

Abstract
Epigenetic mechanisms mediate the acquisition of specialized cellular phenotypes during tissue development, maintenance and repair. When phenotype-committed cells transit through mitosis, chromosomal condensation counteracts epigenetic activation of gene expression. Subsequent post-mitotic re-activation of transcription depends on epigenetic DNA and histone modifications, as well as other architecturally bound proteins that ‘bookmark’ the genome. Osteogenic lineage commitment, differentiation and progenitor proliferation require the bone-related runt-related transcription factor Runx2. Here, we characterized a non-genomic mRNA mediated mechanism by which osteoblast precursors retain their phenotype during self-renewal. We show that osteoblasts produce maximal levels of Runx2 mRNA, but not protein, prior to mitotic cell division. Runx2 mRNA partitions symmetrically between daughter cells in a non-chromosomal tubulin-containing compartment. Subsequently, transcription-independent de novo synthesis of Runx2 protein in early G1 phase results in increased functional interactions of Runx2 with a representative osteoblast-specific target gene (osteocalcin/BGLAP2) in chromatin. Somatic transmission of Runx2 mRNAs in osteoblasts and osteosarcoma cells represents a versatile mechanism for translational rather than transcriptional induction of this principal gene regulator to maintain osteoblast phenotype identity after mitosis.
Keywords: osteoblast, mitosis, mRNA inheritance, RUNX2 transcription factor, lineage commitment
Introduction
Mitotic division during embryonic development and tissue homeostasis in adults results in phenotypically identical or functionally differentiated cells depending on whether cell division results in the symmetrical or asymmetrical distribution of regulatory factors. Inheritance of maternal mRNAs and subsequent distribution of mRNAs during mitotic cleavage stages in the developing zygote controls cell fate during early embryogenesis in both invertebrate and vertebrate species. Upon fertilization, maternal transcripts accumulated in the cytoplasm of mature oocytes are asymmetrically segregated to different embryonic cells during the first cleavages when zygotic transcription is still silenced (Jeffery and Wilson, 1983; Heasman et al., 2001; Schier, 2007; White and Heasman, 2008). This unequal partitioning of transcripts generates a heterogeneous distribution of specific mRNA molecules in progeny cells and regional specialization within the developing embryo (Weeks and Melton, 1987; Mowry and Cote, 1999; Skamagki et al., 2013). Mitotic inheritance of mRNAs organizes cell signaling pathways and localizes transcription factor activities for early embryonic patterning of cell fates (Forristall et al., 1995; Nishida, 2002; Kobayashi et al., 2003; Nakamura et al., 2003; Zhou and Lou King, 2004; Jedrusik et al., 2015).
Asymmetrical segregation of cell fate determinants also generates cellular diversity during stem cell differentiation (Knoblich, 2010; Roubinet and Cabernard, 2014). During neuronal development in Drosophila embryos (Wadsworth et al., 1985; Li et al., 1997) and mammals (Kusek et al., 2012), several pro-neurogenic mRNA determinants (prospero, prox1, Bbs2 and Trim32) are asymmetrically localized in the cytoplasm of neural stem cells (neuroblasts and radial glia precursors). Asymmetric cell division produces both neural stem cells capable of self-renewal, and smaller neural progenitor cells (Broadus et al., 1998). Preferential segregation of specific mRNAs generates uncommitted stem cells or committed neural cells (Matsuzaki et al., 1998; Vessey et al., 2012). While asymmetric partitioning of key regulatory factors is effective in supporting cellular differentiation, when somatic cells have committed to a lineage-specific phenotype at later developmental stages, symmetric division of expanding progenitor cells may supports retention of the parental phenotype. The distribution of mRNAs during symmetric cell division may be selectively mediated or occur via a relatively non-specific mechanism (e.g., binding to microtubules). However, because many mRNAs are cell cycle regulated in proliferating cells, cells appears to be selective in what mRNAs are produced during G2 phase in anticipation of mitotic division. Although any mRNAs that is transmitted from a precursor cell would facilitate the translation of the protein in the two progeny cells, one key point is that many mRNAs encoding regulatory proteins are degraded prior to mitosis and that mRNAs which are transmitted do not necessarily have a regulatory function. A priori, it is not evident whether any regulatory mRNAs that are transmitted would either be symmetrically or asymmetrically distributed upon completion of cell division, but either event is likely to have different functional consequences.
Osteogenic lineage commitment of immature mesenchymal cells and proliferative expansion of osteoprogenitors are fundamental for bone formation, bone regeneration and stem cell-based bone tissue engineering approaches. The osteogenic cell fate is determined by the orchestrated biological effects of extracellular signaling ligands (Bellido et al., 1997, 2003; He et al., 2011; Lin and Hankenson, 2011; Zhu et al., 2011; Almeida and O`Brien, 2013; Canalis, 2013; Greenblatt et al., 2013; Kobayashi and Kronenberg, 2014; Marie, 2013; Tang and Alliston, 2013; Shimizu et al., 2014; Sims and Civitellis, 2014; van de Peppel and van Leeuwen, 2014), bone-related transcription factors (Xiao et al., 2005; Danciu et al., 2012; Li et al., 2012; Zhu et al., 2012) and epigenetic regulators (Thomas and Kansara, 2006; Jensen et al., 2007; Stein et al., 2009; Hesse et al., 2010), as well as microRNAs (Zhang et al., 2011; van der Deen et al., 2013; van Wijnen et al., 2013). Osteogenic transcription factors and co-factors together organize the regulatory machinery for execution of bone phenotype-specific gene expression programs by epigenetic modifications of chromatin (Thomas and kansara, 2006; Jense et al., 2007; Young et al., 2007a, b; Stein et al., 2009; Stein et al., 2010; Tai, 2014). During proliferative expansion of pre-osteoblasts, specific gene expression patterns related to bone-lineage commitment must resume in proliferating osteoprogenitors following mitosis.
The program of gene expression required for lineage determination and differentiation of immature mesenchymal cells to osteoblasts is activated by the runt-related transcription factor Runx2 (Komori, 2010, 2011). Runx2 also controls the proliferative expansion of pre-osteoblastic cells (Pratap et al., 2003; Galindo et al., 2005) and Runx2 gene expression is regulated during the cell cycle to accommodate its cell growth regulatory functions. Runx2 protein levels are highest in the G1 phase (Pratap et al., 2003; Galindo et al., 2005, 2007; San Martin et al., 2009), but low basal levels of Runx2 protein remain associated with mitotic chromosomes as part of an architectural epigenetic mechanism (‘mitotic bookmarking’) that is linked to post-translational modifications of chromatin (Young et al., 2007a, b). Strikingly, Runx2 mRNA levels maximally accumulate at mitosis prior to the up-regulation of Runx2 protein in early G1 phase (Galindo et al., 2005). Because this newly synthesized Runx2 mRNA does not appear to be translated during G2 phase, the biological importance of this accumulation may be related to a post-mitotic function of Runx2. This accumulation is particularly unusual, because other genes (e.g., cyclin A mRNA) are immediately translated into protein during G2 phase and not necessarily transmitted to progeny cells.
In this study, we address the functional significance of this mitotic accumulation of Runx2 mRNA, and address whether Runx2 mRNA is either equally or unequally distributed. Using in situ mRNA hybridization, we show that Runx2 transcripts are segregated symmetrically during cell division into progeny cells. Furthermore, using protein metabolic labeling, immuno-precipitation, and inhibition of RNA polymerase II-dependent transcription of mitotically synchronized cells, we demonstrate that mitotically inherited Runx2 mRNA is rapidly translated to maximize Runx2 protein levels early after mitosis. We propose that post-mitotic segregation of mRNAs encoding the osteogenic transcription factor Runx2 contributes to the maintenance of phenotype commitment to the osteoblast lineage during cell division.
Materials and Methods
Cell culture
Mouse pre-osteoblastic MC3T3-E1 cells, human osteoblastic hFOB cells and human osteosarcoma cells (U2OS, G292, and HOS) were maintained as indicated in αMEM or DMEM culture medium (Gigco, Life Technologies, Grand Island, NY, USA) supplemented with 10–15% fetal bovine serum (FBS) plus 2 mM L-glutamine and a penicillin-streptomycin cocktail at 37°C and 5% CO2 according to ATCC recommendations. MC3T3-E1 and hFOB cells were maintained in αMEM supplemented with 10% FBS. U2OS and G292 cells were cultured in McCoy`s medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% FBS. HOS cells were grown in DMEM medium with 10% FBS. Cells were seeded in either 6-well or 100-mm plates at 0.08 × 106 cells/well or 0.4 × 106 cells/plate, respectively, and grown in a sub-confluent state for 24–72 h until the onset of exponential growth. The growth medium was changed every 2 days.
Cell synchronization
Experiments were performed with the mouse pre-osteoblastic cell line MC3T3-E1. Exponentially growing cell cultures were treated with the indicated cell cycle inhibitors to arrest cells at different cell cycle stages (Galindo et al., 2005). Cells were treated for 24 h with 400 µM mimosine (Sigma-Aldrich, St. Louis, MO, USA) to arrest cell in the late G1 phase (Krude, 1999). Cell cycle arrest in mitosis was achieved by nocodazole treatment. Cells grown in medium plus FBS were treated with 100 ng/ml nocodazole (Sigma-Aldrich) for 16 h, followed by shake-off of mitotic cells. Cells arrested in late G1 (mimosine) or in mitosis (nocodazole) were released by three washes in serum-free medium and stimulated to progress, respectively, to S or G1 phase by the addition of fresh medium without drug containing FBS plus 2 mM L-glutamine and antibiotics. After serum stimulation, cells were harvested at selected time points for Western blot, RT-PCR analysis and fluorescence-activated cell sorting (FACS) analysis.
Flow cytometric analysis
The distribution of cells at specific cell cycle stages was evaluated by assessment of DNA content by flow cytometry, as previously described (Teplyuk et al., 2008). Cells were trypsinized, washed with phosphate-buffered saline (PBS), and fixed in 70% ethanol at −20 °C overnight. Cells were then treated with RNAse A (10 µg/ml) at 37 °C for 15 min. Subsequently, cells were stained with propidium iodide and subjected to FACS analysis based on DNA content. Samples (1 × 106 cells) were analyzed using the FACStar cell sorter and Consort 30 software (Becton Dickinson, San Jose, CA, USA).
