

Abstract
Background
Changes in glycosylation of the constant domain (Fc) of monoclonal antibodies (mAbs) enhance antibody-dependent cell-mediated cytotoxicity independently of downstream effects following receptor blockade by the antibody, thus extending their indication. We investigated the safety, pharmacokinetics, pharmacodynamics and antitumour activity of tomuzotuximab, an IgG1 glycoengineered mAb against the epidermal growth factor receptor with enhanced tumour cytotoxicity in a phase I dose-escalation study (NTC01222637).
Patients and methods
Forty-one patients with advanced solid tumours refractory to standard therapies received tomuzotuximab weekly (12–1370 mg) or two-weekly (990 mg) on a three-plus-three dose escalation design.
Results
A maximum tolerated dose was not reached. The most frequent treatment-related adverse events were infusion-related reactions in 31 (76%) patients (grade 3, 12%), mainly confined to the first dose, and skin toxicities (grade 1 or 2) in 30 (73%) patients. Hypomagnesaemia was observed in 9 out of 23 evaluable patients (39%). Similar to cetuximab, tomuzotuximab concentrations increased proportionally to dose from doses≥480 mg with a median terminal half life (t½) of 82 hours, range 55–113 hours. Antitumour activity included one complete response ongoing since more than 4.5 years in a patient with non-small-cell lung cancer and one partial response lasting 353 days in a patient with colorectal cancer. Twelve patients achieved stable disease (median, 166 days, range, 71–414 days) and two patients had prolonged control (>1 year) of their non-measurable disease.
Conclusion
Tomuzotuximab was safe and showed promising antitumour activity in heavily pretreated patients with advanced metastatic disease. A phase IIb trial of chemotherapy and weekly tomuzotuximab or cetuximab followed with maintenance therapy with the corresponding mAb in patients with recurrent or metastatic head and neck squamous cell carcinoma is ongoing.
Keywords: tomuzotuximab, phase I, egfr, monoclonal antibody, glycoengineered
Key questions.
What is already known about this subject?
Current EFGR antibodies have limited efficacy in Ras mutated patients in spite of their immunological effector functions.
Antibody-dependent cell-mediated cytotoxicity (ADCC) contributes to the antitumour effect of monoclonal antibodies (mAbs).
ADCC best results are observed in patients with the V/V Fc gamma receptor IIIa (FcγRIIIa) allotype (only 20% of the population) and are very poor to intermediate with the F/F and F/V allotypes, respectively (each 40% of the population).
Defucosylation of the constant (Fc) domain of an antibody enhances ADCC to all three FcγRIIIa allotypes in vitro and in preclinical models.
What does this study add?
Tomuzotuximab is an IgG1 glycoengineered mAb of cetuximab with the same binding properties to epidermal growth factor receptor (EGFR) as cetuximab but with enhanced ADCC.
In this phase I study in patients with solid tumours and progressive advanced disease, it was safe and well tolerated.
Pharmacokinetics characteristics were similar to those of cetuximab.
Promising antitumour activity was observed.
How might this impact on clinical practice?
Tomuzotuximab and other glycoengineered mAbs may target a wider population replacing the parent antibody in combination therapies.
Introduction
Cetuximab, a chimeric IgG1 monoclonal antibody (mAb) that targets and binds to the extracellular domain of the epidermal growth factor receptor (EGFR) inhibiting its dimerisation and activation, is approved in the treatment of metastatic colorectal cancer (CRC) and head and neck squamous cell carcinoma (HNSCC).1 2 However, 40% of patients with CRC have tumours expressing mutations in the oncogene KRAS that invalidate the effect of EGFR blockade and do not fully benefit from cetuximab treatment as do patients with tumours expressing KRAS wild-type.3
Tumour cell killing through antibody-dependent cell-mediated cytotoxicity (ADCC), which relies on the constant (Fc) domain of the antibody engaging natural killer cells, also contributes to the clinical activity of cetuximab, but its efficacy in vivo is influenced by Fc gamma receptor IIIa (FcγRIIIa) polymorphism.4–6 Changes in glycosylation of the Fc domain promote ADCC by enhancing FcγRIIIa binding, thus increasing cytotoxicity that is independent of downstream effects following receptor blockade by the antibody.7
Tomuzotuximab (former development name: CetuGEX) is a glycoengineered second generation antibody of cetuximab produced in the human GlycoExpress expression system (Glycotope GmbH, Berlin, Germany). Tomuzotuximab has a fully human glycosylation pattern and is glyco-optimised at its Fc domain to improve its efficacy and reduce it side effects, while fully retaining the affinity, specificity, EGFR inhibition and induction of apoptosis of cetuximab (data on file; Glycotope GmbH). Elimination of fucose leads to a mean ADCC increase of tomuzotuximab compared with cetuximab of 10–50-fold depending on the Fc receptor variant (online supplementary figure S1), while elimination of the α-gal epitope, which can cause severe IgE-mediated hypersensitive reactions to cetuximab, will improve its tolerability (data on file; Glycotope GmbH8 9).
esmoopen-2017-000303supp001.jpg (538.3KB, jpg)
{kind=link}
We investigated the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics of tomuzotuximab and evaluated its preliminary antitumour activity in patients with advanced solid tumours to define dose and regimen for phase II studies.
