within the first 6 months is consistent with published studies.30,34,35 In the present study, the kinetics of immune reconstitution was similar between patients given TCRαβ+/CD19+-depleted and CD34+-selected haplo-HCT. However, we cannot rule out a negative impact of posttransplant immune suppression on slow immune recovery.
Haploidentical transplantation using T-cell–replete grafts and posttransplant cyclophosphamide (PT-Cy) is another recently developed alternative transplant platform.36 Bolanos-Meade et al reported a high graft failure rate (46%) among 14 SCD patients who underwent haploidentical transplantation following nonmyeloablative conditioning regimen and PT-Cy.37 An intensive preconditioning immunoablation followed by a myeloablative conditioning regimen and PT-Cy in the haploidentical setting for thalassemia showed promising results; among 31 patients transplanted, 2 had rejection and 1 patient died, with an OS and DFS of 95% and 94%, respectively.38 Further studies and long-term follow-up are necessary to determine whether this approach results in significant improvement of transplant outcomes.
This study has some inherent limitations, including its retrospective design and single-center setting. The follow-up period for the TCRαβ+/CD19+-depleted group was short. However, all patients had at least 6 months of follow-up, enabling us to make reliable conclusions of early outcomes. Furthermore, this study was conducted in homogeneous groups in terms of disease and treatment, allowing us to draw useful conclusions about the efficacy of TCRαβ+/CD19+-depleted grafts in reducing graft failure rate after haplo-HCT for hemoglobinopathies. In addition, this is the longest follow-up reported to date for outcomes of haploidentical transplantation using CD34+-selected grafts for hemoglobinopathies.
In summary, we have shown that the use of TCRαβ+/CD19+-depleted grafts is associated with significantly reduced graft failure in patients with hemoglobinopathies. CD34+-selected haploidentical transplantation for patients with hemoglobinopathies is associated with a high graft failure rate and low DFS; therefore, the use of such grafts in these patients should be discouraged. Delayed immune reconstitution and associated morbidity and mortality remain a significant problem in this setting. Novel pharmacologic and cell therapy approaches to enhance immune reconstitution and improve outcome after T-cell–depleted haplo-HCT are warranted.
Acknowledgments
The authors thank Guido Lucarelli, the former Scientific Director of the Mediterranean Institute of Hematology, who pioneered haploidentical hematopoietic stem cell transplantation for thalassemia and has run the haploidentical transplant program for thalassemia at our institute. The authors thank the nursing staff for their dedication to patient care and the patients and their families for participating in this study. Special thanks go to the Department of Pediatric Hematology/Oncology and Transfusion Medicine, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy, for graft manipulation.
Authorship
Contribution: J.G. designed the study, performed the transplantations, interpreted data, performed statistical analyses, and wrote the article; A.I., P.S., K.P., G.D.A., M.M., M.R., and C.A. performed the transplantations and followed patients; A.L. and G.A. performed PBSC collection; and T.G. and M.A. performed HLA typing, chimerism, and anti-HLA antibody studies.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Javid Gaziev, International Center for Transplantation in Thalassemia and Sickle Cell Anemia, Mediterranean Institute of Hematology, Policlinico Tor Vergata, Viale Oxford, 81, 00133 Rome, Italy; e-mail: j.gaziev@fondazioneime.it.
References
- 1.Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22(2):53-63. [DOI] [PubMed] [Google Scholar]
- 2.Shenoy S, Eapen M, Panepinto JA, et al. . A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. 2016;128(21):2561-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gragert L, Eapen M, Williams E, et al. . HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. 2014;371(4):339-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aversa F, Tabilio A, Velardi A, et al. . Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med. 1998;339(17):1186-1193. [DOI] [PubMed] [Google Scholar]
- 5.Robinson TM, O’Donnell PV, Fuchs EJ, Luznik L. Haploidentical bone marrow and stem cell transplantation: experience with post-transplantation cyclophosphamide. Semin Hematol. 2016;53(2):90-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Federmann B, Bornhauser M, Meisner C, et al. . Haploidentical allogeneic hematopoietic cell transplantation in adults using CD3/CD19 depletion and reduced intensity conditioning: a phase II study. Haematologica. 2012;97(10):1523-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Handgretinger R. Negative depletion of CD3(+) and TcRαβ(+) T cells. Curr Opin Hematol. 2012;19(6):434-439. [DOI] [PubMed] [Google Scholar]
- 8.Diaz MA, Pérez-Martínez A, Herrero B, et al. . Prognostic factors and outcomes for pediatric patients receiving an haploidentical relative allogeneic transplant using CD3/CD19-depleted grafts. Bone Marrow Transplant. 2016;51(9):1211-1216. [DOI] [PubMed] [Google Scholar]
- 9.Sodani P, Isgrò A, Gaziev J, et al. . Purified T-depleted, CD34+ peripheral blood and bone marrow cell transplantation from haploidentical mother to child with thalassemia. Blood. 2010;115(6):1296-1302. [DOI] [PubMed] [Google Scholar]
- 10.Willasch A, Hoelle W, Kreyenberg H, et al. . Outcome of allogeneic stem cell transplantation in children with non-malignant diseases. Haematologica. 2006;91(6):788-794. [PubMed] [Google Scholar]
- 11.Chaleff S, Otto M, Barfield RC, et al. . A large-scale method for the selective depletion of alphabeta T lymphocytes from PBSC for allogeneic transplantation. Cytotherapy. 2007;9(8):746-754. [DOI] [PubMed] [Google Scholar]