Western blot analysis
Runx2 and cell cycle markers were analyzed by immuno-blot analysis as described previously (Galindo et al., 2005; Galindo et al., 2007). Briefly, equal amounts of total cellular protein collected in the presence of the proteasome inhibitor MG132 (Calbiochem, San Diego, CA, USA) and Complete® cocktail of protease inhibitor (Roche Diagnostics, Mannhein, Germany) were resolved in 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore, Billerica, MA.). Blots were incubated with a 1:2,000 dilution of each primary antibody for 1 h. Rabbit polyclonal antibodies (Cdk4, sc-260; cyclin A, sc-596), mouse monoclonal antibody (cyclin D1, sc-20044), and goat polyclonal antibody (actin, sc-1615) were acquired commercially (Santa Cruz Biotechnology, Inc.). Runx2-specific mouse monoclonal 8G5 antibody was obtained from MBL International (Woburn, MA). Membranes with bound primary antibodies were incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, Inc.) for 1 h. Immuno-reactive protein bands were visualized on a film (BioMax, Kodak) using a chemiluminescence detection kit (PerkinElmer Life Sciences), and signal intensities were quantitated by densitometry. Each experiment was repeated at least three times.
cDNA synthesis and PCR
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s specifications. Total RNA (5 µg/lane) was separated in a 1% agarose-formaldehyde gel. Ethidium bromide staining of the gels was used to assess equal loading and the RNA quality of samples. Purified RNA (1 µg) was subjected to reverse transcription using random hexamer primers (Promega) with M-MLV reverse transcriptase (Promega) according to the manufacturer’s recommendations to produce cDNA. Gene expression was assessed by PCR using the following specific mouse gene primers (0.5 pmol/µl): Runx2: F 5'-CCG CAC GAC AAC CGC ACC AT-3'; R 5'-CGC TCC GGC CCA CAA ATC TC-3'; Cyclin A: F 5'-GAA GAC CAA GAG AAT GTC AA-3'; R 5'-CCT CAT GCT GTT AGT GAT GTC-3'; Cyclin B1: F 5'-TGC AGC ACT ACC TAT CCT AC-3'; R 5'-TGG AGT TAT GCC TTT GTC-3'; Cyclin D1: F 5'-GGC GGA TGA GAA CAA GCA GA-3'; R 5'-ACC AGC CTC TTC CTC CAC TT-3'; Cyclin E: F 5'-TAA GCC CTC TGA CCA TTG-3'; R 5'-GGA ACC ATC CAT TTG ACA C-3'; GADPH: F 5'-CCT TCA TTG ACC TCA ACT A-3'; R 5'-GGC CAT CCA CAG TCT TCT-3'. Aliquots of cDNA (1 µl) were amplified with 0.3 µl recombinant DNA polymerase Thermus aquaticus (Taq) 5 U/µl (Invitrogen Corporation) by incubation for 5 min at 94°C and 20–30 amplification cycles of synthesis were applied to avoid product saturation (1 min at 94°C, 1 min at 52–62°C, and 1 min at 72°C), followed by a final extension step at 72°C for 6 min. Aliquots of the resulting product (5 µl) were visualized in 1% agarose gels by ethidium bromide staining.
Luciferase reporter assays
For reporter assays, MC3T3-E1 cells were seeded at 0.08 × 106 cells/well in a six-well plate and transiently transfected 24 h after plating at a cell density of 60–70% with 1 µg of a previously described construct of the Runx2 promoter/pGL3 luciferase reporter plasmid which contains the 0.6-kb mouse Runx2 promoter (upstream P1 promoter; MASNS isoform) fused to the firefly luciferase reporter (Drissi et al., 2000). Cells were co-transfected with 10 ng SV40/Renilla construct as an internal control. The promoterless pGL3 luciferase parent vector was used as a negative control. Lipofectamine 2000® (Invitrogen) was used as a transfection agent according to the manufacturer’s protocol and transfections were performed in the absence of FBS and antibiotics. Medium was changed after 4 h to normal growth medium with FBS and cells were allowed to growth for 12 h. Cells were then synchronized with mimosine and nocodazole as described above. After serum stimulation, cells were harvested at selected time points in 1x passive lysis buffer for promoter activity studies. The luciferase activity was measured in cell lysates using the Dual Luciferase Reporter Assay System® kit (Promega, Madison, WI) following the manufacturer’s instructions. Luminescent signal was quantified by a luminometer (Synergy® 2SL BioTek), and each measurement from the firefly luciferase construct was normalized using Renilla luciferase values.
RNA in situ hybridization (RISH)
A 933 bp KpnI DNA fragment containing part of the mouse Runx2 coding region, spanning region between positions 703 and 1636 (NCBI identifiers: NM_001146038.2 & GI:410110911), was subcloned in Bluescript KS II plasmid to generate antisense digoxigenin (DIG)-UTP-labeled RNA-probes using T7 RNA polymerase. In situ hybridization on culture cells was performed using a modification of a published procedure (Jin and Lloyd, 1997). Briefly, cells were fixed in 1% paraformaldehyde for 10 minutes and washed twice in 1 × PBST (100 mM phosphate, pH 7.5; 150 mM NaCl; 0.05% Tween 20) for 5 minutes. Cells were incubated with pre-hybridization buffer (dimethyl formamide 50%) for 1 h at 42 °C. For hybridization, cells were incubated with antisense probes for 24 h at 42°C and washed sequentially with 75, 50, 25 and 0 % formamide in SSC 2X at 42°C for 10 minutes. Cells were then rinsed three times for 5 minutes in MABT (100 mM maleic acid, 150 mM NaCl, pH 7.5, 0.1% Tween®) at room temperature, blocked for 1 h at room temperature in GS/BMB/MAB (10% goat serum, 2% Blocking reagent [Roche catalog #1096176] in MABT), and incubated for 24 hours at 4°C in 1% goat serum in MABT with 1:100 alkaline phosphatase-coupled anti-Digoxigenin antibody (Roche Diagnostics). Finally, cells were washed twice with 1x PBS for 5 minutes, three times with alkaline phosphate buffer and then mounted for microscopy. Cells were analyzed in a Zeiss Axiostar Plus light microscope.
Immunofluorescence and fluorescent RNA in situ hybridization (IF-FRISH)
Cells were seeded at low confluence in sterile chamber slide (Lab-Tek, Nalge Nunc Int.) in a total volume of 350 µl. Cells were incubated for 24 h at 37°C to allow cells to adhere to the cover slip. Adherent cells were rinsed with PBS and fixed with 4% paraformaldehyde for 15 min at 4°C, rinsed 3 times with PBS for 5 min and permeabilized for 15 to 30 min with PBS/Triton-x100. Cells were treated with NH4Cl for 5 to 10 min, and blocked with PBS/BSA/1% glycine for 1 h. The cells were then incubated with anti-tubulin β (Sigma-Aldrich) antibody diluted 1:50 overnight at 4°C. After incubation, the preparations were washed 3 times with PBS for 5 min and then incubated with an Alexa Fluor 488 conjugated secondary antibody (1:100) for one hour at room temperature. The preparations were finally washed 3 times with PBS for 5 min. Immunofluorescence was followed by in situ-hybridization that was initiated by pre-incubation with hybridization buffer (dimethyl formamide 50%) supplemented with blockers of nonspecific hybridization (yeast tRNA, and heparin) for 1 h at 42°C. Samples were then incubated with a 1:100 dilution of anti-sense Runx2 (DIG)-UTP-labeled RNA-probe for 24 h at 42°C. Three consecutive washes were then applied for 10 min at 42°C, with formamide at decreasing concentrations (75%, 50% and 25%) in SSC buffer (sodium citrate, saline, pH 7.0) 2X to remove excess probe. Samples were then incubated with MABT blocking solution supplemented with 10% calf serum. Probes were visualized using a rhodamine-conjugated anti-digoxigenin antibody (1:50) by incubation for 1 h at room temperature. Excess antibody was removed by two consecutive rinses with PBS for 5 min and 10 min before DNA staining by incubation with DAPI (1:10,000) for 10 min. Preparations were washed three more times with PBS for 5 min prior to the addition of fluorescence mounting medium (Dako Omnis). Cells were analyzed in a spinning disk confocal microscope system (which combines inverted IX81 motorized microscope Olympus /DSU/MT20 and Cell^R imaging software).
Protein and RNA metabolic labeling
MC3T3-E1 cells synchronized by 16 h nocodazole treatment, as described above, were additionally pre-treated at 14 h with or without the RNA polymerase II inhibitor α-amanitin at 10 µM (A2263, Sigma-Aldrich) and then stimulated to progress into G1 phase in the presence or absence of α-amanitin 10 µM, respectively. Inhibition of RNA synthesis by α-amanitin (10 µM) was confirmed by metabolic labelling of MC3T3-E1 cells using [3H]-uridine (40 Ci mmol−1; New England Nuclear, Boston, MA). Protein metabolic labelling experiments were carried out in mitotically arrested cells, as well as in cells progressing from mitosis into G1 phase in the presence or absence of α-amanitin. Specifically, cells were harvested at selected time points and incubated with [35S]methionine 100 µCi (Amersham SJ 1015, in vivo cell labelling grade, 1,000 Ci mmol−1, 10 µCi µl−1) in methionine-free MEM for 1 h at 37°C. The radioactive medium was removed and cells were rinsed with ice-cold PBS. Cells were subjected to immuno-precipitation using a specific antibody to analyze radiolabeled Runx2 proteins.
Immunoprecipitation
Cells labeled with [35S]methionine were lysed in 1,000 µl of ice-cold lysis buffer (50 mM Tris HCl pH 7.4, 200 mM NaCl, 1 mM EDTA, 1% NP-40, 25 µM MG132, and 1X Complete® cocktail of protease inhibitor for 15 min at 4°C, followed by centrifugation at 16,000 × g. The supernatant was precleared with 30 µl of protein A/G plus agarose beads (sc-2003, Santa Cruz Biotechnology) at 4°C for 30 min. The beads were collected by centrifugation at 1,000 × g for 5 min at 4°C. The precleared supernatant was adjusted to a final protein concentration of 1,000 ug/ml and incubated with 1 µg of Runx2 rabbit polyclonal antibody (M-70, sc-10758, Santa Cruz Biotechnology) or normal IgG rabbit (sc-2027, Santa Cruz Biotechnology) for 2 h at 4°C with agitation before being incubated with Protein A/G plus agarose beads for 1 h at 4°C with agitation. Immuno-precipitated Runx2 was resolved by SDS-PAGE. Gels containing radiolabeled Runx2 protein were dried and data were analyzed using a Storm 840 PhosphorImager (Molecular Dynamics, Inc.). Immuno-precipitated Runx2 was also subjected to western blot analysis with Runx2 antibody.
Chromatin immunoprecipitation (ChIP) analysis
ChIP studies were performed as described previously (van der Deen et al., 2008). Pre-cleared chromatin fragments (200–300 bp) obtained from MC3T3 cell were immunoprecipitated overnight with agitation using the Runx2 M-70 polyclonal antibody (Santa Cruz Biotechnology). The PCR primers used to evaluate the up-stream region of the mouse Osteocalcin gene (−276/−5) by Q-PCR were F 5'-CTG AGA GAG AGA GAG CAC ACA G-3' (forward) and R 5'-CCT CCA GCA TCC AGT AGC AT-3' (reverse).
Statistics
All quantitative data are presented as mean ± SD with a minimum of three independent samples. Statistical significance is determined by two-tailed Student’s t test. A p value less than 0.05 is considered statistically significant.