Patients and methods
Study design and patients
The study was conducted from 25 August 2010 to 14 November 2013 in five European centres as an open-label, multicentre, dose-escalating phase I study to investigate the safety and PK, define dose and regimen of subsequent phase II studies and preliminarily evaluate clinical activity of single-agent tomuzotuximab in patients with locally advanced and/or metastatic carcinomas for whom no further standard therapy was available. Patients were required to have measurable or clinically evaluable and progressive disease (see eligibility criteria in online supplementary text). The study was approved by local ethics committees prior to its initiation.
esmoopen-2017-000303supp002.pdf (313.4KB, pdf)
Dose escalation and tomuzotuximab administration
Patients were sequentially enrolled to receive tomuzotuximab (flat doses of 12, 60, 120, 240, 480, 720, 990 and 1370 mg) every week (QW, N 35) and 990 mg tomuzotuximab every 2 weeks (Q2W, N 6) following a three-plus-three dose escalation design and treated at their assigned dose until disease progression, unacceptable toxicity or consent withdrawal. If dose-limiting toxicity (DLT) occurred, treatment was discontinued for that patient and the dose level was expanded to six patients. The maximum tolerated dose (MTD) was reached if more than one patient developed a DLT at any dose level in the first cycle. DLTs were graded according to National Cancer Institute Common Toxicity Criteria for Adverse Events (AE) V.3.0 criteria,10 and defined as haematological or non-haematological toxicity grade 3 or 4, with the exception of nausea and vomiting.
Tomuzotuximab was administered intravenously under close clinical supervision. Premedication consisted initially of H1/H2 antagonists and glucocorticosteroids, with the addition of paracetamol from dose 120 mg onwards. Infusions were administered initially at 20 mg/hour the first hour, doubling the rate every hour, but rate and mode of administration was adjusted in the course of the study to minimise the risk of infusion-related reaction (IRR). Second infusions were administered over 1–4.5 hours, depending on the dose level, and subsequent infusions were given in 3 hours.
Pharmacokinetics and pharmacodynamics
Serum samples for PK analysis were collected at prespecified time points and quantification of tomuzotuximab in serum was performed at Glycotope GmbH; the lower limit of quantification was 103 ng/mL (see online supplementary text). PK parameters (online supplementary table S1) were computed from the individual serum concentration-time profiles using non-compartmental methods (FUNCALC 3, Prolytic GmbH, Frankfurt, Germany). Dose linearity and proportionality of PK parameters were evaluated by linear regression analysis, and accumulation of tomuzotuximab was assessed by dividing trough concentrations after the second and subsequent doses by the trough concentration after the first dose. Theoretical trough concentration levels (Cmin) required for adequate efficacy of tomuzotuximab were set at 40–50 µg/mL based on data published for cetuximab,11 and results of non-clinical studies comparing the ADCC and Fab-mediated effects of cetuximab and tomuzotuximab (online supplementary figure S1, data on file; Glycotope GmbH).
esmoopen-2017-000303supp003.pdf (152.6KB, pdf)
Additionally, antidrug antibodies (ADA), interleukin (IL)-6, IL-8, tumour necrosis factor alpha (TNFα), histamine and eosinophilic cationic protein (ECP) were tested at prespecified time-points.
Assessments and evaluation of clinical activity
Medical history and demographics were collected at screening and physical examination, vital signs, Eastern Cooperative Oncology Group (ECOG) performance status and haematology/biochemistry were performed at baseline and throughout the study. Tumour response in patients with measurable disease was evaluated according to RECIST 1.1 guidelines.12 Imaging (CT and/or MRI) was performed within 4 weeks before the first tomuzotuximab dose and then every 8 weeks during the study. Antitumour activity was assessed by measuring the target lesions: stable disease (SD) criteria had to be met a minimum of 8 weeks after study entry, complete response (CR) and partial response (PR) needed confirmation at least 4 weeks after the criteria for response were first met. Duration of response was measured from the time at which criteria were first met for CR and PR until the first date that progressive disease (PD) was objectively documented and duration of SD as the time from the start of treatment until criteria for PD were met. The clinical benefit rate (CBR) was calculated as the proportion of patients with CR, PR or SD.
Statistical analysis
Descriptive statistics were used on the intent-to-treat (ITT) population, which included all patients who received at least 1 dose of the study drug, to summarise patient demographics and baseline characteristics, treatment administration, safety parameters, PK variables and anti tumour activity end points (SAS V.9.1). Distribution of parameters was analysed using the Mann-Whitney U test and in contingency tables (IBM SPSS V.24).
Results
Clinical characteristics of the 41 patients in the study are provided in table 1. All patients but one had progressive locoregional or metastatic disease and had exhausted available standard treatment procedures. The most frequent reason for study termination was PD (29 patients, 70.7%), followed by AE and death (5 patients each, 12.2%). One patient did not meet the inclusion criteria and another continued with tomuzotuximab on a named patient use after the study was closed (4.9%).
Table 1.
