

Abstract
We describe an approach for multiplexed microRNA analysis using silicon photonic microring resonators to detect cDNA reverse transcription products via a subsequent enzymatic signal enhancement strategy. Key to this method is a modified stem loop primer that facilitates downstream signal amplification via enzymatic turnover and improves the sensor signal 20-fold when compared to traditional stem loop primers. This approach facilitates targeted microRNA quantification in only 2.5 hours and without requiring target amplification via the polymerase chain reaction (PCR) . Primers for 7 miRNA targets were orthogonally designed to avoid cross-hybridization between capture probes. This approach was applied to the detection of total RNA from human tissues and found to display differential expression profiles consistent with literature precedent. This development holds promise as an alternative to single-plex RT-qPCR methods and more expensive RNA-seq by offering a cost-effective method to analyze targeted miRNA panels in emerging diagnostic applications.
Graphical Abstract
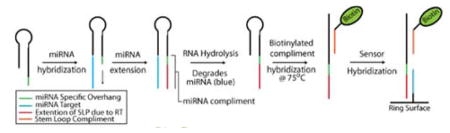
MicroRNAs (miRNAs) constitute an important class of non-coding RNAs that regulate gene expression at the transcriptional and post-transcriptional level. As potent gene regulators, miRNAs have been linked to important developmental processes that establish and maintain tissue differentiation.(1,2) Not surprisingly, miRNA expression in tissue and blood samples is also associated with disease, and has substantial diagnostic utility.(3,4) However, the short sequence lengths, large variability in per-cell copy number, and high sequence similarity within families of expressed miRNAs conspire to make them challenging analytical targets. Furthermore, miRNAs often function in complex regulatory networks whereby many miRNAs work cooperatively to regulate the expression of a single mRNA transcript. However, each miRNA may be involved in many different transcript-targeting regulatory networks. Therefore, the multiplexed detection of many miRNAs simultaneously is an important consideration for both fundamental and translational application of miRNA analysis technologies. For clinical applications, these complications are further exacerbated by technical and practical requirements, including small sample sizes, low cost, and relative ease of use. Therefore, candidate miRNA detection technologies need to offer: high sensitivity; wide dynamic range; high sequence specificity; multiplexing capability; and minimal sample processing and handling.(5)
Current miRNA detection techniques are lacking in one, or more, of these attributes. Specifically, reverse transcription-quantitative polymerase chain reaction (RT-qPCR) methods are incredibly sensitive, relatively rapid, and cost effective; however, they typically measure levels of only a single miRNA sequence per assay. Conversely, microarrays are well-suited to multiplexed analyses but are typically slow, less sensitive, more expensive, and require PCR amplification, which can introduce sequence biases. Next-generation sequencing technologies give a comprehensive picture of miRNA expression levels; however, this global approach, which requires complex library construction and onerous informatics, is both time- and cost-prohibitive for many diagnostic applications. Moreover, advances in database informatics have found that reduced subsets of miRNAs can be identified that offer robust and actionable diagnostic utility.(6) Therefore, technologies that can robustly determine expression levels of targeted panels of miRNAs from a single, clinically-relevant sample could be important in the widespread realization of translational miRNA-based diagnostics.
Silicon photonic microring resonators, which belong to a larger class of whispering gallery biosensors,(7) are an intrinsically multiplexable, array-based technology that has been applied to a range of biomolecular detection applications.(8–13) The operational theory and measurement instrumentation behind the technology has been previously discussed in detail.(14–15) Briefly, a tunable wavelength laser centered around 1550 nm is coupled into linear waveguides via on chip grating couplers. The laser is swept through the appropriate spectral window to determine wavelengths of optical resonance. Changes in the local refractive index near the sensor surface induced by biomolecular binding cause a shift in the resonance, with shifts directly proportional to the amount of surface bound biomolecules, which in turn reflects the solution phase analyte concentration. A more detailed description of this technology is presented in the Supporting Information.
Our group previously demonstrated the detection of miRNAs using microring resonators in both a label-free(16) and capture-agent-enhanced(17) assay format, the latter using a DNA:RNA heteroduplex-specific antibody. Here we report a sandwich-based detection protocol that uses reverse transcription to create cDNA products of targeted miRNAs that are subsequently detected using an enzymatic chemical signal enhancement strategy. Compared to prior work from our group,(17) we report improved limits of detection, increased levels of multiplexing, but most dramatically, an ~85% reduction in analysis time (from 12–15 hours to 2.5 hours) . To our knowledge, this is the first report that directly detects cDNA products resulting from miRNA reverse transcription using specific stem loop primers (SLP) , rather than being integrated with the standard RT-qPCR framework, which has intrinsic limits in terms of multiplexing capacity. We demonstrate the broad applicability of this approach by profiling the expression levels of 7 miRNAs, and an off target control sequence, to differentiate between different tissue types, showing good correlation with previous RT-qPCR analyses.
