

Abstract
MitoNEET is an outer membrane protein whose exact function remains unclear, though a role of this protein in redox and iron sensing as well as in controlling maximum mitochondrial respiratory rates has been discussed. It was shown to contain a redox active and acid labile [2Fe-2S} cluster which is ligated by one histidine and three cysteine residues. Herein we present the first synthetic analogue with biomimetic {SN/S2} ligation which could be structurally characterized in its diferric form, 52−. In addition to being a high fidelity structural model for the biological cofactor, the complex is shown to mediate proton coupled electron transfer (PCET) at the {SN} ligated site, pointing at a potential functional role of the enzyme’s unique His ligand. Full PCET thermodynamic square schemes for the mitoNEET model 52− and a related homoleptic {SN/SN} capped [2Fe-2S] cluster 42− are established, and kinetics of PCET reactivity are investigated by double-mixing stopped-flow experiments for both complexes. While the N-H bond dissociation free energy (BDFE) of 5H2− (230 ± 4 kJ mol−1) and the free energy ΔG°PCET for the reaction with TEMPO (−48.4 kJ mol−1) are very similar to values for the homoleptic cluster 4H2− (232 ± 4 kJ mol−1, −46.3 kJ mol−1) the latter is found to react significantly faster than the mitoNEET model (data for 5H2−: k = 135 ± 27 M−1s−1, ΔH‡ = 17.6 ± 3.0 kJ mol−1, ΔS‡ = −143 ± 11 J mol−1 K−1, ΔG‡ = 59.8 kJ mol−1 at 293 K). Comparison of the PCET efficiency of these clusters emphasizes the relevance of reorganization energy in this process.
TOC image
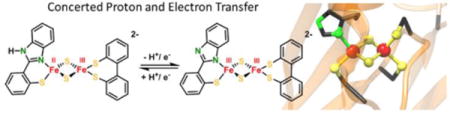
Introduction
Iron-sulfur clusters are ubiquitous cofactors, which exist in a variety of forms and serve a multitude of functions including electron transport, redox reactions and sensing.1 While the majority of iron-sulfur clusters are ligated by four cysteine residues of the surrounding protein, it has been observed that a subset of [2Fe-2S] clusters with different ligands exists. The most commonly observed of those alternative ligands is histidine but in most cases the role of this alternative ligand is not yet fully understood.2
Among clusters featuring non-cysteine ligation, the Rieske center is arguably the most prominent example and has been studied most thoroughly.3 It features a unique coordination environment of two histidine and two cysteine residues (Figure 1) and plays an important role in electron transfer and as a structural gate as well as mediating proton-coupled electron transfer (PCET) in the Q-cycle.4–6 Although synthetic analogues of iron-sulfur clusters have been studied since the 1960s7 and have contributed largely to the understanding of their biological blueprints, first structural model systems for the unique 2Cys 2His coordinated [2Fe-2S] cluster in Rieske proteins have only been reported very recently by our groups (12− and 22− in Figure 2).8,9
Figure 1.
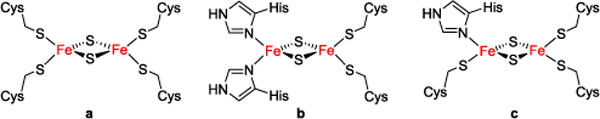
[2Fe-2S] clusters of ferredoxines (a), Rieske centers (b) and mitoNEET and related clusters (c).
Figure 2.
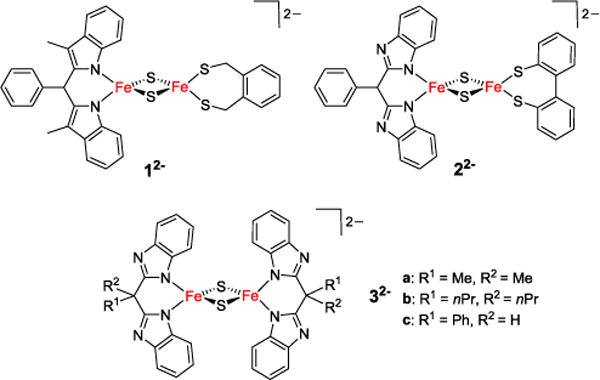
First and second generation Rieske models 12− and 22−, and homoleptic {N}4 coordinated models 32−, all shown in their diferric state.
Proton coupled electron transfer was studied in the high fidelity functional model 22− and related homoleptic clusters 32− (Figure 2), highlighting the importance of the distal nitrogen atom of the His-like ligand as protonation site.9–12 For the first time these systems also allowed for a full characterization of synthetic [2Fe-2S] clusters in their reduced and protonated states;10–13 the first example of a fully characterized all-ferrous [2Fe-2S] cluster has been reported in 2012.14 In a related symmetric diferric cluster 42− with benzimidazolato-based bidentate {SN} capping ligands both iron sites feature a single His-like N-donor.15
Three different classes of [2Fe-2S] clusters with unusual 3Cys 1His coordination are known to date: the bacterial transcription factor IscR, glutaredoxines and CDGSH-proteins including mitoNEET. While the function of the [2Fe-2S] cluster in the first two examples has already been established (modulating the binding of DNA or Atf1, respectively), the exact function of this cluster in mitoNEET remains unknown.2 MitoNEET is an outer mitochondrial membrane protein discovered in 2004 and was shown to contain a redox active and acid labile [2Fe-2S] cluster, ligated by one histidine and three cysteine residues (Figure 3).16–20 It has been identified as the target of pioglitazone and thiazolidinedione drugs (TZDs), which are used in the treatment of diabetes type 2. 21 Among the functions suggested for mitoNEET are a role in redox reactions,20 redox-sensing,22 or as a cluster transfer protein.23 Although the functional relevance of the single His ligand has not been fully understood to date, it has been proposed to be involved in PCET reactivity. Its imidazole backside is positioned at the surface of the protein and is thus easily exposed to protonation upon changes in the environment of the protein (Figure 3).19
Figure 3.