Demographic and baseline clinical characteristics of the study population
| Administration schedule | Total | ||
| Every week | Every 2 weeks | ||
| Number of patients | 35 | 6 | 41 |
| Age in years, median (range) | 61 (36–77) | 58.5 (52–69) | 60 (36–77) |
| Gender, N (%) | |||
| Male | 25 (71.4) | 4 (66.7) | 29 (70.7) |
| Female | 10 (28.6) | 2 (33.3) | 12 (29.3) |
| ECOG performance status, N (%) | |||
| 0 | 21 (60) | 1 (16.7) | 22 (53.7) |
| 1 | 14 (40) | 5 (83.3) | 19 (46.3) |
| Time from diagnosis in months, median (range)* | 25 (3–90) | 41.5 (12–76) | 35 (3–90) |
| Primary tumour site, N (%) | |||
| Colon/rectum | 19 (54.3) | 4 (66.7) | 23 (56.1) |
| Stomach | 4 (11.4) | – | 4 (9.8) |
| Kidney | 4 (11.4) | 1 (16.7) | 5 (12.2) |
| Pancreas | 2 (5.7) | – | 2 (4.9) |
| Other† | 6 (17.1) | 1 (16.7) | 7 (17.1) |
| EGFR expression (N 33)% positive cells, median (range) | 10 (1 – 100) | ||
| Intensity of positive cells, N (%) ‡ | |||
| 1+ | 15 (46) | ||
| 2+ | 10 (30) | ||
| 3+ | 7 (21) | ||
| EGFR expression— cut-off 30 % positive cells § | |||
| < 30% | 22 (66.6) | ||
| ≥ 30 % | 11 (33.3) | ||
| Sum of longest diameter of target lesions in mm at study entry, median (range)¶ | 118 (22–263) | 148 (124–213) | 128 (22–263) |
| Prior chemotherapy regimens, N (%) | |||
| 1–2 | 10 (28.6) | 2 (33.3) | 12 (29,3) |
| 3–4 | 19 (54.3) | 2 (33.3) | 21 (51.2) |
| ≥5 | 6 (17.1) | 2 (33.3) | 8 (19.5) |
| Any prior antibody therapy, N (%)** | 21 (60) | 4 (66.7) | 25 (61) |
| Bevacizumab | 15 (42.9) | 4 (66.7) | 19 (46.3) |
| Cetuximab | 15 (42.9) | – | 15 (36.6) |
| Panitumumab | 3 (8.6) | – | 3 (7.3) |
| Pankomab | 3 (8.6) | 1 (16.7) | 4 (9) |
*Date of first dose of study drug – date of initial diagnosis of the disease + 1.
†Other primary tumours included one each of gall bladder, head and neck, lung, oesophagus, ovary, penis and thyroid cancer.
‡One missing.
§The cut-off is set at the 75% Tukey’s hinges, which is 30.
¶Two patients had no measurable disease according to RECIST criteria.
**12 patients received two antibodies; 15 patients received Cetuximab plus chemotherapy.
ECOG, Eastern Cooperative Oncology Group; EGFR, epidermal growth factor receptor.
Safety
Seven patients (17.1%) died during the study, six due to their underlying disease and one of general physical deterioration eventually leading to death that was assessed by the investigator as possibly related to tomuzotuximab treatment. The drug was withdrawn in one patient because of AE related to the underlying disease and in four patients following an IRR.
Patients in the QW and Q2W dosing scheme received a median of 11 (range: 1–97) and 4 (range: 1–29) doses in the course of the study, respectively. All patients experienced at least one treatment-related AE, the most frequent were IRR and skin toxicity (table 2). IRRs were most frequent with the first infusion (76% of patients), the majority was mild to moderate and all symptoms resolved after medication and/or pausing of the infusion. An IRR grade 3 first observed at the 720 mg dose level and accompanied by an allergic reaction (lip oedema) was rated as a DLT; histamine and ECP serum levels were not elevated. Recovery followed withdrawal of the drug and symptomatic medication. Four and three additional patients were recruited at the same and the previous dose level (480 mg), respectively, and a planned break of 30 min 1 hour after start of infusion, together with a stepwise increase of infusion rate was introduced. Four additional patients experienced a grade 3 IRR; the drug was withdrawn in three cases but continued in the other patient without further complications. The first infusion of dose 1370 mg was administered split over 2 days: 60 mg the first day and 1310 mg the second day over 4.5 hours; IRRs occurred only on the first day. Overall 34 IRRs (4%), all mild to moderate, were observed in the 724 subsequent infusions, 9 of them by the second infusion; 24 (71%) of them occurred in two patients. No further DLTs were observed and the MTD was not reached.
Table 2.
AEs related to the administration of tomuzotuximab with an incidence of ≥than 10% of the total population.
| Dose (mg) | Every week | Every 2 weeks | Total population | ||||||||
| 12 | 60 | 120 | 240 | 480 | 720 | 990 | 1370 | Total | 990 | ||
| No. of patients | 3 | 3 | 4 | 3 | 6 | 7 | 3 | 6 | 35 | 6 | 41 |
| Any AE (total incidence≥10%) | |||||||||||
| No. (%) patients* | |||||||||||
| IRR at first infusion† | 2 (67) | 3 (100) | 4 (100) | 2 (67) | 5 (83) | 4 (57) | 2 (67) | 4 (67) | 26 (74) | 5 (83) | 31 (76) |
| Grade 1 | 0 | 0 | 1 (25) | 1 (33) | 2 (33) | 0 | 1 (33) | 1 (17) | 5 (14) | 1 (17) | 6 (15) |
| Grade 2 | 2 (67) | 3 (100) | 3 (75) | 1 (33) | 2 (33) | 3 (43) | 1 (33) | 3 (50) | 19 (54) | 1 (17) | 20 (49) |
| Grade 3 | 0 | 0 | 0 | 0 | 1 (17) | 1 (14) | 0 | 0 | 2 (8) | 3 (50) | 5 (12) |
| Rash‡ | 1 (33) | 0 | 2 (50) | 1 (33) | 3 (50) | 1 (14) | 3 (100) | 2 (33) | 13 (37) | 2 (66) | 15 (73) |
| Acne‡ | 0 | 2 (66) | 1 (25) | 0 | 0 | 2 (29) | 0 | 2 (33) | 7 (20) | 3 (50) | 10 (24) |
| Dermatitis acneiform‡ | 0 | 0 | 0 | 1 (33) | 0 | 4 (57) | 2 (66) | 3 (50) | 10 (29) | 0 | 10 (24) |
| Dry skin‡ | 0 | 0 | 1 (25) | 0 | 3 (50) | 1 (14) | 1 (33) | 0 | 6 (17) | 0 | 6 (15) |
| Pruritus‡ | 0 | 0 | 1 (25) | 0 | 0 | 1 (14) | 1 (33) | 1 (17) | 4 (11) | 1 (17) | 5 (12) |
| Hypomagnesaemia§ | – | – | – | – | 1 | 1 (14) | 2 (67) | 5 (83) | 8 (44) | 1 (17) | 9 (39) |
| Asthenia‡,§ | 0 | 0 | 2 (50) | 1 (33) | 0 | 0 | 2 (67) | 1 (17) | 6 (17) | 0 | 6 (15) |
| Fatigue‡,§ | 0 | 1 (33) | 0 | 1 (33) | 2 (33) | 0 | 0 | 1 (17) | 5 (14) | 0 | 5 (12) |
*Percentage is calculated using the number of patients in the column heading as denominator. Patients are counted only once within a SOC category.