A schematic of the reverse transcription-horseradish peroxidase (RT-HRP) assay is shown in Figure 1. SLPs are designed with a universal stem loop sequence and a 7–8 nucleotide overhang sequence specific for particular miRNA targets. Hybridization of the miRNA target to the SLP followed by extension via reverse transcription yields the DNA complement to the miRNA sequence. The miRNA is then degraded via base hydrolysis (Figure S1) leaving the RT product accessible to bind to the capture probe attached to the sensor surface. Without degradation, the hybridized miRNA can block the capture probe recognition site on the RT product (Figure S2) .
Figure 1.
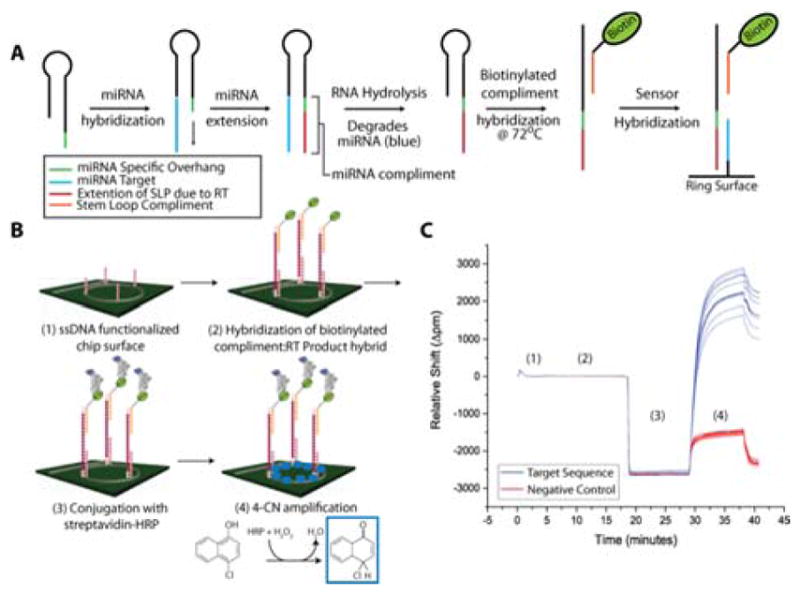
A) Schematic diagram of miRNA processing to prepare for on chip detection. B) Illustration of RT product detection and HRP signal amplification. C) The representative binding curve shows data corresponding to the detection of a 10 picomole miRNA-26a sample subjected to the workflow in A and B. The large signal gain at 30 minutes is obtained from the 4-chloro-1-napthon deposition.
After extension and RNA degradation (RT SLP) , a biotinylated tag sequence of DNA complementary to the conserved stem loop region common to all of SLPs was added to the solution containing RT products. This solution is incubated at 72°C, which allows for the stem loop sequence to linearize and hybridize to the tag. SLPs were designed with a one base pair mismatch in the stem region so that tag-RT SLP complexes are thermodynamically preferred over the secondary structure of the SLPs themselves (Tm of tag-RT SLP > Tm SLP secondary structure) . In this way, the complex remains linear and able to hybridize to surface-bound capture probes on the microring sensors, leading to a 20-fold increase in sensitivity when compared to results obtained using conventional stem loop primers with the RT-HRP assay (Figure S2) . Importantly, this strategy is capable of targeting multiple miRNAs in a single sample volume by adding multiple SLP sequences to the initial RT reaction and presents a streamlined sample preparation process that consists of only 3 incubation steps over the course of ~1.75 hours.
The HRP-enhanced sensing strategy of the target sequences is shown in Figure 1B. While we have previously utilized HRP signal amplification for protein detection,(11) this is the first report to use HRP for the detection of miRNAs. The solution containing tag-RT SLPs is diluted in a high stringency hybridization buffer to ensure complementary binding and then flowed across an array of microring sensors uniquely-functionalized with target-specific DNA capture sequences. Additionally, capture probes were designed to avoid hybridization with non-extended SLPs. After hybridization of tag-RT SLPs to the ssDNA capture probes and a streptavidin-HRP conjugate was flowed across the sensor surface. A solution of 4-chloro-1-napthol (4-CN) was then introduced and enzymatically converted into insoluble 4-chloro-1-napthon (4-CNP) by bound HRP. The deposition of the 4-CNP precipitate causes a dramatic shift in the resonance wavelengths of the microrings (measured in Δpicometers; Δpm) that is directly related to the number of surface bound HRP moieties and thus the concentration of target miRNAs in the original sample matrix.
