

Abstract
Clostridium difficile is a bacterial pathogen that is the leading cause of nosocomial antibiotic-associated diarrhea and pseudomembranous colitis worldwide. The incidence, severity, mortality and healthcare costs associated with C. difficile infection (CDI) are rising, making C. difficile a major threat to public health. Traditional treatments for CDI involve use of antibiotics such as metronidazole and vancomycin, but disease recurrence occurs in about 30% of patients, highlighting the need for new therapies. The pathogenesis of C. difficile is primarily mediated by the actions of two large clostridial glucosylating toxins, toxin A (TcdA) and toxin B (TcdB). Some strains produce a third toxin, the binary toxin C. difficile transferase, which can also contribute to C. difficile virulence and disease. These toxins act on the colonic epithelium and immune cells and induce a complex cascade of cellular events that result in fluid secretion, inflammation and tissue damage, which are the hallmark features of the disease. In this review, we summarize our current understanding of the structure and mechanism of action of the C. difficile toxins and their role in disease.
Keywords: Clostridium difficile, bacterial toxins, glucosyltransferase, pore formation, inflammation, colitis, intestinal epithelium, actin cytoskeleton
This review summarizes the structures, molecular mechanisms and physiological responses to the three toxins associated with disease symptoms in Clostridium difficile infection.
INTRODUCTION
Clostridium difficile is an anaerobic, spore-forming, Gram-positive bacterium that was first described by Hall and O'Toole (1935). While the bacterium (originally named Bacillus difficile) was identified as part of the normal intestinal flora of healthy new-born infants, Hall and O’Toole noted that the organism was capable of causing disease in animals, likely through the production of soluble exotoxin(s). Clostridium difficile gained recognition as an important human pathogen when it was identified as the etiologic agent of antibiotic-associated pseudomembranous colitis (PMC) (Bartlett et al.1978; George et al.1978). PMC is a severe inflammatory disease of the colon, characterized by the formation of pseudomembranes that are composed of necrotic epithelial cells, fibrin, mucous and leukocytes. Since that discovery, it has become clear that C. difficile can cause a spectrum of clinical conditions in humans, collectively known as C. difficile infections (CDI), which range from mild and possibly recurrent diarrhea to life-threatening complications such as PMC, toxic megacolon and colonic perforation (Martin, Monaghan and Wilcox 2016). Clostridium difficile has become a major healthcare problem in the USA with an estimated half a million infections and 29 000 deaths each year (Lessa et al.2015).
Several molecular typing methods, including PCR ribotyping, restriction endonuclease analysis (REA), multilocus sequence typing and pulsed field gel electrophoresis (PFGE), have been developed for C. difficile classification and epidemiological analyses (Smits et al.2016). Owing to the initial lack of a globally standardized typing method for this genetically heterogeneous species, C. difficile isolates were often referred to by multiple typing designations. For example, PCR ribotype 027 strains that have been associated with outbreaks in many countries are often indicated as REA group BI/PFGE type NAP1/PCR ribotype 027 (BI/NAP1/027) (He et al.2013). While PCR ribotyping has gained widespread acceptance for typing C. difficile and an internationally standardized, high-resolution ribotyping protocol has been recently validated (Fawley et al.2015), more and more whole genome sequences are becoming available as the cost of this technology gets less expensive. Recently, Lawson et al. (2016) proposed that Clostridium difficile should be reclassified as Clostridioides difficile based on phenotypic, chemotaxonomic and phylogenetic analyses. This nomenclature has been adopted by the National Center for Biotechnology Information.
Clostridium difficile transmission occurs via the fecal–oral route, primarily in the form of spores. The spores traverse the acidic pH of the stomach and germinate in the small intestine in response to certain primary bile acids (Sorg and Sonenshein 2008; Giel et al.2010). The metabolically active vegetative cells colonize and infect the colon following antibiotic-induced dysbiosis of the gut microbiota (Theriot and Young 2015; Smits et al.2016). While antibiotic exposure, hospitalization, advanced age and immunocompromised status increase the risk for disease, reports of community-acquired infections in otherwise healthy young adults who were not exposed to prior antibiotics are not uncommon (Khanna and Pardi 2012). Although several virulence factors contribute to C. difficile adherence and colonization, the symptoms of CDI correlate with the production of two exotoxins: toxin A (TcdA) and toxin B (TcdB) (Awad et al.2014). TcdA and TcdB are 308 and 270 kDa proteins, respectively. The toxins belong to the family of large clostridial toxins (LCTs), which are a group of homologous, high molecular weight proteins that further include the lethal and hemorrhagic toxins from C. sordellii (TcsL and TcsH, respectively), α-toxin from C. novyi (Tcnα) and a cytotoxin from C. perfringens (TpeL) (Table 1). The LCTs are glycosyltransferases that inactivate specific Rho and Ras GTPases, leading to the disruption of host cell function. Some C. difficile strains, including the epidemic PCR ribotypes 027 and 078, produce a third toxin named C. difficile transferase (CDT; or binary toxin). CDT is an actin-specific ADP-ribosyltransferase that is homologous to iota toxin from C. perfringens (Barth et al.2004) and is thought to enhance C. difficile virulence and disease severity. In this review, we focus on the role of these three toxins in mediating the symptoms associated with CDI. We provide a brief introduction to the genetics and expression of these toxins and their roles in human disease and animal infection models. We then describe the toxins at a molecular and mechanistic level in an effort to relate cellular functions to physiological outcomes. Finally, we provide a brief overview of some toxin-based therapeutic strategies and conclude with key questions for future study.
Table 1.
Sequence comparisons of the large glucosylating toxins.
| TcdA | TcdB | TcsH | TcsL | Tcnα | ||
|---|---|---|---|---|---|---|
| C. difficile | TcdA | |||||
| TcdB | 48 (68) | |||||
| C. sordellii | TcsH | 78 (88) | 48 (68) | |||
| TcsL | 48 (68) | 76 (87) | 49 (69) | |||
| C. novyi | Tcnα | 31 (51) | 31 (51) | 32 (52) | 31 (51) | |
| C. perfringens | TpeL | 42 (62) | 40 (61) | 43 (62) | 41 (62) | 33 (54) |
Values represent the percent identities of amino acids (in parentheses are the percent homologies)
OVERVIEW OF TOXIN GENETICS, EXPRESSION AND SECRETION
The genes encoding TcdA (tcdA) and TcdB (tcdB) are located within a 19.6-kb chromosomal region termed the pathogenicity locus (PaLoc) (Fig. 1A) (Hammond and Johnson 1995; Braun et al.1996). In non-toxigenic strains, the PaLoc is replaced by a 75–115 nucleotide non-coding sequence or a 7.2-kb sequence of unknown function (Braun et al.1996; Elliott et al.2009; Dingle et al.2011; Monot et al.2015). Non-toxigenic Clostridium difficile strains, however, can acquire the PaLoc from toxigenic strains through horizontal gene transfer, resulting in the conversion of non-toxigenic strains to toxin producers (Brouwer et al.2013). Changes in the toxin coding region within the PaLoc, including single nucleotide polymorphisms, insertions or deletions, have been used to classify naturally occurring C. difficile isolates (Rupnik et al.1998). Based on a comparison to a reference strain VPI10463, 34 C. difficile toxinotypes have been defined, highlighting the heterogeneity of the toxin coding region among C. difficile isolates (Rupnik and Janezic 2016).
Figure. 1.
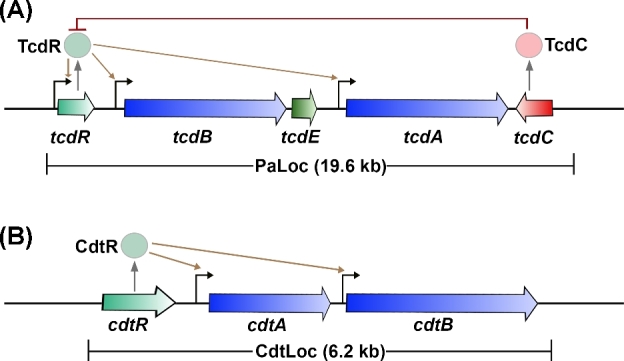
Organization of toxin genes. (A) Schematic representation of the pathogenicity locus (PaLoc). Toxin-encoding genes, tcdA and tcdB, are indicated by blue arrows; regulatory genes are shown in light green (tcdR; positive) or red (tcdC; negative); and holin-encoding gene tcdE is shown in dark green. The direction of the arrows reflects the direction of transcription. TcdR positively regulates its own expression as well as the expression of tcdA and tcdB (indicated by brown arrows). TcdC is an anti-sigma factor that negatively regulates toxin expression by interfering with TcdR function. TcdE is involved in the secretion of toxins. (B) Schematic representation of the binary toxin locus (CdtLoc). CDT-encoding genes, cdtA and cdtB, are shown in blue. The regulatory gene cdtR is shown in light green. CdtR positively regulates the transcription of cdtA and cdtB.
In addition to the toxins, the PaLoc in most pathogenic strains encodes three proteins, TcdR, TcdC and TcdE, which are thought to regulate toxin production and secretion (Fig. 1A) (Bouillaut et al.2015; Monot et al.2015; Smits et al.2016). TcdR is a member of the extracytoplasmic function family of alternative sigma factors and plays a critical role in activating the expression of tcdA and tcdB (Moncrief, Barroso and Wilkins 1997; Mani and Dupuy 2001). Additionally, TcdR positively regulates its own expression (Mani et al.2002). While there have been conflicting reports on the role of TcdC in toxin production, several studies suggest that TcdC functions as an anti-sigma factor that negatively regulates toxin expression (Matamouros, England and Dupuy 2007; Carter et al.2011). The role of TcdE has also been controversial (Govind and Dupuy 2012; Olling et al.2012). The protein shares homology with the bacteriophage holin proteins, which are involved in the release of progeny phages from the host bacterium (Tan, Wee and Song 2001). TcdA and TcdB do not possess any recognizable secretion signal, and toxin export does not require bacterial cell lysis (Mukherjee et al.2002). These observations have led to the suggestion that the toxins might be exported from the bacterial cell by a non-classic secretion pathway involving holin proteins. Several published studies now support this hypothesis (Govind and Dupuy 2012; Govind, Fitzwater and Nichols 2015; Monot et al.2015).
Clostridium difficile cells grown in rich media typically express TcdA and TcdB during the stationary phase (Hundsberger et al.1997; Dupuy and Sonenshein 1998; Darkoh et al.2015). The toxin expression has been reported to be influenced by several environmental stimuli, including temperature (Karlsson et al.2003), subinhibitory concentrations of certain antibiotics (Nakamura et al.1982; Freeman et al.2007; Chilton et al.2012; Aldape et al.2013), quorum signaling (Darkoh et al.2015), short-chain fatty acids such as butyric acid (Karlsson et al.2000), the presence of a rapidly metabolizable carbon source (Dupuy and Sonenshein 1998) and certain amino acids (Karasawa et al.1997; Karlsson, Burman and Akerlund 1999; Karlsson et al.2000). The presence of a rapidly metabolizable carbon source such as glucose in the local environment of the bacterium inhibits toxin production via the carbon catabolite control protein A (CcpA) (Antunes et al.2012). Branched chain amino acids inhibit toxin production via the global transcriptional regulator CodY (Dineen et al.2007; Bouillaut et al.2015). Finally, factors involved in the regulation of motility and sporulation have also been reported to modulate the production of TcdA and TcdB (Underwood et al.2009; Saujet et al.2011; Aubry et al.2012; El Meouche et al.2013; Mackin et al.2013; Martin et al.2013; McKee et al.2013; Edwards, Tamayo and McBride 2016).
The binary toxin CDT, produced by some C. difficile strains, is encoded by two genes, cdtA and cdtB, which are located on a 6.2-kb chromosomal region (distinct from the PaLoc) named the Cdt locus or CdtLoc (Fig. 1B) (Perelle et al.1997; Carter et al.2007). CDT-negative toxigenic strains typically contain a 2-kb deletion within the CdtLoc (Stare, Delmee and Rupnik 2007). The CdtLoc also contains a third gene, cdtR, which encodes an orphan LytTR family response regulator (Carter et al.2007). CdtR positively regulates CDT production and, in epidemic ribotype 027 strains, it also upregulates TcdA and TcdB production (Carter et al.2007; Lyon et al.2016). The environmental signals that regulate CDT production and the mechanism of toxin secretion are not yet known.
ROLE OF TcdA, TcdB AND CDT TOXINS IN DISEASE
The individual role and relative importance of TcdA and TcdB in disease pathogenesis has been a topic of active investigation. TcdA was initially thought to be the key virulence factor in Clostridium difficile pathogenesis based on animal studies performed using purified toxins. The addition of TcdA to rabbit ileal loops and colon recapitulated the hallmark features of CDI including inflammation, increased mucosal permeability, fluid secretion and tissue damage (Lyerly et al.1982; Mitchell et al.1986). TcdB had no effect in these studies. Similarly, when given intragastrically to hamsters and mice, TcdA caused inflammation, diarrhea and eventual death, whereas TcdB caused no symptoms in these animals (Lyerly et al.1985). TcdB was capable of causing death in hamsters, however, if prior intestinal damage was present or if sublethal doses of TcdA were co-administered (Lyerly et al.1985). These findings suggested that TcdA and TcdB might act synergistically. It was proposed that TcdA acts first and disrupts epithelial integrity, which then allows TcdB to enter and mediate toxic effects within the host. Additional evidence supporting the importance of TcdA in disease pathogenesis comes from studies showing that passive immunization with antibodies against TcdA and active immunization with TcdA toxoids or peptides provided protection against CDI in hamsters (Kim, Iaconis and Rolfe 1987; Lyerly et al.1990; Babcock et al.2006). Furthermore, a strong humoral immune response against TcdA has been shown to correlate with reduced disease severity and recurrence in humans (Warny et al.1994; Kyne et al.2000, 2001).
The importance of TcdA in CDI has been questioned following the detection of clinically significant C. difficile strains that produce TcdB, but not TcdA (A−B+) (Drudy, Fanning and Kyne 2007; King, Mackin and Lyras 2015). In humans, these pathogenic A−B+ strains cause the same spectrum of clinical illness that is associated with A+B+C. difficile strains, ranging from mild diarrhea to the more severe outcomes such as PMC and death (Drudy, Fanning and Kyne 2007). Interestingly, the majority of the A−B+ strains produce a modified form of TcdB, whose enzymatic domain shares homology and GTPase substrate specificity with TcsL of C. sordellii (Chaves-Olarte et al.1999). It has been proposed that this variant TcdB, which, like TcdA, is able to modify Ras GTPases, might be able to carry out TcdA-specific glucosylation events in the absence of TcdA (Chaves-Olarte et al.2003). The observation that A−B+ strains are virulent in infected individuals indicates that TcdB is sufficient for pathology in humans. Consistent with this, TcdB has been shown to disrupt epithelial integrity and cause tissue damage in human colon explants and in a chimeric mouse model where human intestinal xenografts were transplanted into immunodeficient mice (Riegler et al.1995; Savidge et al.2003). In the xenograft model, challenge with either TcdA or TcdB elicited the hallmark features of CDI such as increased mucosal permeability and fluid secretion, cytokine production, neutrophil recruitment and tissue damage (Savidge et al.2003). Furthermore, recent phase III clinical trials show that a monoclonal antibody that neutralizes TcdB, bezlotoxumab, can reduce CDI recurrence in human patients (Wilcox et al.2017). Overall, these studies indicate that TcdB plays an important role in C. difficile pathogenesis in humans.
The roles of TcdA and TcdB in disease have also been investigated by using isogenic C. difficile strains with defined toxin deletions in animal infection studies. In the first two studies, clindamycin-treated hamsters were infected with isogenic derivatives of C. difficile strain 630, a low toxin producing clinical isolate. In the first study, the wild-type strain (expressing both toxins) and mutants producing only TcdB were virulent and caused death in hamsters, but mutants producing only TcdA did not cause death in 80% of the infected animals (Lyras et al.2009). These findings suggested that TcdB was the major virulence factor of C. difficile. The second study supported the importance of TcdB in C. difficile virulence by showing that a mutant producing only TcdB was comparable to wild type in its ability to cause fulminant disease and death in hamsters (Kuehne et al.2010). However, in contrast to the earlier study, an isogenic mutant producing only TcdA also resulted in disease and death in hamsters, although the time course of death was delayed compared to wild-type and TcdA−TcdB+ strains (Kuehne et al.2010). This study also showed that an isogenic double mutant that did not produce TcdA and TcdB was avirulent in hamsters, consistent with the observation that naturally occurring TcdA−TcdB−C. difficile strains are typically non-pathogenic in humans. Similar results were obtained in another study performed by the same group, which used isogenic mutants generated from an epidemic PCR ribotype 027 strain, R20291 (Kuehne et al.2014). The first three studies utilized the hamster model of CDI and reported only survival or death of the infected animals. A fourth study used both mouse and hamster models of CDI, and performed detailed analyses of the tissue pathology and the host responses following infection (Carter et al.2015). The wild-type and isogenic single and double toxin knockout strains used in this study were generated from another epidemic PCR ribotype 027 strain, M7404. Results from this study showed that both TcdA and TcdB were capable of inducing host innate immune and proinflammatory responses, but TcdB was the driver of fulminant disease (Carter et al.2015). Strains expressing TcdB (wild type and TcdA−TcdB+) caused significant weightloss and severe systemic disease in both the mouse and hamster models of infection. These findings are consistent with previous observations that purified TcdB causes cardiovascular damage and systemic disease in a zebrafish intoxication model (Hamm, Voth and Ballard 2006), and that only anti-TcdB antibodies prevent systemic disease in piglets infected with C. difficile (Steele et al.2013). In sum, the infection and intoxication studies show that while both TcdA and TcdB play a role in most infections, TcdB may be more important in the severe aspects of the disease.
The role of CDT toxin in disease pathogenesis has until recently remained largely unexplored. Over the last decade, CDT-expressing strains have become increasingly prevalent in the hospital setting (Gerding et al.2014). Strains that produce CDT but not TcdA and TcdB (TcdA−TcdB−CDT+) have been isolated from a few symptomatic patients, but the contribution of CDT to disease is unclear (Geric et al.2003; Androga et al.2015; Eckert et al.2015). Several studies have reported that the production of CDT in addition to TcdA and TcdB by C. difficile is associated with severe disease, higher mortality and an elevated risk of recurrence in humans, suggesting that CDT may play an important role in disease pathogenesis (Stubbs et al.2000; McEllistrem et al.2005; Barbut et al.2007; Bacci et al.2011; Stewart, Berg and Hegarty 2013). Recent in vitro and in vivo studies have shed light on the mechanisms by which CDT may contribute to or enhance C. difficile virulence. Rabbit ileal loops inoculated with supernatants from TcdA−TcdB−CDT+ clinical isolates showed significant fluid accumulation, suggesting that CDT could be enterotoxic (Geric et al.2006). Clindamycin-treated hamsters infected with strains producing CDT, but not TcdA and TcdB, showed some hemorrhage and inflammation in their small intestines but did not develop diarrhea or other CDI symptoms, suggesting that CDT alone may not be sufficient to cause C. difficile disease (Geric et al.2006; Kuehne et al.2014). Kuehne et al. then investigated whether CDT could influence the virulence of C. difficile when TcdA or TcdB is present. They found that a mutant strain that carried both TcdA and CDT (TcdA+TcdB−CDT+), generated from a PCR ribotype 027 isolate (R20291), was more virulent in hamsters than an isogenic TcdA+TcdB−CDT− derivative, suggesting that CDT may act in concert with TcdA to enhance pathogen virulence and disease (Kuehne et al.2014). In support of this hypothesis, purified CDT was found to activate nuclear factor-κB (NF-κB) and induce robust proinflammatory cytokine production in conjunction with TcdA and TcdB in innate immune cells (Cowardin et al.2016a). Infection studies in mice show that CDT can also enhance C. difficile virulence by suppressing a protective host eosinophilic response (Cowardin et al.2016a), or by promoting adherence and colonization of the bacteria (Schwan et al.2009, 2014). Collectively, these studies highlight the emerging understanding of the roles CDT may play in disease.
STRUCTURE AND MECHANISM OF ACTION OF TcdA AND TcdB
TcdA and TcdB are broadly classified as AB toxins, wherein a B subunit is involved in the delivery of an enzymatic A subunit into the cytosol of a target cell. The enzymatic A subunit of TcdA and TcdB is an N-terminal glucosyltransferase domain (GTD) that inactivates members of the Rho family of small GTPases by glucosylation. The B subunit is composed of three regions: a combined repetitive oligopeptides (CROPS) domain, a delivery/pore-forming domain and an autoprocessing domain (APD) (Fig. 2A). The homologous proteins intoxicate host cells through a multistep mechanism that involves (i) receptor binding and endocytosis, (ii) pore formation and translocation of the GTD across the endosomal membrane, (iii) autoprocessing and release of GTD into the cytosol, and (iv) glucosylation of host GTPases (Fig. 2B). These steps are discussed in more detail below.
Figure. 2.