†Symptoms of IRR included chills (12 patients, 39%), tachycardia (eight patients, 26%), nausea (seven patients, 23%), vomiting (seven patients, 23%), dyspnoea (seven patients, 23%), fatigue (six patients, 15%), feeling hot (five patients, 16%), hypertension (four patients, 13%), fever (four patients, 13%) and asthenia (three patients, 7%).
‡All grade 1 or 2.
§Magnesium was measured in 23 patients: in one patient in the 480 mg dose cohort and in all patients from dose 720 mg upwards. One patient had a grade 3, and another patient had three grade 3 and two grade 4 events, both patients received the highest dose. The median time to onset of hypomagnesaemia was 81.5 days, range 22–191 days.
¶Not associated with an IRR.
AE, adverse events; IRR, infusion-related reaction; SOC, System Organ Class.
Skin toxicity was observed in 30 patients (73%); none was grade 3 or 4. Infusion was delayed for 7 days due to skin toxicity in five cases, and no dose reductions were required. Hypomagnesaemia levels were corrected with magnesium supplementation and did not interfere with drug administration, except for an infusion delay of 7 days in one patient (table 2). Treatment related mild to moderate diarrhoea and stomatitis was observed only in three (7%) and two (5%) patients, respectively.
Pharmacokinetics
PK parameters were computed in 37 patients. Non-linear PK evidenced by a dose dependency of terminal half-life and clearance rate was observed for doses lower than 480 mg. Tomuzotuximab exhibited linear PK with respect to dose across the 480–1370 mg dose range, as demonstrated by the dose-proportional increase in area under the curve (AUC0-tlast) and maximum drug concentration (Cmax) and a stable t½ and clearance rate (CL) (online supplementary tables S1, figure S2, S3). For doses≥480 mg, the median (range) of t½ was 82 (55–113) hours. As trough levels (Cmin) were above 50 µg/mL in all patients in the 990 and 1370 mg cohorts after the first infusion (figure 1 and online supplementary table S1), the study was terminated after the 1370 mg dose. The accumulation ratio of Cmin in QW for doses≥480 mg ranged from 1.58 to 2.96 over 4–5 infusions (12 patients); steady state was achieved in 50% of patients.
Figure 1.

Concentration-time profiles of repeated weekly infusions of (A) 720 mg and (B) 990 mg tomuzotuximab measured in individual patients before and at the end of infusion. The dotted line indicates the 50 µg/mL trough level. Tomuzotuximab serum levels were above 50 µg/mL 1 week after the second infusion in four out of five patients who received 720 mg and 1 week after the first infusion in all three patients who received 900 mg tomuzotuximab.
None of the 35 patients tested before the fourth infusion had ADAs. Thereafter, 3 out of 24 patients developed low to medium ADA titers (1.7–6.2) against tomuzotuximab. The patients had received between 15 and 32 infusions of 12–120 mg of tomuzotuximab.
Pharmacodynamics
The majority of patients responded with a significant increase of IL-6, IL-8 and TNFα levels, which was maximal 2 hours after start of first infusion, was dose-independent and returned to near baseline levels after 24 hours. Modifications in drug administration were associated with significantly lower cytokine levels (online supplementary figure S4). Cytokine levels were increased on the first day, but did not differ from preinfusion levels on the second day of infusion with dose 1370 mg (online supplementary figure S5). Cytokine levels in subsequent tomuzotuximab infusions did not differ from baseline levels.