3Cys 1His coordinated [2Fe-2S] cluster of human mitoNEET (PDB entry 2QH7) and conserved hydrogen-bonding residues lysine 55, aspartate 84 and serine 77.
The His residue is also responsible for the observed pH lability of the cluster in its reduced state, suggesting a function of the cluster in redox and/or pH sensing.22 Indeed, reduction of mitoNEET proteins is coupled to proton uptake,24 indicating that the His ligand is crucial in modulating both this reactivity and the redox potential of the cluster. The [2Fe-2S] cluster is embedded in a network of hydrogen-bonding residues, which are conserved in all members of the protein family and are essential in modulating cluster stability and functionality of the protein (Figure 3).22 They may also serve as a gateway for donating/accepting protons during PCET. It has recently been shown that transfer of the [2Fe-2S] cluster from mitoNEET to a respective acceptor protein occurs in the cluster’s oxidized all-ferric state, while no transfer of the cluster in its reduced mixed-valent sate could be observed.25 The presence of the unique His ligand was shown to be crucial for this process by comparing the protein´s reactivity with that of mutants lacking this residue.26 The cluster´s redox potential shifts with pH, and pulsed EPR studies have shown that the unpaired electron is located on the His-ligated iron site in the cluster’s reduced FeIIFeIII form.21,27,28 Their potential as drug targets raises special interest in elucidating the structural and functional properties of the iron-sulfur clusters in mitoNEET proteins.29,30
Herein we present the synthesis and properties of a first high-fidelity structural model system for this unique [2Fe-2S] cluster, 52−. The ability of this model cluster as well as of closely related homoleptic cluster 42− to undergo proton-coupled electron transfer is demonstrated, and the corresponding thermodynamic square schemes are established, showing that involvement in PCET reactivity is one feasible function of the single histidine ligand in mitoNEET proteins. (Figure 5). A comparison of the reactivities of both clusters and previously reported Rieske models provides insight into the factors determining PCET efficiency at biorelevant Fe/S clusters.
Figure 5.
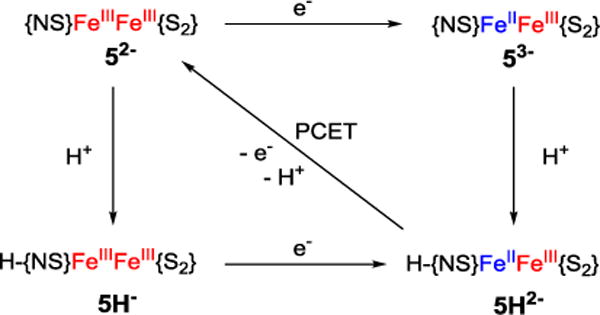
Square scheme of protonation and reduction reactions for a [2Fe–2S] mitoNEET model with {NS} ligation at one Fe site and {S2} ligation at the other Fe site.
Results and Discussion
Cluster synthesis and characterization in solid state
The synthesis of homoleptic 4(NEt4)2 was adapted from literature as described by Beardwood and Gibson.15 The synthesis of heteroleptic [2Fe-2S] clusters with different terminal ligands on the two iron sites is significantly more challenging and is often hampered by ligand scrambling. Diferric complex 5(NEt4)2 could now be prepared via a stepwise ligand exchange pathway starting from [Fe2S2Cl4](NEt4)2 in close analogy to the synthesis of recently reported Rieske models 12− and 22−.8,9 Careful optimization of reaction conditions and several recrystallization steps were found to be necessary in order to obtain pure product, because of difficulties in separating the target compound from homoleptic side products. Diffusion of diethyl ether into a solution of 5(NEt4)2 in MeCN led to growth of crystals suitable for X-ray diffraction. The molecular structure of the diferric cluster anion is shown in Figure 6. 5(NEt4)2 crystallizes in the triclinic space group with 0.5 molecules of MeCN per cluster. Selected geometric parameters and corresponding data for a selected biological mitoNEET cluster are shown in Table 1. While the all-cysteine ligated iron site in the biological mitoNEET cluster shows a nearly ideal tetrahedral coordination sphere, the environment of the His/Cys ligated iron site is more distorted from tetrahedral.20 The heteroleptic model complex 5(NEt4)2 nicely emulates this feature. The Fe···Fe distance in 52− (2.692 Å) is similar to d(Fe∙∙∙Fe) in the Rieske model 22− (2.687 Å). While all these values are somewhat smaller than in the biological mitoNEET systems (2.75 Å) and Rieske proteins (2.71 – 2.72 Å), overall geometric parameters are in good agreement. Strong antiferromagnetic coupling and an S = 0 ground state were observed by SQUID magnetometry for both 4(NEt4)2 (−J = 151 cm−1) and 5(NEt4)2 (−J = 124 cm−1; using a −2JS1·S2 model; see SI for details). These values are all in the range typical for diferric [2Fe-2S] clusters, but comparison with 3c(NEt4)2 (−J = 179 cm−1)13 suggests a significant decrease of −J with increasing number of terminal thiolato ligands.
