

Abstract
Background and Aims
Prader-Willi syndrome (PWS) is a complex, multisystem genetic disorder characterized by endocrine, neurologic and behavioral abnormalities. We report the first case of an unbalanced de-novo reciprocal translocation of chromosome 15 and 19: 45,XY,-15, der (19)t(15;19)(q12;p13.3) resulting in monosomy for the PWS chromosome critical region. We performed high resolution SNP microarray to characterize the breakpoints.
Case report
Our patient had several typical features for PWS including infantile hypotonia, a poor suck and feeding difficulties, tantrums, skin picking, compulsions, small hands and feet and food seeking but not hypopigmentation, a micropenis, cryptorchidism or obesity as common findings seen in PWS at the time of examination at 6 years of age. He had seizures noted from 1 to 3 years of age and marked cognitive delay.
Results
High resolution SNP microarray analysis identified an atypical PWS Type I deletion of chromosome 15 involving proximal breakpoint BP1. The deletion extended beyond the GABRB3 gene but was proximal to the usual distal breakpoint (BP3) within the 15q11-q13 region and GABRA5, GABRG3 and OCA2 genes were intact.
Conclusion
We report a case with atypical features for PWS associated with an unbalanced de-novo reciprocal translocation resulting in monosomy for the 15q11.1–15q12 with intact GABRA5, GABRG3 and OCA2 genes. No deletion of 19p13.3 band was detected therefore the patient was not at an increased risk of tumors from Peutz-Jeghers syndrome associated with a deletion of the STK11 gene.
Keywords: PWS, Prader-Willi syndrome, hyperphagia, hypotonia, atypical 15q11-q13 Type I deletion, unbalanced de novo chromosome t(15;19)
Introduction
Prader-Willi syndrome (PWS) is a complex genetic disorder that affects behavior, cognitive function, growth and metabolism with dysmorphic features. PWS is characterized by hypotonia, growth hormone deficiency with short stature and small hands and feet, hypogonadism/hypogenitalism, neurological and behavioral abnormalities (OCD, skin picking, outbursts, anxiety) delayed development, early-childhood onset of obesity which can be life-threatening, type II diabetes, and unusual craniofacial appearance [Butler 2011; Cassidy et al., 2012; Risheg et al., 2013; Kweh et al., 2014; Angulo et al., 2015; GeneReviews (http://www.ncbi.nlm.nih.gov)]. Dysmorphic features include a narrow forehead with bitemporal narrowing, upslanting almond-shaped eyes, and short nose with downturned corners of the mouth. Some PWS individuals also have hypopigmented hair and skin with features of ocular albinism.
Prader-Willi syndrome affects approximately 1/15,000 to 1/30,000 individuals worldwide and equally occurs across both genders and all races [Butler et al., 2002; Butler et al., 2006; Kweh et al., 2014; Angulo et al., 2015; GeneReviews (http://www.ncbi.nlm.nih.gov) ]. Most cases are suspected to have Prader-Willi syndrome in infancy by their weak cry, poor suck, and feeding problems, hypotonia and hypogonadism/hypogenitalism. PWS is caused by the loss of paternally expressed genes in the imprinted 15q11-q13 chromosome region. About 70% of patients with PWS have a de novo, paternally derived, 15q11-q13 deletion detected by fluorescence in-situ hybridization (FISH) using small nuclear ribonucleoprotein polypeptide N (SNRPN) as a probe, high resolution chromosomal microarray or with methylation specific-multiplex ligation dependent probe amplification (MS-MLPA) studies [e.g., Butler et al., 2008; Hamzi et al., 2010; Henkhaus et al., 2012], while about 25% have uniparental maternal disomy of chromosome 15 (both 15s from the mother) and 2% have epigenetic imprinting defects [Bittel and Butler, 2005; Angulo et al., 2015]. DNA methylation is abnormal in 99% of cases but does not determine the specific PWS genetic subtype. Methylation status is often based on analysis of the SNRPN gene in the critical region [Muralidhar and Butler, 1998; Askree et al., 2012]. Deletion of the oculocutaneous albinism type II (OCA2) gene in the distal area of the 15q11-q13 critical region is also associated with pigmentation defects of skin, hair and eyes in PWS. Additionally, there are small nucleolar RNA (snoRNA) clusters that largely consist of SNORD116 and SNORD115, which are imprinted and appear to play a major role to help regulate biological metabolism and development of PWS. The deletion or loss of SNORD116 appears to be most likely associated with the PWS phenotype [Sahoo et al., 2008].