esmoopen-2017-000303supp006.jpg (875.3KB, jpg)
{kind=link}
esmoopen-2017-000303supp007.jpg (738.6KB, jpg)
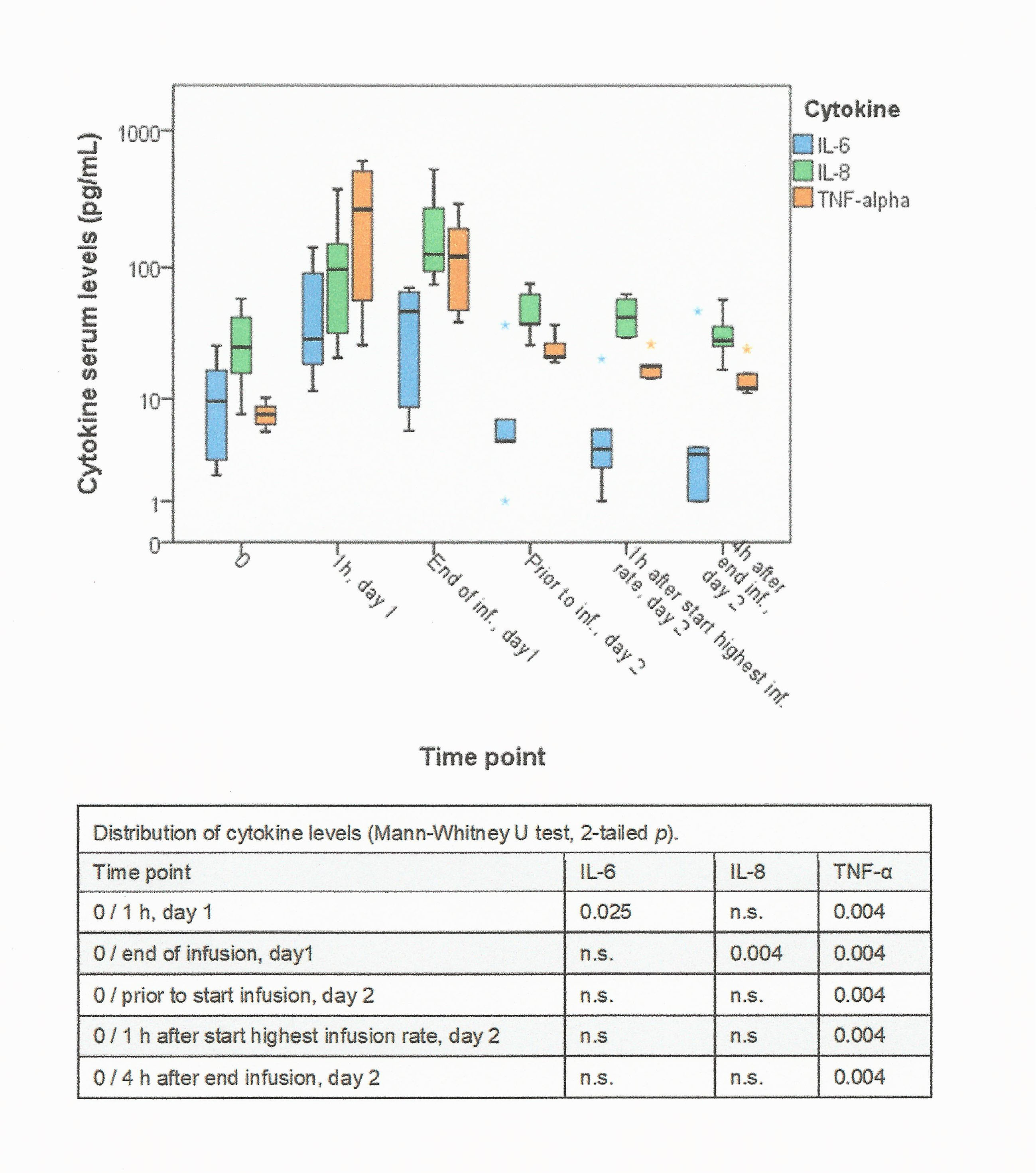{kind=link}
Clinical antitumour activity
Thirty-four patients were evaluable for antitumour activity, 31 and 3, respectively, in the weekly and 2-weekly schedule (figure 2). Seven patients were not evaluable: five due to early withdrawal from the study and two because of lack of tumour assessment. In the ITT population, a clinical benefit was observed in 16 out of 41 (39%) patients: One CR, 1 PR, 12 SD and 2 prolonged control of their non-measurable disease. The overall CBR was 41.2% for the 34 evaluable patients. Tumour shrinkage was observed in nine patients (27%), including one patient with CR, one with PR and one with unconfirmed PR (figure 2). Patients with the largest tumour shrinkage and longest disease control received doses of tomuzotuximab ≥240 mg. Nine out of 11 patients with EGFR expression ≥30% experienced clinical benefit (one CR, one PR, six SD and one patient with prolonged clinical improvement). However, only 4 of 22 patients with an EGFR expression <30% achieved SD (Fisher’s Exact test, P=0.001).
Figure 2.

Waterfall plot of the best per cent change from baseline in SLD of target lesions in 33 patients. Baseline is defined as the last non-missing value before the first dose of tomuzotuximab. Only patients with valid baseline and postbaseline values are included. Tumour assessment was not performed in six patients because of early withdrawal from the study following clinical deterioration or AE; two patients had no measurable disease according to RECIST1.1 criteria. The dotted lines indicate the cut-off for partial response (−30%) and progressive disease (+20%). *Patients with stable target lesions but progression because of new lesions. †Patient (CRC, dose 1370 mg) had a best change from baseline of 32.2%, unconfirmed 50 days later (change from baseline 29.5%). ¶ Patient is still in complete remission (4.5 years) and received tomuzotuximab for 5.2 years. CRC, colorectal cancer; GCA, gastric cancer; NSCLC, non-small cell lung cancer; OVCA, ovarian cancer; PanC, pancreatic cancer; RCC, renal cell cancer; SLD, sum of longest diameters.
SD lasted a median of 166 days (range, 71–414 days). Five out of 12 patients (41.7%) achieved SD for more than 180 days, and 2 patients (1370 mg dose) with oesophageal and gastric cancer and no measurable disease achieved prolonged disease control, assessed on non-target lesions, symptoms and biomarkers, lasting 339 and 443 days, respectively. A patient (1370 mg dose) with CRC and previously treated with cetuximab achieved an unconfirmed PR (−32.7%) after 113 days of treatment. Six patients with CRC and SD had previously received cetuximab.
A PR maximal at day 267 (–53.1%) and lasting 353 days was observed in a patient (990 mg dose Q2W) with CRC metastatic to liver, lung and interaortocaval lymph nodes previously treated with five lines of chemotherapy and bevacizumab.
A patient with squamous non-small-cell lung cancer stage IIIB (720 mg dose) is in CR for already 4.5 years. At study entry, the patient had a metastasis to the left hilus that did not respond to docetaxel and was first detected 4 months after primary treatment with combined radiation and chemotherapy. Tumour shrinkage started on day 169 of treatment and the tumour disappeared by day 337. Except for an acne-like rash grade 2, the patient tolerated well long-lasting treatment with tomuzotuximab (5.2 years). Treatment was discontinued in December 2016 following the patient’s request.