Figure 6.
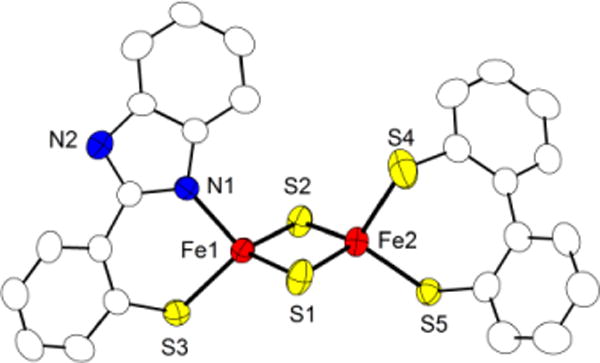
Molecular structure (50% probability thermal ellipsoids) of mitoNEET model 52− determined by X-ray crystallography; hydrogen atoms have been omitted for clarity.
Table 1.
Selected bond lengths (Å) and angles (deg) of diferric cluster 5(NEt4)2 and the mitoNEET [2Fe-2S] cluster.
| 5(NEt4)2 | mitoNEET20 | |
|---|---|---|
| d(Fe∙∙∙Fe) | 2.692(1) | 2.75 |
| d(Fe-μS) | 2.2001(15)-2.2132(17) | 2.20- 2.23 |
| d(Fe-S) | 2.278(2)- 2.2965(15) | 2.21- 2.34 |
| d(Fe-NHis) | 2.009(4) | 2.22-2.18 |
| <(SCys-Fe-SCys) | 103.81(6) | 103.2 (av) |
| <(NHis-Fe-SCys) | 95.65(12) | 98.8 - 99.9 |
| <(Fe-μS-Fe) | 75.19(5) av | 76.7 (av) |
Protonated diferric clusters
4H(NEt4) and 5H(NEt4). To investigate the left part of the square scheme, protonation and deprotonation experiments of both diferric clusters were carried out using [DMPH]BF4 (2,6-dimethylpyridinium tetrafluoroborate, pKa(DMPH+) = 14.13)31 and DBU (1,8-diazabicycloundec-7-ene, pKa(DBUH+) = 24.34 in MeCN)31 as acid/base, and the titrations were monitored by UV-vis spectroscopy. In the heteroleptic mitoNEET model, addition of 1 equiv of DMPH+ leads to the formation of 5H− evidenced by an increase of the band at 375 nm and decrease of bands for 52− at 325, 426, 452 and 525 nm. While protonation proved to be irreversible at room temperature, at −30°C the initial spectrum is fully restored upon addition of DBU. Isosbestic points at 353 nm and 402 nm indicate clean conversion (Figure S10). The pKa value of 5H− was determined by careful backtitration using DBU as a base. Three independent runs were performed and a pKa value of 23.7(2) was derived using mass balance (see SI for details). Likewise, sequential formation of 4H− and 4H2 by addition of 1.0 or 2.0 equivalents of DMPH+ to a MeCN solution of dibasic 42− is accompanied by the disappearance of the band at 434 nm and slight changes of the other absorption maxima in the vis range (see SI for spectra). Three isosbestic points at 340 nm, 392 nm and 576 nm indicate clean conversions. The addition of further equivalents of DMPH+ does not lead to any further spectroscopic changes, suggesting that the diprotonated neutral cluster 4H2 is reasonably stable. Subsequent addition of the base DBU largely restores the initial spectrum of 42−, proving the reversibility of the process. Careful back titration of 4H− with DBU yielded a pKa of 23.0(1) (see SI for details). Therefore the pKa value of the heteroleptic mitoNEET model is almost one unit higher than that of the homoleptic analogue (pKa = 23.7(2) vs 23.0(1)), reflecting a slightly higher proton affinity of 52− compared to 42−. Reversible protonation of 52− to 5H− and of 42− to 4H− was additionally monitored by 1H NMR spectroscopy. In both cases, the single resonances gradually shift upon protonation of the cluster, with chemical shifts proportional to the ratio of protonated vs unprotonated cluster, showing that proton transfer between clusters is fast on the NMR timescale.12 No new signal attributable to an N-H proton has been observed at room temperature, likely because of broadening, rapid exchange, and hydrogen bonding. At 243 K, however, the 1H-NMR spectrum of 4H− shows a relatively broad resonance at 15.68 ppm (see Figure S6), which is in very good agreement with the resonance of the N-H protons observed in 3cH2.10 Although no structural evidence for N protonation could be obtained in this study, crystallographic data for closely related protonated cluster 3cH2 have clearly established the benzimidazole-N as the site of protonation in this type of clusters.10
Mixed valent cluster 43− and 53−
Electrochemical properties of 4(NEt4)2 and 5(NEt4)2 were studied by cyclic voltammetry (CV) in 0.25 M NBu4PF6 solution in MeCN at −15 °C. Potentials for the first and second reduction of 4(NEt4)2, E1/2 = −1.407 V and −2.227 V vs Fc/Fc+, as well as EPR parameters of mixed valent species 43− are in good agreement with those reported by Beardwood and Gibson (see SI).15 The CV of 5(NEt4)2 shows two reduction events (Figure 7, left) that are shifted cathodically by about 150 mV with respect to homoleptic 4(NEt4)2. The first reduction occurring at E1/2 = −1.647 V vs Fc/Fc+ corresponds to the formation of mixed valent 53−, with a peak separation of 59 mV showing the reversibility of the process. The second wave at E1/2 = −2.375 V vs Fc/Fc+ is attributed to the formation of diferrous 54−. Both redox events are separated by 730 mV which reflects a large comproportionation constant of KC = 2.74 × 1012, indicating that mixed-valent 53− should be chemically accessible and relatively stable.