TcdA and TcdB primary structure and mechanism of action. (A) TcdA and TcdB are organized into four functional domains: the glycosyltransferase domain (GTD; pink), the autoprocessing domain (APD; green), the delivery or pore-forming domain (blue) and the combined repetitive oligopeptides domain (CROPS; yellow). (B) The four functional domains contribute to a multistep mechanism of intoxication. TcdA and TcdB bind different cell surface proteins or sugars on the colonic epithelium (step 1) and are internalized by distinct endocytic pathways (step 2). The toxins reach acidified endosomes (step 3) and the low pH triggers a conformational change in the toxin delivery domain, resulting in pore formation and translocation of the GTD (and likely the APD) into the cytosol (step 4). Inositol hexakisphosphate (InsP6) binds and activates the APD, resulting in the cleavage and release of the GTD (step 5). The GTD inactivates Rho family proteins by transferring the glucose moiety (orange squares) from UDP-glucose to the switch I region of the GTPase (step 6). Glucosylation disrupts GTPase signaling and leads to cytopathic ‘rounding’ effects and apoptotic cell death.
Cellular receptors and receptor-binding domains
Historically, receptor binding has been associated with the CROPS domains located at the C-termini of TcdA and TcdB. The CROPS region contains multiple 19–24 amino acid short repeats (SRs) interspersed with four to seven long repeats (LRs) of 31 residues (von Eichel-Streiber and Sauerborn 1990; von Eichel-Streiber et al.1992). While a specific start site for the CROPS is difficult to define, a recent analysis from the Melnyk lab suggests that the CROPS from TcdB strain VPI10463 spans residues 1814–2366 (Gupta et al.2017). The corresponding start site in TcdA is residue 1812, although the TcdA CROPS is longer in that it spans residues 1812–2710 (Fig. 3A). By this analysis, the TcdA CROPS domain contains 33 SRs and 7 LRs, and TcdB CROPS contains 21 SRs and 4 LRs. In 2005, a crystal structure of a fragment of TcdA CROPS (residues 2582–2709) comprising four SRs and one LR was determined (Ho et al.2005). The five repeats form a beta-solenoid fold with each repeat consisting of a beta-hairpin followed by a loop of 7–10 amino acids in SRs and 18 amino acids in LRs. The beta hairpins of adjacent SRs contact each other but are rotated by 120°, resulting in a screw-like structure. In contrast, hydrogen-bonding interactions formed by the LR and the preceding SR lead to a 90° screw-axis transformation. Using this information, the Ng group constructed models of the complete TcdA and TcdB CROPS domains, which predicted an extended S-shaped structure for TcdA CROPS (Fig. 3B) and a horseshoe-shaped structure for the shorter TcdB CROPS domain. Tertiary structures predicted by this model were later confirmed by electron microscopy studies of the holotoxins (Fig. 3C) (Pruitt et al.2010).
Figure. 3.

Structure of the CROPS domain. (A) The CROPS domains of TcdA and TcdB consist of a series of short repeat (SR, yellow) sequences with interspersed long repeat (LR, purple) sequences. (B) A model of the full TcdA CROPS based on a fragment structure (2F6E) is corroborated by (C) negative stain electron microscopy images of TcdA (left) and TcdB (right). (D) The crystal structure of a CROPS fragment from the TcdA C-terminus (2G7C) shows how trisaccharides (orange carbons) bind at the vertices created at the intersection of an SR and LR.
The idea that the CROPS domain can contribute to receptor binding came from studies showing that the TcdA CROPS can bind carbohydrates present in mammalian cell surface glycoconjugates (Krivan et al.1986; von Eichel-Streiber and Sauerborn 1990; Tucker and Wilkins 1991; Pothoulakis et al.1996; Teneberg et al.1996; Greco et al.2006; Dingle et al.2008; El-Hawiet et al.2011). TcdA can bind to the human I, X and Y blood antigens as well as a human glycosphingolipid, all of which have a core β-Gal-(1,4)-β-GlcNAc structure (Tucker and Wilkins 1991; Teneberg et al.1996). TcdA was also shown to bind α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAc, which is not present on human cells (Krivan et al.1986; Tucker and Wilkins 1991). The crystal structure of a derivative of this trisaccharide in complex with a fragment of TcdA CROPS revealed that the sugar binding occurs at the junctions formed between LRs and SRs (Fig. 3D) (Greco et al.2006). TcdA, therefore, has seven putative sugar-binding sites, an observation that suggests a model wherein the toxin can form multivalent, high-avidity interactions with glycosylated receptors on the host cell. Whether TcdA binds multiple glycans simultaneously on host cells and whether such high-avidity interactions are important for toxin binding are not yet known. Additional evidence supporting a role for the TcdA CROPS domain in receptor binding includes observations that (i) the isolated CROPS domain from TcdA can bind to host cells (Frisch et al.2003; Olling et al.2011), (ii) excess TcdA CROPS competes with holotoxin in cell binding and cytopathic assays (Sauerborn, Leukel and von Eichel-Streiber 1997; Frisch et al.2003; Olling et al.2011), and (iii) the TcdA CROPS domain is highly immunogenic, and antibodies against this domain can block TcdA binding to cells and neutralize toxicity (Lyerly et al.1986; Sauerborn, Leukel and von Eichel-Streiber 1997; Babcock et al.2006; Hussack et al.2011; Leuzzi et al.2013; Hernandez et al.2017; Kroh et al.2017).
Two cell surface proteins have been implicated as receptors for TcdA. The first is sucrase-isomaltase (SI), which is a glycoprotein located in the brush border of small intestines. SI was shown to mediate the binding of TcdA to rabbit ileum (Pothoulakis et al.1996). Treatment with alpha-galactosidase inhibited binding of TcdA to SI, indicating that the toxin binds glycosyl modification(s) on the protein. SI, however, is not expressed in many cells and tissues that are sensitive to TcdA, including the human colonic epithelium (Pothoulakis et al.1996; Na et al.2008). A subsequent study performed by the same group identified glycoprotein 96 (gp96), a member of the heat shock protein family, as a binding partner for TcdA in human colonocytes (Na et al.2008). However, cells lacking gp96 are only partially resistant to TcdA intoxication, suggesting that TcdA binds additional receptor structures (Na et al.2008). Interestingly, gp96 is predicted to have five N-linked glycosylation sites but the identities of the glycan moieties are not known. It is possible that TcdA binds the sugar moieties on gp96 rather than the protein itself, but this needs to be investigated.
Unlike the CROPS domain of TcdA, evidence for carbohydrate binding by TcdB CROPS is limited to one study conducted with an electrospray mass spectrometry binding assay (Dingle et al.2008). However, a role for TcdB CROPS in receptor binding is supported by the observation that bezlotoxumab, a TcdB-neutralizing antibody targeting the CROPS domain, blocks toxin binding to the host cell (Orth et al.2014). In line with this, a recent study identified chondroitin sulfate proteoglycan 4 (CSPG4) as a receptor for TcdB (Yuan et al.2015), and the binding interaction was mapped to the N-terminus of the CROPS domain (Yuan et al.2015; Tao et al.2016). In the initial study that identified CSPG4 as a receptor, the Wei group showed that TcdB1500-2366, but not TcdB1852-2366, was able to bind CSPG4 (Yuan et al.2015). A more recent study from the Dong group noted that while full-length TcdB binds CSPG4, TcdB1-1830 does not (Tao et al.2016). Taken together, these studies suggest that the 1831–1851 region within the CROPS domain is important for TcdB binding to CSPG4. A role for the N-terminus of CROPS in CSPG4 binding was recently confirmed by a study performed by the Melnyk group (Gupta et al.2017). This study also showed that bezlotoxumab blocks binding of CSPG4 to TcdB, suggesting a mechanism of neutralization that involves direct receptor blockade. It is important to note, however, that CSPG4 knockout cells are sensitive to high concentrations of TcdB, and NG2 (the rodent homolog of CSPG4) knockout mice succumb to disease induced by TcdB (Yuan et al.2015), highlighting the existence of additional receptors for TcdB.
While the CROPS domain has historically been dubbed the receptor-binding domain, several recent studies have demonstrated that the receptor-binding function is not limited to this region of the toxin (Barroso et al.1994; Genisyuerek et al.2011; Olling et al.2011; LaFrance et al.2015; Manse and Baldwin 2015; Tao et al.2016). Truncated toxins (TcdA1-1849, TcdA1-1874, TcdB1-1811 and TcdB1-1830) lacking most or all of the CROPS domain can still bind, enter and perturb host cellular function (Olling et al.2011; Schorch et al.2014; Tao et al.2016). It appears that the CROPS domain contributes to but is not essential for host cell binding. Recently, Lambert and Baldwin (2016) reported that the region comprising residues 1361–1874 in TcdA is capable of binding and entering the host cell. Interestingly, a study by Olling et al. (2011) observed that the presence of TcdA holotoxin does not affect the binding of TcdA1-1874 to cells in competition assays. This finding implies that the full-length TcdA and the truncated toxin lacking the majority of the CROPS domain do not bind the same cellular receptors. It is possible that, in the presence of the extended CROPS domain, the alternate receptor-binding site is not accessible for interaction with a host receptor. Similar to TcdA, additional binding in TcdB may be mediated by the 300 to 350 residues preceding the CROPS domain. Studies using truncated TcdB toxins show that TcdB1-1500 and TcdB1-1529 are unable to induce cytopathic effects in cells, but the first 1500–1550 residues of TcdB are sufficient for intoxication when tethered with the diphtheria toxin receptor-binding domain (Barroso et al.1994; Genisyuerek et al.2011).
After the identification of CSPG4 as a receptor for TcdB, two other reports were published showing that NECTIN3 (also termed poliovirus receptor-like protein (PVRL3)) and frizzled proteins 1, 2 and 7 function as colonic epithelial receptors for TcdB (LaFrance et al.2015; Tao et al.2016). Both NECTIN3 and frizzled proteins bind TcdB outside the CROPS region (LaFrance et al.2015; Tao et al.2016). It is, however, not known whether NECTIN3 and frizzled proteins bind TcdB at distinct sites or compete for binding to the toxin. NECTIN3 and frizzled proteins are both expressed on the surface of the human colonic epithelium (LaFrance et al.2015; Tao et al.2016), and NECTIN3 has been shown to co-stain with TcdB in tissues resected from a Clostridium difficile-infected patient (LaFrance et al.2015). In contrast, CSPG4 is highly expressed in the intestinal subepithelial myofibroblasts and is not detectable in the surface epithelium (Terada et al.2006; Tao et al.2016). During infection, it is likely that TcdB initially engages NECTIN3 and frizzled proteins to enter and intoxicate the colonic epithelium. Upon damage to the epithelium or loss of tight junctions, the toxin could gain access to CSPG4 in the subepithelial myofibroblasts causing further damage to the mucosa. More work needs to be done to characterize the binding interactions between TcdB and its receptors and to define the role each receptor plays in the context of disease. In particular, frizzled receptors are key players in the Wnt signaling pathway, which regulates self-renewal and proliferation of colonic epithelial cells (Gregorieff and Clevers 2005; MacDonald and He 2012). TcdB binding to frizzled receptors has been shown to block Wnt signaling (Tao et al.2016). The effect of Wnt signaling inhibition on toxin-induced pathology during infection is another important area for future study.
Cellular uptake of toxins
After binding to their receptors, TcdA and TcdB are endocytosed into the host cell (Fig. 2B). Entry into the host cell is critical for the delivery of the GTD and disruption of host cell function. TcdA and TcdB have been shown to utilize a dynamin-dependent entry mechanism for host cell intoxication. Dynamin is a large GTPase that typically facilitates scission and release of newly formed endocytic structures from the plasma membrane into the cytosol. Interfering with the expression or the function of dynamin GTPases, using genetic or pharmacological approaches, prevents TcdA and TcdB entry and inhibits toxin-induced cellular effects in epithelial cells (Papatheodorou et al.2010; Gerhard et al.2013; Chandrasekaran, Kenworthy and Lacy 2016).
Not all endocytic mechanisms, however, require the activity of dynamin for vesicle release (Doherty and McMahon 2009; Howes, Mayor and Parton 2010). Of the entry pathways that depend on dynamin for scission, only perturbations to the clathrin-mediated endocytic pathway affected TcdB intoxication, indicating that this toxin predominantly uses the clathrin route for entry (Papatheodorou et al.2010; Chandrasekaran, Kenworthy and Lacy 2016). In contrast, TcdA uses a clathrin-independent entry mechanism mediated by PACSIN2 (Chandrasekaran, Kenworthy and Lacy 2016). PACSIN2 (also termed Syndapin II) is an F-BAR domain protein previously shown to be important for caveolae-mediated endocytosis; it contributes to the curvature of caveolae and the recruitment of dynamin for caveolae fission (Hansen, Howard and Nichols 2011; Senju et al.2011). Colocalization studies and analyses of toxin uptake and toxin-induced cytopathic and cytotoxic effects under specific knockdown conditions show that PACSIN2 mediates entry of TcdA independent of the caveolae system (Chandrasekaran, Kenworthy and Lacy 2016). The observation that PACSIN2 can function outside of the caveolae system suggests a novel mechanism of entry, one for which TcdA is now a known cargo. Consequently, TcdA could serve as a valuable cell biological tool in furthering our understanding of this uncharacterized host endocytic process.
Despite their homology, TcdA and TcdB utilize distinct endocytic pathways to intoxicate epithelial cells. The entry mechanism is often directed by the cell surface receptor. As discussed above, TcdA and TcdB bind different cell surface proteins and sugars, which may explain internalization by distinct endocytic pathways. It is also possible that the mechanism of receptor binding could affect the internalization process. For instance, the CROPS domain of TcdA is thought to mediate high-avidity interactions with the host cell surface, which could result in receptor clustering and endocytosis via mechanisms distinct from that of TcdB.
Pore formation
Once the toxins are internalized, they are trafficked to acidified endosomal compartments within the cell (Florin and Thelestam 1983). Toxic cellular effects induced by TcdA and TcdB are inhibited by lysosomotropic agents and depend on the vacuolar H+-ATPase, indicating a requirement for low pH in toxin action (Florin and Thelestam 1983; Florin and Thelestam 1986; Henriques, Florin and Thelestam 1987; Qa’Dan, Spyres and Ballard 2000; Olling et al.2011). Other AB toxins that require low pH for action, such as diphtheria and anthrax toxins, have been shown to undergo conformational changes in an acidic environment that lead to the exposure of hydrophobic regions that subsequently insert into the host membrane and form a pore (Collier and Young 2003; Geny and Popoff 2006). Studies with C. difficile toxins A and B support a similar mode of action for these toxins.
Early evidence for pH-dependent conformational change and pore formation came from studies using TcdB. TcdB exhibited differences in native tryptophan fluorescence and protease susceptibility between neutral and acidic pH conditions, suggesting that the toxin undergoes structural changes at low pH. An increase in fluorescence of the probe 2-(p-toluidinyl) naphthalene-6-sulfonic acid, sodium salt (TNS) was also observed when TcdB was exposed to pH 5.0 or lower, indicating the exposure of hydrophobic regions (Qa’Dan, Spyres and Ballard 2000). In addition to undergoing structural changes at acidic pH, TcdB was shown to form pores in cell membranes and artificial lipid bilayers (Barth et al.2001). TcdA also undergoes conformational changes and forms pores at low pH (Giesemann et al.2006; Pruitt et al.2010), but unlike TcdB, pore formation by TcdA requires cholesterol (Giesemann et al.2006).
Pore formation and translocation are thought to be mediated by the central delivery domain (Fig. 2A). A crystallographic structure of TcdA4-1802 was recently published, revealing a structurally unique delivery domain (Chumbler et al.2016b). The domain begins after a three-helix bundle (residues 767–841) at the GTD-APD interface, and consists of a small globular subdomain (residues 850–1025), an extended ‘hydrophobic helical stretch’ containing four α-helices (1026–1135) and a β-scaffold (1136–1802) which ends at the base of the APD (Fig. 4A). Placement of the TcdA4-1809 structure into the EM maps of TcdA holotoxin at neutral and low pH shows conformational flexibility at the junction between the APD, delivery domain and the CROPs region (Fig. 4B and C).
Figure. 4.
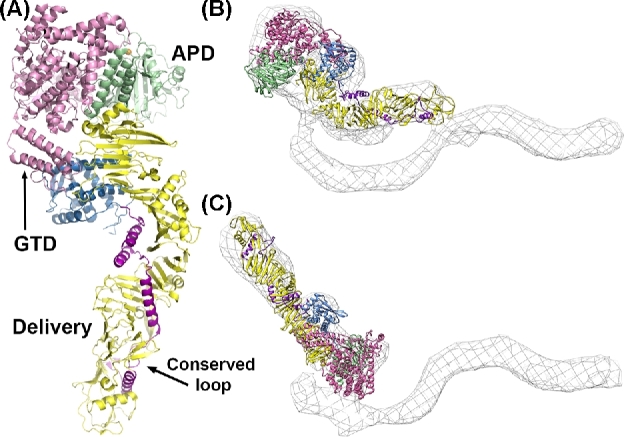
TcdA structure. (A) The crystal structure of residues 4–1802 from TcdA (4R04) with the glucosyltransferase domain in pink and the autoprocessing domain in light green with its zinc atom depicted as an orange sphere. The delivery domain begins with a globular subdomain (sky blue), followed by an extended stretch of four hydrophobic α-helices (magenta). The α-helical stretch is scaffolded by an extended array of β-sheets (yellow). (B) The TcdA4-1802 structure docked into a 3D structure of the TcdA holotoxin obtained by negative stain electron microscopy (EM) at neutral pH. (C) The TcdA4-1802 structure docked into a 3D structure of the TcdA holotoxin obtained by negative stain EM at acidic pH indicates flexibility around the junction with the C-terminal CROPS domain.
The structure of the pore and the mechanism of pore formation by TcdA and TcdB have not yet been determined and remain a priority in the field. The delivery domain of TcdA and TcdB contain hydrophobic sequences (958–1130 in TcdA and 956–1128 in TcdB) that have been predicted to insert into the endosomal membrane with acidic pH von Eichel-Streiber et al.1992 (Mol Gen Genet). Mutational studies have shown that residues within this hydrophobic region, comprising the globular subdomain and four α-helices, are important for TcdA and TcdB pore formation (Genisyuerek et al.2011; Zhang et al.2014).
In the crystal structure of the TcdA delivery domain, the hydrophobic helices appear to wrap around the extended β-sheet structures, which could help maintain solubility while keeping them in a readily accessible conformation for subsequent membrane insertion. Notably, the hydrophobic helical stretch contains a surface loop that is strictly conserved across the LCTs (Fig. 4A). In both TcdA and TcdB, residues within this conserved loop have been shown to be critical for pore formation and cytotoxicity (Zhang et al.2014; Chumbler et al.2016b). These findings suggest that targeting the conserved surface loop with antibodies or small molecules could provide a generalizable strategy for blocking the toxicity of the LCTs.
Translocation and autoproteolysis
Although TcdA and TcdB have been shown to form pores in cellular membranes, how these large, single polypeptide toxins deliver their effector domains to the host cytosol is not understood. The enzymatic domains of other pore-forming AB type toxins, such as diphtheria and anthrax toxin, have been shown to unfold at low pH, and the unfolding is thought be important for the translocation of these domains across the pore (Collier and Young 2003).
In 2003, Pfiefer and colleagues demonstrated that TcdB is proteolytically processed within the host cell, and only the N-terminal GTD domain was released into the cytosol upon translocation. Imaging and fractionation assays showed that the remainder of the toxin localized to endosomes (Pfeifer et al.2003). The cleavage occurs after a conserved leucine residue (542 in TcdA and 543 in TcdB), and results in the release of the GTD into the cytosol (Rupnik et al.2005; Kreimeyer et al.2011). Rupnik et al. also observed that the cleavage reaction in vitro occurred at neutral pH and required the addition of host cell cytosol. Subsequently, Reineke et al. (2007) demonstrated that protein-free cytosolic extracts also induced toxin cleavage, and they identified inositol hexakisphosphate (InsP6) as the inducer of this autocatalytic cleavage event. InsP6 is a highly charged molecule that is abundant (10–60 μM) within mammalian cells (Irvine and Schell 2001). In vitro experiments show efficient autoprocessing of toxins at InsP6 concentrations of 1–10 μM (Egerer et al.2007; Pruitt et al.2009), and support the idea that InsP6 can induce autocatalytic cleavage of the toxins in vivo. The domain adjacent to the GTD in TcdA and TcdB shares sequence homology with the cysteine protease domain of the MARTX family of toxins, which is also activated by InsP6 (reviewed in Egerer and Satchell 2010 and Shen 2010). This APD was subsequently shown to induce the InsP6-dependent cleavage and release of the GTD (Egerer et al.2007).
The APD has been described as a cysteine protease. The active site of the TcdA and TcdB APD has three conserved residues: a cysteine, a histidine and an aspartate (Fig. 5). Individual mutation of these residues (D589, H655 or C700 in TcdA; D587, H653 or C698 in TcdB) inhibits autoprocessing (Egerer et al.2007; Pruitt et al.2009). However, a recent report has shown that the conserved cysteine and histidine residues of TcdA and TcdB help to coordinate a zinc ion that is essential for autoprocessing activity (Chumbler et al.2016b). What serves as the nucleophile in this reaction is currently unclear, and warrants the use of the term autoprotease, instead of cysteine protease, when referring to this domain.
Figure. 5.