Discussion
Tomuzotuximab was safe and well-tolerated after repeated weekly administration and an MTD was not reached after a maximum dose of 1370 mg; ADA responses were negligible. The most frequent drug-related AEs were IRR and skin toxicity, and the majority of them were mild to moderate. Incidence of IRR (76%) was similar to that observed with RG7160 (77%), an anti-EGFR humanised glycoengineered mAb, but, contrary to it, no IRR grade 4 was observed.13 The incidence of IRR reported for cetuximab is 6%–19%,14 but is higher with both glycoengineered mAbs and similar to that observed with mAbs that target antigens on cells of the immune system.15 This may be related to the increased binding of the Fc domain of the glycoengineered antibody to Fcγ receptors on immune cells leading to a higher cytokine release at first infusion. The overall incidence of skin toxicity (73%) was slightly lower than that reported for cetuximab (88%) and RG7160 (80%). All rashes were mild to moderate, whereas 11% and 25% of rashes observed with cetuximab and RG7160, respectively, were grade 3 or 4.13 16 The incidence of hypomagnesaemia was 39%, similar to cetuximab (35%) and lower than RG7160 (56%).13 17
PK characteristics of tomuzotuximab were similar to that of cetuximab,11 18 and adequate trough levels (>50 µg/mL), also for optimal ADCC, were already provided by a weekly administration of 720 mg.
Clinical benefit was observed in 39% of patients; the best responses were observed with the highest doses, and a significant relationship between higher EGFR expression levels and clinical benefit was observed. The strong ADCC mediated by tomuzotuximab may well be behind its observed promising preliminary clinical activity. Tomuzotuximab is more effective than cetuximab in mediating ADCC against EGFR expressing tumour cells independently of the FcγRIIIa allotype of donor blood mononuclear cells (online