Figure 7.
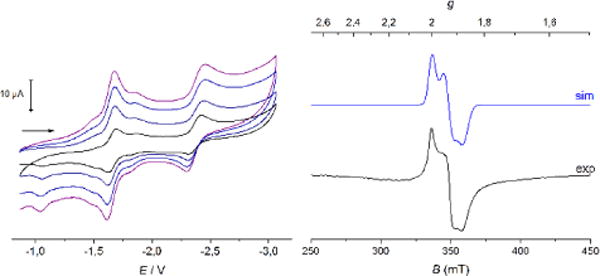
Left: Cyclic voltammogram of 5(NEt4)2 recorded at −15°C in MeCN/0.25 M NBu4PF6 vs Fc at various scan rates (v = 50, 100, 200, 500 mV s−1). Right: EPR spectrum of 53− in MeCN measured as frozen glass at 160 K. The blue line is a powder simulation with g1 = 2.005, g2 = 1.932, g3 = 1.875 and gav = 1.937.
5(NEt4)2 in MeCN solution was chemically reduced by addition of 1.0 equiv of CoCp2* (E1/2 vs Fc/Fc+ = −1.91 V),32 and EPR data of the reduced species were collected. The total spin of S = ½ caused by strong antiferromagnetic coupling between the two iron ions gives rise to a rhombic EPR spectrum; its simulation gives g1 = 2.005, g2 = 1.932, g3 = 1.875 and an average g value gav = 1.937 (Figure 7, right). This latter value is in good agreement with gav = 1.945 found for biological mitoNEET clusters in their reduced form,27 and it is reasonably in between the observed gav of 1.90-1.91 for biological Rieske proteins and gav = 1.96 for [2Fe-2S] ferrodoxines with four cysteine ligands.33,34 As Mouesca showed, the average g value of mixed valent [2Fe-2S] clusters increases towards the free electron value ge = 2.0023 with increasing valence delocalization.35 The average g values of 43− and 53− are both higher than the value observed in a synthetic Rieske model, pointing to slightly more pronounced valence delocalization in line with the symmetric ligation (in 43−) or less pronounced donor asymmetry at the two iron sites (in 53−), if compared with 23−. The dianionic dithiolato ligand is a strong σ and π donor, which stabilizes the higher oxidation state; consequently, this favors localization with ferrous character of the other iron site coordinated by the benzimidazolato ligand. These trends are reflected by a lower gav of the mixed-valent heteroleptic cluster 53− in comparison with 43− (gav = 1.941), although the effect is less pronounced than in models of the Rieske center.8,9
Reduction of 5(NEt4)2 resulted in changes in visible absorption spectra, leading to an overall decrease in absorbance. The original bands disappear while a new band at 562 nm evolves. 43− and 53− can be reoxidized by addition of [CoCp2]PF6 (E1/2 vs Fc/Fc+ = −1.31 V)40 as evidenced by UV-vis spectroscopy, proving the chemical reversibility of the process. Both chemical reduction and oxidation reactions were also studied by stopped flow experiments but were complete already after the initial mixing time of 1 ms.
The zero-field Mößbauer spectrum of diferric 4(NEt4)2 at 13 K displays one doublet with isomer shift of δ = 0.28 mm s−1 and quadrupole splitting of ΔEQ = 0.90 mm s−1. The Mößbauer spectrum of mixed-valent 4(NEt4)2(CoCp2*) displays two doublets at 12K: one doublet corresponding to the FeII site with δ = 0.62/ΔEQ = 3.09 mm s−1 and one doublet corresponding to the FeIII with δ = 0.35/ΔEQ = 1.20 mm s−1. At 200 K the Mößbauer spectrum shows only one doublet with δ = 0.41/ΔEQ = 1.39 mm s−1 due to fast electron hopping between the two sites (see SI for spectra). The zero-field Mößbauer spectrum of solid 5(NEt4)2 shows two overlapping doublets with δ1 = 0.28/ΔEQ = 0.77 mm s−1 for the all-sulfur coordinated FeS and δ2 = 0.29/ΔEQ = 1.01 mm s−1 for the mixed {SN}-capped FeSN site (Figure 8), whereas mixed-valent 5(NEt4)2(CoCp2*) shows two distinct doublets with δ1 = 0.39/ΔEQ = 1.29 mm s−1 (corresponding to FeIII) and δ2 = 0.66/ΔEQ = 3.13 (corresponding to FeII). In contrast to homoleptic 43−, two distinct doublets with different isomer shift are still visible at 200 K, indicating increased localization of the unpaired electron in the heteroleptic cluster.
Figure 8.
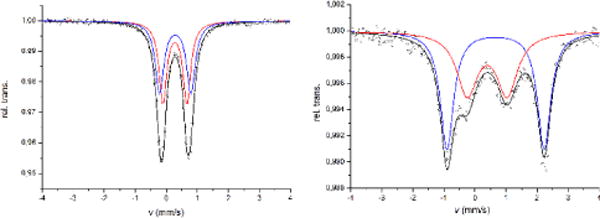
Mößbauer spectra of 5(NEt4)2 at 80 K (left) and 5(NEt4)2(CoCp2*) at 13 K (right). Simulation of the data gave the following parameters: (left) δ1 = 0.28/ΔEQ = 0.77/FWHM = 0.30 (red) and δ2 = 0.29/ΔEQ = 1.01/FWHM = 0.33 mm s−1 (blue); (right) δ1 = 0.39/ΔEQ = 1.29/FWHM = 0.81 (FeIII, red) and δ2 = 0.66/ΔEQ = 3.13/FWHM = 0.52 mm s−1 (FeII, blue).