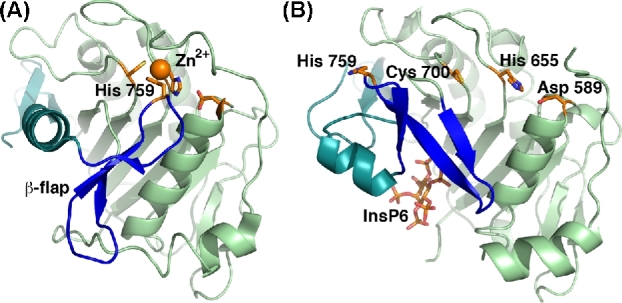
The autoprocessing domain (APD) undergoes a significant conformational change upon binding to InsP6. The crystal structures of the TcdA APD in the (A) absence (4R04) and (B) presence of InsP6 (3HO6) reveal significant changes in the central β-flap structure (blue) and the C-terminal sequence that follows (teal). The structure of the APD in the context of TcdA4-1802 revealed the unexpected requirement for zinc (orange sphere) in TcdA and TcdB autoprocessing activity. Other key residues include Asp 589, His 655 and Cys 700 (side chains depicted with orange carbon atoms). His 759 is located at the tip of the β-flap and is bound to the zinc in the absence of InsP6. It moves significantly upon InsP6 binding.
Crystal structures of the APD in the presence of InsP6 reveal that InsP6 binds a positively charged pocket that is separated from the active site by a structure termed the ‘β-flap’ (Lupardus et al.2008; Pruitt et al.2009; Puri et al.2010). Binding of InsP6 was shown to induce significant conformational changes by NMR and these changes were thought to be linked to protease activation (Pruitt et al.2009). Through mutational analyses Shen et al. (2011) showed that this ‘β-flap’ structure transduces the allosteric change induced by InsP6 binding to the active site. Comparisons of the InsP6-bound isolated TcdA APD structure with that of the APD from TcdA4-1802 (crystallized in the absence of InsP6) reveal that the β-flap (residues 746–765) rotates ∼90°, and there is significant repositioning of the subsequent residues (766–802) following InsP6 binding (Fig. 5) (Chumbler et al.2016b). One effect of this structural change is an increase in positively charged residues at the InsP6-binding site. InsP6 binding also results in a 19 Å movement of H759, located at the tip of the β-flap (also involved in zinc binding), out of the active site (Fig. 5). Mutation of H759 (or H757 in TcdB) leads to autoprocessing that is no longer dependent on InsP6 concentration (Chumbler et al.2016b), suggesting that this residue in the β-flap is a key regulator of InsP6-induced allostery in TcdA and TcdB.
While TcdA and TcdB APDs share the same mechanism of InsP6-induced activation, cleavage is not equivalent between these two toxins. In vitro, TcdB holotoxin is more sensitive than TcdA to InsP6-induced cleavage (Kreimeyer et al.2011). Structural and biochemical studies indicate that autoprocessing of TcdA is repressed in the context of the holotoxin due to interdomain interactions between CROPS and the N-terminus (Olling et al.2014; Zhang et al.2015; Chumbler et al.2016b). This CROPS-mediated repression is alleviated upon acidification of the toxin-containing medium (Olling et al.2014).
Mechanisms that modulate autoprocessing activity of the toxins can affect their virulence properties in the host. A study by Savidge et al. (2011) demonstrated that C. difficle toxins are S-nitrosylated at the conserved cysteine of the APD by the infected host, which inhibits the autoprocessing activity of the toxins in an InsP6-dependent manner and reduces virulence. Nevertheless, it is important to note that autoprocessing-deficient mutants of TcdA and TcdB are still able to inactivate some of their GTPase substrates and cause cytopathic effects in cells, albeit with delayed kinetics (Kreimeyer et al.2011; Chumbler et al.2012; Li et al.2013). In an in vivo intoxication study, virulence of a TcdB autoprocessing mutant is attenuated but not abolished (Li et al.2015). While autoprocessing can affect toxin potency by regulating the rate at which host cells targets are modified, it is not essential for the cytopathic and cytotoxic effects mediated by TcdA and TcdB. Interestingly, a study of the autoprocessing mutant of TcsL (a homologous glucosylating toxin from C. sordellii) showed that this mutant, which is less toxic, can inactivate Rac but is impaired in its ability to glucosylate Ras GTPase (Craven and Lacy 2016). Rac has been reported to cycle to the endosomes where it is activated before trafficking back to the membrane, whereas Ras is trafficked to and remains at the plasma membrane (Apolloni et al.2000; Palamidessi et al.2008). In autoprocessing mutants, the GTD remains tethered to the endosomes and can access substrates that encounter the endosome. These findings suggest that the importance of autoprocessing in mediating toxin virulence in the host may vary depending on the localization of the GTD substrates.
Glucosylation and substrates
TcdA and TcdB encode a 63-kDa GTD at the N-terminus, which inactivates small GTPases from the Rho family Aktories and Barbieri 2005; Jank et al.2007a (Glycobiology). GTPases targeted by TcdA, TcdB and other LCTs are listed in Table 2. GTPases are molecular switches that cycle between active GTP-bound and inactive GDP-bound states. This GTPase cycle is regulated by three classes of proteins: (i) guanine nucleotide exchange factors or GEFs, which activate the GTPases by exchanging GDP for GTP; (ii) GTPase activating proteins or GAPs, which facilitate the inactivation of the GTPases by stimulating their GTP hydrolyzing activity; and (iii) guanine nucleotide dissociation inhibitors or GDIs, which extract the GTPases from the membrane and maintain the inactive GDP-bound form in the cytosol. In their active state, Rho family GTPases can interact with a wide range of effector molecules such as kinases, phosphatases, lipases and scaffolding proteins to regulate many cellular functions including assembly and organization of the actin cytoskeleton (Bishop and Hall 2000; Etienne-Manneville and Hall 2002; Raaijmakers and Bos 2009).
Table 2.
Substrate specificity of the large glucosylating toxins.
| Organism | Toxin | Strain | Targets | Reference |
|---|---|---|---|---|
| C. difficile | TcdA | VPI10463a | Rho, Rac, cdc42, Rap | Chaves-Olarte et al. (1997) |
| C. difficile | TcdB | VPI10463a | Rho, Rac, cdc42 | Just et al. (1995); Chaves-Olarte et al. (1997) |
| 1470b | Rac, cdc42, Rap, Ral, R-Ras | Chaves-Olarte et al. (1999) | ||
| 8864c | Rac, cdc42, Rap, Ral, R-Ras | Muller, von Eichel-Streiber and Moos (1999); Mehlig et al. (2001) | ||
| C34d | Rho, Rac, cdc42, Rap, Ral, R-Ras | Mehlig et al. (2001) | ||
| NAP1/RT027e | RhoA, Rac, cdc42, Rap, R-Ras | Quesada-Gomez et al. (2016) | ||
| NAP1V/RT019 | Rac, cdc42, Rap, R-Ras | Quesada-Gomez et al. (2016) | ||
| C. sordellii | TcsL | VPI9048 | Rac, cdc42, Rap, Ras | Hofmann et al. (1996); Genth et al. (2014) |
| IP 82 | Rac, Rap, Ral, Ras | Popoff et al. (1996); Genth et al. (2014) | ||
| 6018 | Rac, Rap, Ral, Ras | Hofmann et al. (1996) | ||
| C. sordellii | TcsH | VPI9048 | Rho, Rac, cdc42, Ras | Genth et al. (1996, 2014) |
| C. novyi | Tcnα | 590, 19402 | Rho, Rac, cdc42 | Selzer et al. (1996); Genth et al. (2014) |
| C. perfringens | TpeL | MC18 | Rac, Rap, Ral, Ras | Nagahama et al. (2011) |
Reference strain.
This strain has a nonsense mutation in the tcdA gene, which introduces a stop codon at amino acid position 47; classified as TcdA negative (von Eichel-Streiber et al.1999).
This strain has a 5.9-kb deletion in the 3’ end of the tcdA region; classified as TcdA negative (Soehn et al.1998).
This strain has an insertion of ∼2 kb in the tcdA gene that does not hinder expression of a fully active toxin (Braun et al.1996).
Epidemic strain.
GTPases in their GDP-bound, membrane-associated form are the preferred substrates for the LCTs (Just et al.1995a; Genth, Aktories and Just 1999). The toxins modify their targets through monoglucosylation of the conserved threonine in the switch I region of the GTPase (Just et al.1995a,b). This threonine residue is involved in coordination of the Mg2+ ion required for GTP binding (Ihara et al.1998), and conformational changes in the switch I region subsequent to GTP binding affects interactions with regulatory proteins and effectors (Bishop and Hall 2000; Schaefer, Reinhard and Hordijk 2014). Consequently, glucosylation of the GTPases by the LCTs inhibits nucleotide exchange by GEFs (Herrmann et al.1998), GAP-stimulated GTPase activity (Sehr et al.1998), GDI binding and cytosol-membrane cycling (Genth, Aktories and Just 1999) and interaction with effector proteins (Herrmann et al.1998; Sehr et al.1998; Geyer et al.2003). The above-mentioned glucosylation-induced effects disrupt GTPase signaling and have been linked to the cytopathic and cytotoxic effects observed with these toxins (discussed in the next section).
The LCTs use either uridine diphosphate (UDP)-glucose or UDP-GlcNac as the co-substrate for GTPase modification. Glucosylating toxins from C. difficile (TcdA/B) and C. sordellii (TcsH/L) use UDP-glucose as the glycosyl donor (Just et al.1995a,b, 1996; Popoff et al.1996; Genth et al.2014). TpeL from C. perfringens can use either UDP-glucose or UDP-GlcNAc (Nagahama et al.2011), and α-toxin (Tcnα) from C. novyi uses UDP-GlcNAc as the sugar source (Selzer et al.1996). Mutational studies show that two amino acids near the catalytic cleft dictate the co-substrate specificity of the LCTs. TcdA, TcdB and TcsL have isoleucine and glutamine in equivalent positions (Ile383/Gln385 in TcdB and TcsL; Ile382/Gln384 in TcdA), whereas TpeL and Tcnα have amino acids with smaller side chains at one or both positions (Ala383/Gln385 in TpeL and Ser385/Ala387 Tcnα), which can accommodate bulkier UDP-GlcNAc into the catalytic pocket. Replacing the bulky side chains with smaller groups and vice versa changes the donor substrate specificity (Jank et al.2005; Nagahama et al.2011).
Crystal structures of the GTDs from TcdA, TcdB, TcsL and Tcnα have been determined and have helped our understanding of the enzymatic mechanism (Reinert et al.2005; Ziegler et al.2008; D’Urzo et al.2012; Pruitt et al.2012; Alvin and Lacy 2017). At the core of the structure is a Rossman fold, which is similar to that of the glucosyltransferase type A (GT-A) family of enzymes (Fig. 6) (Unligil and Rini 2000; Reinert et al.2005). Within this core is an Asp-X-Asp (DXD) motif, which is conserved in all LCTs and other GT-A members, and is essential for the enzymatic activity (Busch et al.1998; Wiggins and Munro 1998). The DXD motif (Asp285/ Asp287 in TcdA and Asp286/ Asp288 in TcdB) is important for the coordination of the manganese cofactor, and the first Asp residue of the DXD motif also binds the 3΄-hydroxyl group of the UDP-ribose and the glucose. UDP binding involves additional residues that are conserved, including two tryptophans (Trp101/Trp519 in TcdA and Trp102/Trp520 in TcdB) that stabilize the uracil ring of the UDP by aromatic stacking or by binding the glycosidic oxygen. Mutation of these conserved residues results in significantly attenuated glycosyltransferase activity (Busch et al.2000; Jank et al.2007b).
Figure. 6.
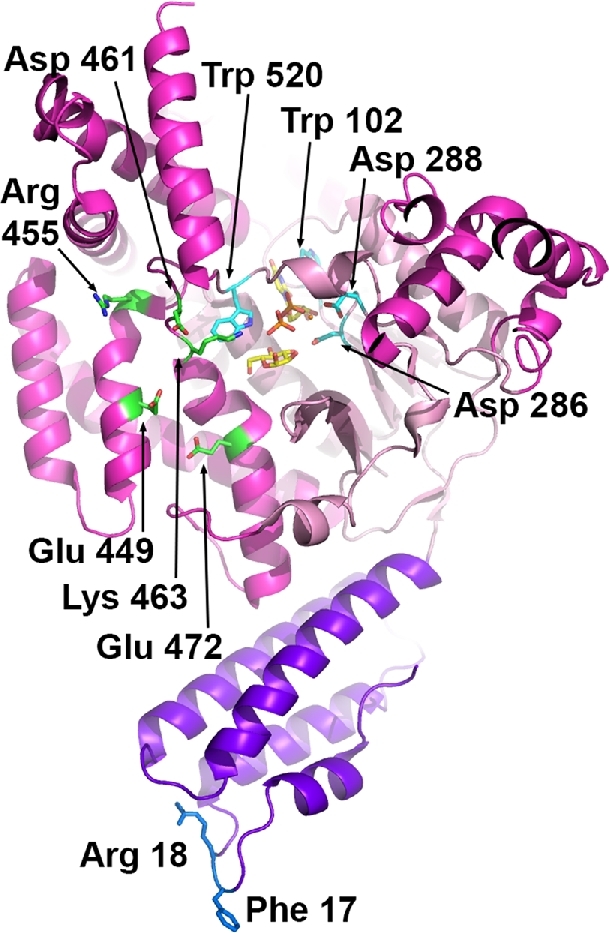
The glucosyltransferase domain. The crystal structure of the TcdB glucosyltransferase domain (2BVL) with hydrolyzed UDP-glucose (yellow carbon atoms) bound in the core GT-A fold (light pink). The side chains of key catalytic residues (Trp 102, Asp 286, Asp 288 and Trp 520; aqua carbon atoms) are indicated along with the manganese atom (orange sphere). Residues that have been implicated in GTPase substrate recognition include Glu 449, Arg 455, Asp 461, Lys 463 and Glu 472 (green carbon atoms). The membrane localization domain corresponds to the four α-helices at the base of the structure (in purple) with the Phe 17 and Arg 18 residues implicated in membrane binding indicated with blue carbon atoms.
In addition to the common GT-A family fold, LCTs have four α-helical subdomains. The N-terminal subdomain, also called the membrane localization domain (MLD), is a four-helix bundle formed by the first ∼90 residues of the GTD (Fig. 6). The MLD has been shown to target the GTD to the cytosolic leaflets of cell membranes (such as the plasma membrane), where the enzyme can access its membrane-bound GTPase substrates (Mesmin et al.2004; Geissler, Tungekar and Satchell 2010; Varela Chavez et al.2015). Mutation of conserved residues (particularly Phe17/Arg18) extending from the tip of the MLD four-helix bundle has been shown to impair localization of the TcdB and TcsL GTDs to membrane lipids in vitro and in cells (Geissler, Ahrens and Satchell 2012; Varela Chavez et al.2016). Additionally, MLD mutants of TcsL are defective in their ability to glucosylate GTPase substrates and cause cytotoxicity (Craven and Lacy 2016; Varela Chavez et al.2016), suggesting that localization to membranes is important for GTD-dependent cellular effects of LCTs.
The role of the other α-helical subdomains is not known but they have been proposed to be involved in substrate binding (Reinert et al.2005). Specificity for Rho and Ras substrates varies among the LCTs (Table 2), but the molecular basis for these differences is not fully understood. In general, TcdB (from reference strains) and Tcnα exclusively modify Rho subfamily proteins, while TcdA and TcsH can also modify Ras GTPases (albeit to a lesser extent). TpeL and TcsL modify Ras GTPases and Rac but not Rho. Through mutagenesis and generation of chimeric GTDs, Jank et al. identified several charged residues located near the sugar binding pocket (Glu449, Arg455, Asp461, Lys463 and Glu472) that are important for substrate modification by TcdB and demonstrated that helix 17 contributes to RhoA recognition by TcdB (Fig. 6) (Jank et al.2007b). Interestingly, TcdB from reference (VPI10463) and variant (1470, 8864, C34 and NAP1/RT027) strains shows significant differences in the GTPase substrate specificity (Table 2). TcdB sequences from variant strains have accumulated several mutations on the proposed substrate-binding surface compared to the classical TcdB, which likely contributes to recognition of Ras GTPases in addition to the Rho subfamily proteins (Reinert et al.2005). Of these variants strains, 1470 and 8864 lack functional TcdA and are classified as TcdA-TcdB+ (Soehn et al.1998; von Eichel-Streiber et al.1999). TcdB isoforms from TcdA-TcdB+ strains have GTPase substrate specificity similar to that of TcsL in that they modify Ras and Rac but not Rho GTPases (Table 2). It has been proposed that these GTD variants may have arisen to compensate for the lack of functional TcdA, but the impact of the broader substrate specificity observed with TcdA is unclear.
CELLULAR EFFECTS OF TcdA AND TcdB
TcdA and TcdB disrupt the epithelial tight junctions and induce epithelial cell death, thereby causing direct injury to the colonic epithelium. Additionally, the toxins stimulate colonic epithelial cells to release proinflammatory cytokines and neutrophil chemoattractants, which lead to an acute innate inflammatory response with neutrophil recruitment, a key characteristic of the clinical pathophysiology of CDI (Fig. 7) (Voth and Ballard 2005; Kelly and Kyne 2011). An impaired barrier in association with an active inflammation leads to increased intestinal and vascular permeability, and likely promotes fluid secretion. The loss of a protective barrier may also permit entry of toxins and/or bacteria into the lamina propria, resulting in further intestinal inflammation (Fig. 7) (Feltis et al.2000). Prolonged exposure of the mucosal innate immune system to proinflammatory mediators can amplify the tissue damage, and may lead to severe disease outcomes (Steiner et al.1997; El Feghaly et al.2013; Yu et al.2017). Disruption of the epithelial barrier, an intense inflammatory response with neutrophil infiltration into the lumen and associated tissue damage are thought to contribute to the formation of pseudomembrane, which is observed in severe cases of CDI (Pothoulakis and Lamont 2001; Voth and Ballard 2005; Sun and Hirota 2015). The toxin-induced cellular effects and their underlying mechanisms are discussed below.
Figure. 7.

Cellular effects of C. difficile toxins. The toxins act on colonic epithelial cells and immune cells to induce inflammation and tissue damage. TcdA and TcdB disrupt the tight junctions and induce epithelial cell death, causing direct damage to the colonic epithelium. Additionally, the toxins stimulate epithelial cells to release inflammatory mediators that recruit neutrophils to the colonic mucosa. TcdA and TcdB can also enter the lamina propria following the disruption of the epithelial barrier and directly stimulate macrophages, dendritic cells, and mast cells to release inflammatory mediators, which further contribute to inflammation and neutrophil recruitment. Intoxication also results in the activation of enteric neurons and increased production of substance P (SP). SP can induce mast cell degranulation and can stimulate the lamina propria macrophages to release inflammatory cytokines. Prolonged intestinal inflammation can amplify tissue damage and contribute to neutrophil infiltration into the lumen, a key clinical feature of pseudomembranous colitis. The binary toxin CDT, expressed by some C. difficile strains, also induces cytopathic effects that lead to disruption of the tight junctions. Additionally, CDT can suppress a protective host eosinophilic response in the colon and can act synergistically with TcdA and TcdB to increase proinflammatory cytokine production by innate immune cells. Finally, CDT also contributes to C. difficile virulence by inducing the formation of microtubule-based cell protrusions that increase adherence of the bacteria.
Glucosylation-dependent cytopathic and cytotoxic effects in epithelial cells
Rho GTPases regulate the assembly and organization of the actin cytoskeleton. RhoA induces the assembly of focal adhesions and actin stress fibers, whereas Rac1 and Cdc42 induce the formation of actin-rich surface protrusions lamellipodia and filopodia, respectively (Nobes and Hall 1995; Etienne-Manneville and Hall 2002). Additionally, Rho GTPases are important for the establishment of epithelial cell morphology and polarity (Etienne-Manneville and Hall 2002). Consequently, GTPase inactivation by TcdA and TcdB results in the loss of the cytoskeletal structure, disassembly of focal adhesions and disruption of tight junctions (Hecht et al.1988, 1992; Moore et al.1990; Riegler et al.1995; May et al.2013). In tissue culture cells, these effects result in the characteristic cell rounding phenotype (also termed the cytopathic effect). The glucosylation-dependent cytopathic effect is thought to play an important role in the context of disease; toxin-induced disruption of tight junctions could result in impaired barrier function, increased intestinal permeability and inflammation.
GTPase inactivation by TcdA and TcdB also affects cell cycle progression. Intoxication by TcdA and TcdB is associated with reduced expression of cyclin D1, which is required for progression through the G1 phase of the cell cycle, resulting in G1-S arrest (Welsh et al.2001; D’Auria et al.2012). Additionally, Rac1 has been shown to promote the activation of the mitotic kinase Aurora A and cyclin B/cyclin-dependent kinase (Cdk) I complex, which are required for mitotic entry, through its effector protein p21-activated kinase (PAK) (Ando et al.2007; May et al.2014). Consequently, inactivation of Rac1 by C. difficile toxins delays activation of Aurora A and the CyclinB/Cdk1 complex in G2 phase and results in delayed G2-M transition (Kim et al.2005a; Ando et al.2007; Nottrott et al.2007; Gerhard et al.2008; D’Auria et al.2012; May et al.2014). Finally, RhoA inactivation by these toxins inhibits contractile ring formation and subsequent cytokinesis, resulting in the formation of binucleated cells (Huelsenbeck et al.2009). Thus, the toxins are capable of interfering with host cell proliferation by inactivating GTPases that regulate various stages of the cell cycle.