supplementary figure S1). This is particularly important in view of the fact that less than 20% of the population express the CD16 allotype with high IgG binding affinity, whereas 40% express that of low binding affinity. Furthermore, because the mechanism of action of ADCC is independent of downstream effects of EFGR blockade and is independent of the mutational status of KRAS and BRAF,19 tomuzotuximab may benefit a wider population than cetuximab. RG7160 also has enhanced ADCC and showed clinical efficacy at high doses in a phase I/II trial, but its development has been discontinued.7 13 20
Based on the safety, PK and promising preliminary efficacy data of the study, a phase IIb study is ongoing with a loading dose of 990 mg followed by a weekly dose of 720 mg tomuzotuximab or cetuximab combined with chemotherapy and maintenance therapy with the corresponding antibody in patients with recurrent or metastatic HNSCC.
esmoopen-2017-000303supp004.jpg (414KB, jpg)
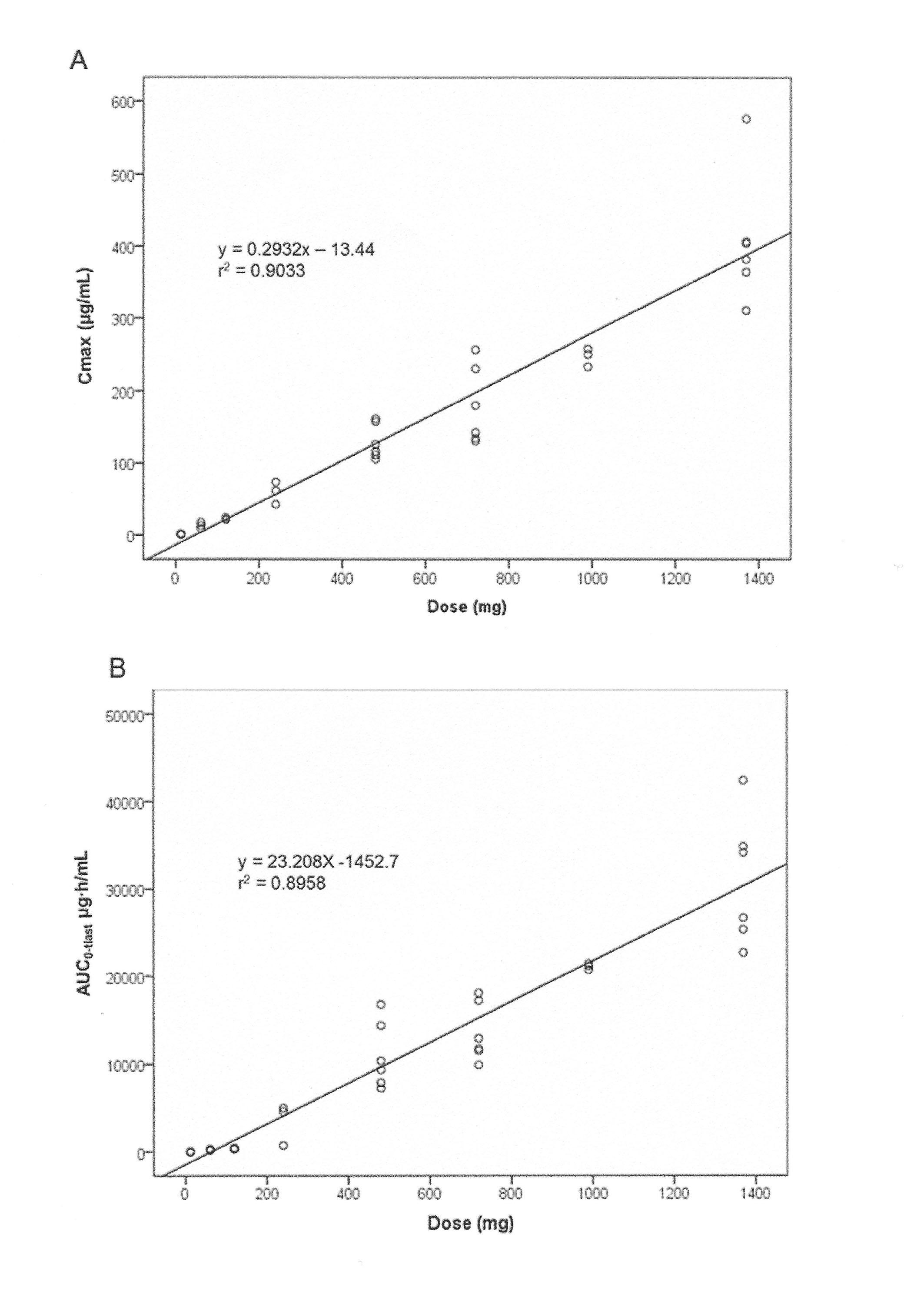{kind=link}
esmoopen-2017-000303supp005.jpg (376.4KB, jpg)
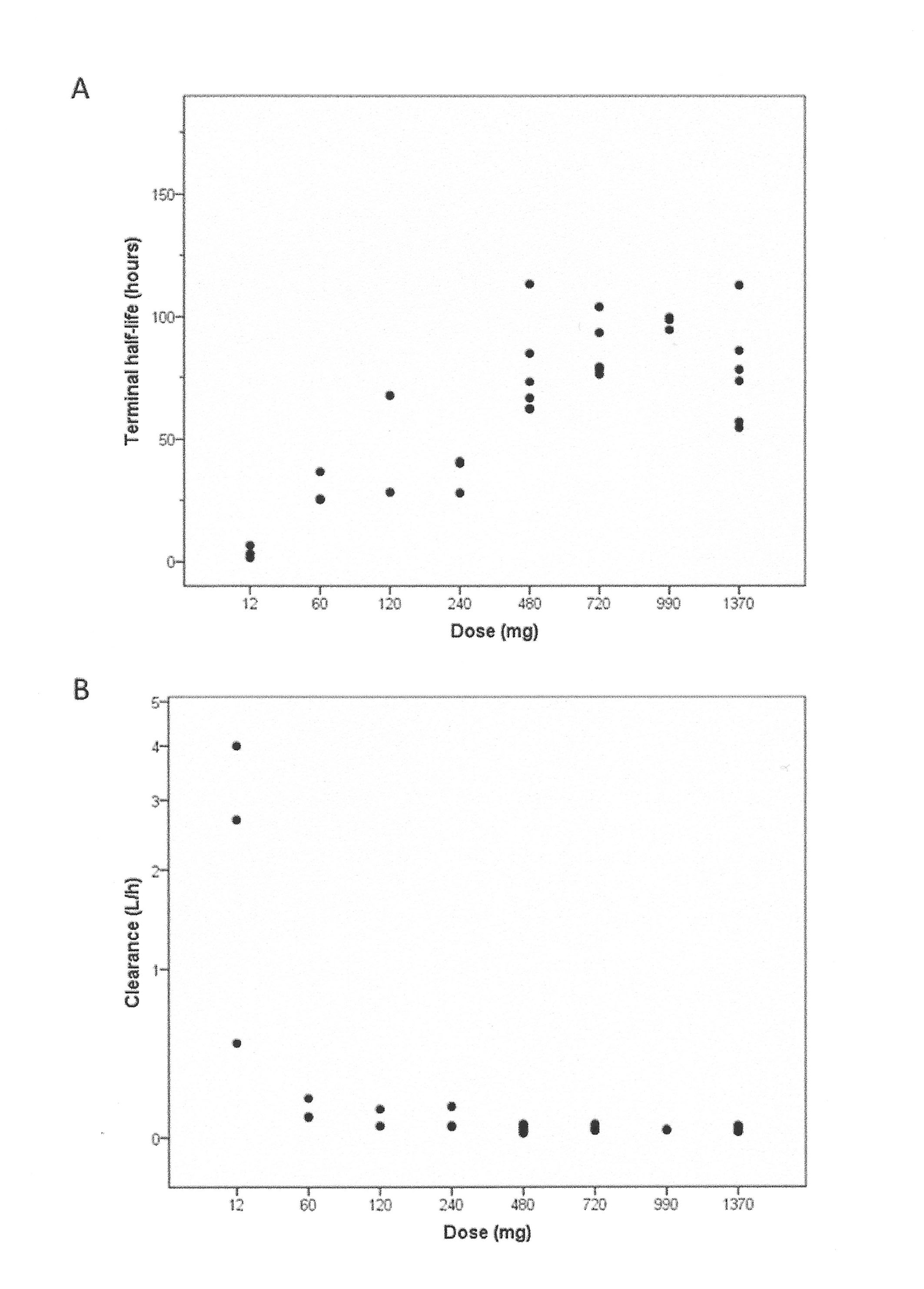{kind=link}
Acknowledgments
The authors thank Dr Silvia von Mensdorff-Pouilly for editorial assistance and critical reading of the manuscript.
Footnotes
Contributors: Conception and design: WF, HB, BD, SG, AD, CS, LG. Provision of study materials or patients: WF, SC, HS-B, JW, SDD, AT, HB, BD, AD, CS, LG. Collection and assembly of data: WF, SC, HS-B, JW, SDD, AT, HB, CS, LG. Data analysis and interpretation: WF, HB, BD, AD, SG, AZ, CS, LG. Reading and approval of the final manuscript: all authors.
Funding: This work was supported by Glycotope GmbH, which provided the drug and financial support for the conduct of the trial. The authors were fully responsible for all content and editorial decisions, were involved in all stages of manuscript preparation and have approved the final version.