For fully localized FeIIFeIII clusters a difference in isomer shifts of Δδ = 0.4 mm s−1 has been predicted for FeS4 sites based on an empirical correlation.36 The partial mixing of FeII and FeIII characters for 43− and 53− appears to be similar (Δδ = 0.27 mm s−1) and stronger than the mixing in the reduced Rieske model 23− (Δδ = 0.36 mm s−1 at 6 K) but much less pronounced than in a related symmetric mixed-valent [2Fe-2S] model cluster with two {N2} capping ligands, 3c3− (Δδ = 0.22 mm s−1 at 4.2 K).13 Isomer shift differences Δδ in the range 0.42−0.48 mm s−1 have been reported for biological Rieske clusters that are assumed to feature full valence localization, while Δδ = 0.36 mm s−1 has been observed for the reduced mitoNEET cluster.25
Protonation of mixed valent clusters
Protonation of the mixed-valent clusters 43− and 53− proved to be challenging because of the low stability of the involved species and their limited solubility. Protonation was followed by UV-vis spectroscopy at −25° C. For the homoleptic model 43− addition of 1 equiv of [DMPH]BF4 leads to a broadening of the band at 558 nm along with a blue shift of about 5 nm and the formation of a shoulder at 510 nm. Subsequent addition of base reverses those changes accompanied by an overall decrease of intensity probably due to the instability of the protonated species. In the heteroleptic mitoNEET model 53− similar spectroscopic changes are observed (see SI for spectra). However, in both cases the changes in visible absorption spectra upon protonation are only minor.
The effect of protonation on the redox potential of both model systems was studied by cyclic voltammetry. For the homoleptic cluster 42− in 0.25 M NBu4PF6, addition of 1 equiv of DMPH+ leads to a surprisingly small anodic shift of the cathodic peak potential by about +65 mV corresponding to E1/2 = −1.342 for 4H−. Addition of DBU restores the initial position of the redox wave although its intensity can not be completely recovered due to partial precipitation of a decomposition product. Addition of 0.9 equiv of DMPH+ to a solution of the heteroleptic cluster 52− leads to emergence of a new cathodic peak at −1.437 V which is anodically shifted by +240 mV in comparison with the cathodic peak potential of parent 52− (corresponding to E1/2 = −1.407 for 5H−), as expected for protonation of a benzimidazole-N. Similar shifts of E1/2 upon single protonation have been observed for Rieske model 22− (+230 mV)9 and the all-N ligated homoleptic 3b2− (+245 mV).11 In the case of 52−, however, subsequent addition of base does not lead back completely to the initial cyclic voltammogram. Both experiments suggest only partial reversibility of the protonation of the mixed-valent species, along with some decomposition.
Comparability to biological systems is limited as those are usually studied in water where a network of hydrogen bonds from solvent and protein environment tunes the cluster’s redox properties. In wild type mitoNEET, the redox potential changes from +40 mV vs SHE to −160 mV vs SHE when changing the pH from 6 to 11, the pKa values being pKa,red > 11.5 and pKa,ox = 6.7 in the mixed-valent and all ferric forms, respectively, from which a bond dissociation free energy (BDFE) of the imidazole N-H bond of 286 kJ/mol has been derived.12,18 The importance of the surrounding environment within the protein is reflected by a change in the pKa of the coordinating histidine of about 3 units when the neighboring lysine residue is replaced by a non-hydrogen bonding residue.22 The heteroleptic complex 52− presented in this work shows a similar dependence of its redox potential upon protonation and thus appears to be a promising model to investigate the reactivity of the [2Fe-2S] cluster in mitoNEET proteins. The cause of the much smaller shift of E1/2 upon protonation in case of 42− remains unclear.
Reaction with TEMPO and thermodynamic square scheme
Full square schemes for proton and electron transfer in MeCN solution could be established for both clusters based on the thermodynamic parameters derived above (Schemes 1 and S1). BDFEs of the N-H bonds calculated from those data are 232 ± 4 kJ mol−1 for homoleptic 4H2− and 230 ± 4 kJ mol−1 for the heteroleptic model 5H2−. These values are lower than the N-H BDFEs of 252 ± 2 kJ mol−1 reported for Rieske model 2H2− and 253 ± 2 kJ mol−1 for homoleptic 3bH2−, both featuring bis(benzimidazolato) ligands as proton acceptor sites, instead of the thiolato/benzimidazolato ligands in the present complexes (Table 3). The pKa values of the protonated mixed-valent species were then calculated according to Hess’ law giving pKa = 24.1 for 4H2− and pKa = 27.7 for 5H2−. The higher pKa value in the heteroleptic cluster is possibly due to increased valence localization and hence a more pronounced ferrous character at the protonation site.
Scheme 1.