To demonstrate the quantitative ability of the developed scheme, we exposed the microring sensors functionalized with a target specific capture probe to 7 cDNA product solutions of the miRNA target ranging in initial concentrations from 2 μM to 2 nM, as well as a blank (no input RNA) . Calibration curves were generated by diluting synthetic miRNA sequences to the different concentrations, subjecting these solutions to the RT-HRP protocol, and quantitating the initial slope of the HRP amplification response. Figure 2A shows representative binding curves that were used to compile the calibration curves in Figure 2B. Clear concentration-dependent responses were seen across a 4 order of magnitude dynamic range, as shown in Figure 2B, which also lists the determined limits of detection for each target sequence.
Figure 2.

A) Overlay of the signal responses achieved for each concentration dilution of target miRNA cDNA product. Initial concentrations utilized were: 2 μM (black) , 1 μM (red) , 200 nM (blue) , 20 nM (pink) , 2 nM (green) , and a blank (purple) . B) Calibration curves for the HRP response of each miRNA target. The red curves represent linear fits of the initial slope of the HRP amplification step. Error bars represent the standard deviation of between 8 and 20 technical replicates at each concentration.
Next, we show the applicability of this approach for multiplexed measurements of seven different miRNA targets. Notably, the use of the conserved region on the biotinylated stem loop primers as a universal recognition element reduces assay complexity by eliminating the need for multiple tagging sequences. Microring resonators were spatially arrayed via functionalization with eight unique capture probes (7 specific to miRNA targets and one negative control) . Seven chips were identically functionalized and each exposed to solutions containing the RT-product of individual miRNAs at a constant input of 10 picomoles. This process was repeated for each of the seven miRNA targets, and the compiled results are shown in Figure 3. Each column in the figure represents a different sensor array incubated with the RT product of the target on the column heading. As can be seen, this detection approach had high sequence specificity for the targeted miRNAs and minimal cross-reactive response.
Figure 3.

Detection of specific miRNA target products with minimal off target response from non-complementary capture probes. Each column represents a sensor chip arrayed with different capture probes and exposed to the miRNA RT product listed as the column heading. Each row represents the response at the target-specific microring exposed to the different RT-products in different experiments. Importantly, sensors only show responses at the when exposed to the specifically-targeted miRNA RT product, demonstrating the potential for multiplexed miRNA measurements.
To demonstrate that this approach is able to probe more complex samples, and also to benchmark the assay against gold standard techniques (i.e. RT-qPCR) , we simultaneously profiled the expression of seven miRNAs from brain and lung total RNA samples (Figure S3) .
The detected concentrations are shown in Figure 4A. Likewise, we used RT-qPCR to compile expression profiles from the same sample (see Table S4 for C(t) values) . After normalizing both data sets by dividing by the concentration measured for miR-26a, which corrects for sampling differences,(17) the fold change of brain:lung miRNA expression was determined and plotted in Figure 4B, together with ratios calculated based upon a previous study.(18)
Figure 4.
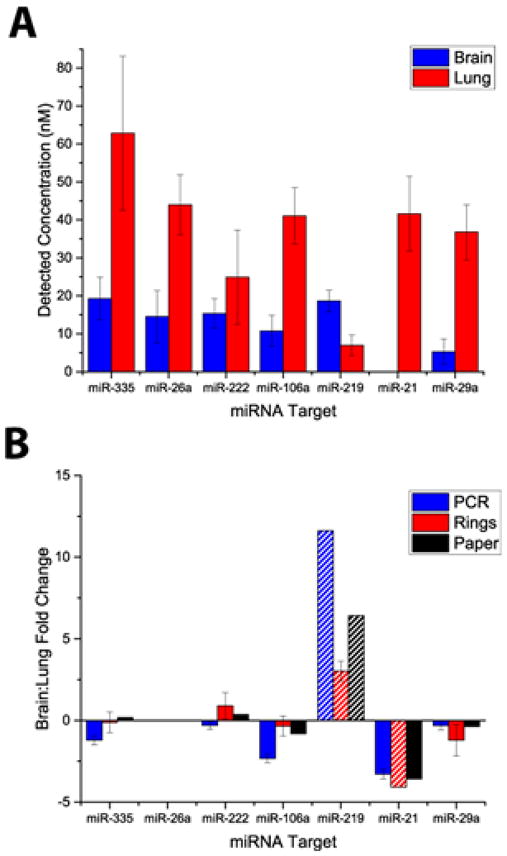
A) Comparison of the concentrations for each of the 7 targets (n=8–16 technical replicates) . B) Comparison of miRNA expression profiles obtained using microrings and RT-qPCR normalized to miR-26a expression.