Results
Cell cycle dependent activation of the Runx2 promoter during interphase supports mitotic accumulation of Runx2 mRNA in MC3T3-E1 osteoblasts
Runx2 gene expression is modulated when cells transit through the cell cycle and mitosis, as reflected by temporal regulation of the protein and mRNA levels in mimosine-synchronized MC3T3-E1 osteoblasts progressing through the cell cycle (Fig. 1). Cell cycle progression upon release from mimosine-arrest was monitored by flow cytometric analysis (Figs. 1A and 1B) and sequential expression of the classical cell cycle markers cyclin D1 (G1 phase), cyclin E (S phase), cyclin A (G2/M phases) and cyclin B1 (M phase)(Fig. 1C). MC3T3-E1 cells that are arrested with mimosine in late G1 phase exhibit low levels of Runx2 mRNA and protein (Figs. 1D and 1E). However, upon release from the mimosine blockade, maximal mRNA accumulation precedes elevation of Runx2 protein levels during subsequent cell cycle progression towards mitosis (Figs. 1D and 1E). Runx2 mRNA levels are initially maintained at low levels during late G1 until at least the G1/S phase transition (at 12 h), but these levels reach a maximum in G2 phase and mitosis by 18 h (Fig. 1E; P=0.001). In parallel, Runx2 protein remains relatively low in the late G1 and S phases during the initial 12 h after release. A pronounced post-mitotic increase in Runx2 protein levels is observed as cells progress beyond mitosis (G2/M) and enter the ensuing early G1 phase by 24 h (Fig. 1D; P=0.018). The selective mitotic accumulation of Runx2 mRNA and the post-mitotic expression of Runx2 protein (Figs. 1D and 1E) is firmly supported by our prior studies that examined Runx2 mRNA and protein regulation (Galindo et al., 2005; San Martin et al., 2009).
Fig. 1. Runx2 promoter activity controls Runx2 mRNA accumulation prior to mitosis in MC3T3 osteoblasts.

Runx2 protein and mRNA levels as well as Runx2 gene promoter activity were assessed during progression through the cell cycle in MC3T3-E1 osteoblasts to determine specific transition stages when Runx2 mRNA levels are modulated. Cells were synchronized by incubation for 24 h with mimosine to generate a late G1 phase block. (A) Cells were then released from late G1 phase arrest and stimulated to progress through the cell cycle by the addition of fresh culture medium without mimosine and harvested after 0, 6, 12, 18, 24, 30 and 36 h. Progression through successive cell cycle phases (G1 late, S, G2/M and G1 early) was monitored by flow cytometry. (B) Graphic representation of cell cycle stage data presented in panel A. (C) Expression of cell cycle markers was evaluated by western blot analysis (cyclins D, A and Cdk4) and RT-PCR (cyclins D, E, A and B). (D) Cell cycle-dependent modulations in Runx2 protein and mRNA levels were assessed by western blot and RT-PCR. (E) Graphic representation of cell cycle-related changes in Runx2 protein (between: 0 h & 12 h, P = 0.017; 0 h & 24 h, P = 0.018) and mRNA levels (between: 0 h & 6 h, P = 0.049; 18 h & 24 h, P = 0.001; 24 h & 36 h, P = 0.014). Datapoints represent the averages and standard deviation of multiple experiments. Protein and mRNA values were normalized to actin and GAPDH, respectively. (F) Relative promoter activity of Runx2 promoter/luciferase reporter gene construct (mouse 0.6 kb/LUC) is shown. Luciferase values were normalized to SV40/Ranilla construct activity. Values are means of three single experiments. Cell cycle phases as determined by flow cytometry are indicated at the bottom of the gels and graphs.
The gene expression analyses presented in Fig. 1 and elsewhere in the paper were performed using semi-quantitative PCR with three distinct sets of biological samples that were derived from three independent cell cycle synchronies (Fig. 1E). This technique was selected instead of real-time qPCR, because it permits direct visualization of amplified cDNAs and because it is a cost-effective method that shows reproducible cell cycle changes in Runx2 mRNA in relation to cyclin mRNA expression. Our mRNA data were obtained with the same samples that exhibit temporal changes in cyclin protein accumulation thus further strengthening our results. In the three synchronization experiments, each of the gene expression analyses in synchronized cells showed essentially very similar results as evidenced by standard deviations and P-values of results obtained during different cell cycle stages. In all synchronization experiments, we observed that Runx2 mRNA accumulates before Runx2 protein, and that Runx2 mRNA accumulation coincides with increased expression of Cyclin A mRNA (S/G2 markers), while Runx2 protein is only upregulated when cyclin A mRNA levels decline. Furthermore, our results corroborate similar cell synchronization experiments that were performed previously (Galindo et al., 2005).
Cell cycle dependent modulations in Runx2 mRNA may be regulated by both transcriptional and post-transcriptional mechanisms. Previous studies have shown that Runx2 transcription is regulated by multiple distinct transcription factors, including proteins interacting in a cell cycle dependent manner with an AP1 binding site (Hovhannisyan et al., 2013), while other proteins (e.g., Runx2, Sp1, Ets-1, Hes-1) may have additional roles (Drissi et al., 2000; Zhang et al., 2009a; Zhang et al., 2009b). Although dissecting precise contributions of individual transcription factors is beyond the focus of this study, measurements of promoter activity during the cell cycle may reveal whether transcriptional modulations contribute to selective accumulation of Runx2 mRNA. Therefore, we examined luciferase activity driven by the principal 0.6 kbp regulatory region of the Runx2 gene promoter (Fig. 1F). This promoter region (designated P1) contains two sites of in vivo protein/DNA interactions, one of which involves cell growth regulated binding of factors to an AP1 element (Hovhannisyan et al., 2013). Luciferase activity driven by the Runx2 P1 gene promoter gradually increases in tandem with the accumulation of Runx2 mRNA as cells progress through G2 towards and beyond mitosis. Hence, accumulation on Runx2 mRNA at mitosis is at least in part supported by a G2-related transcriptional mechanism. Furthermore, we have previously estimated that the average half-life of Runx2 mRNA in asynchronously growing cells is approximately 2 h (Zhang et al., 2011). Thus, both mRNA degradation and transcription may account for cell cycle changes in Runx2 gene expression.
Post-mitotic increase in Runx2 protein during interphase in MC3T3-E1 osteoblasts
Runx2 promoter activity and Runx2 mRNA levels were examined in more detail during the M/G1 phase transition. Cells were synchronized in mitosis using nocodazole and then released into G1. Mitotic synchronization and progression into G1 was confirmed by FACS analysis based on DNA content and immunoblotting of cell cycle markers (Figs. 2A and B). Upon nocodazole treatment, 92% of cells are arrested in mitosis (0 h). After drug withdrawal and serum stimulation, the percentage of mitotic cells decreases to 7%, while the percentage of cells in G1 increases to 82% between 2 and 10 h after release from mitotic arrest. Modulations in cell cycle markers further reflect synchronized progression from mitosis into early G1 phase. For example, protein or mRNA levels of cyclins A and B decrease abruptly during early G1, whereas cyclin D protein levels increase progressively during interphase (Fig. 2C).
Fig. 2. Regulation of Runx2 gene promoter activity and mRNA levels after mitosis.
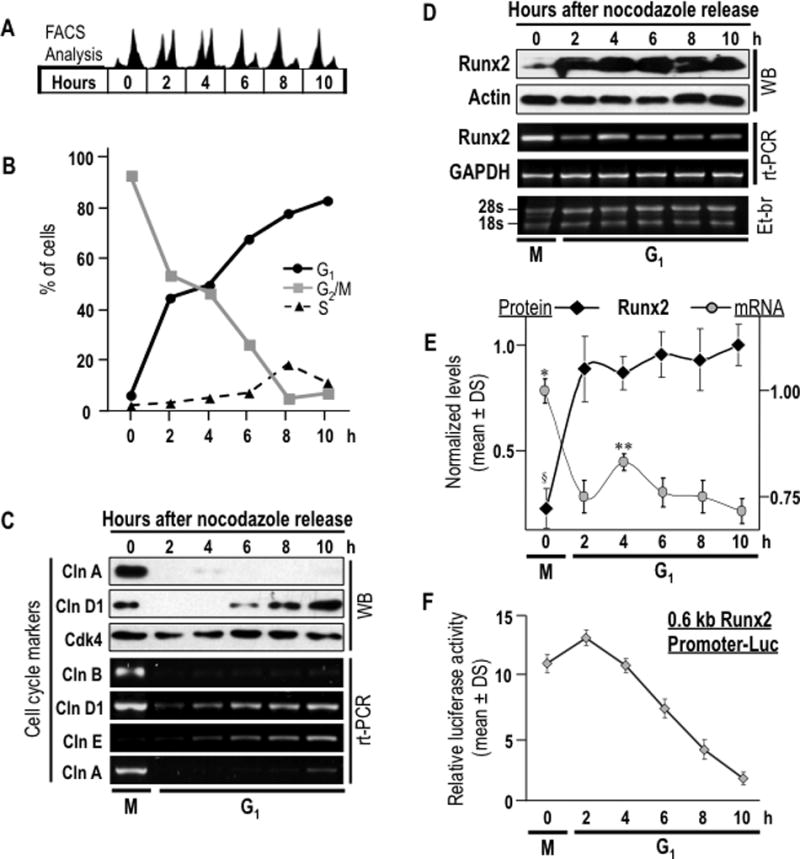
Runx2 protein and mRNA levels as well as Runx2 promoter activity were assessed during M/G1 phase transition in pre-osteoblasts MC3T3-E1 cells to analyze post-mitotic modulation of Runx2 mRNA levels. (A) Cells were synchronized by incubation for 16 h with nocodazole to generate a mitotic block. Mitotic cells were collected by gentle agitation (‘mitotic shake-off’), replated and released from mitosis into G1 by addition of fresh culture medium. Cells were harvested after 0, 2, 4, 6, and 10 h. Progression through mitosis into the next G1 phase was monitored by flow cytometry. (B) Graphic representation of cell cycle stage data presented in panel A. (C) Expression of cell cycle markers was evaluated by western blot analysis (cyclins A, D and Cdk4) and RT-PCR (cyclins D, E, A and B). (D) Post-mitotic modulations in Runx2 protein and mRNA levels were assessed by western blot and RT-PCR analyses. (E) Graphic representation of post-mitotic changes in Runx2 protein (§P = 0,003 between time 0 h, mitosis, and times 2 to 10 h, G1 phase) and mRNA levels (*P = 0.001 between 0 h, mitosis, and times 2 to 10 h, G1 phase; **P = 0.030 between 4 h and 2 h, and P = 0.03 between 4 h and 6 h). Datapoints represent the averages and standard deviation of multiple experiments. Protein and mRNA values were normalized to actin and GAPDH, respectively. (F) Relative promoter activity of Runx2 promoter/luciferase reporter gene construct (mouse 0.6 kB/LUC) is plotted. Luciferase values were normalized to SV40/Ranilla construct activity. Values are the means of three distinct experiments. Onset of the M/G1 phase transition as determined by flow cytometry is indicated at the bottom of the gels and graphs.
Importantly, the minimal levels of Runx2 protein observed in mitotically arrested cells (0 h) increase acutely (P=0.003) within two hours during early G1 and remain high until late G1 (2–10 h), (Figs. 2 D and E). The minimal levels in mitosis are not due to proteasomal degradation, because proteasomal inhibition does not elevate Runx2 protein levels (galindo et al., 2005). In contrast to the low levels of Runx2 protein, the elevated Runx2 mRNA levels in mitotic cells (0 h) decrease (P=0.001) when cells progress into early G1 (2 h) and these levels remain lower than observed in mitosis until at least late G1 (Figs. 2 D and E). Decreased Runx2 promoter activity parallels the down regulation of the Runx2 mRNA during post-mitotic cell cycle progression (Fig. 2F). Decreased gene promoter activity concomitant with decreased mRNA levels during G1 phase indicates that Runx2 gene expression is in part controlled by a transcriptional mechanism. However, the acute increase in Runx2 protein levels within two hours following mitosis suggests that expression is also translationally controlled.