In addition to the cytopathic effect, inactivation of Rho GTPases by TcdA and TcdB can promote epithelial cell death (referred to as a cytotoxic effect). In tissue culture models, the glucosylation-dependent cell death induced by TcdA and TcdB is evident after 18–48 h of intoxication and occurs by an apoptotic mechanism, with intoxicated cells exhibiting hallmark features including cell shrinkage, phosphatidylserine externalization, caspase activation and DNA fragmentation (Fiorentini et al.1998; Brito et al.2002, 2005; Qa’Dan et al.2002; Carneiro et al.2006; Nottrott et al.2007; Gerhard et al.2008; Chumbler et al.2016a). Apoptosis can occur via caspase-dependent and independent mechanisms (Broker, Kruyt and Giaccone 2005; Chipuk and Green 2006; Pradelli, Beneteau and Ricci 2010; Tait and Green 2010). Investigations with TcdA and TcdB show that the toxins induce the activation of executioner caspases 3 and 7 in a variety of cell lines (Brito et al.2002; Qa’Dan et al.2002; Kim et al.2005a; Carneiro et al.2006; Matarrese et al.2007; Nottrott et al.2007; Gerhard et al.2008; Matte et al.2009; Chumbler et al.2016a). Activation of executioner caspases can occur via a death receptor-dependent extrinsic pathway (involving caspase 8) or by a mitochondria-dependent intrinsic pathway (involving caspase 9). TcdA and TcdB have been shown to induce mitochondrial outer membrane permeabilization (MOMP), cytochrome c release and activation of caspase 9, and TcdA has also been shown to activate caspase 8 (Brito et al.2002; Carneiro et al.2006; Matarrese et al.2007; Nottrott et al.2007; Gerhard et al.2008; Matte et al.2009). Although the toxin treatment induces caspase activation, the role of caspases in the apoptotic cell death caused by TcdA and TcdB is currently unclear. Experiments using caspase inhibitors and glucosyltransferase-deficient mutants have yielded conflicting results with both caspase-dependent and independent apoptotic mechanisms having been reported for TcdA and TcdB (Brito et al.2002; Qa’Dan et al.2002; Matarrese et al.2007; Nottrott et al.2007; Gerhard et al.2008; Matte et al.2009; Chumbler et al.2016a). It is important to note that MOMP, which is regulated by pro- and anti-apoptotic B-cell lymphoma 2 (Bcl-2) family members, can also promote apoptosis in a caspase-independent manner (Pradelli, Beneteau and Ricci 2010). MOMP can be induced by the cleavage and activation of Bid, a pro-apoptotic Bcl-2 family protein. Bid can be cleaved by caspase 8 or by non-caspase proteases such as cathepsins and calpains (Broker, Kruyt and Giaccone 2005; Tait and Green 2010). Interestingly, intoxication by TcdA also results in the cleavage of Bid, which was inhibited by a cathepsin/calpain inhibitor but not by caspase inhibitors, suggesting a role for Bid in the caspase-independent apoptosis mechanism (Brito et al.2002; Carneiro et al.2006; Nottrott et al.2007).
Glucosylation-independent cytotoxic effects in epithelial cells
TcdB has been shown to induce a bimodal cell death mechanism that is dependent on the toxin concentration (Chumbler et al.2016a). While at lower concentrations, TcdB induces apoptosis in a glucosylation-dependent manner, at higher concentrations (100 pM or above), TcdB causes a necrotic form of cell death that does not require either the autoprocessing or glucosyltransferase activities of the toxin (Chumbler et al.2012, 2016a; Farrow et al.2013; Wohlan et al.2014). The necrotic death can be observed in both cell culture and colon explant models after 2–4 h of intoxication and is marked by rapid ATP depletion, early breakdown of the plasma membrane and cellular leakage, and chromatin condensation (Chumbler et al.2012, 2016a; Farrow et al.2013; Wohlan et al.2014). TcdB induces necrosis by triggering an aberrant production of reactive oxygen species (ROS) through the assembly of the NADPH oxidase (NOX) complex on endosomes (Farrow et al.2013; Wohlan et al.2014; Chumbler et al.2016a) (Fig. 8). High levels of ROS promote cellular necrosis likely though DNA damage, lipid peroxidation, protein oxidation and/or mitochondrial dysfunction (Yu 1994; Temple, Perrone and Dawes 2005; Daiber 2010). Interestingly, a TcdB mutant that is defective in pore formation is unable to induce cell death even at high nanomolar concentrations, suggesting that pore formation is important for the glucosylation-independent necrotic cell death caused by TcdB (Zhang et al.2014). Unlike TcdB, TcdA does not trigger ROS production via the NOX complex and causes a glucosylation-dependent apoptosis at all concentrations tested (Chumbler et al.2016a). The ability of TcdB, but not TcdA, to induce a necrotic cell death may explain why both a wild-type (TcdA+TcdB+) epidemic strain and an isogenic TcdA−TcdB+ mutant cause significantly more colonic tissue damage than an isogenic TcdA+TcdB− mutant strain in animal models of infection (Carter et al.2015). The glucosylation-independent mechanism of TcdB may play a similar role in the context of human disease; TcdB-induced necrosis likely contributes to the extensive gut damage observed in patients with severe forms of CDI.
Figure. 8.

TcdB-induced necrosis. At higher concentrations (100 pM and above), TcdB causes a necrotic form of cell death that does not require the autoprocessing and glucosyltransferase activities of the toxin. TcdB induces necrosis by promoting the assembly of the NADPH oxidase (NOX) complex on endosomes (step 1). The fully assembled NOX complex in the redox-active endosome mediates the transfer of an electron from NADPH to molecular oxygen, resulting in the generation of superoxide (step 2) and production of reactive oxygen species (ROS) (step 3). High levels of ROS promote cellular necrosis likely though DNA damage, lipid peroxidation, protein oxidation and/or mitochondrial dysfunction.
Cytokine production by epithelial cells
In addition to disrupting the epithelial barrier and causing epithelial cell death, TcdA and TcdB trigger the release of a variety of inflammatory mediators from epithelial cells (Fig. 7). Chemokines that promote neutrophil recruitment to the colonic mucosa, including interleukin-8 (IL-8), growth-related oncogene-alpha and monocyte chemoattractant protein-1, are released from human colonic epithelial cells upon intoxication with TcdA and TcdB (Mahida et al.1996; Branka et al.1997; Kim et al.2002; Savidge et al.2003; Bobo et al.2013). Neutrophil infiltration has been shown to promote fluid secretion and enhance mucosal inflammation and injury in animal intoxication studies and is considered a hallmark feature of C. difficile-associated enterocolitis (Kelly et al.1994a; Qiu et al.1999; Kelly and Kyne 2011). TcdA and TcdB induce the secretion of neutrophil chemotactic factors from epithelial cells through the activation of the NF-κB signaling pathway and the mitogen-activated protein kinase (MAPK) pathways, including p38 MAPK, c-Jun N-terminal Kinase (JNK), and extracellular signal-regulated kinases (ERKs) (He et al.2002; Na et al.2005; Kim et al.2006; Lee et al.2007; Bobo et al.2013; Hansen et al.2013). These pathways result in chemokine and cytokine production through the activation of transcription factors NF-κB and activator protein-1.
TcdA and TcdB have been shown to stimulate IL-8 secretion through both glucosylation-dependent and independent mechanisms. IL-8 release from HT-29 human colonic epithelial cells is induced through the activation of p38 MAPK and its substrate MAPK-activated protein kinase 2 (MK2) (Bobo et al.2013). Toxin-induced disruption of the actin cytoskeleton is thought to trigger the p38/MK2-dependent response, suggesting that this mechanism is glucosylation dependent (Bobo et al.2013). Interestingly, Bobo et al. observed that p38 MAPK and MK2 are activated in the colons of mice and hamsters infected with C. difficile, suggesting that this pathway might play an important role in the toxin-induced cytokine production and intestinal inflammation during CDI. TcdA has also been shown to cause mitochondrial damage in HT-29 cells and trigger the generation of ROS, which mediates NF-κB activation and downstream IL-8 expression (He et al.2000, 2002). Presumably, ROS production resulting from TcdA-induced mitochondrial damage is not occurring at levels comparable to that observed downstream of NOX activation by TcdB in epithelial cells, and may explain the lack of a necrotic phenotype with TcdA. Interestingly, He et al. showed that NF-κB activation precedes Rho glucosylation by TcdA and can be blocked by pretreatment of cells with an antioxidant, suggesting that mitochondrial ROS generation drives a glucosylation-independent mechanism of IL-8 production in TcdA-treated HT-29 cells (He et al.2002). In primary colonic epithelial cells, TcdB has been shown to activate the ERK-MAP kinase pathway and stimulate IL-8 release through transactivation of epidermal growth factor receptor (EGFR) (Na et al.2005). TcdB treatment induces the release of transforming growth factor-alpha (TGF-α) from human colonocytes in a matrix metalloproteinase-dependent manner. The secreted TGF-α activates EGFR and the ERK-MAP kinase pathway, and mediates IL-8 secretion in TcdB-treated primary colonocytes (Na et al.2005). Furthermore, Na et al. reported that TGF-α release preceded GTPase glucosylation, suggesting that this mechanism of IL-8 production by TcdB is glucosylation independent. TcdB has also been shown to induce NF-κB activation and IL-8 release from Caco-2 human epithelial cells by triggering cellular stress and damage. The underlying mechanism involves activation of P2Y6, a G-protein coupled receptor, by extracellular nucleotides that are released from stressed and dying Caco-2 cells (Hansen et al.2013). Interestingly, TcdA does not induce IL-8 release from Caco-2 cells, indicating that the toxin-induced cytokine production and the underlying mechanisms may vary depending on the cell type (Mahida et al.1996; Hansen et al.2013). In addition to the production of neutrophil chemotactic factors, activation of p38 MAPK results in the induction of cyclooxygenase-2 and the release of prostaglandin E2 (PGE2), a known mediator of intestinal inflammation (Kim et al.2005b). PGE2 secretion was stimulated by TcdA, but not TcdB, and involves mitogen- and stress-activated protein kinase and transcription factors cAMP response element-binding protein and activating transcription-factor 1 (Kim et al.2005b).
Effects on innate immune cells and the enteric nervous system
Following the disruption of the colonic epithelial barrier, TcdA and TcdB can enter the lamina propria and directly stimulate macrophages and dendritic cells to release inflammatory mediators (Fig. 7). Monocytes and macrophages are a major source of inflammatory cytokines and secrete tumor necrosis factor alpha (TNF-α), IL-1β, IL-6, IL-8, PGE2 and leukotriene B4 upon exposure to TcdA and TcdB (Flegel et al.1991; Linevsky et al.1997; Melo Filho et al.1997; Rocha et al.1997; Souza et al.1997; Alcantara et al.2001; Sun et al.2009). Macrophage-derived IL-8, TNF-α and IL-1β are thought to play an important role in the recruitment of neutrophils into the colonic mucosa (Linevsky et al.1997; Rocha et al.1997; Souza et al.1997; Warny et al.2000). The mechanisms that promote toxin-induced cytokine production in macrophages are not fully understood. Sun et al. (2009) showed that TcdA-induced secretion of TNF-α by murine macrophages is dependent on the glucosyltransferase activity of the toxin. TcdA has been shown to induce IL-8 secretion through the activation of p38 and ERK MAP kinases, calmodulin and NF-κB (Jefferson et al.1999; Warny et al.2000). Activation of the apoptosis-associated speck-like protein (ASC)-containing inflammasome by TcdA and TcdB, and p38 and ERK MAP kinases by TcdA have been shown to result in IL-1β release from human and murine macrophages (Warny et al.2000; Ng et al.2010; Hirota et al.2012; Xu et al.2014).
The mechanism by which TcdA triggers inflammasome activation in macrophages remains to be elucidated. Ng et al. (2010) reported that TcdA requires cellular entry and endosomal acidification to induce inflammasome activation in differentiated human THP-1 monocytes. Cowardin et al. (2016b) observed reduced caspase-1 activation in THP-1 cells treated with a glucosyltransferase-deficient mutant of TcdA, suggesting that the inflammasome activation by TcdA is mediated, at least in part, by the glucosyltransferase activity. Two mechanisms have been reported for inflammasome activation by TcdB. Ng et al. (2010) implicated the NOD-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome and proposed a glucosylation-independent mechanism for inflammasome activation and IL-1β release by TcdB in mouse peritoneal macrophages and differentiated THP-1 cells. In contrast, a recent study by Xu et al. (2014) showed that TcdB-induced inflammasome activation and IL-1β release in differentiated THP-1 cells and mouse primary bone marrow-derived macrophages require the enzymatic activity of the toxin and pyrin. Pyrin is an inflammasome sensor that responds to the inactivation of Rho GTPases by bacterial toxins (Xu et al.2014) and may also mediate the glucosylation-dependent inflammasome activation by TcdA.
Intoxication of monocytes and macrophages by TcdA and TcdB can also lead to cell death (Melo Filho et al.1997; Mahida et al.1998; Warny and Kelly 1999; Warny et al.2000; Ng et al.2010; Xu et al.2014; D’Auria et al.2015). In addition to stimulating IL-1β production, toxin-induced activation of the inflammasome and MAP kinases have been shown to result in cell death by pyroptosis and necrosis, respectively (Warny et al.2000; Ng et al.2010; Xu et al.2014). TcdA and TcdB can also activate the ASC-containing inflammasome in dendritic cells (Jafari et al.2013; Cowardin et al.2015, 2016b). The IL-1β that is secreted in response to inflammasome activation can signal via the IL-1 receptor and enhance IL-23 production in dendritic cells (Cowardin et al.2015). IL-23 signaling has been shown to promote colonic neutrophil recruitment and amplify intestinal inflammation, and is associated with increased disease severity in mouse models of CDI (Buonomo et al.2013; McDermott et al.2016). Additionally, IL-1β can stimulate the release of IL-6 and IL-8 from human colonic epithelial cells and IL-8 from submucosal enteric neurons (Kelly et al.1994b; Ng et al.2003; Tixier et al.2005). Thus, IL-1β secreted from activated lamina propria macrophages and dendritic cells can propagate the inflammatory cascade through both autocrine and paracrine signaling. In addition to IL-1β, TcdA induces the production of IL-6, CXCL1 (KC/murine IL-8 homolog) and CXCL2 from murine bone marrow-derived dendritic cells (Lee et al.2009; Cowardin et al.2016b). Both in vitro and in vivo intoxication studies suggest that TcdA-induced secretion of IL-1β, IL-6 and CXCL1 requires the glucosyltransferase activity of the toxin (Cowardin et al.2016b).
In addition to macrophages and dendritic cells, intestinal mast cells and neurons may also be involved in the innate inflammatory response to C. difficile toxins A and B (Fig. 7). Mast cells can be directly activated by TcdA and TcdB, resulting in degranulation and the release of inflammatory mediators, including TNF-α, IL-8 and histamine (Kurose et al.1994; Calderon et al.1998; Meyer et al.2007). TcdB-induced mast cell degranulation is mediated, in part, by a p38 MAP kinase-dependent mechanism. Activation of p38 MAP kinase upregulates the production of prostaglandins D2 and E2, which promote mast cell degranulation through autocrine signaling (Meyer et al.2007). The mechanism underlying direct activation of mast cells by TcdA is not known. Interestingly, administration of TcdA into rat or mouse ileum has been shown to result in the activation of intestinal neurons and increased production of substance P (SP), a small peptide with proinflammatory properties secreted by nerves and inflammatory cells (Mantyh et al.1996b; Castagliuolo et al.1997; Anton et al.2004). SP signaling has been shown to cause mast cell degranulation in ileal loops exposed to TcdA, and provides an indirect mechanism by which TcdA can activate mast cells (Castagliuolo et al.1994, 1999; Pothoulakis et al.1994; Wershil, Castagliuolo and Pothoulakis 1998). Additionally, SP can stimulate the lamina propria macrophages to release TNFα, furthering the intestinal inflammation (Castagliuolo et al.1997). The proinflammatory responses mediated by SP are thought to contribute to the secretory and inflammatory effects of TcdA (Castagliuolo et al.1994, 1998; Pothoulakis et al.1994; Mantyh et al.1996a; Kirkwood et al.2001). The enteric nervous system is also involved in TcdB-mediated inflammatory responses (Neunlist et al.2003). Tixier et al. (2005) showed that treatment of intact human intestinal tissues and human intestinal mucosa/submucosa co-cultures with TcdB results in IL-8 production in submucosal enteric neurons, and suggested that the interactions between mucosa and submucosa regulate IL-8 secretion during TcdB-induced inflammation. TcdB has also been shown to induce caspase-3 activation and apoptosis in enteric glial cells via a mechanism involving NOX-mediated cytosolic ROS generation and subsequent JNK activation (Fettucciari et al.2017; Macchioni et al.2017). Collectively, these studies highlight the complexity of the innate immune responses triggered by C. difficile toxins A and B.
Role of toxin-induced inflammation in Clostridium difficile disease
The role of toxin-induced inflammation in disease is a topic of active investigation. The effects of TcdA and TcdB on innate immune cells and their contribution to the pathophysiology of C. difficile disease have been primarily studied using intoxication models. Evidence from in vivo intoxication studies suggests that toxin-induced inflammatory responses likely play an important role in mediating C. difficile-associated diarrhea and colitis. Administration of drugs that inhibit the release of inflammatory mediators such as leukotriene B4, PGE2 and SP has been shown to reduce neutrophil recruitment, fluid secretion, and mucosal damage in mice and rat ileal loops treated with TcdA (Pothoulakis et al.1993, 1994; Wershil, Castagliuolo and Pothoulakis 1998; Warny et al.2000; Kim et al.2005b). Depletion of mast cells or inhibition of neutrophil recruitment via an antibody against leukocyte adhesion molecule CD18 also provided protection against TcdA-induced enteritis (Kelly et al.1994a; Wershil, Castagliuolo and Pothoulakis 1998). Furthermore, blockade of MAP kinase or IL-β signaling or preventing inflammasome activation through the deletion of the adaptor protein ASC reduced TcdA- and TcdB-induced inflammation and tissue damage in animal intoxication studies (Warny et al.2000; Kim et al.2005b; Chen et al.2006; Ng et al.2010; Hirota et al.2012). It is important to note that certain innate inflammatory responses, which enhance fluid secretion and mucosal damage in ileal loop intoxication studies, have protective roles during infection. Infection studies in mice show that ASC-mediated inflammasome signaling and neutrophil recruitment to the site of infection are important for containing luminal commensal bacteria and preventing systemic dissemination during CDI (Hasegawa et al.2011, 2012; Jarchum et al.2012). The studies also highlight the importance of using infection models for understanding the precise roles played by immune cells and the innate inflammatory responses in disease vs host defense. Although host innate responses are important in the early defense against CDI, a dysregulated inflammatory response that leads to continuous recruitment of inflammatory cells and prolonged intestinal inflammation is likely pathogenic. Consistent with this notion, elevated levels of inflammatory cytokines and fecal lactoferrin (a marker for fecal leukocytes and an indicator of intestinal inflammation) correlate with increased disease severity and poor outcomes in patients with CDI, while C. difficile bacterial burden does not (Steiner et al.1997; El Feghaly et al.2013; Yu et al.2017). Furthermore, IL-23, a proinflammatory cytokine that is capable of inducing sustained intestinal inflammation, is elevated in colon biopsies of patients with CDI and has been shown to promote disease in mouse models of CDI (Buonomo et al.2013). Mice lacking IL-23 signaling through genetic knockout or monoclonal antibody-mediated neutralization had reduced disease severity and enhanced overall survival compared with wild-type controls, suggesting a role for IL-23 in mediating C. difficile-associated colitis (Buonomo et al.2013). Future infection studies may help identify additional pathogenic inflammatory mechanisms and determine if modulating these signaling mechanisms could lead to successful resolution of disease. Furthermore, infection studies using C. difficile strains with defined toxin deletions will provide a better understanding of the individual roles of TcdA and TcdB in mediating pathogenic inflammation.
STRUCTURE AND MECHANISM OF ACTION OF CDT
CDT belongs to a family of toxins, called binary toxins, which are produced by certain pathogenic Clostridium and Bacillus species. Other members of this family include Clostridium perfringens iota toxin, C. spiroforme toxin (CST), C. botulinum C2 toxin, Bacillus cereus vegetative insecticidal proteins (VIP), and B. anthracis lethal and edema toxins (Barth et al.2004). The binary toxins, like the LCTs, are AB type toxins. However, unlike the LCTs, which engage host cells as a contiguous polypeptide, binary toxins are secreted as two separate protein components (A and B) that associate on the surface of host cells following receptor binding by the B component (Barth et al.2004; Simon, Aktories and Barbieri 2014). In addition to receptor binding and uptake, the B component also mediates pore formation and translocation of an enzymatic A component into the cytosol (Gerding et al.2014). The enzyme moiety of CDT (CDTa) is an ADP-ribosyltransferase that modifies actin, similar to that of iota toxin, CST, C2 toxin and VIP (Barth et al.2004). The individual components of CDT (CDTa and CDTb) are non-toxic, but when combined, they cause toxic effects in host cells (Popoff and Boquet 1988; Barth et al.2004; Sundriyal et al.2010). The proposed mechanism of CDT intoxication is shown in Fig. 9A.