Competing interests: Employment or leadership position: HB, BD, SG, AZ, AD. Stock ownership: HB, SG, AD. MS was employee of Pharma Brains AG, which received funding for the study from Glycotope GmbH. Other remuneration: WF, Meeting participation to present the data at the American Society of Clinical Oncology 2013 Annual Meeting.
Patient consent: Obtained.
Ethics approval: Ethik-Kommission der Ärztekammer Hamburg and the Ethics Committees of each Center.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Van Cutsem E, Köhne CH, Hitre E, et al. . Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408–17. 10.1056/NEJMoa0805019 [DOI] [PubMed] [Google Scholar]
- 2. Vermorken JB, Mesia R, Rivera F, et al. . Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008;359:1116–27. 10.1056/NEJMoa0802656 [DOI] [PubMed] [Google Scholar]
- 3. Van Cutsem E, Köhne CH, Láng I, et al. . Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 2011;29:2011–9. 10.1200/JCO.2010.33.5091 [DOI] [PubMed] [Google Scholar]
- 4. Kurai J, Chikumi H, Hashimoto K, et al. . Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res 2007;13:1552–61. 10.1158/1078-0432.CCR-06-1726 [DOI] [PubMed] [Google Scholar]
- 5. Bibeau F, Lopez-Crapez E, Di Fiore F, et al. . Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol 2009;27:1122–9. 10.1200/JCO.2008.18.0463 [DOI] [PubMed] [Google Scholar]
- 6. Rodríguez J, Zarate R, Bandres E, et al. . Fc gamma receptor polymorphisms as predictive markers of Cetuximab efficacy in epidermal growth factor receptor downstream-mutated metastatic colorectal cancer. Eur J Cancer 2012;48:1774–80. 10.1016/j.ejca.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 7. Gerdes CA, Nicolini VG, Herter S, et al. . GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res 2013;19:1126–38. 10.1158/1078-0432.CCR-12-0989 [DOI] [PubMed] [Google Scholar]
- 8. Shields RL, Lai J, Keck R, et al. . Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem 2002;277:26733–40. 10.1074/jbc.M202069200 [DOI] [PubMed] [Google Scholar]
- 9. Chung CH, Mirakhur B, Chan E, et al. . Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008;358:1109–17. 10.1056/NEJMoa074943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. National Cancer Institute. Common terminology criteria for adverse events and common toxicity criteria version 3.0. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.
- 11. Fracasso PM, Burris H, Arquette MA, et al. . A phase 1 escalating single-dose and weekly fixed-dose study of cetuximab: pharmacokinetic and pharmacodynamic rationale for dosing. Clin Cancer Res 2007;13:986–93. 10.1158/1078-0432.CCR-06-1542 [DOI] [PubMed] [Google Scholar]
- 12. Eisenhauer EA, Therasse P, Bogaerts J, et al. . New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 13. Paz-Ares LG, Gomez-Roca C, Delord JP, et al. . Phase I pharmacokinetic and pharmacodynamic dose-escalation study of RG7160 (GA201), the first glycoengineered monoclonal antibody against the epidermal growth factor receptor, in patients with advanced solid tumors. J Clin Oncol 2011;29:3783–90. 10.1200/JCO.2011.34.8888 [DOI] [PubMed] [Google Scholar]
- 14. Song X, Long SR, Barber B, et al. . Systematic review on infusion reactions associated with chemotherapies and monoclonal antibodies for metastatic colorectal cancer. Curr Clin Pharmacol 2012;7:56–65. 10.2174/157488412799218806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kimby E. Tolerability and safety of rituximab (MabThera). Cancer Treat Rev 2005;31:456–73. 10.1016/j.ctrv.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 16. Su X, Lacouture ME, Jia Y, et al. . Risk of high-grade skin rash in cancer patients treated with cetuximab–an antibody against epidermal growth factor receptor: systemic review and meta-analysis. Oncology 2009;77:124–33. 10.1159/000229752 [DOI] [PubMed] [Google Scholar]
- 17. Fakih MG, Wilding G, Lombardo J. Cetuximab-induced hypomagnesemia in patients with colorectal cancer. Clin Colorectal Cancer 2006;6:152–6. 10.3816/CCC.2006.n.033 [DOI] [PubMed] [Google Scholar]
- 18. Tan AR, Moore DF, Hidalgo M, et al. . Pharmacokinetics of cetuximab after administration of escalating single dosing and weekly fixed dosing in patients with solid tumors. Clin Cancer Res 2006;12:6517–22. 10.1158/1078-0432.CCR-06-0705 [DOI] [PubMed] [Google Scholar]
- 19. Seo Y, Ishii Y, Ochiai H, et al. . Cetuximab-mediated ADCC activity is correlated with the cell surface expression level of EGFR but not with the KRAS/BRAF mutational status in colorectal cancer. Oncol Rep 2014;31:2115–22. 10.3892/or.2014.3077 [DOI] [PubMed] [Google Scholar]
- 20. Williams R. Discontinued in 2013: oncology drugs. Expert Opin Investig Drugs 2015;24:95–110. 10.1517/13543784.2015.971154 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
esmoopen-2017-000303supp001.jpg (538.3KB, jpg)
esmoopen-2017-000303supp002.pdf (313.4KB, pdf)
esmoopen-2017-000303supp003.pdf (152.6KB, pdf)
esmoopen-2017-000303supp006.jpg (875.3KB, jpg)
esmoopen-2017-000303supp007.jpg (738.6KB, jpg)
esmoopen-2017-000303supp004.jpg (414KB, jpg)
esmoopen-2017-000303supp005.jpg (376.4KB, jpg)