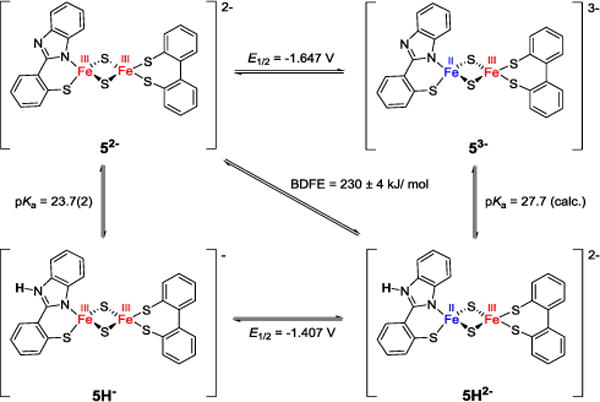
Square scheme summarizing thermodynamic parameters for mitoNEET model 52− in MeCN, potentials referenced vs Fc/Fc+.
Table 3.
Thermodynamic parameters for the reaction of 4H2− and 5H2− and previously reported Rieske models 2H2− and 3bH2− with TEMPO.
| 3bH2− 11 | 2H2− 9 | 4H2− | 5H2− | |
|---|---|---|---|---|
| ΔH‡ [kJ/mol] | 6.7 ±1.3 | 8.7 ±1.0 | 14.2 ± 3.3 | 17.6 ± 3.0 |
| ΔS‡ [J/mol K] | −159 ± 10 | −120 ±5 | −132 ± 12 | −143 ± 11 |
| kobs at 293K [M−1 s−1] | 2200 ± 350 | 95000 ± 12000 | 1280 ± 120 | 135 ± 27 |
| BDFE [kJ/mol] | 253 ± 4 | 252 ± 2 | 232 ± 4 | 230 ± 4 |
| ΔG‡ at 293 K [kJ/mol] | 54.0 | 43.8 | 54.3 | 59.8 |
| ΔG0CPET [kJ/mol] | −25.1 | −26.4 | −46.3 | −48.4 |
To investigate PCET reactivity, mixed valent protonated clusters 4H2− and 5H2− were treated with the nitroxyl radical TEMPO. The free energies for the concerted proton and electron transfer reactions, ΔG°CPET, were calculated to be −46.3 kJ mol−1 for the homoleptic and −48.4 kJ mol−1 for the heteroleptic model (for calculations of all thermodynamic parameters see SI),37 which is significantly higher than ΔG°CPET determined for 2H2− and 3bH2− (around −26 kJ mol−1; see Table 3).9,11 To gain mechanistic insight, double mixing stopped flow experiments were performed at varying temperatures under pseudo-first order conditions using different amounts of excess TEMPO. Even though the BDFE and thus the driving forces for the reaction of both clusters are very similar, the heteroleptic mitoNEET model 5H2− reacts much more slowly than the homoleptic analogue 4H2−.
Instead of a concerted pathway, a stepwise process of subsequent electron and proton transfer might also be feasible. To examine this possibility more closely, the single initial steps of such a process were considered. Proton transfer as an initial step would lead to 43− or 53− (and TEMPO•-H+) from 4H2− or 5H2− and TEMPO with ΔG°PT= 161 and 182 kJ mol−1, respectively (see Supporting Information). Since these values are considerably higher than the activation free energies for the reactions of 4H2− and 5H2− with TEMPO determined in this work (ΔG‡ = 54.3 and 59.8 kJ mol−1, respectively), initial proton transfer is not possible. From a similar analysis, initial electron transfer to give 4H− or 5H− and TEMPO− has ΔG°ET values of 58.8 kJ mol−1 and 52.1 kJ mol−1, respectively, which are essentially the same as the ΔG‡ measured for the reaction with TEMPO. Thus initial ET is thermodynamically possible, but unlikely because it would require ΔG°ET to be equal to the ET intrinsic barrier [from Marcus Theory, ΔG‡ = (ΔG°ET + λ)2/4λ]. Values of λ for ET reactions in MeCN are typically larger than 60 kJ mol−1.38,39 The reactions thus likely follow a concerted rather than a stepwise pathway.
These results can be interpreted using again Marcus theory, which was originally developed for electron transfer reactions, but has been shown to be applicable to PCET reactions.37,40,41 Thus the rate of the PCET reaction depends on both the driving force and the reorganization energy. It is surprising that the rate constants are slower for mitoNEET models 4H2− and 5H2− than for the prior Rieske models, since the ΔG0CPET is more favorable. We suggest that this difference lies at least in part in the different reorganization energies. As predicted and evidenced by EPR, in the heteroleptic mitoNEET model the unpaired electron is somewhat more localized on the {SN} ligated iron site, and hence the structural changes of the iron-sulfur core upon removal of an electron are expected to be more severe than in the homoleptic case. This would result in a higher reorganization energy and consequently a lower rate constant, as it has been observed in this work (see kobs entries in Table 3). kobs for the PCET reaction of 4H2− is in a similar range as the one reported for 3bH2−,11 both featuring homoleptic ligation of the [2Fe-2S] core. Comparability with kobs reported for the Rieske model 2H2− is somewhat limited as the ligand in this case was shown to undergo tautomerization upon protonation;9 the very high rate constant was attributed to a small reorganization energy in this case.