Figure. 9.

The CDT binary toxin. (A) Schematic representation of the mechanism of action of CDT. CDT consists of an enzymatic component (CDTa; blue) and a transport component (CDTb; yellow). The monomeric form of CDTb binds to its cell surface receptor, the lipolysis-stimulated lipoprotein receptor (LSR). Thereafter, CDTb undergoes proteolytic activation and oligomerization on the cell surface. Alternatively, the monomeric form of CDTb may undergo proteolytic activation and oligomer formation before binding to LSR. The oligomeric prepore binds CDTa, and the receptor-CDT complex is subsequently internalized into cells. As the endosome matures, the acidic pH triggers conformational changes in CDTb, resulting in membrane insertion and formation of a pore through which CDTa is translocated into the cytosol. Translocation of CDTa is facilitated by heat shock protein 70 (Hsp70), Hsp90 and cyclophilin A (CypA). In the cytosol, CDTa ADP-ribosylates actin at arginine-177. ADP-ribosylation of actin results in the eventual breakdown of the actin cytoskeleton and cytopathic cell rounding, and the formation of microtubule-based protrusions that enhance bacterial adherence. (B) The structure of the CDTa ADP-ribosyltransferase (2WN7). The N-terminal domain (purple) is responsible for binding CDTb, while the C-terminal domain (light blue) is responsible for the enzymatic activity. The side chains of residues within the RSE motif are depicted with blue carbons and are important for catalysis. Other residues important for binding of NAD (yellow carbons) are depicted with aqua carbons.
Receptor binding
The transport (or B) component of CDT (strain CD196), which is involved in receptor binding, consists of 876 amino acids and has a molecular mass of 98.8 kDa. The N-terminal 42 amino acids of the transport component (CDTb) correspond to a signal peptide, which is cleaved to release a precursor protein comprising 834 amino acids (Perelle et al.1997). As known for other binary toxins, the precursor form is proteolytically processed by serine-type proteases to yield a mature, active protein (Perelle et al.1997; Barth et al.2004). The cleavage likely occurs between Lys167 and Leu168 in the precursor form (or Lys209 and Leu210 in the full length) and releases a propeptide (∼20 kDa) and an activated CDTb (∼75 kDa) (Perelle et al.1997). Both the precursor and mature forms of CDTb are able to interact with the cellular receptor, indicating that the proteolytic activation of CDTb is not a prerequisite for binding to the host cell (Papatheodorou et al.2013).
To intoxicate host cells, CDTb engages lipolysis-stimulated lipoprotein receptor (LSR) (Papatheodorou et al.2011), a cell surface receptor that is highly expressed in many tissues including the small intestine and colon (Mesli et al.2004). Notably, LSR was also shown to function as the cellular receptor for C. perfringens iota toxin and CST (Papatheodorou et al.2011, 2012). The precursor form of CDTb shares 91% and 89% sequence homology with the transport components of iota toxin (Ib) and C. spiroforme toxin (Sb), respectively. Both Ib and Sb are capable of delivering the enzyme component of CDT into the target cell, highlighting the functional conservation between these transport components (Popoff and Boquet 1988; Gulke et al.2001). Cell-binding studies using wild-type and truncated variants of Ib have shown that the C-terminal residues 637 to 836 (precursor form) contain the receptor-binding region of iota toxin (Marvaud et al.2001). The corresponding region in CDTb (residues 635–834 in the precursor form) was later tested and shown to bind the extracellular Ig-like domain of LSR (Hemmasi et al.2015). Furthermore, by generating a series of truncations in this region, amino acids 715 to 824 of CDTb (precursor form) were shown to be sufficient for interaction with the receptor (Hemmasi et al.2015). Recently, another cell surface protein, CD44, has been proposed to play a role in CDT intoxication, but the role of CD44 in CDT binding and/or uptake remains unclear (Wigelsworth et al.2012).
Cellular uptake of CDT
Once bound to LSR, CDTb induces clustering and accumulation of the receptor into lipid rafts (Papatheodorou et al.2013). Based on studies with iota toxin, a model for CDT uptake has been proposed wherein the local accumulation of receptor-bound CDTb monomers in lipid rafts promotes oligomerization of CDTb on the cell surface, which is followed by binding of the enzymatic component (CDTa) and internalization of this complex into cells (Blocker et al.2001; Hale et al.2004; Nagahama et al.2004). Although proteolytic activation of the transport component is not essential for receptor binding and clustering into lipid rafts, it is required for oligomerization and subsequent intoxication of host cells (Perelle et al.1997; Gibert et al.2000; Stiles et al.2002; Barth et al.2004; Sundriyal et al.2010; Papatheodorou et al.2013).
The mechanism of CDTb oligomerization is currently unknown. Much of our understanding of this process comes from studies with protective antigen (PA), the transport component of anthrax toxin. PA is organized into four domains, as defined by X-ray crystallography (Petosa et al.1997). Domain 3 (residues 488–595) mediates oligomerization of PA and contains a loop of critical residues (510–518) that contacts the neighboring monomer to stabilize the oligomeric structure (Mogridge, Mourez and Collier 2001; Lacy et al.2004). Domain 3 of PA shares 54% sequence homology with a region spanning residues 472–576 in CDTb (precursor form). This region of CDTb may be involved in oligomerization, a hypothesis that remains to be tested.
The oligomer–receptor complex acts as a docking platform for the enzymatic component. Studies with iota toxin show that Ib oligomer, but not the monomer, binds the enzyme moiety (Ia) (Nagahama et al.2004), and the N-terminal 27 residues of activated Ib are important for interaction with Ia (Marvaud et al.2001). Given the sequence and functional conservation between Ib and CDTb, it is likely that the N-terminus of activated CDTb serves as the docking site for CDTa. Following binding of CDTa, the toxin–receptor complexes are internalized and transported to acidified endosomal compartments within cells. This is deduced from the observation that bafilomycin A1, which blocks endosomal acidification by specifically inhibiting the vacuolar H+-ATPase, protects epithelial cells from CDT intoxication (Kaiser et al.2011).
Pore formation and translocation
The finding that bafilomycin A1 inhibits CDT intoxication suggests a requirement for low pH in toxin action. Acid activation of surface-bound CDT was shown to be sufficient in allowing intoxication of host cells by CDTa, indicating that low pH is important for CDTa delivery into the cytosol (Kaiser et al.2011). The low pH of endosomes is proposed to trigger conformational changes in the transport component that lead to membrane insertion and formation of a transmembrane channel through which CDTa can be translocated into the cytosol. Support for this model comes from studies with anthrax toxin, C. botulinum C2 toxin and iota toxin (Collier and Young 2003; Knapp, Benz and Popoff 2016).
The structure of the pore and the underlying mechanisms involved in pore formation by the CDTb oligomer remain to be elucidated. In PA, prepore-to-pore conversion involves pH-dependent conformational changes in domain 2 (residues 259–487) (Petosa et al.1997; Benson et al.1998; Qa’dan et al.2005), which shares 62% sequence homology with amino acids 255–468 of CDTb (precursor form). Domain 2 of PA contains a large amphipathic loop (residues 302–323) that forms a β-hairpin. The β-hairpins assemble to generate a transmembrane β-barrel pore, with hydrophobic residues facing the lipid and hydrophilic residues facing the lumen of the channel (Petosa et al.1997; Benson et al.1998; Qa’dan et al.2005; Jiang et al.2015). A similar amphipathic stretch is present in Ib (amino acids 295–320 in the precursor form) and CDTb (amino acids 293–318 in the precursor form). The amphipathic sequence of Ib has been shown to be involved in membrane insertion and channel formation (Knapp et al.2002, 2015).
Following pore formation, the enzymatic component is translocated across the membrane into the host cytosol. Although the mechanisms underlying CDTa translocation are not fully understood, productive translocation of CDTa has been shown to rely on host proteins, the chaperone heat shock proteins 70 and 90 (Hsp70 and 90) and cyclophilin A (CypA), a peptidyl-prolyl cis/trans isomerase (Kaiser et al.2011; Ernst et al.2017). CDTa binds Hsp70, Hsp90 and CypA in vitro, and inhibitors of Hsp70, Hsp90 and CypA block pH-dependent membrane translocation of CDTa and downstream cytopathic effects in cells (Kaiser et al.2011; Ernst et al.2015, 2017). The Hsp70/Hsp90/CypA-dependent translocation mechanism is also shared by iota toxin and C. botulinum C2 toxin (Haug et al.2003; Haug, Aktories and Barth 2004; Kaiser et al.2009, 2011; Ernst et al.2017).
Actin-ADP-ribosyltransferase
The enzyme component of CDT (strain CD196) consists of 463 amino acids and has a molecular mass of 53 kDa (Perelle et al.1997; Chang and Song 2001). The N-terminal 43 amino acids likely correspond to a signal peptide that is cleaved to release a mature protein (CDTa) with a mass of ∼48 kDa (Perelle et al.1997). CDTa is an ADP-ribosyltransferase (ADPRT), which catalyzes the transfer of ADP-ribose from nicotinamide adenine dinucleotide (NAD) to Arg-177 of monomeric G-actin (Popoff et al.1988; Schwan et al.2009). The enzyme components of C. perfringens iota toxin and C. botulinum C2 toxin also ADP-ribosylate actin at Arg-177 (Vandekerckhove et al.1987, 1988).
CDTa shares 86%, 30% and 33% sequence identity with the enzyme components of iota toxin (Ia), C2 toxin (C2I) and B. cereus VIP2, respectively. Crystal structures of CDTa, Ia, C2I and VIP2 have been determined and reveal structural similarities among these toxin components (Han et al.1999; Tsuge et al.2003; Schleberger et al.2006; Sundriyal et al.2009). Like other binary actin ADPRTs, CDTa is a mixed α/β protein and is composed of two domains that are linked by a loop. The domains are structurally homologous and are thought to have arisen by gene duplication from a common ancestral ADPRT gene (Han et al.1999). In CDTa, the N-terminal domain spans residues 1–215 and the C-terminal domain spans residues 224–420. Both domains are composed of five α-helices and eight β-strands (Sundriyal et al.2009). The N-domain is catalytically inactive and mediates interaction of CDTa with the transport component, whereas the C-domain possesses the ADPRT activity (Gulke et al.2001).
The active site of CDTa has an RSE motif that is conserved in many bacterial ADPRTs (Fig. 9B) (Simon, Aktories and Barbieri 2014). The RSE motif consists of an arginine residue (Arg302), a Ser-Thr-Ser motif (Ser345-Thr346-Ser347) and a Glu-X-Glu motif (Glu385 and Glu387). Of these conserved residues, Arg302, Ser345, Glu385 and Glu387 have been shown to be critical for ADP-ribosylation of actin by CDTa (Gulke et al.2001). The crystal structure of CDTa in complex with NAD shows that Arg302 and Ser345 (from the Ser-Thr-Ser motif) interact directly with the ligand (Sundriyal et al.2009). NAD binding involves additional residues within the catalytic cleft that include Gln307, Asn342 and Arg359. The first Glu residue of the Glu-X-Glu motif (Glu385) has been predicted to promote the transfer of ADP-ribose to the substrate based on structural studies of actin-Ia complex (Tsuge et al.2003). The second Glu residue (Glu387) forms a strong hydrogen bond with Ser345 and likely promotes catalysis.
CELLULAR EFFECTS OF CDT INTOXICATION
CDT intoxication leads to multiple pathogenic cellular effects, which include loss of the actin-based cytoskeleton of the host cell, formation of microtubule (MT)-based cell protrusions that increase pathogen adherence, enhanced production of inflammatory cytokines and suppression of innate immune cells (Fig. 7). The CDT-induced cellular effects and the underlying mechanisms are discussed below.
Destruction of the actin cytoskeleton of epithelial cells
Actin is a 43-kDa globular protein which under physiological conditions polymerizes into helical filaments (F-actin) (Pollard and Cooper 2009). Association of three globular-actin (G-actin) monomers generates a filament nucleus, which is a prerequisite for polymerization. Actin filaments are polarized, with a fast-growing (plus or barbed) end and a slow-growing (minus or pointed) end. At steady state, there is a net polymerization at the barbed end and a net depolymerization at the pointed end (Wegner 1976; Pollard and Borisy 2003). Actin filaments contribute to the mechanical structure and shape of cells, which are critical for cellular function (Lanzetti 2007; Pollard and Cooper 2009).
ADP-ribosylation of G-actin at Arg-177 by CDT and other binary actin-ADPRTs changes the functional properties of actin. ADP-ribosylated G-actin is unable to nucleate polymerization or assemble into filaments (Aktories et al.1986; Wegner and Aktories 1988; Perieteanu et al.2010). Additionally, ADP-ribosylated G-actin acts like a capping protein and binds the barbed end of non-modified F-actin and inhibits polymerization (Wegner and Aktories 1988; Weigt et al.1989). Actin depolymerization at the pointed end, however, is not affected. The G-actin monomers released from the pointed end are substrates for the toxin. Thus, ADP-ribosylation of actin inhibits G-actin polymerization and promotes F-actin depolymerization, leading to the eventual breakdown of the actin cytoskeleton and the generation of cytopathic cell rounding (Schwan et al.2009; Papatheodorou et al.2011).
Formation of MT-based epithelial cell protrusions
ADP-ribosylation of G-actin by CDT also affects the MT system. At moderate toxin concentrations (5 ng/mL CDTa + 10 ng/mL CDTb and 20 ng/mL CDTa + 40 ng/mL CDTb), CDT induces the formation of MT-based cell protrusions, which are 0.05–0.5 μm in diameter and can extend up to a few hundred micrometers in length (Schwan et al.2009). Formation of MT-based protrusions predominantly occurs in cholesterol- and sphingolipid-rich microdomains (or lipid rafts) and requires ADP-ribosylation of actin (Schwan et al.2009, 2011). Interestingly, the cellular protrusions can be observed at time points where only a small fraction of actin was modified by the toxin and the cell morphology was not significantly altered, suggesting that complete depolymerization of the actin cytoskeleton is not necessary for this process (Schwan et al.2009). ADP-ribosylation of actin is thought to promote the formation of MT-based protrusions by causing redistribution of the MT capture proteins, which regulate MT plus-end dynamics, from the cell cortex into the cell interior (Schwan et al.2009). The protrusions induced by CDT create a dense meshwork at the cell surface and embed clostridia, thereby increasing their adherence and colonization (Schwan et al.2009). Additionally, CDT-induced ADP-ribosylation of actin causes re-routing of Rab11-positive vesicles containing fibronectin from the basolateral to the apical side of epithelial cells, where the protrusions are formed (Schwan et al.2014). Fibronectin is then released at the base and distal parts of MT protrusions. Secreted fibronectin and the meshwork formed by protrusions together contribute to increased adherence of the pathogenic bacteria. Notably, other binary actin-ADP-ribosylating toxins also induced the formation of similar MT-based cellular protrusions, suggesting that this may be a common strategy employed by this family of toxins to promote pathogen adherence and virulence (Schwan et al.2009).
Effects on immune cells
CDT enhances pathogenic host inflammation and subverts the host immune response during infection, resulting in enhanced bacterial virulence and severe disease. CDT can activate NF-κB in a RAW murine macrophage-derived cell line and can act synergistically with TcdA and TcdB to increase proinflammatory cytokine production by bone marrow-derived dendritic cells generated from C57BL6 mice (Cowardin et al.2016a). CDT-induced inflammatory cytokine production by innate immune cells requires signaling via Toll-like Receptor 2 (TLR2). Recognition of CDT by TLR2 was also shown to indirectly induce apoptosis of eosinophils, thereby suppressing a CDT-specific host eosinophilic response in the colon (Cowardin et al.2016a). Furthermore, adoptive transfer of TLR2–/– bone marrow-derived eosinophils (BM Eos), but not TLR2+/+ BM Eos, protected mice against an epidemic, CDT-producing C. difficile strain, suggesting that an eosinophilic response is protective during CDI and that CDT-mediated TLR2 signaling on eosinophils suppresses this protective response (Cowardin et al.2016a).
THERAPEUTIC STRATEGIES THAT TARGET THE TOXINS
Given that CDI is a toxin-mediated disease, there has been considerable interest in developing toxin-based therapies to extend the treatment and prevention options for CDI. The use of toxin-centric, non-antibiotic therapeutic strategies could allow for the recovery of the host microbiota while preventing toxin-induced damage. Strategies that neutralize Clostridium difficile toxins include active and passive immunotherapies and small molecule-based inhibition of toxin function. A brief overview of these approaches is provided below, but some of them are reviewed in greater detail elsewhere (Hussack and Tanha 2016; Kociolek and Gerding 2016; Martin and Wilcox 2016; Feher, Soriano and Mensa 2017).
Active vaccination
Inactivated toxoid and recombinant toxin-based vaccines are promising approaches for CDI prevention, with three vaccine candidates currently in phase II or III clinical trials. These include a toxoid-based vaccine containing formalin-inactivated TcdA and TcdB from VPI10463 (called Cdiffense) developed by Sanofi Pasteur (Phase III clinical trial identifier: NCT01887912), a genetically modified and formalin-inactivated toxoid vaccine developed by Pfizer (Phase II NCT02117570 and NCT02561195), and a recombinant fusion protein of TcdA and TcdB C-terminal domains (VLA84 or IC84) developed by Valneva Austria GmbH (Phase II NCT02316470) (Martin and Wilcox 2016; Feher, Soriano and Mensa 2017). In addition to protein-based vaccines, DNA-based immunizations against C. difficile toxins have also garnered interest (Gardiner et al.2009; Jin et al.2013; Baliban et al.2014; Zhang et al.2016). DNA vaccines encoding the C-terminal CROPS domains of TcdA and TcdB are immunogenic and protect against disease in mice and hamster models of CDI (Baliban et al.2014; Zhang et al.2016). The DNA- and protein-based vaccine formulations are typically administered intramuscularly. Orally administered mucosal CDI vaccine is another possibility, potentially using Bacillus subtilis spores as a vehicle for mucosal delivery of toxin-derived antigens (Permpoonpattana et al.2011; Hong et al.2017).
Passive immunizations using antitoxin antibodies
The presence of circulating antitoxin antibodies or an ability to mount a strong humoral immune response to toxins A and B correlates with natural protection against severe or recurrent CDI (Warny et al.1994; Kyne et al.2000, 2001; Leav et al.2010; Monaghan et al.2013). Consequently, passive immunizations with neutralizing antibodies that target different toxin domains have been tested by numerous investigators for their efficacy in treatment and prevention of CDI and subsequent recurrences (Kociolek and Gerding 2016; Feher, Soriano and Mensa 2017). Several human or humanized monoclonal antibodies and some camelid single-domain antibodies have been shown to confer significant protection against CDI in animal models, but the majority of them are not in clinical trials yet (Hussack and Tanha 2016; Feher, Soriano and Mensa 2017). One monoclonal TcdB-neutralizing antibody, bezlotoxumab, recently completed phase III clinical trials and was approved by the Food and Drug Administration for use as a supportive therapy to prevent recurrent CDI in humans (Mullard 2016; Wilcox et al.2017). Bezlotoxumab was shown to bind the C-terminal CROPs domain of TcdB and neutralize toxicity in vitro by blocking cell surface binding (Orth et al.2014). It is important to note that two of the three receptor binding sites in TcdB are located outside the CROPS. It will be interesting to determine if antibodies that target other regions of the toxins, such as the GTD or the conserved surface loop that is critical for pore formation, will be equally or more efficacious in the prevention or treatment of CDI in humans.
Small molecule-based inhibition of toxin function
The multistep mechanism of toxin action depicted in Fig. 2 provides several potential avenues for therapeutic development. Screens for small molecules that inhibit toxin function have identified several compounds, including those that can block the enzymatic activities of the toxins (Bender et al.2015; Tam et al.2015). One such small molecule, ebselen, reduced intestinal pathology in mice infected with C. difficile strain 630 (Bender et al.2015). Ebselen has a well-characterized pharmacology profile; is known to be clinically safe; and has been shown to have anti-oxidative, anti-inflammatory and anti-microbial properties (Thangamani, Younis and Seleem 2015a,b). It is unclear whether these added functional properties of ebselen also contribute to observed reduction in CDI pathology. Finally, investigations of the mechanisms underlying aberrant NOX activation and unregulated ROS production by TcdB could also lead to identification of additional targets and inhibitors of therapeutic value. The observation that N-acetylcysteine, an FDA approved antioxidant, prevented TcdB-induced tissue damage in an explant model suggests that such mechanism-based inhibitors might be effective in limiting toxin-mediated tissue damage during infection (Farrow et al.2013).