- 12.Schumm M, Lang P, Bethge W, et al. . Depletion of T-cell receptor alpha/beta and CD19 positive cells from apheresis products with the CliniMACS device. Cytotherapy. 2013;15(10):1253-1258. [DOI] [PubMed] [Google Scholar]
- 13.Przepiorka D, Weisdorf D, Martin P, et al. . 1994 consensus conference on acute GVHD grading. Bone Marrow Transplant. 1995;15(6):825-828. [PubMed] [Google Scholar]
- 14.Filipovich AH, Weisdorf D, Pavletic S, et al. . National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11(12):945-956. [DOI] [PubMed] [Google Scholar]
- 15.Li Pira G, Malaspina D, Girolami E, et al. . Selective depletion of αβ T cells and B cells for human leukocyte antigen-haploidentical hematopoietic stem cell transplantation. A three-year follow-up of procedure efficiency. Biol Blood Marrow Transplant. 2016;22(11):2056-2064. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan EL, Meier P. Non parametric estimation from incomplete observations. J Am Stat Assoc. 1958;53(282):457-481. [Google Scholar]
- 17.Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18(6):695-706. [DOI] [PubMed] [Google Scholar]
- 18.Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48(3):452-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreani M, Testi M, Sodani P, et al. . Impact of donor-specific anti-HLA antibodies and donor KIR characteristics in haploidentical HSCT for beta-thalassemia. Mediterr J Hematol Infect Dis. 2017;9(1):e2017020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dallas MH, Triplett B, Shook DR, et al. . Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2013;19(5):820-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonough CH, Jacobsohn DA, Vogelsang GB, Noga SJ, Chen AR. High incidence of graft failure in children receiving CD34+ augmented elutriated allografts for nonmalignant diseases. Bone Marrow Transplant. 2003;31(12):1073-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin PJ, Akatsuka Y, Hahne M, Sale G. Involvement of donor T-cell cytotoxic effector mechanisms in preventing allogeneic marrow graft rejection. Blood. 1998;92(6):2177-2181. [PubMed] [Google Scholar]
- 23.Jiang Z, Adams GB, Hanash AM, Scadden DT, Levy RB. The contribution of cytotoxic and noncytotoxic function by donor T-cells that support engraftment after allogeneic bone marrow transplantation. Biol Blood Marrow Transplant. 2002;8(11):588-596. [DOI] [PubMed] [Google Scholar]
- 24.Gilman AL, Eckrich MJ, Epstein S, et al. . Alternative donor hematopoietic stem cell transplantation for sickle cell disease. Blood Adv. 2017;16(1):1217-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertaina A, Merli P, Rutella S, et al. . HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood. 2014;124(5):822-826. [DOI] [PubMed] [Google Scholar]
- 26.Blazar BR, Taylor PA, Bluestone JA, Vallera DA. Murine γ/δ-expressing T cells affect alloengraftment via the recognition of nonclassical major histocompatibility complex class Ib antigens. Blood. 1996;87(10):4463-4472. [PubMed] [Google Scholar]
- 27.Drobyski WR, Majewski D. Donor γ δ T lymphocytes promote allogeneic engraftment across the major histocompatibility barrier in mice. Blood. 1997;89(3):1100-1109. [PubMed] [Google Scholar]
- 28.Balashov D, Shcherbina A, Maschan M, et al. . Single-center experience of unrelated and haploidentical stem cell transplantation with TCRαβ and CD19 depletion in children with primary immunodeficiency syndromes. Biol Blood Marrow Transplant. 2015;21(11):1955-1962. [DOI] [PubMed] [Google Scholar]
- 29.Ciurea SO, de Lima M, Cano P, et al. . High risk of graft failure in patients with anti-HLA antibodies undergoing haploidentical stem-cell transplantation. Transplantation. 2009;88(8):1019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lang P, Feuchtinger T, Teltschik HM, et al. . Improved immune recovery after transplantation of TCRαβ/CD19-depleted allografts from haploidentical donors in pediatric patients. Bone Marrow Transplant. 2015;50(suppl 2):S6-S10. [DOI] [PubMed] [Google Scholar]
- 31.Landgren O, Gilbert ES, Rizzo JD, et al. . Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood. 2009;113(20):4992-5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drobyski WR, Potluri J, Sauer D, Gottschall JL. Autoimmune hemolytic anemia following T cell-depleted allogeneic bone marrow transplantation. Bone Marrow Transplant. 1996;17(6):1093-1099. [PubMed] [Google Scholar]
- 33.Faraci M, Zecca M, Pillon M, et al. ; Italian Association of Paediatric Haematology and Oncology. Autoimmune hematological diseases after allogeneic hematopoietic stem cell transplantation in children: an Italian multicenter experience. Biol Blood Marrow Transplant. 2014;20(2):272-278. [DOI] [PubMed] [Google Scholar]
- 34.Janossy G, Prentice HG, Grob JP, et al. . T lymphocyte regeneration after transplantation of T cell depleted allogeneic bone marrow. Clin Exp Immunol. 1986;63(3):577-586. [PMC free article] [PubMed] [Google Scholar]
- 35.Daley JP, Rozans MK, Smith BR, Burakoff SJ, Rappeport JM, Miller RA. Retarded recovery of functional T cell frequencies in T cell-depleted bone marrow transplant recipients. Blood. 1987;70(4):960-964. [PubMed] [Google Scholar]
- 36.Luznik L, O’Donnell PV, Symons HJ, et al. . HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14(6):641-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolaños-Meade J, Fuchs EJ, Luznik L, et al. . HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285-4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anurathapan U, Hongeng S, Pakakasama S, et al. . Hematopoietic stem cell transplantation for homozygous β-thalassemia and β-thalassemia/hemoglobin E patients from haploidentical donors. Bone Marrow Transplant. 2016;51(6):813-818. [DOI] [PMC free article] [PubMed] [Google Scholar]