Conclusions
In conclusion, we have presented the first structural model mimicking the unique Cys3His coordination of the recently discovered [2Fe-2S] cluster in mitoNEET proteins, which also emulates well the spectroscopic properties of the biological cofactor. Electrochemical properties are shown to depend on the protonation state of the cluster (most likely on the distal N atom of the imidazole type ligand) as is the case in the biological archetype. Thermodynamic parameters for protonation and reduction of both systems 42− and 52− have been determined and full square schemes have been established. We have also examined the ability of the mitoNEET model system and its homoleptic analogue to undergo PCET reactions with TEMPO. Kinetic studies by double mixing stopped flow experiments show that the homoleptic cluster 4H2− reacts about ten times faster than the heteroleptic model system 5H2−. Comparison with thermodynamic parameters of the single ET and PT steps prove that the reaction follows a concerted pathway. Because the driving force for this reaction is very similar in both cases, this difference in rate is attributed to higher reorganization energy in the heteroleptic model, which is slowing down the reaction by about an order of magnitude. This increase in reorganization energy is likely caused by more pronounced electron localization in the reduced form of the heteroleptically ligated cluster. These findings suggest a potential role of the Cys3His ligated [2Fe-2S] cluster of mitoNEET proteins in proton coupled electron transfer. Furthermore, the proposed correlation between PCET rates and reorganizational energies may indicate a strategy how Fe/S proteins gate PCET reactivity.
Supplementary Material
Figure 4.
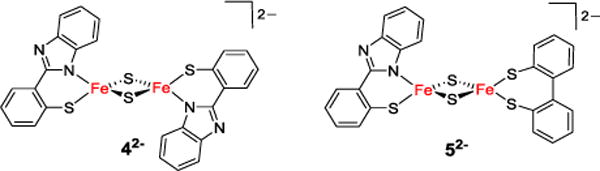
Homoleptic model 42− and mitoNEET model 52− in their diferric state.
Figure 9.
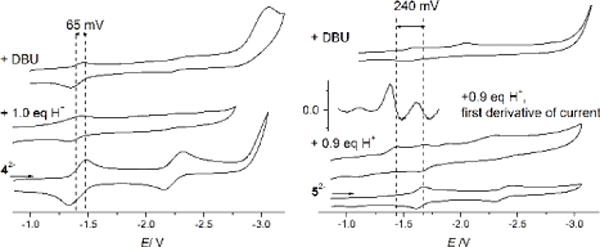
Effect of protonation on cyclic voltammograms of 4(NEt4)2 (left) and 5(NEt4)2 (right); 0.25 M NBu4PF6 at −15°C vs Fc/Fc+ at a scan rate of 400 mV/s. Left: the first reduction of 42− (bottom) is shifted upon addition of 1.0 equiv. of acid (middle); addition of DBU partly restores the initial spectrum (top). Right: the first reduction of 52− (bottom) is shifted upon addition of 0.9 equiv of acid (second from bottom). The initial redox event is still visible in this spectrum since protonation is not complete. The first derivative of this spectrum shows that these two redox events are separated by 240 mV (second from top); addition of DBU does not fully restore the initial voltammogram (top).
Table 2.
EPR Data of Model Complexes 43−, 53− and 23− and mitoNEET Protein
| 43− | 53− | 23− | mitoNEET27 | |
|---|---|---|---|---|
| g1 | 2.010 | 2.005 | 2.017 | 2.005 |
| g2 | 1.932 | 1.932 | 1.934 | 1.937 |
| g3 | 1.882 | 1.875 | 1.854 | 1.895 |
| gav | 1.941 | 1.937 | 1.935 | 1.945 |
Acknowledgments
We would like to thank Dr. Wesley Morris for assistance with stopped-flow measurements. Financial support by the DFG (International Research Training Group 1422 “Metal Sites in Biomolecules: Structures, Regulation and Mechanisms,” and project ME 1313/13-1 in the framework of the SPP 1927 “Iron-Sulfur for Life”) as well as from the U.S. NIH (grant 5R01GM050422 to JMM) is gratefully acknowledged.
Footnotes
Supporting Information. Synthetic procedures, full experimental details, 1H NMR and UV-vis titrations, additional Mößbauer spectra, details of magnetic measurements, details of kinetic investigations and crystallographic details (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Beinert H, Holm RH, Münck E. Science. 1997;277:653. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 2.Bak DW, Elliott SJ. Curr Opin Chem Biol. 2014;19:50. doi: 10.1016/j.cbpa.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 3.Fontecave M. Nat Chem Biol. 2006;2:171. doi: 10.1038/nchembio0406-171. [DOI] [PubMed] [Google Scholar]
- 4.Link TA. Adv Inorg Chem. 1999;47:83. [Google Scholar]
- 5.Ferraro DJ, Gakhar L, Ramaswamy S. Biochem Biophys Res Commun. 2005;338:175. doi: 10.1016/j.bbrc.2005.08.222. [DOI] [PubMed] [Google Scholar]
- 6.Crofts AR. Biochim Biophys Acta, Bioenerg. 2004;1655:77. doi: 10.1016/j.bbabio.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Rao PV, Holm RH. Chem Rev. 2004;104:527. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 8.Ballmann J, Albers A, Dechert S, Bill E, Bothe E, Ryde U, Meyer F. Angew Chem Int Ed. 2008;47:9537. doi: 10.1002/anie.200803418. [DOI] [PubMed] [Google Scholar]
- 9.Albers A, Demeshko S, Dechert S, Saouma CT, Mayer JM, Meyer F. J Am Chem Soc. 2014;136:3946. doi: 10.1021/ja412449v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albers A, Bayer T, Demeshko S, Dechert S, Meyer F. Chem- Eur J. 2013;19:10101. doi: 10.1002/chem.201301760. [DOI] [PubMed] [Google Scholar]