Mitotically accumulated Runx2 mRNA partitions symmetrically into progeny cells
Our data show that cells mitotically accumulate Runx2 mRNA even though the protein is not fully expressed until the next cell cycle (Figs. 1 and 2). We assessed whether this mitotic mRNA may be functionally important by examining if Runx2 transcripts are transmitted to daughter cells upon cell division (Fig. 3). The cellular distribution of Runx2 mRNA was analysed during proliferative expansion of osteoprogenitors by in situ hybridization of probes with Runx2 mRNA in MC3T3-E1 cells both during interphase and mitosis. Interphase cells exhibit a perinuclear distribution of Runx2 mRNA (Fig. 3A). However, the distribution and intensity of Runx2 mRNA hybridization signal changes as cell progress from interphase into mitosis (Fig. 3A). The latter is consistent with alterations in nuclear structure (e.g., nuclear envelope breakdown) and the progressive accumulation of Runx2 mRNA observed during G2/M transition (see Fig. 1).
Fig. 3. Runx2 mRNA accumulates during mitosis and segregates to progeny cells following cell division.
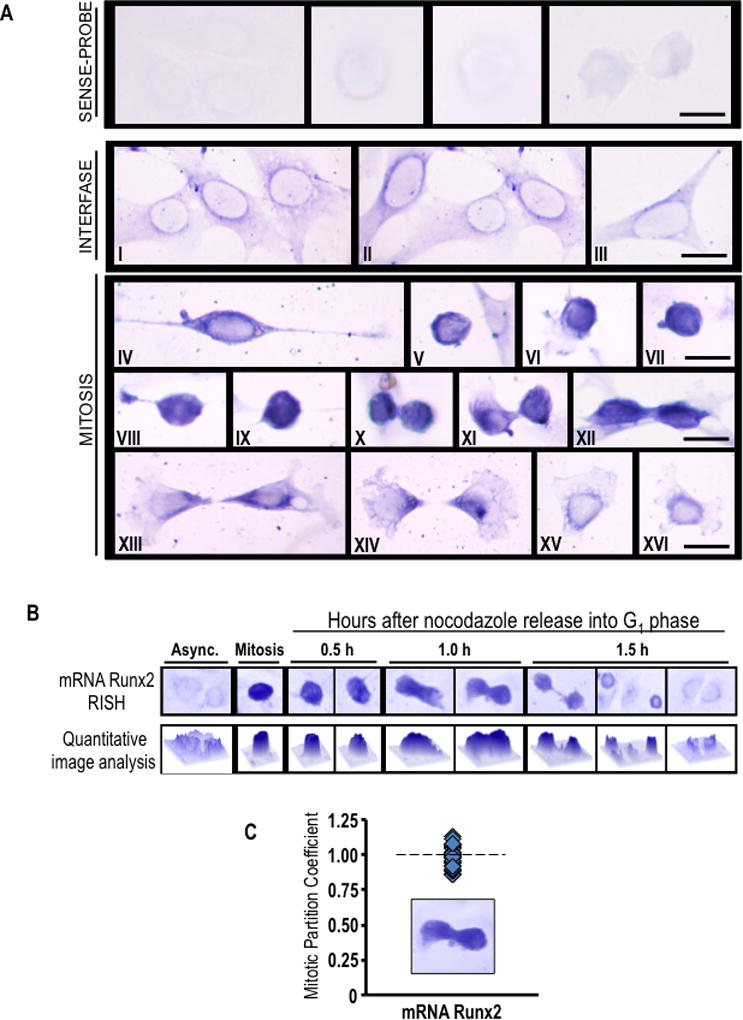
(A) Asynchronously growing MC3T3-E1 cells were fixed and subjected to in situ hybridization (RISH) analysis of Runx2 mRNA using DIG-labeled antisense probe. Sense probe was used as a control. Cytoplasmic/perinuclear blue staining denotes presence of Runx2 mRNA in pre-osteoblast cells during interphase (I-III). As interphase cells progressing into mitosis, Runx2 mRNA is concentrated as strong blue-black staining that is observed throughout the cell cortex (IV-IX). At cytokinesis, most of Runx2 mRNA persists in a cortical distribution (X-XII). After cell division, Runx2 mRNA appears to be segregated and evenly distributed between daughter cells (XIII-XIV) showing a polarized distribution at the region of the cleavage furrow. When the remaining cytoplasm of the midbody is retracted into nascent cells, Runx2 mRNA relocalizes around the nucleus (XV-XVI). Interphase and mitotic cells were identified by cell morphology. Scale bars: 20 µm, 100X oil objective, numerical aperture = 1.25. (B and C) RISH studies were also performed during mitotic block-release experiments at specific time points (0.5, 1.0 and 1.5 h) after nocodazole release. Runx2 RISH signal intensity was examined in situ by microscopy and quantitative image analysis (B) of progeny cells at the last step of cytokinesis (n=10) (C). We defined the partition coefficient, which reflects the ratio of integrated signal intensities between progeny cells. Cells progressing into G1 exhibit symmetrical partitioning of Runx2 mRNA between daughter cells.
We also analysed the distribution of Runx2 mRNA in greater detail during successive mitotic sub-stages. As mitosis progresses through metaphase to anaphase and telophase, Runx2 mRNA segregates to progeny cells (Fig. 3A). During mitotic exit and cytokinesis, Runx2 mRNA redistributes in a perinuclear pattern in daughter cells concomitant with nuclear envelope reassembly and cell division (Fig. 3A). We also quantified in situ RNA hybridization with DNA staining in telophase cells, and observed that Runx2 mRNA is similarly distributed to progeny cells per unit DNA during mitosis (Fig. 3B and C). Therefore, Runx2 mRNA is symmetrically segregated to progeny cells during mitosis.
We also assessed whether mitotic Runx2 mRNA also segregates in other osseous cells types, including human fetal osteoblasts (hFOB) and osteosarcoma cells (U2OS, G292, and HOS)(Fig. 4A). The results from in situ RNA hybridization show that Runx2 mRNA also segregates similarly in progeny of both normal and cancer cells (Fig. 4B). Interestingly, Runx2 mRNA is localized during metaphase in an extra-chromosomal microtubule-containing compartment based on results obtained by triple-label fluorescence microscopy that simultaneously visualizes Runx2 mRNA, DNA and microtubules (Fig. 5). Thus, unlike Runx2 protein which remains at least in part associated with metaphase chromosomes (Stein et al., 2009; Young et al., 2007a; Young et al., 2007b), Runx2 mRNA may distribute during mitosis in association with or in the vicinity of the mitotic spindle.
Fig. 4. Runx2 mRNA partitions equivalently in progeny of osseous cells following cell division.
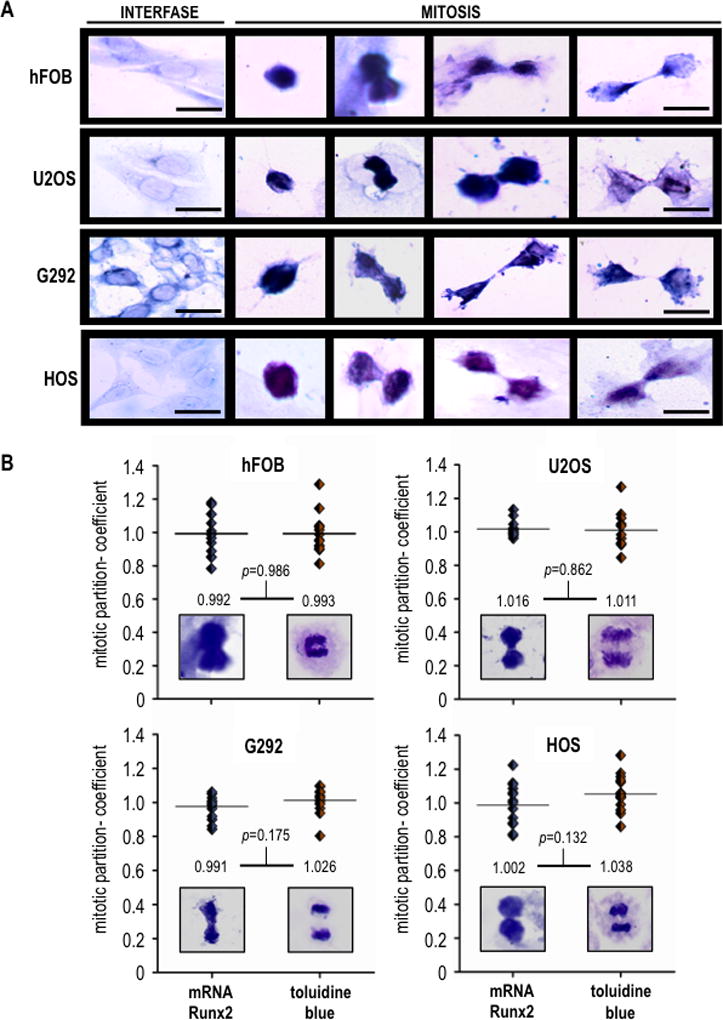
(A) Osteoblastic cell (hFOB) and osteosarcoma cells (U2OS, G292 and HOS) were subjected to RISH analysis. Scale bars: 20 µm, 100X oil objective, numerical aperture = 1.25. (B) Runx2 RISH signal intensity was determinede by quantitative image analysis in progeny cells at the last step of cytokinesis (n=10). Runx2 mRNA (RISH, left image) exhibits a partitioning coefficient equivalent to that of DNA (mitotic chromosomes staining, right image), demonstrating that this transcript segregates symmetrically in progeny cells upon cell division. Student’s t test was performed to assess the significance of the observed differences.
Fig. 5. Runx2mRNA co-localizes with β-tubulin during interphase and mitosis.
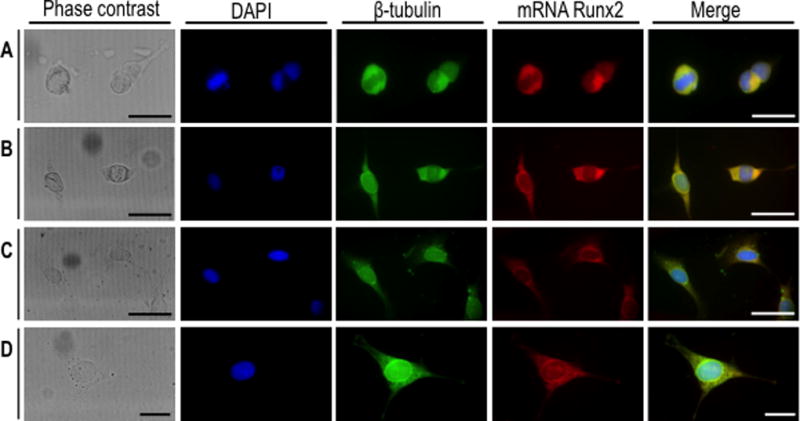
Asynchronously growing MC3T3-E1 cells were fixed and subjected to β-tubulin immunofluorescence (IF) (green) combined with Runx2 mRNA fluorescence in situ hybridization (FRISH) analysis by using DIG-labeled antisense probe (red). DAPI blue staining was used to visualize DNA. Mitotic cells (A-B) and interphase cells (C-D) were analyzed by confocal microscopy. (A, B and C) Scale bars: 50 µm, 60X oil objective, numerical aperture = 1.42; (D) Scale bars: 20 µm, 100X oil objective, numerical aperture = 1.40.