Clinical Report
We report a male child who was delivered via Cesarean section due to concerns in fetal heart tracing. At delivery, the baby required resuscitation with bag mask ventilation and oxygen. The baby had a weight of 3.572 kg (10–25th percentile), length of 54.5 cm (10–25th percentile) and head circumference of 39.5 cm (50th percentile). The anterior fontanel was large with a slightly overriding sagittal suture. He was noted to have hypotonia, bradycardia, a weak cry and poor suck with inability to initiate and sustain enteral feeding requiring a normal suck and swallow mechanism. He had a primitive grasp, an incomplete Moro response and a positional contracture of his left hip. A modified barium swallow showed that he had very poor endurance resulting in a gastrostomy tube placed at three weeks of age with removal at 11 months. He had normal pigment pattern for the family background and did not have a micropenis or cryptorchidism. There was a family history of a paternal nephew with cleft palate, deafness and developmental delay and a maternal cousin with hypoglycemia and seizures. Consequently, his mother underwent fragile X DNA testing which was negative. At age 11 months he developed seizure activity associated with upper respiratory infections or after exposure to excessive heat. Seizures were characterized by eye rolling to the right, slumping over, body stiffening and tonic-clonic activities. He was treated with valproic acid and memantine HCl.
An EKG revealed sinus bradycardia and minor right ventricular conduction delay. An EEG showed an abnormal 7–8 Hz pattern background with slower activity and occipital low amplitude background rhythms with intermittent burst of generalized paradoxical delta or 3 Hz spike and wave activity intermittent with drowsiness. The EEG was considered abnormal for what appeared to be a primary epileptic pattern with occasional right temporal asymmetry in sleep. The epilepsy was stabilized with medication by the age of 5 years.
He had frequent tantrums and began severe skin picking at the age of 3 years leading to chronic bleeding sites. At the age of 4 years, he exhibited anxiety, would become emotional and cry easily with occasional head banging episodes. Beginning at age 5 years he developed an attention deficit hyperactivity disorder with autistic features including social impairments and repetitive movements/stereotypies. He had a compulsion for arranging his belongings and liked to play with strings. He had delayed language development and exhibited food seeking with hiding behaviors. A brain MRI revealed adequate gray/white differentiation and symmetry; however, there was prominence of the subarachnoid spaces in the middle cranial fossa bilateral with temporal lobes being small. The frontal horns were also prominent with respect to the ventricular system. The anterior pituitary gland was slightly small for the patient’s age and correlation with pituitary function was recommended.
At the age of 5 years and 9 months he participated in a multisite, NIH funded natural history study [RDCRN (Rare Diseases Clinical Research Network)]. The physical exam revealed a flat occiput, bitemporal narrowing with almond-shaped eyes, strabismus, hypotelorism, upslanting of the eyes and narrow nasal bridge, relatively small hands and feet, dark pigmentation of hair and skin, and a thin habitus with an abnormal posture (Figure 1). He had a hypernasal voice and pes planus. His weight was 23 kg (60th percentile), height was 47.3 cm (90th percentile) and head circumference was 52 cm (65th percentile). Medications included valproic acid, memantine HCl, Genotropin, coenzyme Q10, multivitamins, and fish oil. The KBIT2 IQ test was administered and verbal and nonverbal IQ scores were 65 (1st percentile) and 66 (0.5th percentile), respectively with a composite IQ score of 61. He was attending special education classes and was in the third grade.
Figure 1.