- 11.Saouma CT, Kaminsky W, Mayer JM. J Am Chem Soc. 2012;134:7293. doi: 10.1021/ja3019324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saouma CT, Pinney MM, Mayer JM. Inorg Chem. 2014;53:3153. doi: 10.1021/ic403131p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albers A, Demeshko S, Dechert S, Bill E, Bothe E, Meyer F. Angew Chem, Int Ed. 2011;50:9191. doi: 10.1002/anie.201100727. [DOI] [PubMed] [Google Scholar]
- 14.Albers A, Demeshko S, Pröpper K, Bill E, Meyer F. J Am Chem Soc. 2013;135:1704. doi: 10.1021/ja311563y. [DOI] [PubMed] [Google Scholar]
- 15.Beardwood P, Gibson JF. J Chem Soc, Dalton Trans. 1992:2457. [Google Scholar]
- 16.Wiley SE, Paddock ML, Abresch EC, Gross L, van der Geer P, Nechushtau R, Murphy AN, Jennings PA, Dixon JE. J Biol Chem. 2007;282(33):23745. doi: 10.1074/jbc.C700107200. [DOI] [PubMed] [Google Scholar]
- 17.Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE, Jennings PA. Proc Natl Acad Sci USA. 2007;104:14342. doi: 10.1073/pnas.0707189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bak DW, Zuris JA, Paddock ML, Jennings PA, Elliott AJ. Biochemistry. 2009;48:10193. doi: 10.1021/bi9016445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamir S, Paddock ML, Darah-Yahana-Baram M, Holt SH, Sohn YS, Agranat L, Michaeli D, Stofleth JT, Lipper CH, Morcos F, Cabantchik IZ, Ouchic JN, Jennings PA, Mittler R, Nechustai R. Biochim et Biophys Acta. 2015;1853(6):1294. doi: 10.1016/j.bbamcr.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Lin J, Zhou T, Ye K, Wang J. Proc Natl Acad Sci U S A. 2007;104(37):14640. doi: 10.1073/pnas.0702426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, Lund ET, Mathews WR. Am J Physiol Endocrinol Metab. 2004;286:E252. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- 22.Bak DW, Elliott SJ. Biochemistry. 2013;52:4687. doi: 10.1021/bi400540m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuris JA, Harir Y, Conlan AR, Shvartsman M, Michaeli D, Tamir S, Paddock ML, Onuchic JN, Mittler R, Cabantchik ZI, Jennings PA, Nechushtai R. Proc Natl Acad Sci USA. 2011;108:13047. doi: 10.1073/pnas.1109986108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuris JA, Halim DA, Conlan AR, Abresch EC, Nechushtai R, Paddock ML, Jennings PA. J Am Chem Soc. 2010;132:13120. doi: 10.1021/ja103920k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golinelli-Cohen MP, Lescop E, Mons C, Goncalves S, Clémancey M, Santolini J, Guittet E, Blondin G, Latour JM, Bouton C. J Biol Chem. 2016;291:7583. doi: 10.1074/jbc.M115.711218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan G, Liu D, Pan F, Zhao J, Li T, Ma Y, Shen B, Lyu J. Biochem Biophys Res Com. 2016;470:226. doi: 10.1016/j.bbrc.2016.01.040. [DOI] [PubMed] [Google Scholar]
- 27.Iwasaki T, Samilova RI, Kounosu A, Ohmori D, Dikanov SA. J Am Chem Soc. 2009;131:13659. doi: 10.1021/ja903228w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dicus MM, Conlan A, Nechushtai R, Jennings PA, Paddock ML, Britt RD, Stoll S. J Am Chem Soc. 2010;132:2037. doi: 10.1021/ja909359g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, Scherer PE. Nat Med. 2012;18:1539. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sohn YS, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, Harir Y, Holt SH, Shulaev V, Paddock ML, Hochberg A, Cabanchick IZ, Onuchic JN, Jennings PA, Nechushtai R, Mittler R. Proc Natl Acad Sci USA. 2013;110:14676. doi: 10.1073/pnas.1313198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. J Org Chem. 2005;70:1019. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- 32.Connelly NG, Geiger WE. Chem Rev. 1996;96:877. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 33.Link TA. In: Handbook of Metalloproteins. Messerschmidt A, Huber R, Wieghardt K, Poulos T, editors. Vol. 1. John Wiley & Sons; Chichester, UK: 2001. p. 518. [Google Scholar]
- 34.Boxhammer S, Glaser S, Kühl A, Wagner AK, Schmidt CL. Biometals. 2008;21:459. doi: 10.1007/s10534-008-9134-y. [DOI] [PubMed] [Google Scholar]
- 35.Orio M, Mouesca JM. Inorg Chem. 2008;47:5394. doi: 10.1021/ic701730h. [DOI] [PubMed] [Google Scholar]
- 36.Hoggins JT, Steinfink H. Inorg Chem. 1976;15:1682. [Google Scholar]
- 37.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eberson L. Electron Transfer Reactions in Organic Chemistry. Springer-Verlag; Berlin: 1987. p. 48. [Google Scholar]
- 39.Grampp G, Rasmussen K. Phys Chem Chem Phys. 2002;4:5546. doi: 10.1039/c4cp04581e. [DOI] [PubMed] [Google Scholar]
- 40.Roth JP, Yoder JC, Won TJ, Mayer JM. Science. 2001;294:2524. doi: 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- 41.Mader EA, Larsen AS, Mayer JM. J Am Chem Soc. 2004;126:8066. doi: 10.1021/ja049246k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.