Post-mitotic inheritance of Runx2 mRNA supports de novo Runx2 protein synthesis in early G1
To assess whether post-mitotically inherited Runx2 mRNA is able to support induction of Runx2 protein expression during early G1 in progeny cells, we examined Runx2 protein synthesis by metabolic labelling with [35S]-methionine and immuno-precipitation in cells synchronously progressing from M into G1 phase (Fig. 6). Mitotic cells were released into G1 in the presence or absence of the transcriptional inhibitor α-amanitin to inhibit synthesis of novel transcripts and to ensure that protein synthesis changes are translationally controlled (Figs. 6A). FACS analysis reveals that progression into G1 after mitotic shake-off and removal of nocodazole proceeds normally upon inhibition of RNA Pol II-dependent transcription in the presence of α-amanitin (Figs. 6B and 6C). The cellular effectiveness of α-amanitin as a transcriptional inhibitor is evidenced at different cell cycle time-points by modest inhibition of Runx2 protein accumulation in mid-G1, but no significant differences (Fig. 6D and E), and changes in cyclin D1 and Cdk4 protein accumulation (Fig. 6D and F). Because cyclin D1 is a very labile protein, its levels closely mirror changes in α-amanitin dependent mRNA accumulation (Fig. 7A).
Fig. 6. Restitution of Runx2 protein levels during early G1 phases of cell cycle is independent of transcriptional activity for mRNA synthesis.
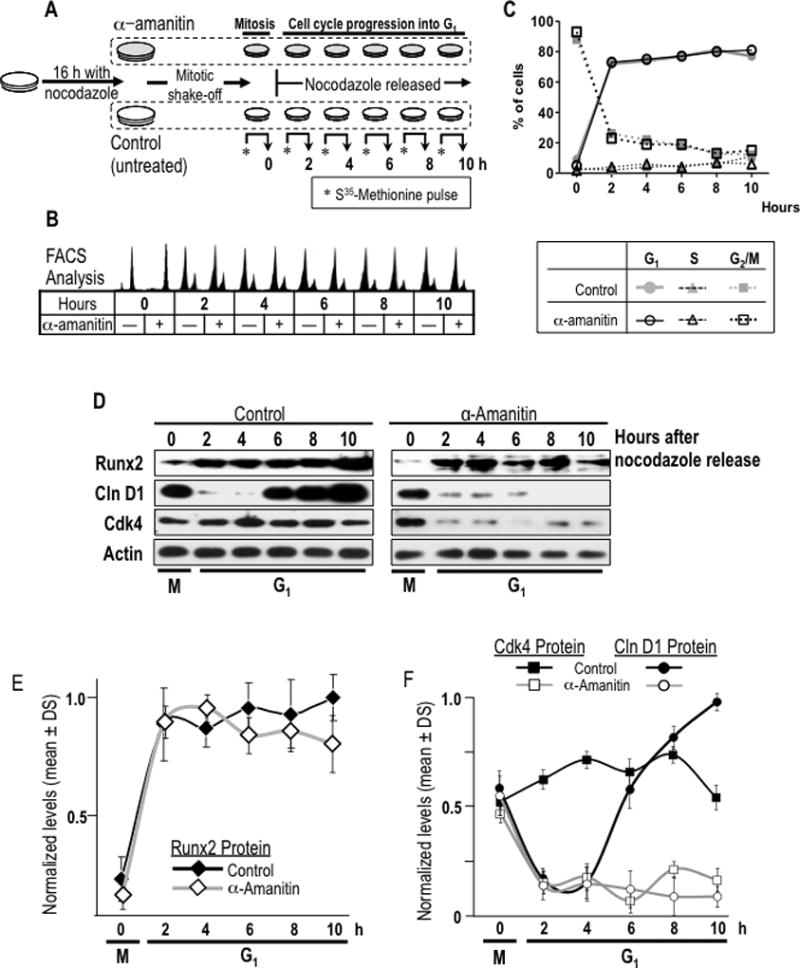
(A) A specific experimental strategy using the RNA polymerase II inhibitor α-amanitin was developed to block the post-mitotic contribution of mRNA synthesis to Runx2 protein synthesis during G1. MC3T3-E1 cells were synchronized by incubation for 16 h with nocodazole to generate a mitotic block. Mitotic cells were pre-treated with or without α-amanitin for 2 h and harvested by gentle agitation (0 h) and then released through mitosis into G1 by washing and the addition of fresh culture medium supplemented with (+) or without (−) α-amanitin. (B) Cells were harvested at selected time points during the M/G1 phase transition and interphase (0, 2, 4, 6, and 10 h). Progression through mitosis to G1 phase in presence or absence of α-amanitin was monitored by flow cytometry. (C) Graphic representation of cell cycle stage data presented in panel B shown that α-amanitin treatment does not affect normal cell cycle progression of mitotic cells into G1 phase. (D) Resumption of Runx2 protein levels at the M/G1 transition in α-amanitin-treated cells or untreated control cells was analysed by western blotting. Cell cycle progression into G1 was defined by monitoring cyclin D1 and Cdk4 protein levels. (E and F) Graphic representations of α-amanitin-treatment affecting post-mitotic change in Runx2 (no statistical differences between control and treated group), cyclin D1 (significant statistical differences between control and treated group from 6 h, P = 0.001) and Cdk4 (significant statistical differences between control and treated group from 2 h, P = 0.001). Protein levels normalized to actin.
Fig. 7. Post-mitotic inheritance of Runx2 mRNA supports de novo Runx2 protein synthesis during early G1 phase.
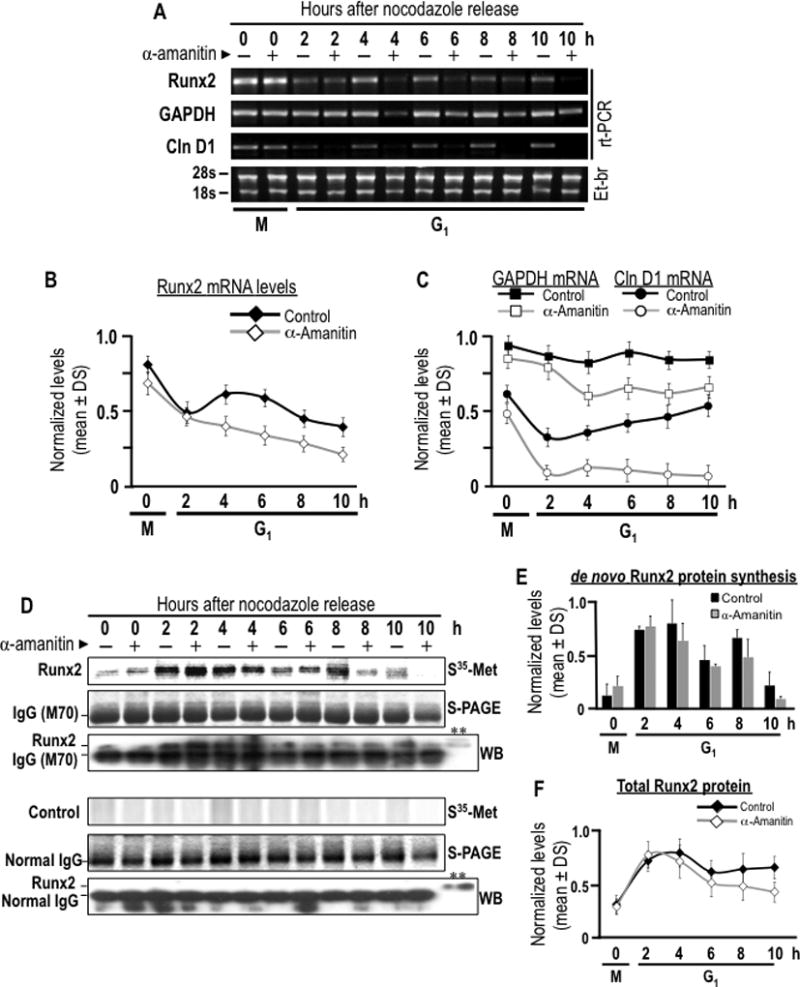
The contribution of post-mitotically inherited Runx2 mRNA to Runx2 protein synthesis during G1 phase was assessed by combining mitotic cell synchronization with metabolic labeling. (A) MC3T3-E1 cells were synchronized with nocodazole to generate a mitotic block. Mitotic cells were pre-treated with or without α-amanitin for 2 h and harvested by gentle agitation (0 h) and then released through mitosis into G1 by washing and the addition of fresh culture medium supplemented with (+) or without (−) α-amanitin. Cells were harvested at selected time course during the M/G1 phase transition (0, 2, 4, 6, and 10 h). Runx2, cyclin D1 and Gapdh mRNA levels were analyzed by RT-PCR to evaluated α-amanitin effect on RNA polymerase II inhibition and transcripts stability. (B and C) Runx2, cyclin D1 and Gapdh mRNA levels showed in panel A are graphically represented. Significant statistically differences between the control and the treated group: (B) Runx2, for 4, 6, 8 and 10 h (P = 0.028, 0.013, 0.047 and 0.031, respectively), (C) cyclin D1, for 2 to 10 h (P = 0.04, 0.032, 0.018, 0.009 and 0.003, respectively), and Gapdh, from 4 h (P = 0.026, 0.021, 0.017 and 0.036, respectively). Cells were metabolically labeled by 2 h pulse with [35S]-methionine and harvested at selected time course during the M/G1 phase transition (0, 2, 4, 6, and 10 h). (D) Cells pulse-labeled with [35S]-methionine were subjected to immuno-precipitation using a Runx2 polyclonal antibody or non-specific IgG control. Immuno-precipitates of endogenous Runx2 were separated in by SDS-PAGE (S-PAGE) and de novo synthesis of Runx2 protein was assessed by autoradiographic (35S-Met-Arg) analysis. Immuno-precipitations were analyzed by western blot analysis with a Runx2 monoclonal antibody or non-specific IgG control. A representative input was included to validate immuno-reactive Runx2 bands (**). Note that Runx2 is observed immediately above the immunoglobulin heavy chain. The data shown is representative of three experiments with similar outcomes. (E and F) Bar and line graphs show autoradiographic and western blot signals for immuno-precipitated Runx2 showed in panel D, no statistical differences between the control group and treated.
As cells exit mitosis and progress into early G1 phase (2–4 h after mitosis), Runx2 mRNA levels decrease after mitosis both in α-amanitin treated cells and in untreated cells, but the effect is more pronounced when transcription is inhibited (Figs. 7A and 7B). Thus, Runx2 mRNA levels during G1 phase are determined by the equilibrium between new mRNA synthesis and degradation. Furthermore, α-amanitin inhibits new synthesis of cyclin D1 mRNA indicating that α-amanitin effectively blocks post-mitotic new transcription of this gene (Fig. 7A and C). Strikingly, although Runx2 mRNA levels decline modestly after mitosis, initiation of de novo Runx2 protein synthesis as measured by [35S]-methionine labeling is robust, even in the presence of α-amanitin (Figs. 7D and 7E). Furthermore, restitution of total cellular protein levels as determined by immuno-blotting of whole lysates (Fig. 6E) and immuno-precipitates (Fig. 7F) occurs relatively unimpeded by transcriptional inhibition. These data together demonstrate that Runx2 protein synthesis in interphase is supported by translation of post-mitotically inherited Runx2 mRNAs during early G1 phase.