CONCLUDING REMARKS
In the last decade, significant progress has been made in elucidating structural and molecular mechanisms of TcdA, TcdB and CDT and in defining their role in disease. In this section, we highlight some of the long-standing questions that remain and new questions that have arisen from recent discoveries. Receptor binding is the first step in the intoxication mechanism. While TcdB and CDT receptors are known, the epithelial cell receptor for TcdA has yet to be identified and represents an important question for future study. Three receptors have been recently identified for TcdB but their relative contributions to TcdB-mediated disease pathologies remain to be elucidated. How the Clostridium difficile toxins deliver their effector domains to the host cytosol is also not known. Understanding the mechanisms underlying pore formation and cargo translocation continue to be a priority in the field. While in vitro studies have identified the GTPase targets for TcdA, TcdB and TcdB variants, the determinants of GTPase substrate specificity and its impact on the observed pathologies are not known and need to be investigated. Furthermore, the discovery that TcdB is capable of causing a necrotic cell death independent of its glucosyltransferase activity also raises some exciting questions for future study. Elucidating how TcdB is able to induce aberrant NOX activation and ROS production could lead to identification of strategies that block TcdB-induced tissue damage. Finally, most studies investigating mechanism of action and cellular effects of C. difficile toxins have been performed by intoxicating cells or animals with individual toxins. We are just beginning to appreciate how the three C. difficile toxins work synergistically to enhance pathology. Future studies in this area will be critical toward understanding C. difficile pathogenesis and will have important implications for therapeutic development.
Acknowledgements
We thank the Lacy lab members Dave Anderson, Joe Alvin, and Mike Sheedlo for their helpful suggestions.
FUNDING
This work was supported by the National Institutes of Health [R01 AI095755 to DBL] and the Department of Veterans Affairs [BX002943 to DBL].
Conflict of interest. None declared.
REFERENCES
- Aktories K, Barbieri JT. Bacterial cytotoxins: targeting eukaryotic switches. Nat Rev Microbiol 2005;3:397–410. [DOI] [PubMed] [Google Scholar]
- Aktories K, Barmann M, Ohishi I et al. Botulinum C2 toxin ADP-ribosylates actin. Nature 1986;322:390–2. [DOI] [PubMed] [Google Scholar]
- Alcantara C, Stenson WF, Steiner TS et al. Role of inducible cyclooxygenase and prostaglandins in Clostridium difficile toxin A-induced secretion and inflammation in an animal model. J Infect Dis 2001;184:648–52. [DOI] [PubMed] [Google Scholar]
- Aldape MJ, Packham AE, Nute DW et al. Effects of ciprofloxacin on the expression and production of exotoxins by Clostridium difficile. J Med Microbiol 2013;62:741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvin JW, Lacy DB. Clostridium difficile toxin glucosyltransferase domains in complex with a non-hydrolyzable UDP-glucose analogue. J Struct Biol 2017;198:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando Y, Yasuda S, Oceguera-Yanez F et al. Inactivation of Rho GTPases with Clostridium difficile toxin B impairs centrosomal activation of Aurora-A in G2/M transition of HeLa cells. Mol Biol Cell 2007;18:3752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androga GO, Hart J, Foster NF et al. Infection with toxin A-negative, toxin B-negative, binary toxin-positive Clostridium difficile in a young patient with ulcerative colitis. J Clin Microbiol 2015;53:3702–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton PM, Gay J, Mykoniatis A et al. Corticotropin-releasing hormone (CRH) requirement in Clostridium difficile toxin A-mediated intestinal inflammation. P Natl Acad Sci USA 2004;101:8503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes A, Camiade E, Monot M et al. Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res 2012;40:10701–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni A, Prior IA, Lindsay M et al. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol 2000;20:2475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry A, Hussack G, Chen W et al. Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect Immun 2012;80:3521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad MM, Johanesen PA, Carter GP et al. Clostridium difficile virulence factors: Insights into an anaerobic spore-forming pathogen. Gut Microbes 2014;5:579–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock GJ, Broering TJ, Hernandez HJ et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun 2006;74:6339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci S, Molbak K, Kjeldsen MK et al. Binary toxin and death after Clostridium difficile infection. Emerg Infect Dis 2011;17:976–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliban SM, Michael A, Shammassian B et al. An optimized, synthetic DNA vaccine encoding the toxin A and toxin B receptor binding domains of Clostridium difficile induces protective antibody responses in vivo. Infect Immun 2014;82:4080–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbut F, Gariazzo B, Bonne L et al. Clinical features of Clostridium difficile-associated infections and molecular characterization of strains: results of a retrospective study, 2000–2004. Infect Cont Hosp Ep 2007;28:131–9. [DOI] [PubMed] [Google Scholar]
- Barroso LA, Moncrief JS, Lyerly DM et al. Mutagenesis of the Clostridium difficile toxin B gene and effect on cytotoxic activity. Microbial Pathog 1994;16:297–303. [DOI] [PubMed] [Google Scholar]
- Barth H, Aktories K, Popoff MR et al. Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol R 2004;68:373–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H, Pfeifer G, Hofmann F et al. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J Biol Chem 2001;276:10670–6. [DOI] [PubMed] [Google Scholar]
- Bartlett JG, Moon N, Chang TW et al. Role of Clostridium difficile in antibiotic-associated pseudomembranous colitis. Gastroenterology 1978;75:778–82. [PubMed] [Google Scholar]
- Bender KO, Garland M, Ferreyra JA et al. A small-molecule antivirulence agent for treating Clostridium difficile infection. Sci Transl Med 2015;7:306ra148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson EL, Huynh PD, Finkelstein A et al. Identification of residues lining the anthrax protective antigen channel. Biochemistry 1998;37:3941–3948. [DOI] [PubMed] [Google Scholar]
- Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J 2000;348 Pt 2:241–55. [PMC free article] [PubMed] [Google Scholar]
- Blocker D, Behlke J, Aktories K et al. Cellular uptake of the Clostridium perfringens binary iota-toxin. Infect Immun 2001;69:2980–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobo LD, El Feghaly RE, Chen YS et al. MAPK-activated protein kinase 2 contributes to Clostridium difficile-associated inflammation. Infect Immun 2013;81:713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillaut L, Dubois T, Sonenshein AL et al. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol 2015;166:375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branka JE, Vallette G, Jarry A et al. Early functional effects of Clostridium difficile toxin A on human colonocytes. Gastroenterology 1997;112:1887–94. [DOI] [PubMed] [Google Scholar]
- Braun V, Hundsberger T, Leukel P et al. Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene 1996;181:29–38. [DOI] [PubMed] [Google Scholar]
- Brito GA, Carneiro-Filho B, Oria RB et al. Clostridium difficile toxin A induces intestinal epithelial cell apoptosis and damage: role of Gln and Ala-Gln in toxin A effects. Digest Dis Sci 2005;50:1271–8. [DOI] [PubMed] [Google Scholar]
- Brito GA, Fujji J, Carneiro-Filho BA et al. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis 2002;186:1438–47. [DOI] [PubMed] [Google Scholar]
- Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res 2005;11:3155–62. [DOI] [PubMed] [Google Scholar]
- Brouwer MS, Roberts AP, Hussain H et al. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat Commun 2013;4:2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonomo EL, Madan R, Pramoonjago P et al. Role of interleukin 23 signaling in Clostridium difficile colitis. J Infect Dis 2013;208:917–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch C, Hofmann F, Gerhard R et al. Involvement of a conserved tryptophan residue in the UDP-glucose binding of large clostridial cytotoxin glycosyltransferases. J Biol Chem 2000;275:13228–34. [DOI] [PubMed] [Google Scholar]
- Busch C, Hofmann F, Selzer J et al. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J Biol Chem 1998;273:19566–72. [DOI] [PubMed] [Google Scholar]
- Calderon GM, Torres-Lopez J, Lin TJ et al. Effects of toxin A from Clostridium difficile on mast cell activation and survival. Infect Immun 1998;66:2755–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro BA, Fujii J, Brito GA et al. Caspase and bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect Immun 2006;74:81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GP, Chakravorty A, Pham Nguyen TA et al. Defining the roles of TcdA and TcdB in localized gastrointestinal disease, systemic organ damage, and the host response during Clostridium difficile infections. mBio 2015;6:e00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GP, Douce GR, Govind R et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 2011;7:e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GP, Lyras D, Allen DL et al. Binary toxin production in Clostridium difficile is regulated by CdtR, a LytTR family response regulator. J Bacteriol 2007;189:7290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagliuolo I, Keates AC, Qiu B et al. Increased substance P responses in dorsal root ganglia and intestinal macrophages during Clostridium difficile toxin A enteritis in rats. P Natl Acad Sci USA 1997;94:4788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagliuolo I, LaMont JT, Letourneau R et al. Neuronal involvement in the intestinal effects of Clostridium difficile toxin A and Vibrio cholerae enterotoxin in rat ileum. Gastroenterology 1994;107:657–65. [DOI] [PubMed] [Google Scholar]
- Castagliuolo I, Riegler M, Pasha A et al. Neurokinin-1 (NK-1) receptor is required in Clostridium difficile- induced enteritis. J Clin Invest 1998;101:1547–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagliuolo I, Wang CC, Valenick L et al. Neurotensin is a proinflammatory neuropeptide in colonic inflammation. J Clin Invest 1999;103:843–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran R, Kenworthy AK, Lacy DB. Clostridium difficile toxin A undergoes clathrin-independent, PACSIN2-dependent endocytosis. PLoS Pathog 2016;12:e1006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SY, Song KP. ADP-ribosylating binary toxin genes of Clostridium difficile strain CCUG 20309. DNA Seq 2001;12:115–20. [DOI] [PubMed] [Google Scholar]
- Chaves-Olarte E, Freer E, Parra A et al. R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J Biol Chem 2003;278:7956–63. [DOI] [PubMed] [Google Scholar]
- Chaves-Olarte E, Low P, Freer E et al. A novel cytotoxin from Clostridium difficile serogroup F is a functional hybrid between two other large clostridial cytotoxins. J Biol Chem 1999;274:11046–52. [DOI] [PubMed] [Google Scholar]
- Chaves-Olarte E, Weidmann M, Eichel-Streiber C et al. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J Clin Invest 1997;100:1734–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Kokkotou EG, Mustafa N et al. Saccharomyces boulardii inhibits ERK1/2 mitogen-activated protein kinase activation both in vitro and in vivo and protects against Clostridium difficile toxin A-induced enteritis. J Biol Chem 2006;281:24449–54. [DOI] [PubMed] [Google Scholar]
- Chilton CH, Freeman J, Crowther GS et al. Co-amoxiclav induces proliferation and cytotoxin production of Clostridium difficile ribotype 027 in a human gut model. J Antimicrob Chemoth 2012;67:951–4. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ 2006;13:994–1002. [DOI] [PubMed] [Google Scholar]
- Chumbler NM, Farrow MA, Lapierre LA et al. Clostridium difficile toxins TcdA and TcdB cause colonic tissue damage by distinct mechanisms. Infect Immun 2016a;84:2871–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumbler NM, Farrow MA, Lapierre LA et al. Clostridium difficile Toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 2012;8:e1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumbler NM, Rutherford SA, Zhang Z et al. Crystal structure of Clostridium difficile toxin A. Nat Microbiol 2016b;1. [DOI] [PubMed] [Google Scholar]
- Collier RJ, Young JA. Anthrax toxin. Annu Rev Cell Dev Bi 2003;19:45–70. [DOI] [PubMed] [Google Scholar]
- Cowardin CA, Buonomo EL, Saleh MM et al. The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat Microbiol 2016a;1:16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowardin CA, Jackman BM, Noor Z et al. Glucosylation drives the innate inflammatory response to Clostridium difficile toxin A. Infect Immun 2016b;84:2317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowardin CA, Kuehne SA, Buonomo EL et al. Inflammasome activation contributes to interleukin-23 production in response to Clostridium difficile. mBio 2015;6:e02386–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven R, Lacy DB. Clostridium sordellii lethal-toxin autoprocessing and membrane localization activities drive GTPase glucosylation profiles in endothelial cells. mSphere 2016;1:e00012–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Auria KM, Bloom MJ, Reyes Y et al. High temporal resolution of glucosyltransferase dependent and independent effects of Clostridium difficile toxins across multiple cell types. BMC Microbiol 2015;15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Auria KM, Donato GM, Gray MC et al. Systems analysis of the transcriptional response of human ileocecal epithelial cells to Clostridium difficile toxins and effects on cell cycle control. BMC Syst Biol 2012;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Urzo N, Malito E, Biancucci M et al. The structure of Clostridium difficile toxin A glucosyltransferase domain bound to Mn2+ and UDP provides insights into glucosyltransferase activity and product release. FEBS J 2012;279:3085–97. [DOI] [PubMed] [Google Scholar]
- Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta 2010;1797:897–906. [DOI] [PubMed] [Google Scholar]
- Darkoh C, DuPont HL, Norris SJ et al. Toxin synthesis by Clostridium difficile is regulated through quorum signaling. mBio 2015;6:e02569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineen SS, Villapakkam AC, Nordman JT et al. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 2007;66:206–19. [DOI] [PubMed] [Google Scholar]
- Dingle KE, Griffiths D, Didelot X et al. Clinical Clostridium difficile: clonality and pathogenicity locus diversity. PLoS One 2011;6:e19993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle T, Wee S, Mulvey GL et al. Functional properties of the carboxy-terminal host cell-binding domains of the two toxins, TcdA and TcdB, expressed by Clostridium difficile. Glycobiology 2008;18:698–706. [DOI] [PubMed] [Google Scholar]
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem 2009;78:857–902. [DOI] [PubMed] [Google Scholar]
- Drudy D, Fanning S, Kyne L. Toxin A-negative, toxin B-positive Clostridium difficile. Int J Infect Dis 2007;11:5–10. [DOI] [PubMed] [Google Scholar]
- Dupuy B, Sonenshein AL. Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 1998;27:107–20. [DOI] [PubMed] [Google Scholar]
- Eckert C, Emirian A, Le Monnier A et al. Prevalence and pathogenicity of binary toxin-positive Clostridium difficile strains that do not produce toxins A and B. New Microbes New Infect 2015;3:12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AN, Tamayo R, McBride SM. A novel regulator controls Clostridium difficile sporulation, motility and toxin production. Mol Microbiol 2016;100:954–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerer M, Giesemann T, Jank T et al. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J Biol Chem 2007;282:25314–21. [DOI] [PubMed] [Google Scholar]
- Egerer M, Satchell KJ. Inositol hexakisphosphate-induced autoprocessing of large bacterial protein toxins. PLoS Pathog 2010;6:e1000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Feghaly RE, Stauber JL, Deych E et al. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin Infect Dis 2013;56:1713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hawiet A, Kitova EN, Kitov PI et al. Binding of Clostridium difficile toxins to human milk oligosaccharides. Glycobiology 2011;21:1217–27. [DOI] [PubMed] [Google Scholar]
- El Meouche I, Peltier J, Monot M et al. Characterization of the SigD regulon of C. difficile and its positive control of toxin production through the regulation of tcdR. PLoS One 2013;8:e83748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Reed R, Chang BJ et al. Bacteremia with a large clostridial toxin-negative, binary toxin-positive strain of Clostridium difficile. Anaerobe 2009;15:249–51. [DOI] [PubMed] [Google Scholar]
- Ernst K, Langer S, Kaiser E et al. Cyclophilin-facilitated membrane translocation as pharmacological target to prevent intoxication of mammalian cells by binary clostridial actin ADP-ribosylated toxins. J Mol Biol 2015;427:1224–38. [DOI] [PubMed] [Google Scholar]
- Ernst K, Schmid J, Beck M et al. Hsp70 facilitates trans-membrane transport of bacterial ADP-ribosylating toxins into the cytosol of mammalian cells. Sci Rep 2017;7:2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002;420:629–35. [DOI] [PubMed] [Google Scholar]
- Farrow MA, Chumbler NM, Lapierre LA et al. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. P Natl Acad Sci USA 2013;110:18674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawley WN, Knetsch CW, MacCannell DR et al. Development and validation of an internationally-standardized, high-resolution capillary gel-based electrophoresis PCR-ribotyping protocol for Clostridium difficile. PLoS One 2015;10:e0118150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher C, Soriano A, Mensa J. A review of experimental and off-label therapies for Clostridium difficile infection. Infect Dis Therapy 2017;6:1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltis BA, Wiesner SM, Kim AS et al. Clostridium difficile toxins A and B can alter epithelial permeability and promote bacterial paracellular migration through HT-29 enterocytes. Shock 2000;14:629–34. [DOI] [PubMed] [Google Scholar]
- Fettucciari K, Ponsini P, Gioe D et al. Enteric glial cells are susceptible to Clostridium difficile toxin B. Cell Mol Life Sci 2017;74:1527–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini C, Fabbri A, Falzano L et al. Clostridium difficile toxin B induces apoptosis in intestinal cultured cells. Infect Immun 1998;66:2660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegel WA, Muller F, Daubener W et al. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect Immun 1991;59:3659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin I, Thelestam M. Internalization of Clostridium difficile cytotoxin into cultured human lung fibroblasts. Biochim Biophys Acta 1983;763:383–392. [DOI] [PubMed] [Google Scholar]
- Florin I, Thelestam M. Lysosomal involvement in cellular intoxication with Clostridium difficile toxin B. Microb Pathogenesis 1986;1:373–85. [DOI] [PubMed] [Google Scholar]
- Freeman J, Baines SD, Saxton K et al. Effect of metronidazole on growth and toxin production by epidemic Clostridium difficile PCR ribotypes 001 and 027 in a human gut model. J Antimicrob Chemoth 2007;60:83–91. [DOI] [PubMed] [Google Scholar]
- Frisch C, Gerhard R, Aktories K et al. The complete receptor-binding domain of Clostridium difficile toxin A is required for endocytosis. Biochem Bioph Res Co 2003;300:706–11. [DOI] [PubMed] [Google Scholar]
- Gardiner DF, Rosenberg T, Zaharatos J et al. A DNA vaccine targeting the receptor-binding domain of Clostridium difficile toxin A. Vaccine 2009;27:3598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Ahrens S, Satchell KJ. Plasma membrane association of three classes of bacterial toxins is mediated by a basic-hydrophobic motif. Cell Microbiol 2012;14:286–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Tungekar R, Satchell KJ. Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. P Natl Acad Sci USA 2010;107:5581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genisyuerek S, Papatheodorou P, Guttenberg G et al. Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol Microbiol 2011;79:1643–54. [DOI] [PubMed] [Google Scholar]
- Genth H, Aktories K, Just I. Monoglucosylation of RhoA at threonine 37 blocks cytosol-membrane cycling. J Biol Chem 1999;274:29050–6. [DOI] [PubMed] [Google Scholar]
- Genth H, Hofmann F, Selzer J et al. Difference in protein substrate specificity between hemorrhagic toxin and lethal toxin from Clostridium sordellii. Biochem Bioph Res Commun 1996;229:370–4. [DOI] [PubMed] [Google Scholar]
- Genth H, Pauillac S, Schelle I et al. Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell Microbiol 2014;16:1706–21. [DOI] [PubMed] [Google Scholar]
- Geny B, Popoff MR. Bacterial protein toxins and lipids: pore formation or toxin entry into cells. Biol Cell 2006;98:667–78. [DOI] [PubMed] [Google Scholar]
- George WL, Sutter VL, Goldstein EJ et al. Aetiology of antimicrobial-agent-associated colitis. Lancet 1978;1:802–3. [DOI] [PubMed] [Google Scholar]
- Gerding DN, Johnson S, Rupnik M et al. Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 2014;5:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard R, Frenzel E, Goy S et al. Cellular uptake of Clostridium difficile TcdA and truncated TcdA lacking the receptor binding domain. J Med Microbiol 2013;62:1414–22. [DOI] [PubMed] [Google Scholar]
- Gerhard R, Nottrott S, Schoentaube J et al. Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J Med Microbiol 2008;57:765–70. [DOI] [PubMed] [Google Scholar]
- Geric B, Carman RJ, Rupnik M et al. Binary toxin-producing, large clostridial toxin-negative Clostridium difficile strains are enterotoxic but do not cause disease in hamsters. J Infect Dis 2006;193:1143–50. [DOI] [PubMed] [Google Scholar]
- Geric B, Johnson S, Gerding DN et al. Frequency of binary toxin genes among Clostridium difficile strains that do not produce large clostridial toxins. J Clin Microbiol 2003;41:5227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer M, Wilde C, Selzer J et al. Glucosylation of Ras by Clostridium sordellii lethal toxin: consequences for effector loop conformations observed by NMR spectroscopy. Biochemistry 2003;42:11951–9. [DOI] [PubMed] [Google Scholar]
- Gibert M, Petit L, Raffestin S et al. Clostridium perfringens iota-toxin requires activation of both binding and enzymatic components for cytopathic activity. Infect Immun 2000;68:3848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giel JL, Sorg JA, Sonenshein AL et al. Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS One 2010;5:e8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesemann T, Jank T, Gerhard R et al. Cholesterol-dependent pore formation of Clostridium difficile toxin A. J Biol Chem 2006;281:10808–15. [DOI] [PubMed] [Google Scholar]
- Govind R, Dupuy B. Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog 2012;8:e1002727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind R, Fitzwater L, Nichols R. Observations on the role of TcdE isoforms in Clostridium difficile toxin secretion. J Bacteriol 2015;197:2600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco A, Ho JG, Lin SJ et al. Carbohydrate recognition by Clostridium difficile toxin A. Nat Struct Mol Biol 2006;13:460–1. [DOI] [PubMed] [Google Scholar]
- Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Gene Dev 2005;19:877–90. [DOI] [PubMed] [Google Scholar]
- Gulke I, Pfeifer G, Liese J et al. Characterization of the enzymatic component of the ADP-ribosyltransferase toxin CDTa from Clostridium difficile. Infect Immun 2001;69:6004–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Zhang Z, Sugiman-Marangos SN et al. Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor binding determinants. J Biol Chem 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale ML, Marvaud JC, Popoff MR et al. Detergent-resistant membrane microdomains facilitate Ib oligomer formation and biological activity of Clostridium perfringens iota-toxin. Infect Immun 2004;72:2186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall IC, O’Toole E. Intestinal flora in new-born infants: with a description of a new pathogenic anaerobe, Bacillus difficilis. Am J Dis Child 1935;49:390–402. [Google Scholar]
- Hamm EE, Voth DE, Ballard JD. Identification of Clostridium difficile toxin B cardiotoxicity using a zebrafish embryo model of intoxication. P Natl Acad Sci USA 2006;103:14176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GA, Johnson JL. The toxigenic element of Clostridium difficile strain VPI 10463. Microb Pathogenesis 1995;19:203–13. [DOI] [PubMed] [Google Scholar]
- Han S, Craig JA, Putnam CD et al. Evolution and mechanism from structures of an ADP-ribosylating toxin and NAD complex. Nat Struct Biol 1999;6:932–6. [DOI] [PubMed] [Google Scholar]
- Hansen A, Alston L, Tulk SE et al. The P2Y6 receptor mediates Clostridium difficile toxin-induced CXCL8/IL-8 production and intestinal epithelial barrier dysfunction. PLoS One 2013;8:e81491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Howard G, Nichols BJ. Pacsin 2 is recruited to caveolae and functions in caveolar biogenesis. J Cell Sci 2011;124:2777–85. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kamada N, Jiao Y et al. Protective role of commensals against Clostridium difficile infection via an IL-1beta-mediated positive-feedback loop. J Immunol 2012;189:3085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Yamazaki T, Kamada N et al. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 2011;186:4872–80. [DOI] [PubMed] [Google Scholar]
- Haug G, Aktories K, Barth H. The host cell chaperone Hsp90 is necessary for cytotoxic action of the binary iota-like toxins. Infect Immun 2004;72:3066–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug G, Leemhuis J, Tiemann D et al. The host cell chaperone Hsp90 is essential for translocation of the binary Clostridium botulinum C2 toxin into the cytosol. J Biol Chem 2003;278:32266–74. [DOI] [PubMed] [Google Scholar]
- He D, Hagen SJ, Pothoulakis C et al. Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology 2000;119:139–50. [DOI] [PubMed] [Google Scholar]
- He D, Sougioultzis S, Hagen S et al. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 2002;122:1048–57. [DOI] [PubMed] [Google Scholar]
- He M, Miyajima F, Roberts P et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 2013;45:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht G, Koutsouris A, Pothoulakis C et al. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 1992;102:416–23. [DOI] [PubMed] [Google Scholar]
- Hecht G, Pothoulakis C, LaMont JT et al. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest 1988;82:1516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmasi S, Czulkies BA, Schorch B et al. Interaction of the Clostridium difficile binary toxin CDT and its host cell receptor, lipolysis-stimulated lipoprotein receptor (LSR). J Biol Chem 2015;290:14031–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques B, Florin I, Thelestam M. Cellular internalisation of Clostridium difficile toxin A. Microb Pathogenesis 1987;2:455–63. [DOI] [PubMed] [Google Scholar]
- Hernandez LD, Kroh HK, Hsieh E et al. Epitopes and mechanism of action of the Clostridium difficile toxin A-neutralizing antibody actoxumab. J Mol Biol 2017;429:1030–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C, Ahmadian MR, Hofmann F et al. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J Biol Chem 1998;273:16134–9. [DOI] [PubMed] [Google Scholar]
- Hirota SA, Iablokov V, Tulk SE et al. Intrarectal instillation of Clostridium difficile toxin A triggers colonic inflammation and tissue damage: development of a novel and efficient mouse model of Clostridium difficile toxin exposure. Infect Immun 2012;80:4474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JG, Greco A, Rupnik M et al. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. P Natl Acad Sci USA 2005;102:18373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Rex G, Aktories K et al. The ras-related protein Ral is monoglucosylated by Clostridium sordellii lethal toxin. Biochem Bioph Res Commun 1996;227:77–81. [DOI] [PubMed] [Google Scholar]
- Hong HA, Hitri K, Hosseini S et al. Mucosal antibodies to the C terminus of toxin A prevent colonization of Clostridium difficile. Infect Immun 2017;85:e01060–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes MT, Mayor S, Parton RG. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr Opin Cell Biol 2010;22:519–27. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck SC, May M, Schmidt G et al. Inhibition of cytokinesis by Clostridium difficile toxin B and cytotoxic necrotizing factors–reinforcing the critical role of RhoA in cytokinesis. Cell Motil Cytoskel 2009;66:967–75. [DOI] [PubMed] [Google Scholar]
- Hundsberger T, Braun V, Weidmann M et al. Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur J Biochem 1997;244:735–42. [DOI] [PubMed] [Google Scholar]
- Hussack G, Arbabi-Ghahroudi M, van Faassen H et al. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J Biol Chem 2011;286:8961–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussack G, Tanha J. An update on antibody-based immunotherapies for Clostridium difficile infection. Clin Exp Gastroenterol 2016;9:209–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara K, Muraguchi S, Kato M et al. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J Biol Chem 1998;273:9656–66. [DOI] [PubMed] [Google Scholar]
- Irvine RF, Schell MJ. Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Bio 2001;2:327–38. [DOI] [PubMed] [Google Scholar]
- Jafari NV, Kuehne SA, Bryant CE et al. Clostridium difficile modulates host innate immunity via toxin-independent and dependent mechanism(s). PLoS One 2013;8:e69846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jank T, Giesemann T, Aktories K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology 2007a;17:15r–22r. [DOI] [PubMed] [Google Scholar]
- Jank T, Giesemann T, Aktories K. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J Biol Chem 2007b;282:35222–31. [DOI] [PubMed] [Google Scholar]
- Jank T, Reinert DJ, Giesemann T et al. Change of the donor substrate specificity of Clostridium difficile toxin B by site-directed mutagenesis. J Biol Chem 2005;280:37833–8. [DOI] [PubMed] [Google Scholar]
- Jarchum I, Liu M, Shi C et al. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun 2012;80:2989–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson KK, Smith MF Jr, Bobak DA. Roles of intracellular calcium and NF-kappa B in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J Immunol 1999;163:5183–91. [PubMed] [Google Scholar]
- Jiang J, Pentelute BL, Collier RJ et al. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 2015;521:545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Wang S, Zhang C et al. Protective antibody responses against Clostridium difficile elicited by a DNA vaccine expressing the enzymatic domain of toxin B. Hum Vacc Immunother 2013;9:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just I, Selzer J, Hofmann F et al. Inactivation of Ras by Clostridium sordellii lethal toxin-catalyzed glucosylation. J Biol Chem 1996;271:10149–53. [DOI] [PubMed] [Google Scholar]
- Just I, Selzer J, Wilm M et al. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995a;375:500–503. [DOI] [PubMed] [Google Scholar]
- Just I, Wilm M, Selzer J et al. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 1995b;270:13932–6. [DOI] [PubMed] [Google Scholar]
- Kaiser E, Kroll C, Ernst K et al. Membrane translocation of binary actin-ADP-ribosylating toxins from Clostridium difficile and Clostridium perfringens is facilitated by cyclophilin A and Hsp90. Infect Immun 2011;79:3913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser E, Pust S, Kroll C et al. Cyclophilin A facilitates translocation of the Clostridium botulinum C2 toxin across membranes of acidified endosomes into the cytosol of mammalian cells. Cell Microbiol 2009;11:780–95. [DOI] [PubMed] [Google Scholar]
- Karasawa T, Maegawa T, Nojiri T et al. Effect of arginine on toxin production by Clostridium difficile in defined medium. Microbiol Immunol 1997;41:581–5. [DOI] [PubMed] [Google Scholar]
- Karlsson S, Burman LG, Akerlund T. Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 1999;145(Pt 7):1683–93. [DOI] [PubMed] [Google Scholar]
- Karlsson S, Dupuy B, Mukherjee K et al. Expression of Clostridium difficile toxins A and B and their sigma factor TcdD is controlled by temperature. Infect Immun 2003;71:1784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S, Lindberg A, Norin E et al. Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun 2000;68:5881–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CP, Becker S, Linevsky JK et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest 1994a;93:1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CP, Keates S, Siegenberg D et al. IL-8 secretion and neutrophil activation by HT-29 colonic epithelial cells. Am J Physiol 1994b;267:G991–7. [DOI] [PubMed] [Google Scholar]
- Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol 2011;60:1070–9. [DOI] [PubMed] [Google Scholar]
- Khanna S, Pardi DS. Community-acquired Clostridium difficile infection: an emerging entity. Clin Infect Dis 2012;55:1741–2. [DOI] [PubMed] [Google Scholar]
- Kim H, Kokkotou E, Na X et al. Clostridium difficile toxin A-induced colonocyte apoptosis involves p53-dependent p21(WAF1/CIP1) induction via p38 mitogen-activated protein kinase. Gastroenterology 2005a;129:1875–88. [DOI] [PubMed] [Google Scholar]
- Kim H, Rhee SH, Kokkotou E et al. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J Biol Chem 2005b;280:21237–45. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kim JS, Jun HC et al. Differential expression and polarized secretion of CXC and CC chemokines by human intestinal epithelial cancer cell lines in response to Clostridium difficile toxin A. Microbiol Immunol 2002;46:333–42. [DOI] [PubMed] [Google Scholar]
- Kim JM, Lee JY, Yoon YM et al. NF-kappa B activation pathway is essential for the chemokine expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. Scand J Immunol 2006;63:453–60. [DOI] [PubMed] [Google Scholar]
- Kim PH, Iaconis JP, Rolfe RD. Immunization of adult hamsters against Clostridium difficile-associated ileocecitis and transfer of protection to infant hamsters. Infect Immun 1987;55:2984–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AM, Mackin KE, Lyras D. Emergence of toxin A-negative, toxin B-positive Clostridium difficile strains: epidemiological and clinical considerations. Future Microbiol 2015;10:1–4. [DOI] [PubMed] [Google Scholar]
- Kirkwood KS, Bunnett NW, Maa J et al. Deletion of neutral endopeptidase exacerbates intestinal inflammation induced by Clostridium difficile toxin A. Am J Physiol–Gastr L 2001;281:G544–51. [DOI] [PubMed] [Google Scholar]
- Knapp O, Benz R, Gibert M et al. Interaction of Clostridium perfringens iota-toxin with lipid bilayer membranes. Demonstration of channel formation by the activated binding component Ib and channel block by the enzyme component Ia. J Biol Chem 2002;277:6143–52. [DOI] [PubMed] [Google Scholar]
- Knapp O, Benz R, Popoff MR. Pore-forming activity of clostridial binary toxins. Biochim Biophys Acta 2016;1858:512–25. [DOI] [PubMed] [Google Scholar]
- Knapp O, Maier E, Waltenberger E et al. Residues involved in the pore-forming activity of the Clostridium perfringens iota toxin. Cell Microbiol 2015;17:288–302. [DOI] [PubMed] [Google Scholar]
- Kociolek LK, Gerding DN. Breakthroughs in the treatment and prevention of Clostridium difficile infection. Nat Rev Gastro Hepat 2016;13:150–60. [DOI] [PubMed] [Google Scholar]
- Kreimeyer I, Euler F, Marckscheffel A et al. Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A. N-S Arch Pharmacol 2011;383:253–62. [DOI] [PubMed] [Google Scholar]
- Krivan HC, Clark GF, Smith DF et al. Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1–3Gal beta 1–4GlcNAc. Infect Immun 1986;53:573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroh HK, Chandrasekaran R, Rosenthal K et al. Use of a neutralizing antibody helps identify structural features critical for binding of Clostridium difficile toxin TcdA to the host cell surface. J Biol Chem 2017;292:14401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne SA, Cartman ST, Heap JT et al. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010;467:711–3. [DOI] [PubMed] [Google Scholar]
- Kuehne SA, Collery MM, Kelly ML et al. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis 2014;209:83–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurose I, Pothoulakis C, LaMont JT et al. Clostridium difficile toxin A-induced microvascular dysfunction. Role of histamine. J Clin Invest 1994;94:1919–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyne L, Warny M, Qamar A et al. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. New Engl J Med 2000;342:390–7. [DOI] [PubMed] [Google Scholar]
- Kyne L, Warny M, Qamar A et al. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 2001;357:189–93. [DOI] [PubMed] [Google Scholar]
- Lacy DB, Wigelsworth DJ, Melnyk RA et al. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. P Natl Acad Sci USA 2004;101:13147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFrance ME, Farrow MA, Chandrasekaran R et al. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. P Natl Acad Sci USA 2015;112:7073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert GS, Baldwin MR. Evidence for dual receptor-binding sites in Clostridium difficile toxin A. FEBS Lett 2016;590:4550–63. [DOI] [PubMed] [Google Scholar]
- Lanzetti L. Actin in membrane trafficking. Curr Opin Cell Biol 2007;19:453–8. [DOI] [PubMed] [Google Scholar]
- Lawson PA, Citron DM, Tyrrell KL et al. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prevot 1938. Anaerobe 2016;40:95–9. [DOI] [PubMed] [Google Scholar]
- Leav BA, Blair B, Leney M et al. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 2010;28:965–9. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kim H, Cha MY et al. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J Mol Med 2009;87:169–80. [DOI] [PubMed] [Google Scholar]
- Lee JY, Park HR, Oh YK et al. Effects of transcription factor activator protein-1 on interleukin-8 expression and enteritis in response to Clostridium difficile toxin A. J Mol Med 2007;85:1393–404. [DOI] [PubMed] [Google Scholar]
- Lessa FC, Mu Y, Bamberg WM et al. Burden of Clostridium difficile infection in the United States. New Engl J Med 2015;372:825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzzi R, Spencer J, Buckley A et al. Protective efficacy induced by recombinant Clostridium difficile toxin fragments. Infect Immun 2013;81:2851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Shi L, Yang Z et al. Cytotoxicity of Clostridium difficile toxin B does not require cysteine protease-mediated autocleavage and release of the glucosyltransferase domain into the host cell cytosol. Pathog Dis 2013;67:11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Shi L, Yang Z et al. Critical roles of Clostridium difficile toxin B enzymatic activities in pathogenesis. Infect Immun 2015;83:502–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linevsky JK, Pothoulakis C, Keates S et al. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am J Physiol 1997;273:G1333–40. [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Shen A, Bogyo M et al. Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 2008;322:265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyerly DM, Johnson JL, Frey SM et al. Vaccination against lethal Clostridium difficile enterocolitis with a nontoxic recombinant peptide of toxin A. Curr Microbiol 1990;21:29–32. [Google Scholar]
- Lyerly DM, Lockwood DE, Richardson SH et al. Biological activities of toxins A and B of Clostridium difficile. Infect Immun 1982;35:1147–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyerly DM, Phelps CJ, Toth J et al. Characterization of toxins A and B of Clostridium difficile with monoclonal antibodies. Infect Immun 1986;54:70–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyerly DM, Saum KE, MacDonald DK et al. Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun 1985;47:349–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon SA, Hutton ML, Rood JI et al. CdtR regulates TcdA and TcdB production in Clostridium difficile. PLoS Pathog 2016;12:e1005758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyras D, O’Connor JR, Howarth PM et al. Toxin B is essential for virulence of Clostridium difficile. Nature 2009;458:1176–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchioni L, Davidescu M, Fettucciari K et al. Enteric glial cells counteract Clostridium difficile Toxin B through a NADPH oxidase/ROS/JNK/caspase-3 axis, without involving mitochondrial pathways. Sci Rep 2017;7:45569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott AJ, Falkowski NR, McDonald RA et al. Interleukin-23 (IL-23), independent of IL-17 and IL-22, drives neutrophil recruitment and innate inflammation during Clostridium difficile colitis in mice. Immunology 2016;147:114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, He X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harbor Persp Biol 2012;4:a007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEllistrem MC, Carman RJ, Gerding DN et al. A hospital outbreak of Clostridium difficile disease associated with isolates carrying binary toxin genes. Clin Infect Dis 2005;40:265–72. [DOI] [PubMed] [Google Scholar]
- McKee RW, Mangalea MR, Purcell EB et al. The second messenger cyclic Di-GMP regulates Clostridium difficile toxin production by controlling expression of sigD. J Bacteriol 2013;195:5174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackin KE, Carter GP, Howarth P et al. Spo0A differentially regulates toxin production in evolutionarily diverse strains of Clostridium difficile. PloS One 2013;8:e79666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahida YR, Galvin A, Makh S et al. Effect of Clostridium difficile toxin A on human colonic lamina propria cells: early loss of macrophages followed by T-cell apoptosis. Infect Immun 1998;66:5462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahida YR, Makh S, Hyde S et al. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut 1996;38:337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani N, Dupuy B. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. P Natl Acad Sci USA 2001;98:5844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani N, Lyras D, Barroso L et al. Environmental response and autoregulation of Clostridium difficile TxeR, a sigma factor for toxin gene expression. J Bacteriol 2002;184:5971–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manse JS, Baldwin MR. Binding and entry of Clostridium difficile toxin B is mediated by multiple domains. FEBS Lett 2015;589:3945–51. [DOI] [PubMed] [Google Scholar]
- Mantyh CR, Maggio JE, Mantyh PW et al. Increased substance P receptor expression by blood vessels and lymphoid aggregates in Clostridium difficile-induced pseudomembranous colitis. Digest Dis Sci 1996a;41:614–20. [DOI] [PubMed] [Google Scholar]
- Mantyh CR, Pappas TN, Lapp JA et al. Substance P activation of enteric neurons in response to intraluminal Clostridium difficile toxin A in the rat ileum. Gastroenterology 1996b;111:1272–80. [DOI] [PubMed] [Google Scholar]
- Martin J, Wilcox M. New and emerging therapies for Clostridium difficile infection. Curr Opin Infect Dis 2016;29:546–54. [DOI] [PubMed] [Google Scholar]
- Martin JS, Monaghan TM, Wilcox MH. Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nat Rev Gastro Hepat 2016;13:206–16. [DOI] [PubMed] [Google Scholar]
- Martin MJ, Clare S, Goulding D et al. The agr locus regulates virulence and colonization genes in Clostridium difficile 027. J Bacteriol 2013;195:3672–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvaud JC, Smith T, Hale ML et al. Clostridium perfringens iota-toxin: mapping of receptor binding and Ia docking domains on Ib. Infect Immun 2001;69:2435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matamouros S, England P, Dupuy B. Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol Microbiol 2007;64:1274–88. [DOI] [PubMed] [Google Scholar]
- Matarrese P, Falzano L, Fabbri A et al. Clostridium difficile toxin B causes apoptosis in epithelial cells by thrilling mitochondria. Involvement of ATP-sensitive mitochondrial potassium channels. J Biol Chem 2007;282:9029–41. [DOI] [PubMed] [Google Scholar]
- Matte I, Lane D, Cote E et al. Antiapoptotic proteins Bcl-2 and Bcl-XL inhibit Clostridium difficile toxin A-induced cell death in human epithelial cells. Infect Immun 2009;77:5400–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May M, Schelle I, Brakebusch C et al. Rac1-dependent recruitment of PAK2 to G2 phase centrosomes and their roles in the regulation of mitotic entry. Cell Cycle 2014;13:2211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May M, Wang T, Muller M et al. Difference in F-actin depolymerization induced by toxin B from the Clostridium difficile strain VPI 10463 and toxin B from the variant Clostridium difficile serotype F strain 1470. Toxins 2013;5:106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlig M, Moos M, Braun V et al. Variant toxin B and a functional toxin A produced by Clostridium difficile C34. FEMS Microbiol Lett 2001;198:171–6. [DOI] [PubMed] [Google Scholar]
- Melo Filho AA, Souza MH, Lyerly DM et al. Role of tumor necrosis factor and nitric oxide in the cytotoxic effects of Clostridium difficile toxin A and toxin B on macrophages. Toxicon 1997;35:743–52. [DOI] [PubMed] [Google Scholar]
- Mesli S, Javorschi S, Berard AM et al. Distribution of the lipolysis stimulated receptor in adult and embryonic murine tissues and lethality of LSR-/- embryos at 12.5 to 14.5 days of gestation. EurJ Biochem 2004;271:3103–14. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Robbe K, Geny B et al. A phosphatidylserine-binding site in the cytosolic fragment of Clostridium sordellii lethal toxin facilitates glucosylation of membrane-bound Rac and is required for cytotoxicity. J Biol Chem 2004;279:49876–82. [DOI] [PubMed] [Google Scholar]