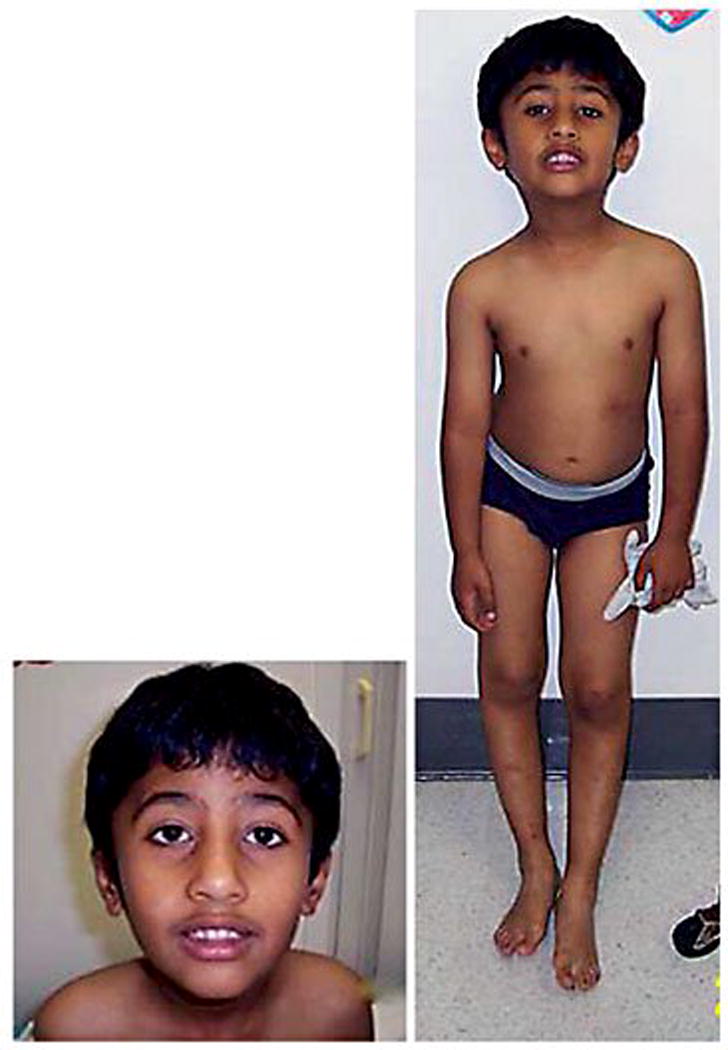
Frontal and facial view of our patient at 6yrs of age with the 15q11.1-q12 deletion resulting from a translocation involving chromosomes 15 and 19. Craniofacial features included a narrow forehead, triangular shaped facies with a pointed chin and almond shaped upslanting eyes. Other physical features included a lean habitus with abnormal posture, hypotonia and darkly pigmented hair and skin with small hands and feet.
Material and Methods
Cytogenetic analysis was performed and showed an unbalanced de-novo reciprocal translocation between the proximal long arm of chromosome 15 and the distal short arm of chromosome 19 resulting in monosomy for the 15q11.1–15q12 region which is associated with Prader-Willi syndrome. DNA methylation analysis was performed and showed an abnormal pattern consistent with Prader-Willi syndrome. His karyotype was 45,XY,-15,der(19)t(15;19)(q12;p13.3). Subsequently, a high resolution SNP 6.0 version microarray (Affymetrix, Santa Clara, California) consisting of 1.8 million copy number and single nucleotide polymorphic probes confirmed an atypical PWS Type I deletion of chromosome 15 involving the proximal 15q11-q13 breakpoint (BP1) but terminated after the GABRB3 gene and more proximal than the typical distal breakpoint (BP3) at genomic coordinates 22,973,542bp – 27,001,01bp (hg19) or 4.0 Mb in size. No deletion of the 19p13.3 band was found using the standard genome filter parameters for high resolution microarray analysis. With less restrictive genome filter parameters, a small deletion was found encompassing only the PPAP2C gene.
Results and Discussion
We report an unusual case of PWS with an atypical Type I deletion of the 15q11-q13 region resulting from a de novo unbalanced reciprocal translocation: 45,XY,-15,der (19)t(15;19)(q12;p13.3) resulting in monosomy for the PWS chromosome critical region at 15q11.1–15q12 of 4.0Mb in size including 18 genes/transcripts recognized in OMIM (http://www.omim.org) using high resolution microarray analysis (Figure 2). The deletion of the 15q11.1-q12 region contained the following list of genes (http://www.omim.org): TUBGCP5, CYFIP1, NIPA2, NIPA1, MRKN3, MAGEL2, NDN, SNRPN, SNURF, SNORD107, SNORD64, SNORD109A, SNORD116, SNORD115, SNORD109B, UBE3A, ATP10A and GABRB3.
Figure 2.