We addressed the functional relevance of mitotically transmitted mRNAs and the concomitant post-mitotic translation of Runx2 protein that occurs during early G1 (2–4 h) by analysing the binding of this transcription factor to the osteocalcin (OC/BGLAP2) gene promoter. This gene is a classical transcriptional target of Runx2 in osteoblasts (Merriman et al., 1995). Chromatin immuno-precipitation analyses reveal that Runx2 binds to the OC/BGLAP2 promoter of mitotic cells (0 h) and occupancy increases in progeny cells progressing through early G1 (2 to 6 h) after mitosis (Fig. 8). As expected, α-amanitin permits increased interactions of Runx2 protein translation despite transcriptional inhibition (Fig. 7). Taken together, these findings demonstrate that resumption of Runx2 protein translation in G1 phase (from mitotically inherited transcripts) supports post-mitotic transcriptional control of bone-phenotypic genes. This capability is consistent with our model that Runx2 maintains osteoblast-lineage commitment during proliferative expansion of osteoprogenitors in part by mitotic transmission of its mRNA.
Fig. 8. Runx2 binding to OC/BGLAP2 gene promoter during early G1 is independent of α-amanitin.
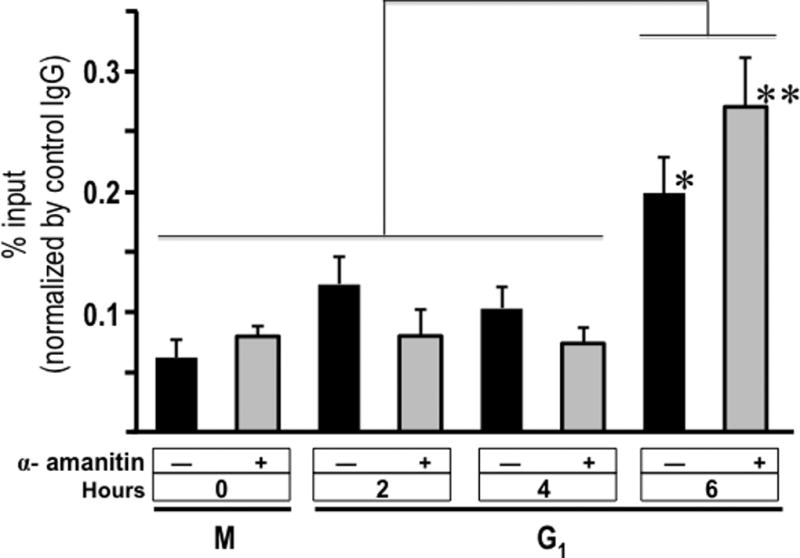
MC3T3-E1 cells were synchronized by incubation for 16 h with nocodazole to generate a mitotic block. Mitotic cells were pre-treated with or without the transcriptional inhibitor α-amanitin for 2 h and harvested by gentle agitation and then released through mitotic into G1 the addition of fresh culture medium supplemented with (+) or without (−) α-amanitin, and cells were harvested after 0, 2, 4, and 6 h. Cells were crosslinked with 1% formaldehyde and sonicated chromatin fragments were immune-precipitated with a Runx2 polyclonal antibody. The enrichment levels of OC/BGLAP2 gene promoter sequences (−276/−5 pb) in the precipitated chromatin were determined by QPCR using specific primers. Student’s t test was performed to assess the significance of the observed differences. *P = 0.024; **P = 0.002.
Discussion
The accumulation of mRNAs in mitosis is of major biological importance during development, because such mRNAs can support regional specialization within the embryo or maintenance of cell identity in expanding progenitor cell populations. Our studies show that mRNA for the osteoblast lineage-specific transcription factor Runx2, which determines osteogenic cell fate and controls osteoblast growth, accumulates maximally in mitosis while the protein is present at minimal levels. Our data indicate that mitotic Runx2 mRNA partitions symmetrically into progeny cells of either osteoblastic or osteosarcoma cells, while localized in a non-chromosomal tubulin-containing compartment. This mitotically inherited Runx2 mRNA is actively translated by progeny cells within 2 h after mitosis to resume maximal Runx2 protein expression during interphase. Transcription-independent translation of Runx2 protein in early G1 phase results in increased functional interactions of this de novo synthesized Runx2 with a representative osteoblast-specific target gene (osteocalcin/BGLAP2) in chromatin. Therefore, we propose that post-mitotic symmetric segregation of Runx2 mRNA to progeny cells supports maintenance of osteogenic lineage commitment and retention of the osteoblast phenotype.
These findings for mammalian Runx2 during cell division of somatic osteoblasts complement observations for the zebrafish homolog runx2b, which is a maternal determinant of dorsoventral patterning (van der Meulen et al., 2005; Flores et al., 2008). Maternal runx2b transcripts are localized in the blastodisc of zebrafish embryos at the 1-cell stage, continue to be expressed ubiquitously in the blastoderm, but are excluded from the presumptive dorsal embryonic shield (van der Meulen et al., 2005; Flores et al., 2008). The presence of the maternally-inherited runx2b isoform activates early zygotic expression of transcriptional repressors of dorsal genes that specify dorsoventral polarity (Flores et al., 2008). This partitioning of maternally inherited runx2b/Runx2 mRNA during early embryogenesis biologically complements the symmetrical somatic inheritance of Runx2 during mitotic division that supports osteogenic lineage-committed during later developmental stages when the skeleton ossifies.
Our finding that Runx2 mRNA symmetrically segregates during mitosis mirrors previous studies from our laboratories which show that Runx2 protein persist at minimal basal levels during mitosis and is retained on mitotic chromatin through sequence-specific DNA binding at the promoter regions of several target genes including bone phenotypic and rRNA genes (Zaidi et al., 2003, 2011, 2014; Young et al., 2007a, b; Stein et al., 2009). This mitotic occupancy of Runx2 at gene promoters by Runx2 represents an architectural epigenetic mechanism (‘mitotic bookmarking’) that retains chromatin-encoded gene regulatory instructions for the osteoblast phenotype during cell division (Zaidi et al., 2003, 2011). Our study indicates that osteoblasts have a second mechanism that involves the post-mitotic translational up-regulation of Runx2 protein using mitotically inherited mRNAs. One attractive model that emerges from these studies is that cells remain a ‘sentry’ level of Runx2 protein at its target genes during mitosis. However, cells require rapid occupancy of additional Runx2 binding sites and this demand for Runx2 protein is mediated by translational induction using mitotically transmitted mRNAs for Runx2 that are ‘front-loaded’ in G2 phase. One biological advantage of this translational mechanism would be that it permits rapid post-mitotic reactivation of gene expression patterns required for bone cell identity in early G1.
Beyond Runx2, a number of other phenotype-specific transcription factors may be involved in mitotic bookmarking (Stein et al., 2009; Zaidi et al., 2011, 2014; Kadauke and Blobel, 2012, 2013), including MLL in various cell types (Blobel et al., 2009), GATA1 in erythroid precursors (G1E cells)(Kadauke et al., 2012) and FoxA1 in hepatoma cell (HUH7 cells)(Caravaca et al., 2013). While these factors are retained during mitosis, they also exhibit reduced promoter occupancy in mitotic chromosomes. Thus, re-activation of transcription (or reinforced suppression) in early G1 may require the rapid re-emergence gene regulatory factors on chromatin to attain optimal binding occupancy (Kadauke and Blobel, 2013; Zaidi et al., 2014). It is conceivable that mitotic (‘protein & mRNA’) transmission of both sentry levels of Runx2 protein on chromatin during mitosis and anticipatory front-loading of Runx2 mRNA during G2 phase could be extrapolated to other transcription factors associated with mitotic bookmarking.
One question that arises from our study is how osteoblasts transmit comparable amounts of Runx2 mRNA during mitosis. The transcriptome is not located randomly within the cell (Johnston, 1995). Rather, mRNAs functionally interact with specific cellular structures and molecules to accomplish stabilization, transportation, localization and translational activation or repression (Suprenant, 2004; Blower, 2013; Romasko et al., 2013), as well as inheritance to progeny cells after cell division (Lambert and Nagy, 2002; Blower et al., 2007). We find that mitotically transmitted Runx2 mRNA resides in a non-chromosomal microtubule containing compartment associated with the mitotic spindle. Interestingly, previous studies suggest specific mRNAs are targeted to microtubules at mitotic spindles, thus suggesting that mitotic spindles may serve as a mechanism for their segregation during cell division (Blower et al., 2007; Sharp et al., 2011). Post-mitotic inheritance of Runx2 mRNAs to daughter cells may also involve interactions with RNA-binding proteins (RBPs). For example, specific mRNAs are trapped by RBPs and selectively retained in one of the daughter cells by asymmetric segregation during neural stem cell division, a mechanism that promotes lineage progression during embryonic development of Drosophila (Li et al., 1997; Benoit et al., 2009; Lerit and Gavis, 2011) and development of the mammalian central nervous system (Kusek et al., 2012; Vessey et al., 2012).
In summary, we show that Runx2 mRNA is transmitted through mitosis and translated immediately after mitosis in early osteoprogenitors to control a program of gene expression required for reinforcement of cell fate decisions in committed pre-osteoblasts. Our findings combined with other studies support a working model in which microtubule-facilitated symmetric segregation of Runx2 mRNA during mitosis supports transcription-independent induction of Runx2 protein in G1 phase to support retention of the osteogenic cell fate during proliferative expansion.
Acknowledgments
This work was supported by “Fondo Nacional de Desarrollo Científico y Tecnológico” FONDECYT 1060772 (to MG) and “Iniciativa Científica Milenio, Ministerio de Economía, Fomento y Turismo” grant P09/016-F (to MG), FONDAP 15090007 (to MM), as well as by National Institutes of Health Grants R01 AR049069 (to AJvW), R01 AR039588 and P01 CA082834 (to GSS). We thank the members of our research groups, including Mariana Osorio, Francisco Villanueva and Hector Araya (University of Chile), Scott Riester and Amel Dudakovic (Mayo Clinic), as well as Janet Stein (University of Vermont) for assistance with the experimentation, critical comments, technical advice, sharing of reagents and/or general support.
Contract grant sponsor: Iniciativa Científica Milenio (to MG); Contract grant number: P09/016-F.
Contract grant sponsor: FONDECYT (to MG); Contract grant number: 1060772.
Contract grant sponsor: FONDAP (to MM); Contract grant number: 15090007.
Contract grant sponsor: National Institutes of Health (to AJvW); Contract grant number: R01AR049069.
Contract grant sponsor: National Institutes of Health (to GSS); Contract grant number: R01 AR039588 and P01 CA082834.