The relatively large fold-change deviations for miR-219 might be explained by the fact that the low overall expression levels cause the analysis to approach the C(t) cutoff threshold for reliable detection using RT-qPCR, and also increase the microring measurements susceptibility to any analytical errors. That said, both platforms are in agreement with the literature that miRNA-219 is more abundant in brain tissue.(19) Similarly, the detection of miR-21 could also be unreliable since it is an oncogene that is upregulated during cancer progression,(20) whereas the samples analyzed were not from cancer patients. Interestingly though, if the expression is assumed to be one order of magnitude lower than our limit of detection, the brain:lung fold change correlates well with literature precedent,(18) as well as our in-house RT-qPCR measurements.
In conclusion, we have developed a new approach for the sensitive and multiplexed analysis of miRNAs using a reverse-transcription-enabled enzymatic signal enhancement strategy coupled with detection using silicon photonic microring resonators. While only shown for seven targets here, this technology is capable of delivering significantly higher levels of multiplexing, which exceed that of many other emerging miRNA detection strategies.(5) Moreover, this type of platform offers the capacity to analyze for panels of miRNAs not easily accessible via RT-qPCR while avoiding the cost and informatics required for RNA sequencing, and therefore may be well-positioned to fit an important niche in helping translate miRNA-based diagnostics to the clinic. Future efforts to further improve the analytical performance metrics of the methods will be required in addition to more clinically-relevant demonstrations more closely-targeted to specific disease diagnoses and from expanded patient cohorts.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support from the National Cancer Institute of the Institutes of Health through Grant CA1774 and the National Science Foundation through Grant CHE 12-14081. RMG acknowledges support from the National Cancer Institute Alliance for Nanotechnology in Cancer ‘Midwest Cancer Nanotechnology Training Center’ Grant R25 CA154015A. We also appreciate the support provided through UIUC’s Institute for Genomic Biology.
Footnotes
ASSOCIATED CONTENT
SI includes experimental details and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Garcia I, Miska EA. Development. 2005;132:4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 3.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 4.Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, Zepeniuk M, Benjamin H, Shabes N, Tabak S, Levy A, Lebanony D, Goren Y, Silberschein E, Targan N, Ben-Ari A, Gilad S, Sion-Vardy N, Tobar A, Feinmesser M, Kharenko O, Nativ O, Nass D, Perelman M, Yosepovich A, Shalmon B, Polak-Charcon S, Fridman E, Avniel A, Bentwich I, Bentwich Z, Cohen D, Chajut A, Barshack I. Nat Biotech. 2008;26I:462–469. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 5.Graybill RM, Bailey RC. Anal Chem. 2016;88:431–450. doi: 10.1021/acs.analchem.5b04679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz NA, Dehlendorff C, Jensen BV, et al. JAMA. 2014;311:392–404. doi: 10.1001/jama.2013.284664. [DOI] [PubMed] [Google Scholar]
- 7.Wade JH, Bailey RC. Ann Rev Anal Chem. 2016;9:1–25. doi: 10.1146/annurev-anchem-071015-041742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qavi AJ, Mysz TM, Bailey RC. Anal Chem. 2011;83:6827–6833. doi: 10.1021/ac201659p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luchansky MS, Bailey RC. J Am Chem Soc. 2011;133:20500–20506. doi: 10.1021/ja2087618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sloan CDK, Marty MT, Sligar SG, Bailey RC. Anal Chem. 2013;85:2970–2976. doi: 10.1021/ac3037359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kindt JT, Luchansky MS, Qavi AJ, Lee SH, Bailey RC. Anal Chem. 2013;85:10653–10657. doi: 10.1021/ac402972d. [DOI] [PubMed] [Google Scholar]
- 12.Wade JH, Alsop AT, Vertin NR, Yang H, Johnson MD, Bailey RC. ACS Central Science. 2015;1:374–382. doi: 10.1021/acscentsci.5b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valera E, Shia WW, Bailey RC. Clin Biochem. 2016;49:121–126. doi: 10.1016/j.clinbiochem.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vollmer F, Arnold S. Nat Methods. 2008;5:591–596. doi: 10.1038/nmeth.1221. [DOI] [PubMed] [Google Scholar]
- 15.Iqbal M, Gleeson MA, Spaugh B, Tybor F, Gunn WG, Hochberg M, Baehr-Jones T, Bailey RC, Gunn LC. J Sel Topics Quantum Electron. 2010;16:654–661. [Google Scholar]
- 16.Qavi AJ, Bailey RC. Angew Chem Intl Ed. 2010;49:4608–4611. doi: 10.1002/anie.201001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qavi AJ, Kindt JT, Bailey RC. Anal Chem. 2011;83:5949–5956. doi: 10.1021/ac201340s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang Y, Ridzon D, Wong L, Chen C. BMC Genomics. 2007;8:166. doi: 10.1186/1471-2164-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Si M-L, Zhu S, Wu H, Lu Z, Wu F, Mo Y-Y. Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 20.Asangani IA, Rasheed SAK, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.