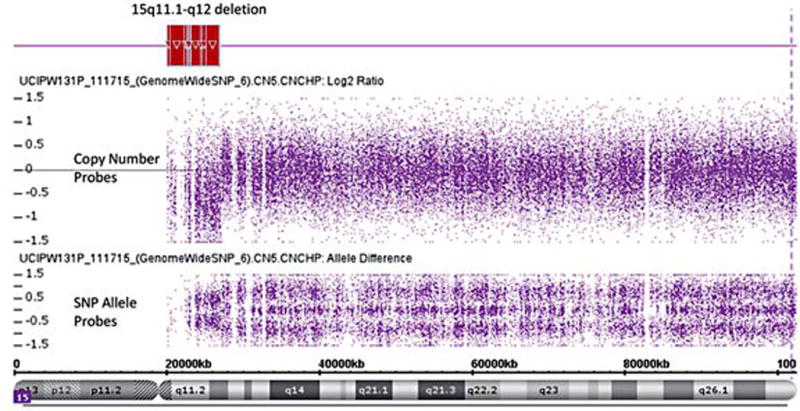
High resolution SNP microarray representing both copy number and SNP probe signals revealed an atypical deletion of the chromosome 15q11.1-q12 region (highlighted in red at top). The copy number and SNP allele probe tracks show the loss of signal within the PWS critical 15q11-q13 region.
We reviewed the literature and noted subjects with PWS reported with atypical 15q11-q13 deletions. For example, Kim et al. [2012] reported two cases with PWS having an atypical Type I deletion of the 15q11-q13 region. A high resolution SNP microarray revealed a deletion extending from NIPA1 to GABRB3 genes while the OCA2 gene was intact in both patients (Figure 3). Case 1 had microcephaly and normal pigmentation but no skin picking and had a pleasant disposition without aggressive behavior. Case 2 had microcephaly, later age onset of obesity, and was reported to be unusually high functioning. These two cases and our patient had intact the OCA2 gene and did not display hypopigmentation which is generally seen in PWS with the typical 15q11-q13 deletion The OCA2 gene encodes a protein that corresponds to the ‘pink-eyed dilution’ (p) mouse mutant. The human cDNA DN10, linked to the p locus in mice, identifies the human homolog (P) of the mouse p gene, and appears to encode an integral membrane transporter protein. The human P protein is an 838-amino acid polypeptide that contains 12 putative transmembrane domains and exhibits structural homology to transporters of small organic molecules. Our case also had the GABRB3 (gamma-amino butyric acid - GABA) gene deleted but GABRA5 and GABRG3 receptor genes located within the 15q11-q13 region were intact using standard genome filter parameters for segment length and marker count (i.e., 50 markers per 100kb segment length) for high resolution chromosome microarray analysis. GABA plays a major inhibitory neurotransmitter function at the brain level. Using standard genome filter parameters, no genes were deleted from the chromosome 19p13.3 band. However, when lowering the genome filter parameters to 20 markers and 35 kb segment length, PPAP2C (phosphatidic acid phosphatase type 2C) gene was deleted. This gene encodes a protein member of the phosphatidic acid phosphatase (PAP) family involved in the conversion of phosphatidic acid to diacylglycerol needed for the de novo synthesis of glycerophospholipids and triacylglycerols and the hydrolysis of lysophosphatidic acid, ceramide and sphingosine which may impact neurodevelopment and brain function (http://www.omim.org).
Figure 3.
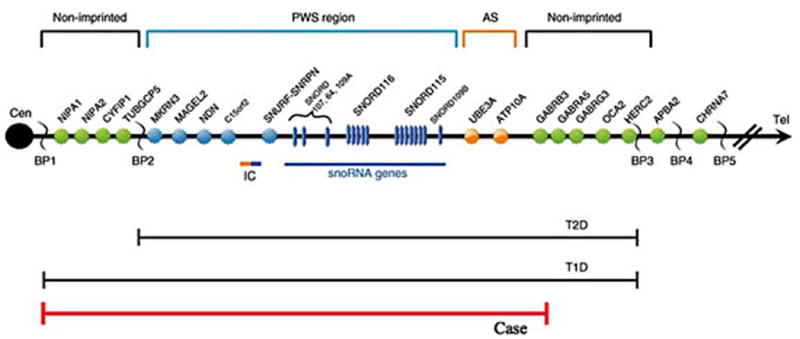
Schematic representation of the 15q11-q13 chromosome region displaying the order of genes in the region. Genes in the non-imprinted region are shown in green while genes in the Prader-Willi syndrome (PWS) region are shown in blue. Genes in the Angelman syndrome (AS) region are shown in orange. The 15q11.1-q12 deletion seen in our patient included NIPA1 to GABRB3 genes and illustrated by the red line. GABRA5, GABRG3 and OCA2 or P (pigment) genes located distally in the 15q11-q13 region, commonly deleted in PWS, were intact. BP= Breakpoints BP1, BP2, BP3, BP4 and BP5 are designated; IC=imprinting center; T1D=Typical 15q11-q13 Type I deletion, T2D=Typical 15q11-q13 Type II deletion.