Literature Cited
- Almeida M, O’Brien CA. Basic biology of skeletal aging: role of stress response pathways. J Gerontol A Biol Sci Med Sci. 2013;68:1197–1208. doi: 10.1093/gerona/glt079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido T, Borba VZ, Roberson P, Manolagas SC. Activation of the Janus kinase/STAT (signal transducer and activator of transcription) signal transduction pathway by interleukin-6-type cytokines promotes osteoblast differentiation. Endocrinology. 1997;138:3666–3676. doi: 10.1210/endo.138.9.5364. [DOI] [PubMed] [Google Scholar]
- Bellido T, Ali AA, Plotkin LI, Fu Q, Gubrij I, Roberson PK, Weinstein RS, O’Brien CA, Manolagas SC, Jilka RL. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem. 2003;278:50259–50272. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- Benoit B, He CH, Zhang F, Votruba SM, Tadros W, Westwood JT, Smibert CA, Lipshitz HD, Theurkauf WE. An essential role for the RNA-binding protein Smaug during the Drosophila maternal-to-zygotic transition. Development. 2009;136:923–932. doi: 10.1242/dev.031815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel GA, Kadauke S, Wang E, Lau AW, Zuber J, Chou MM, Vakoc CR. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell. 2009;36:970–983. doi: 10.1016/j.molcel.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower M, Feric E, Weis K, Heald R. Genome-wide analysis demonstrates conserved localization of messenger RNAs to mitotic microtubules. The Journal of Cell Biology. 2007;179:1365–1373. doi: 10.1083/jcb.200705163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD. Molecular insights into intracellular RNA localization. Int Rev Cell Mol Biol. 2013;302:1–39. doi: 10.1016/B978-0-12-407699-0.00001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadus J, Fuerstenberg S, Doe CQ. Staufen-dependent localization of prospero mRNA contributes to neuroblast daughter-cell fate. Nature. 1998;391:792–795. doi: 10.1038/35861. [DOI] [PubMed] [Google Scholar]
- Canalis E. Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol. 2013;9:575–583. doi: 10.1038/nrendo.2013.154. [DOI] [PubMed] [Google Scholar]
- Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013;27:251–260. doi: 10.1101/gad.206458.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danciu TE, Li Y, Koh A, Xiao G, McCauley LK, Franceschi RT. The basic hélix loop helix transcription factor Twist1 is a novel regulator of ATF4 in osteoblasts. J Cell Biochem. 2012;113:70–79. doi: 10.1002/jcb.23329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drissi H, Luc Q, Shakoori R, Chuva De Soussa Lopes S, Choi JY, Terry A, Hu M, Jones S, Neil JC, Lian JB, Stein JL, Van Wijnen AJ, Stein GS. Transcriptional autoregulation of the bone related CBFA1/RUNX2 gene. J Cell Physiol. 2000;184:341–350. doi: 10.1002/1097-4652(200009)184:3<341::AID-JCP8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Flores MV, Lam EY, Crosier KE, Crosier PS. Osteogenic transcription factor Runx2 is a maternal determinant of dorsoventral patterning in zebrafish. Nat Cell Biol. 2008;10:346–352. doi: 10.1038/ncb1697. [DOI] [PubMed] [Google Scholar]
- Forristall C, Pondel M, Chen L, King ML. Patterns of localization and cytoskeletal association of two vegetally localized RNAs, Vg1 and Xcat-2. Development. 1995;121:201–208. doi: 10.1242/dev.121.1.201. [DOI] [PubMed] [Google Scholar]
- Galindo M, Pratap J, Young DW, Hovhannisyan H, Im HJ, Choi JY, Lian JB, Stein JL, Stein GS, van Wijnen AJ. The bone-specific expression of Runx2 oscillates during the cell cycle to support a G1-related antiproliferative function in osteoblasts. J Biol Chem. 2005;280:20274–20285. doi: 10.1074/jbc.M413665200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo M, Kahler RA, Teplyuk NM, Stein JL, Lian JB, Stein GS, Westendorf JJ, van Wijnen AJ. Cell cycle related modulations in Runx2 protein levels are independent of lymphocyte enhancer-binding factor 1 (Lef1) in proliferating osteoblasts. J Mol Histol. 2007;38:501–506. doi: 10.1007/s10735-007-9143-0. [DOI] [PubMed] [Google Scholar]
- Greenblatt MB, Shim JH, Glimcher LH. Mitogen-activated protein kinase pathways in osteoblasts. Annu Rev Cell Dev Biol. 2013;29:63–79. doi: 10.1146/annurev-cellbio-101512-122347. [DOI] [PubMed] [Google Scholar]
- He N, Xiao Z, Yin T, Stubbs J, Li L, Quarles LD. Inducible expression of Runx2 results in multiorgan abnormalities in mice. J Cell Biochem. 2011;112:653–665. doi: 10.1002/jcb.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman J, Wessely O, Langland R, Craig EJ, Kessler DS. Vegetal localization of maternal mRNAs is disrupted by VegT depletion. Dev Biol. 2001;240:377–386. doi: 10.1006/dbio.2001.0495. [DOI] [PubMed] [Google Scholar]
- Hesse E, Kiviranta R, Wu M, Saito H, Yamana K, Correa D, Atfi A, Baron R. Zinc finger protein 521, a new player in bone formation. Ann N Y Acad Sci. 2010;1192:32–37. doi: 10.1111/j.1749-6632.2009.05347.x. [DOI] [PubMed] [Google Scholar]
- Hovhannisyan H, Zhang Y, Hassan MQ, Wu H, Glackin C, Lian JB, Stein JL, Montecino M, Stein GS, van Wijnen AJ. Genomic occupancy of HLH, AP1 and Runx2 motifs within a nuclease sensitive site of the Runx2 gene. J Cell Physiol. 2013;228:313–321. doi: 10.1002/jcp.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrusik A, Cox A, Wicher K, Glover DM, Zernicka-Goetz M. Maternal-zygotic knockout reveals a critical role of Cdx2 in the morula to blastocyst transition. Dev Biol. 2015;398:147–152. doi: 10.1016/j.ydbio.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery W, Wilson L. Localization of messenger RNA in the cortex of Chaetopterus eggs and early embryos. J. Embryol. Exp. Morph. 1983;75:225–239. [PubMed] [Google Scholar]
- Jensen ED, Nair AK, Westendorf JJ. Histone deacetylase co-repressor complex control of Runx2 and bone formation. Crit Rev Eukaryot Gene Expr. 2007;17:187–196. doi: 10.1615/critreveukargeneexpr.v17.i3.20. [DOI] [PubMed] [Google Scholar]
- Jin L, Lloyd RV. In situ hybridization: methods and applications. J Clin Lab Anal. 1997;11:2–9. doi: 10.1002/(SICI)1098-2825(1997)11:1<2::AID-JCLA2>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D. The intracellular localization of messenger RNAs. Cell. 1995;81:161–170. doi: 10.1016/0092-8674(95)90324-0. [DOI] [PubMed] [Google Scholar]
- Kadauke S, Blobel GA. “Remembering” tissue-specific transcription patterns through mitosis. Cell Cycle. 2012;11:3911–3912. doi: 10.4161/cc.22237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, Blobel GA. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012;150:725–737. doi: 10.1016/j.cell.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, Blobel GA. Mitotic bookmarking by transcription factors. Epigenetics Chromatin. 2013;6:6–15. doi: 10.1186/1756-8935-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11:849–860. doi: 10.1038/nrm3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Sawada K, Yamamoto H, Wada S, Saiga H, Nishida H. Maternal macho-1 is an intrinsic factor that makes cell response to the same FGF signal differ between mesenchyme and notochord induction in ascidian embryos. Development. 2003;130:5179–5190. doi: 10.1242/dev.00732. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Kronenberg HM. Overview of skeletal development. Methods Mol Biol. 2014;1130:3–12. doi: 10.1007/978-1-62703-989-5_1. [DOI] [PubMed] [Google Scholar]
- Komori T. Regulation of osteoblast differentiation by Runx2. Adv Exp Med Biol. 2010;658:43–49. doi: 10.1007/978-1-4419-1050-9_5. [DOI] [PubMed] [Google Scholar]
- Komori T. Signaling networks in RUNX2-dependent bone development. J Cell Biochem. 2011;112:750–755. doi: 10.1002/jcb.22994. [DOI] [PubMed] [Google Scholar]
- Krude T. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp Cell Res. 1999;247:148–159. doi: 10.1006/excr.1998.4342. [DOI] [PubMed] [Google Scholar]
- Kusek G, Campbell M, Doyle F, Tenenbaum SA, Kiebler M, Temple S. Asymmetric segregation of the double-stranded RNA binding protein Staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell. 2012;11:505–516. doi: 10.1016/j.stem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JD, Nagy LM. Asymmetric inheritance of centrosomally localized mRNAs during embryonic cleavages. Nature. 2002;420:682–686. doi: 10.1038/nature01241. [DOI] [PubMed] [Google Scholar]
- Lerit DA, Gavis ER. Transport of germ plasm on astral microtubules directs germ cell development in Drosophila. Curr Biol. 2011;21:439–448. doi: 10.1016/j.cub.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Yang X, Wasser M, Cai Y, Chia W. Inscuteable and Staufen mediate asymmetric localization and segregation of prospero RNA during Drosophila neuroblast cell divisions. Cell. 1997;90:437–447. doi: 10.1016/s0092-8674(00)80504-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Ge C, Long JP, Begun DL, Rodriguez JA, Goldstein SA, Franceschi RT. Biomechanical stimulation of osteoblast gene expression requires phosphorylation of the RUNX2 transcription factor. J Bone Miner Res. 2012;27:1263–1274. doi: 10.1002/jbmr.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin GL, Hankenson KD. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J Cell Biochem. 2011;112:3491–3501. doi: 10.1002/jcb.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie PJ. Targeting integrins to promote bone formation and repair. Nat Rev Endocrinol. 2013;9:288–295. doi: 10.1038/nrendo.2013.4. [DOI] [PubMed] [Google Scholar]
- Matsuzaki F, Ohshiro T, Ikeshima-Kataoka H, Izumi H. miranda localizes staufen and prospero asymmetrically in mitotic neuroblasts and epithelial cells in early Drosophila embryogenesis. Development. 1998;125:4089–4098. doi: 10.1242/dev.125.20.4089. [DOI] [PubMed] [Google Scholar]
- Merriman HL, van Wijnen AJ, Hiebert S, Bidwell JP, Fey E, Lian J, Stein J, Stein GS. The tissue-specific nuclear matrix protein, NMP-2, is a member of the AML/CBF/PEBP2/runt domain transcription factor family: interactions with the osteocalcin gene promoter. Biochemistry. 1995;34:13125–13132. doi: 10.1021/bi00040a025. [DOI] [PubMed] [Google Scholar]
- Mowry KL, Cote CA. RNA sorting in Xenopus oocytes and embryos. FASEB J. 1999;13(3):435–445. doi: 10.1096/fasebj.13.3.435. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Makabe KW, Nishida H. Localization and expression pattern of type I postplasmic mRNAs in embryos of the ascidian Halocynthia roretzi. Gene Expr Patterns. 2003;3:71–75. doi: 10.1016/s1567-133x(02)00069-8. [DOI] [PubMed] [Google Scholar]