Although we report a patient with PWS and a deletion associated with an unbalanced de novo chromosomal translocation, only a few reports have been noted in the medical literature [Butler, 1990; Butler and Thompson, 2000; Butler et al., 2006]. Heidemann et al. [2010] summarized 14 individuals with PWS having chromosome translocations but only one case involved chromosomes 15 and 19 [Sun et al., 1996]. This case described a male child with the PWS phenotype due to a de novo balanced translocation between chromosomes 15 and 19 involving the PWS critical region on chromosome 15 and the long arm of chromosome 19 and not the short arm as seen in our patient. This patient also did not have a recognized deletion of the 15q11-q13 region. Our patient had marked cognitive delays, seizures and ADHD in addition to the typical features of PWS including hyperphagia and skin picking but not hypogonadism/hypogenitalism or hypopigmentation, as the OCA2 gene in the 15q11-q13 region was not deleted. Peutz-Jeghers disease associated with deletion of STK11 on chromosome 19p13.3 [Peddibhotla et al., 2013] was not present in our patient as this gene was not deleted. Hence, our patient with a translocation involving the 15q11-q13 region did not have a recognized deletion of the 19p13.3 band detectable with a high resolution SNP microarray using standard genome filter parameters. However, when lower genome filter parameters were set, PPAP2C gene was deleted.
The cause of our patient’s atypical features with the t(15;19)(q12;p13.3) is not clear although the size of the 15q11-q13 deletion was unusual or not typical for PWS. There are other factors that might contribute to the clinical outcome, such as epigenetic factors related to the 15/19 chromosomal translocation or process and as yet undefined environmental or genetic factors. Additional research is needed to further explain the role of factors contributing to hyperphagia and obesity in PWS [Hochberg and Hochberg, 2010]; those patients with an unusual presentation of the PWS phenotype due to atypical genetic causes such as chromosome translocations may shed light on the causation of the key features in this syndrome.
Summary
We report a unique case with an atypical Type I deletion of the 15q11-q13 region secondary to a translocation involving chromosomes 15 and 19 and intact GABRA5, GABRG3 and OCA2 genes. The common features of PWS were seen including hypotonia, developmental delay, strabismus, behavioral problems, intellectual disability, speech impairment and small hands and feet. Atypical features included epilepsy, a lean habitus with an abnormal posture, and marked cognitive delay. Peutz-Jeghers disease associated with a deletion of the STK11 gene seen in patients with the 19p13.3 deletion was not found in our patient. High resolution SNP microarray analysis was useful in confirming the absence of the deletion of chromosome 19p to rule out Peutz-Jeghers disease and associated tumors. Studying individuals with atypical deletions may offer further opportunities to explain the spectrum and causation of PWS. We encourage other authors to report subjects with PWS and unique chromosomal anomalies including translocations involving the 15q11-q13 region to increase understanding of causation and unique function of overlapping genetic regions on two separate translocated chromosomes.
Acknowledgments
We would like to thank the patient and family for participating in this study. We would like to thank the Prader-Willi Syndrome Association (USA) and the Angelman, Rett and Prader-Willi Syndromes Consortium (U54 HD06122), which was part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN) supported through collaboration between the NIH Office of Rare Disease Research (ORDR) at the National Center of Advancing Translational Science (NCATS) and the National Institute of Child Health and Human Development (NICHD).
Footnotes
Conflict of Interest:
The authors declare no conflicts of interest
Statement of Ethics:
The study subject and family have given their informed written consent and the University of California, Irvine ethics committee have strictly approved the study protocol.