- Nishida H. Patterning the marginal zone of early ascidian embryos: localized maternal mRNA and inductive interactions. Bioessays. 2002;24:613–624. doi: 10.1002/bies.10099. [DOI] [PubMed] [Google Scholar]
- Pratap J, Galindo M, Zaidi SK, Vraidii D, Bhat BM, Robinson JA, Choi JY, Komori T, Stein JL, Lian JB, Stein GS, van Wijnen AJ. Cell growth regulatory role of Runx2 during proliferative expansion of pre-osteoblasts. Cancer Res. 2003;63:5357–5362. [PubMed] [Google Scholar]
- Romasko EJ, Amarnath D, Midic U, Latham KE. Association of maternal mRNA and phosphorylated EIF4EBP1 variants with the spindle in mouse oocytes: localized translational control supporting female meiosis in mammals. Genetics. 2013;195:349–358. doi: 10.1534/genetics.113.154005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubinet C, Cabernard C. Control of asymmetric cell division. Curr Opin Cell Biol. 2014;31:84–91. doi: 10.1016/j.ceb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- San Martin IA, Varela N, Gaete M, Villegas K, Osorio M, Tapia JC, Antonelli M, Mancilla EE, Pereira BP, Nathan SS, Lian JB, Stein JL, Stein GS, van Wijnen AJ, Galindo M. Impaired cell cycle regulation of the osteoblast-related heterodimeric transcription factor Runx2-Cbfbeta in osteosarcoma cells. J Cell Physiol. 2009;221:560–571. doi: 10.1002/jcp.21894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier AF. The maternal-zygotic transition: death and birth of RNAs. Science. 2007;316:406–407. doi: 10.1126/science.1140693. [DOI] [PubMed] [Google Scholar]
- Sharp JA, Plant JJ, Ohsumi TK, Borowsky M, Blower MD. Functional analysis of the microtubule-interacting transcriptome. Mol Biol Cell. 2011;22:4312–4323. doi: 10.1091/mbc.E11-07-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu E, Nakatani T, He Z, Partridge NC. Parathyroid hormone regulates histone deacetylase (HDAC) 4 through protein kinase A-mediated phosphorylation and dephosphorylation in osteoblastic cells. J Biol Chem. 2014;289:21340–21350. doi: 10.1074/jbc.M114.550699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NA, Civitelli R. Cell-cell signaling: broadening our view of the basic multicellular unit. Calcif Tissue Int. 2014;94:2–3. doi: 10.1007/s00223-013-9766-y. [DOI] [PubMed] [Google Scholar]
- Skamagki M, Wicher KB, Jedrusik A, Ganguly S, Zernicka-Goetz M. Asymmetric localization of Cdx2 mRNA during the first cell-fate decision in early mouse development. Cell Rep. 2013;3:442–457. doi: 10.1016/j.celrep.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein GS, Zaidi SK, Stein JL, Lian JB, van Wijnen AJ, Montecino M, Young DW, Javed A, Pratap J, Choi JY, Ali SA, Pande S, Hassan MQ. Transcription-factor-mediated epigenetic control of cell fate and lineage commitment. Biochem Cell Biol. 2009;87:1–6. doi: 10.1139/o08-094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein GS, van Wijnen AJ, Imbalzano AN, Montecino M, Zaidi SK, Lian JB, Nickerson JA, Stein JL. Architectural genetic and epigenetic control of regulatory networks: compartmentalizing machinery for transcription and chromatin remodeling in nuclear microenvironments. Crit Rev Eukaryot Gene Expr. 2010;20:149–155. doi: 10.1615/critreveukargeneexpr.v20.i2.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprenant KA. Microtubules, ribosomes, and RNA: evidence for cytoplasmatic localization and translational regulation. Cell Motil. Cytoskeleton. 2004;25:1–9. doi: 10.1002/cm.970250102. [DOI] [PubMed] [Google Scholar]
- Tai PW, Zaidi SK, Wu H, Grandy RA, Montecino M, van Wijnen AJ, Lian JB, Stein GS, Stein JL. The dynamic architectural and epigenetic nuclear landscape: developing the genomic almanac of biology and disease. J Cell Physiol. 2014;229:711–727. doi: 10.1002/jcp.24508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SY, Alliston T. Regulation of postnatal bone homeostasis by TGFβ. Bonekey Rep. 2013;2:1–5. doi: 10.1038/bonekey.2012.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplyuk NM, Galindo M, Teplyuk VI, Pratap J, Young DW, Lapointe D, Javed A, Stein JL, Lian JB, Stein GS, van Wijnen AJ. Runx2 regulates G protein-coupled signaling pathways to control growth of osteoblast progenitors. J Biol Chem. 2008;283:27585–27597. doi: 10.1074/jbc.M802453200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Kansara M. Epigenetic modifications in osteogenic differentiation and transformation. J Cell Biochem. 2006;98:757–769. doi: 10.1002/jcb.20850. [DOI] [PubMed] [Google Scholar]
- van de Peppel J, van Leeuwen JP. Vitamin D and gene networks in human osteoblasts. Front Physiol. 2014;5:1–10. doi: 10.3389/fphys.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Deen M, Hassan MQ, Pratap J, Teplyuk NM, Young DW, Javed A, Zaidi SK, Lian JB, Montecino M, Stein JL, Stein GS, van Wijnen AJ. Chromatin immunoprecipitation assays: application of ChIP-on-chip for defining dynamic transcriptional mechanisms in bone cells. Methods Mol Biol. 2008;455:165–176. doi: 10.1007/978-1-59745-104-8_13. [DOI] [PubMed] [Google Scholar]
- van der Deen M, Taipaleenmäki H, Zhang Y, Teplyuk NM, Gupta A, Cinghu S, Shogren K, Maran A, Yaszemski MJ, Ling L, Cool SM, Leong DT, Dierkes C, Zustin J, Salto-Tellez M, Ito Y, Bae SC, Zielenska M, Squire JA, Lian JB, Stein JL, Zambetti GP, Jones SN, Galindo M, Hesse E, Stein GS, van Wijnen AJ. MicroRNA-34c inversely couples the biological functions of the runt-related transcription factor RUNX2 and the tumor suppressor p53 in osteosarcoma. J Biol Chem. 2013;288:21307–21319. doi: 10.1074/jbc.M112.445890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Kranenbarg S, Schipper H, Samallo J, van Leeuwen JL, Franssen H. Identification and characterisation of two runx2 homologues in zebrafish with different expression patterns. Biochim Biophys Acta. 2005;1729:105–117. doi: 10.1016/j.bbaexp.2005.03.008. [DOI] [PubMed] [Google Scholar]
- van Wijnen AJ, van de Peppel J, van Leeuwen JP, Lian JB, Stein GS, Westendorf JJ, Oursler MJ, Im HJ, Taipaleenmäki H, Hesse E, Riester S, Kakar S. MicroRNA functions in osteogenesis and dysfunctions in osteoporosis. Curr Osteoporos Rep. 2013;11:72–82. doi: 10.1007/s11914-013-0143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey JP, Amadei G, Burns SE, Kiebler MA, Kaplan DR, Miller FD. An asymmetrically localized Staufen2-dependent RNA complex regulates maintenance of mammalian neural stem cells. Cell Stem Cell. 2012;11:517–528. doi: 10.1016/j.stem.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Wadsworth S, Madhavan K, Bilodeau-Wentworth D. Maternal inheritance of transcripts from three Drosophila src-related genes. Nucleic Acids Research. 1985;13:2153–2170. doi: 10.1093/nar/13.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks DL, Melton DA. A maternal mRNA localized to the animal pole of Xenopus eggs encodes a subunit of mitochondrial ATPase. Proc Natl Acad Sci U S A. 1987;84:2798–2802. doi: 10.1073/pnas.84.9.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JA, Heasman J. Maternal control of pattern formation in Xenopus laevis. J Exp Zool B Mol Dev Evol. 2008;310:73–84. doi: 10.1002/jez.b.21153. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Awad HA, Liu S, Mahlios J, Zhang S, Guilak F, Mayo MS, Quarles LD. Selective Runx2-II deficiency leads to low-turnover osteopenia in adult mice. Dev Biol. 2005;283:345–356. doi: 10.1016/j.ydbio.2005.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DW, Hassan MQ, Yang X-Q, Galindo M, Javed A, Zaidi SK, Furcinitti P, Lapointe D, Montecino M, Lian J, Stein J, van Wijnen A, Stein G. Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2. Proc. Natl. Acad. Sci. USA. 2007a;104:3189–3194. doi: 10.1073/pnas.0611419104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee S, Yang X, Xie R, Javed A, Underwood JM, Furcinitti P, Imbalzano AN, Penman S, Jeffrey AN, Montecino M, Lian JB, Stein J, van Wijnen A, Stein G. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 2007b;445:442–445. doi: 10.1038/nature05473. [DOI] [PubMed] [Google Scholar]
- Zaidi SK, Young D, Pockwinse S, Javed A, Lian J, Stein J, van Wijnen A, Stein G. Mitotic partitioning and selective reorganization of tissue-specific transcription factors in progeny cells. Proc. Natl. Acad. Sci. USA. 2003;100:14852–14857. doi: 10.1073/pnas.2533076100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Young DW, Montecino M, van Wijnen AJ, Stein JL, Lian JB, Stein GS. Bookmarking the genome: maintenance of epigenetic information. J Biol Chem. 2011;286:18355–18361. doi: 10.1074/jbc.R110.197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Grandy RA, Lopez-Camacho C, Montecino M, van Wijnen AJ, Lian JB, Stein JL, Stein GS. Bookmarking target genes in mitosis: a shared epigenetic trait of phenotypic transcription factors and oncogenes? Cancer Res. 2014;74:420–425. doi: 10.1158/0008-5472.CAN-13-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hassan MQ, Xie RL, Hawse JR, Spelsberg TC, Montecino M, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Co-stimulation of the bone-related Runx2 P1 promoter in mesenchymal cells by SP1 and ETS transcription factors at polymorphic purine-rich DNA sequences (Y-repeats) J Biol Chem. 2009a;284(5):3125–3135. doi: 10.1074/jbc.M807466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lian JB, Stein JL, van Wijnen AJ, Stein GS. The Notch-responsive transcription factor Hes-1 attenuates osteocalcin promoter activity in osteoblastic cells. J Cell Biochem. 2009b;108(3):651–659. doi: 10.1002/jcb.22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xie RL, Croce CM, Stein JL, Lian JB, van Wijnen AJ, Stein GS. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci U S A. 2011;108:9863–9868. doi: 10.1073/pnas.1018493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lou King M. Sending RNAs into the future: RNA localization and germ cell fate. IUBMB Life. 2004;56:19–27. doi: 10.1080/15216540310001658886. [DOI] [PubMed] [Google Scholar]
- Zhu F, Friedman MS, Luo W, Woolf P, Hankenson KD. The transcription factor osterix (SP7) regulates BMP6-induced human osteoblast differentiation. J Cell Physiol. 2012;227:2677–2685. doi: 10.1002/jcp.23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Shimizu E, Zhang X, Partridge NC, Qin L. EGFR signaling suppresses osteoblast differentiation and inhibits expression of master osteoblastic transcription factors Runx2 and Osterix. J Cell Biochem. 2011;112:1749–1760. doi: 10.1002/jcb.23094. [DOI] [PMC free article] [PubMed] [Google Scholar]