References
- Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38:1249–1263. doi: 10.1007/s40618-015-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askree SH, Dharamrup S, Hjelm LN, Coffee B. Parent-of-origin testing for 15q11-q13 gains by quantitative DNA methylation analysis. J Mol Diagn. 2012;14(3):192–198. doi: 10.1016/j.jmoldx.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005;7(14):1–20. doi: 10.1017/S1462399405009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am J Med Genet. 1990;35(3):319–332. doi: 10.1002/ajmg.1320350306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Lee PDK, Whitman BY. Management of Prader-Willi Syndrome. Springer; New York: 2006. [Google Scholar]
- Butler MG. Prader-Willi syndrome: Obesity due to genomic imprinting. Curr Genomics. 2011;12(3):204–215. doi: 10.2174/138920211795677877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: A population-based study. Dev Med Child Neurol. 2002;44(4):248–255. doi: 10.1017/s001216220100202x. [DOI] [PubMed] [Google Scholar]
- Butler MG, Thompson T. Prader Willi syndrome: Clinical and genetic findings. Endocrinol. 2000;10:3S–16S. doi: 10.1097/00019616-200010041-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Fischer W, Kibiryeva N, Bittel DC. Array comparative genomic hybridization (aCGH) analysis in Prader-Willi syndrome. Am J Med Genet A. 2008;146A(7):854–60. doi: 10.1002/ajmg.a.32249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- Hamzi K, Itto AB, Nassereddine S, Nadifi S. Prader-Willi syndrome: Methylation study of fluorescence in situ hybridization first? Indian J Hum Genet. 2010;16(3):172–174. doi: 10.4103/0971-6866.73417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidemann S, Plendl H, Vater I, Gesk S, Exeler-Telker JR, Grote W, Siebert R, Caliebe A. Maternal uniparental disomy 15 in a fetus resulting from a balanced familial translocation t(2;15)(p11;q11.2) Prenat Diagn. 2010;30(2):183–185. doi: 10.1002/pd.2436. [DOI] [PubMed] [Google Scholar]
- Henkhaus RS, Kim SJ, Kimonis VE, Gold JA, Dykens EM, Driscoll DJ, Butler MG. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet Test Mol Biomarkers. 2012;16(3):178–186. doi: 10.1089/gtmb.2011.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg I, Hochberg Z. Expanding the definition of hypothalamic obesity. Obes Rev. 2010;11(10):709–721. doi: 10.1111/j.1467-789X.2010.00727.x. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Miller JL, Kuipers PJ, German JR, Beaudet AL, Sahoo T, Driscoll DJ. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. 2012;20(3):283–90. doi: 10.1038/ejhg.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweh FA, Miller JL, Sulsona CR, Wasserfall C, Atkinson M, Shuster JJ, Goldstone AP, Driscoll DJ. Hyperghrelinemia in Prader-Willi syndrome begins in early infancy long before the onset of hyperphagia. Am J Med Genet. 2015;A167A(1):69–79. doi: 10.1002/ajmg.a.36810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidhar B, Butler MG. Methylation PCR analysis of Prader-Willi syndrome, Angelman syndrome, and control subjects. Am J Med Genet. 1998;80(3):263–265. [PMC free article] [PubMed] [Google Scholar]
- Peddibhotla S, Khalifa M, Probst FJ, Stein J, Harris LL, Kearney DL, Vance GH, Bull MJ, Grange DK, Scharer GH, Kang SH, Stankiewicz P, Bacino CA, Cheung SW, Patel A. Expanding the genotype-phenotype correlation in subtelomeric 19p13.3 microdeletions using high resolution clinical chromosomal microarray analysis. Am J Med Genet A. 2013;161A(12):2953–63. doi: 10.1002/ajmg.a.35886. [DOI] [PubMed] [Google Scholar]
- Risheg H, Pasion R, Sacharow S, Proud V, Immken L, Schwartz S, Tepperberg JH, Papenhausen P, Tan TY, Andrieux J, Plessis G, Amor DJ, Keitges EA. Clinical comparison of overlapping deletions of 19p13.3. Am J Med Genet A. 2013;161A(5):1110–1116. doi: 10.1002/ajmg.a.35923. [DOI] [PubMed] [Google Scholar]
- Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40(6):719–721. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Nicholls RD, Butler MG, Saitoh S, Hainline BE, Palmer CG. Breakage in the SNRPN locus in a balanced 46,XY,t(15;19) Prader-Willi syndrome patient. Hum Mol Genet. 1996;5(4):517–524. doi: 10.1093/hmg/5.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]