- Meyer GK, Neetz A, Brandes G et al. Clostridium difficile toxins A and B directly stimulate human mast cells. Infect Immun 2007;75:3868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TJ, Ketley JM, Haslam SC et al. Effect of toxin A and B of Clostridium difficile on rabbit ileum and colon. Gut 1986;27:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogridge J, Mourez M, Collier RJ. Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J Bacteriol 2001;183:2111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan TM, Robins A, Knox A et al. Circulating antibody and memory B-Cell responses to C. difficile toxins A and B in patients with C. difficile-associated diarrhoea, inflammatory bowel disease and cystic fibrosis. PLoS One 2013;8:e74452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncrief JS, Barroso LA, Wilkins TD. Positive regulation of Clostridium difficile toxins. Infect Immun 1997;65:1105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monot M, Eckert C, Lemire A et al. Clostridium difficile: new insights into the evolution of the pathogenicity locus. Sci Rep 2015;5:15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Pothoulakis C, LaMont JT et al. C. difficile toxin A increases intestinal permeability and induces Cl- secretion. Am J Physiol 1990;259:G165–72. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Karlsson S, Burman LG et al. Proteins released during high toxin production in Clostridium difficile. Microbiology 2002;148:2245–53. [DOI] [PubMed] [Google Scholar]
- Mullard A. FDA approves antitoxin antibody. Nat Rev Drug Discov 2016;15:811. [DOI] [PubMed] [Google Scholar]
- Muller S, von Eichel-Streiber C, Moos M. Impact of amino acids 22–27 of Rho-subfamily GTPases on glucosylation by the large clostridial cytotoxins TcsL-1522, TcdB-1470 and TcdB-8864. Eur J Biochem 1999;266:1073–80. [DOI] [PubMed] [Google Scholar]
- Na X, Kim H, Moyer MP et al. gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect Immun 2008;76:2862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na X, Zhao D, Koon HW et al. Clostridium difficile toxin B activates the EGF receptor and the ERK/MAP kinase pathway in human colonocytes. Gastroenterology 2005;128:1002–11. [DOI] [PubMed] [Google Scholar]
- Nagahama M, Ohkubo A, Oda M et al. Clostridium perfringens TpeL glycosylates the Rac and Ras subfamily proteins. Infect Immun 2011;79:905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahama M, Yamaguchi A, Hagiyama T et al. Binding and internalization of Clostridium perfringens iota-toxin in lipid rafts. Infect Immun 2004;72:3267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Mikawa M, Tanabe N et al. Effect of clindamycin on cytotoxin production by Clostridium difficile. Microbiol Immunol 1982;26:985–92. [DOI] [PubMed] [Google Scholar]
- Neunlist M, Barouk J, Michel K et al. Toxin B of Clostridium difficile activates human VIP submucosal neurons, in part via an IL-1beta-dependent pathway. Am J Physiol-Gastro L 2003;285:G1049–55. [DOI] [PubMed] [Google Scholar]
- Ng EK, Panesar N, Longo WE et al. Human intestinal epithelial and smooth muscle cells are potent producers of IL-6. Mediat Inflamm 2003;12:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng J, Hirota SA, Gross O et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 2010;139:542–52, 552.e541-543. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995;81:53–62. [DOI] [PubMed] [Google Scholar]
- Nottrott S, Schoentaube J, Genth H et al. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis 2007;12:1443–53. [DOI] [PubMed] [Google Scholar]
- Olling A, Goy S, Hoffmann F et al. The repetitive oligopeptide sequences modulate cytopathic potency but are not crucial for cellular uptake of Clostridium difficile toxin A. PLoS One 2011;6:e17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olling A, Huls C, Goy S et al. The combined repetitive oligopeptides of Clostridium difficile toxin A counteract premature cleavage of the glucosyl-transferase domain by stabilizing protein conformation. Toxins 2014;6:2162–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olling A, Seehase S, Minton NP et al. Release of TcdA and TcdB from Clostridium difficile cdi 630 is not affected by functional inactivation of the tcdE gene. Microb Pathogenesis 2012;52:92–100. [DOI] [PubMed] [Google Scholar]
- Orth P, Xiao L, Hernandez LD et al. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J Biol Chem 2014;289:18008–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamidessi A, Frittoli E, Garre M et al. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell 2008;134:135–47. [DOI] [PubMed] [Google Scholar]
- Papatheodorou P, Carette JE, Bell GW et al. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). P Natl Acad Sci USA 2011;108:16422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatheodorou P, Hornuss D, Nolke T et al. Clostridium difficile binary toxin CDT induces clustering of the lipolysis-stimulated lipoprotein receptor into lipid rafts. mBio 2013;4:e00244–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatheodorou P, Wilczek C, Nolke T et al. Identification of the cellular receptor of Clostridium spiroforme toxin. Infect Immun 2012;80:1418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatheodorou P, Zamboglou C, Genisyuerek S et al. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 2010;5:e10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelle S, Gibert M, Bourlioux P et al. Production of a complete binary toxin (actin-specific ADP-ribosyltransferase) by Clostridium difficile CD196. Infect Immun 1997;65:1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perieteanu AA, Visschedyk DD, Merrill AR et al. ADP-ribosylation of cross-linked actin generates barbed-end polymerization-deficient F-actin oligomers. Biochemistry 2010;49:8944–54. [DOI] [PubMed] [Google Scholar]
- Permpoonpattana P, Hong HA, Phetcharaburanin J et al. Immunization with Bacillus spores expressing toxin A peptide repeats protects against infection with Clostridium difficile strains producing toxins A and B. Infect Immun 2011;79:2295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petosa C, Collier RJ, Klimpel KR et al. Crystal structure of the anthrax toxin protective antigen. Nature 1997;385:833–8. [DOI] [PubMed] [Google Scholar]
- Pfeifer G, Schirmer J, Leemhuis J et al. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J Biol Chem 2003;278:44535–41. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003;112:453–65. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science 2009;326:1208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff MR, Boquet P. Clostridium spiroforme toxin is a binary toxin which ADP-ribosylates cellular actin. Biochem Bioph Res Co 1988;152:1361–8. [DOI] [PubMed] [Google Scholar]
- Popoff MR, Chaves-Olarte E, Lemichez E et al. Ras, Rap, and Rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J Biol Chem 1996;271:10217–24. [DOI] [PubMed] [Google Scholar]
- Popoff MR, Rubin EJ, Gill DM et al. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect Immun 1988;56:2299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothoulakis C, Castagliuolo I, LaMont JT et al. CP-96,345, a substance P antagonist, inhibits rat intestinal responses to Clostridium difficile toxin A but not cholera toxin. P Natl Acad Sci USA 1994;91:947–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothoulakis C, Gilbert RJ, Cladaras C et al. Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J Clin Invest 1996;98:641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothoulakis C, Karmeli F, Kelly CP et al. Ketotifen inhibits Clostridium difficile toxin A-induced enteritis in rat ileum. Gastroenterology 1993;105:701–7. [DOI] [PubMed] [Google Scholar]
- Pothoulakis C, Lamont JT. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol-Gastro L 2001;280:G178–83. [DOI] [PubMed] [Google Scholar]
- Pradelli LA, Beneteau M, Ricci JE. Mitochondrial control of caspase-dependent and -independent cell death. Cell Mol Life Sci 2010;67:1589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt RN, Chagot B, Cover M et al. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J Biol Chem 2009;284:21934–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt RN, Chambers MG, Ng KK et al. Structural organization of the functional domains of Clostridium difficile toxins A and B. P Natl Acad Sci USA 2010;107:13467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt RN, Chumbler NM, Rutherford SA. et al, Structural determinants of Clostridium difficile toxin A glucosyltransferase activity. J Biol Chem 2012;287:8013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri AW, Lupardus PJ, Deu E et al. Rational design of inhibitors and activity-based probes targeting Clostridium difficile virulence factor TcdB. Chem Biol 2010;17:1201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qa’dan M, Christensen KA, Zhang L et al. Membrane insertion by anthrax protective antigen in cultured cells. Mol Cell Biol 2005;25:5492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qa’Dan M, Ramsey M, Daniel J et al. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol 2002;4:425–34. [DOI] [PubMed] [Google Scholar]
- Qa’Dan M, Spyres LM, Ballard JD. pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun 2000;68:2470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu B, Pothoulakis C, Castagliuolo I et al. Participation of reactive oxygen metabolites in Clostridium difficile toxin A-induced enteritis in rats. Am J Physiol 1999;276:G485–90. [DOI] [PubMed] [Google Scholar]
- Quesada-Gomez C, Lopez-Urena D, Chumbler N et al. Analysis of TcdB proteins within the hypervirulent clade 2 reveals an impact of RhoA glucosylation on Clostridium difficile proinflammatory Aativities. Infect Immun 2016;84:856–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers JH, Bos JL. Specificity in Ras and Rap signaling. J Biol Chem 2009;284:10995–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke J, Tenzer S, Rupnik M et al. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 2007;446:415–9. [DOI] [PubMed] [Google Scholar]
- Reinert DJ, Jank T, Aktories K et al. Structural basis for the function of Clostridium difficile toxin B. J Mol Biol 2005;351:973–81. [DOI] [PubMed] [Google Scholar]
- Riegler M, Sedivy R, Pothoulakis C et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest 1995;95:2004–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha MF, Maia ME, Bezerra LR et al. Clostridium difficile toxin A induces the release of neutrophil chemotactic factors from rat peritoneal macrophages: role of interleukin-1beta, tumor necrosis factor alpha, and leukotrienes. Infect Immun 1997;65:2740–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik M, Avesani V, Janc M et al. A novel toxinotyping scheme and correlation of toxinotypes with serogroups of Clostridium difficile isolates. J Clin Microbiol 1998;36:2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik M, Janezic S. An update on Clostridium difficile toxinotyping. J Clin Microbiol 2016;54:13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik M, Pabst S, Rupnik M et al. Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 2005;151:199–208. [DOI] [PubMed] [Google Scholar]
- Sauerborn M, Leukel P, von Eichel-Streiber C. The C-terminal ligand-binding domain of Clostridium difficile toxin A (TcdA) abrogates TcdA-specific binding to cells and prevents mouse lethality. FEMS Microbiol Lett 1997;155:45–54. [DOI] [PubMed] [Google Scholar]
- Saujet L, Monot M, Dupuy B et al. The key sigma factor of transition phase, SigH, controls sporulation, metabolism, and virulence factor expression in Clostridium difficile. J Bacteriol 2011;193:3186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidge TC, Pan WH, Newman P et al. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 2003;125:413–20. [DOI] [PubMed] [Google Scholar]
- Savidge TC, Urvil P, Oezguen N et al. Host S-nitrosylation inhibits clostridial small molecule-activated glucosylating toxins. Nat Med 2011;17:1136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, Reinhard NR, Hordijk PL. Toward understanding RhoGTPase specificity: structure, function and local activation. Small GTPases 2014;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleberger C, Hochmann H, Barth H et al. Structure and action of the binary C2 toxin from Clostridium botulinum. J Mol Biol 2006;364:705–15. [DOI] [PubMed] [Google Scholar]
- Schorch B, Song S, van Diemen FR et al. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. P Natl Acad Sci USA 2014;111:6431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan C, Kruppke AS, Nolke T et al. Clostridium difficile toxin CDT hijacks microtubule organization and reroutes vesicle traffic to increase pathogen adherence. P Natl Acad Sci USA 2014;111:2313–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan C, Nolke T, Kruppke AS et al. Cholesterol- and sphingolipid-rich microdomains are essential for microtubule-based membrane protrusions induced by Clostridium difficile transferase (CDT). J Biol Chem 2011;286:29356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan C, Stecher B, Tzivelekidis T et al. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog 2009;5:e1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehr P, Joseph G, Genth H et al. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry 1998;37:5296–304. [DOI] [PubMed] [Google Scholar]
- Selzer J, Hofmann F, Rex G et al. Clostridium novyi alpha-toxin-catalyzed incorporation of GlcNAc into Rho subfamily proteins. J Biol Chem 1996;271:25173–7. [DOI] [PubMed] [Google Scholar]
- Senju Y, Itoh Y, Takano K et al. Essential role of PACSIN2/syndapin-II in caveolae membrane sculpting. J Cell Sci 2011;124:2032–40. [DOI] [PubMed] [Google Scholar]
- Shen A. Allosteric regulation of protease activity by small molecules. Mol Biosyst 2010;6:1431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A, Lupardus PJ, Gersch MM et al. Defining an allosteric circuit in the cysteine protease domain of Clostridium difficile toxins. Nat Struct Mol Biol 2011;18:364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon NC, Aktories K, Barbieri JT. Novel bacterial ADP-ribosylating toxins: structure and function. Nat Rev Microbiol 2014;12:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits WK, Lyras D, Lacy DB et al. Clostridium difficile infection. Nat Rev Dis Primers 2016;2:16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehn F, Wagenknecht-Wiesner A, Leukel P et al. Genetic rearrangements in the pathogenicity locus of Clostridium difficile strain 8864–implications for transcription, expression and enzymatic activity of toxins A and B. Mol Gen Genet 1998;258:222–32. [DOI] [PubMed] [Google Scholar]
- Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 2008;190:2505–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza MH, Melo-Filho AA, Rocha MF et al. The involvement of macrophage-derived tumour necrosis factor and lipoxygenase products on the neutrophil recruitment induced by Clostridium difficile toxin B. Immunology 1997;91:281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stare BG, Delmee M, Rupnik M. Variant forms of the binary toxin CDT locus and tcdC gene in Clostridium difficile strains. J Med Microbiol 2007;56:329–35. [DOI] [PubMed] [Google Scholar]
- Steele J, Mukherjee J, Parry N et al. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J Infect Dis 2013;207:323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner TS, Flores CA, Pizarro TT et al. Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin Diagn Lab Immun 1997;4:719–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DB, Berg A, Hegarty J. Predicting recurrence of C. difficile colitis using bacterial virulence factors: binary toxin is the key. J Gastrointest Surg 2013;17:118–24; discussion 124-115. [DOI] [PubMed] [Google Scholar]
- Stiles BG, Hale ML, Marvaud JC et al. Clostridium perfringens iota toxin: characterization of the cell-associated iota b complex. Biochem J 2002;367:801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs S, Rupnik M, Gibert M et al. Production of actin-specific ADP-ribosyltransferase (binary toxin) by strains of Clostridium difficile. FEMS Microbiol Lett 2000;186:307–12. [DOI] [PubMed] [Google Scholar]
- Sun X, He X, Tzipori S et al. Essential role of the glucosyltransferase activity in Clostridium difficile toxin-induced secretion of TNF-alpha by macrophages. Microb Pathogenesis 2009;46:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Hirota SA. The roles of host and pathogen factors and the innate immune response in the pathogenesis of Clostridium difficile infection. Mol Immunol 2015;63:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundriyal A, Roberts AK, Ling R et al. Expression, purification and cell cytotoxicity of actin-modifying binary toxin from Clostridium difficile. Protein Expres Purif 2010;74:42–8. [DOI] [PubMed] [Google Scholar]
- Sundriyal A, Roberts AK, Shone CC et al. Structural basis for substrate recognition in the enzymatic component of ADP-ribosyltransferase toxin CDTa from Clostridium difficile. J Biol Chem 2009;284:28713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Bio 2010;11:621–32. [DOI] [PubMed] [Google Scholar]
- Tam J, Beilhartz GL, Auger A et al. Small molecule inhibitors of Clostridium difficile toxin B-induced cellular damage. Chem Biol 2015;22:175–85. [DOI] [PubMed] [Google Scholar]
- Tan KS, Wee BY, Song KP. Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile. J Med Microbiol 2001;50:613–9. [DOI] [PubMed] [Google Scholar]
- Tao L, Zhang J, Meraner P et al. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 2016;538:350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple MD, Perrone GG, Dawes IW. Complex cellular responses to reactive oxygen species. Trends Cell Biol 2005;15:319–26. [DOI] [PubMed] [Google Scholar]
- Teneberg S, Lonnroth I, Torres Lopez JF et al. Molecular mimicry in the recognition of glycosphingolipids by Gal alpha 3 Gal beta 4 GlcNAc beta-binding Clostridium difficile toxin A, human natural anti alpha-galactosyl IgG and the monoclonal antibody Gal-13: characterization of a binding-active human glycosphingolipid, non-identical with the animal receptor. Glycobiology 1996;6:599–609. [DOI] [PubMed] [Google Scholar]
- Terada N, Ohno N, Murata S et al. Immunohistochemical study of NG2 chondroitin sulfate proteoglycan expression in the small and large intestines. Histochem Cell Biol 2006;126:483–90. [DOI] [PubMed] [Google Scholar]
- Thangamani S, Younis W, Seleem MN. Repurposing clinical molecule ebselen to combat drug resistant pathogens. PLoS One 2015a;10:e0133877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangamani S, Younis W, Seleem MN. Repurposing ebselen for treatment of multidrug-resistant staphylococcal infections. Sci Rep 2015b;5:11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CM, Young VB. Interactions between the gastrointestinal microbiome and Clostridium difficile. Annu Rev Microbiol 2015;69:445–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tixier E, Lalanne F, Just I et al. Human mucosa/submucosa interactions during intestinal inflammation: involvement of the enteric nervous system in interleukin-8 secretion. Cell Microbiol 2005;7:1798–810. [DOI] [PubMed] [Google Scholar]
- Tsuge H, Nagahama M, Nishimura H et al. Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens iota-toxin. J Mol Biol 2003;325:471–83. [DOI] [PubMed] [Google Scholar]
- Tucker KD, Wilkins TD. Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect Immun 1991;59:73–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood S, Guan S, Vijayasubhash V et al. Characterization of the sporulation initiation pathway of Clostridium difficile and its role in toxin production. J Bacteriol 2009;191:7296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unligil UM, Rini JM. Glycosyltransferase structure and mechanism. Curr Opin Struct Biol 2000;10:510–7. [DOI] [PubMed] [Google Scholar]
- Vandekerckhove J, Schering B, Barmann M et al. Clostridium perfringens iota toxin ADP-ribosylates skeletal muscle actin in Arg-177. FEBS Lett 1987;225:48–52. [DOI] [PubMed] [Google Scholar]
- Vandekerckhove J, Schering B, Barmann M et al. Botulinum C2 toxin ADP-ribosylates cytoplasmic beta/gamma-actin in arginine 177. J Biol Chem 1988;263:696–700. [PubMed] [Google Scholar]
- Varela Chavez C, Haustant GM, Baron B et al. The tip of the four N-terminal alpha-helices of Clostridium sordellii lethal toxin contains the interaction site with membrane phosphatidylserine facilitating small GTPases glucosylation. Toxins 2016;8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela Chavez C, Hoos S, Haustant GM et al. The catalytic domains of Clostridium sordellii lethal toxin and related large clostridial glucosylating toxins specifically recognize the negatively charged phospholipids phosphatidylserine and phosphatidic acid. Cell Microbiol 2015;17:1477–93. [DOI] [PubMed] [Google Scholar]
- von Eichel-Streiber C, Laufenberg-Feldmann R, Sartingen S et al. Comparative sequence analysis of the Clostridium difficile toxins A and B. Mol Gen Genet 1992;233:260–8. [DOI] [PubMed] [Google Scholar]
- von Eichel-Streiber C, Sauerborn M. Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene 1990;96:107–13. [DOI] [PubMed] [Google Scholar]
- von Eichel-Streiber C, Sauerborn M, Kuramitsu HK. Evidence for a modular structure of the homologous repetitive C-terminal carbohydrate-binding sites of Clostridium difficile toxins and Streptococcus mutans glucosyltransferases. J Bacteriol 1992;174:6707–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eichel-Streiber C, Zec-Pirnat I, Grabnar M et al. A nonsense mutation abrogates production of a functional enterotoxin A in Clostridium difficile toxinotype VIII strains of serogroups F and X. FEMS Microbiol Lett 1999;178:163–8. [DOI] [PubMed] [Google Scholar]
- Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 2005;18:247–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warny M, Keates AC, Keates S et al. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest 2000;105:1147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warny M, Kelly CP. Monocytic cell necrosis is mediated by potassium depletion and caspase-like proteases. Am J Physiol 1999;276:C717–24. [DOI] [PubMed] [Google Scholar]
- Warny M, Vaerman JP, Avesani V et al. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect Immun 1994;62:384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner A. Head to tail polymerization of actin. J Mol Biol 1976;108:139–50. [DOI] [PubMed] [Google Scholar]
- Wegner A, Aktories K. ADP-ribosylated actin caps the barbed ends of actin filaments. J Biol Chem 1988;263:13739–42. [PubMed] [Google Scholar]
- Weigt C, Just I, Wegner A et al. Nonmuscle actin ADP-ribosylated by botulinum C2 toxin caps actin filaments. FEBS Lett 1989;246:181–4. [DOI] [PubMed] [Google Scholar]
- Welsh CF, Roovers K, Villanueva J et al. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol 2001;3:950–7. [DOI] [PubMed] [Google Scholar]
- Wershil BK, Castagliuolo I, Pothoulakis C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology 1998;114:956–64. [DOI] [PubMed] [Google Scholar]
- Wigelsworth DJ, Ruthel G, Schnell L et al. CD44 Promotes intoxication by the clostridial iota-family toxins. PLoS One 2012;7:e51356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins CA, Munro S. Activity of the yeast MNN1 alpha-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. P Natl Acad Sci USA 1998;95:7945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox MH, Gerding DN, Poxton IR et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. New Engl J Med 2017;376:305–17. [DOI] [PubMed] [Google Scholar]
- Wohlan K, Goy S, Olling A et al. Pyknotic cell death induced by Clostridium difficile TcdB: chromatin condensation and nuclear blister are induced independently of the glucosyltransferase activity. Cell Microbiol 2014;16:1678–92. [DOI] [PubMed] [Google Scholar]
- Xu H, Yang J, Gao W et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014;513:237–41. [DOI] [PubMed] [Google Scholar]
- Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol Rev 1994;74:139–62. [DOI] [PubMed] [Google Scholar]
- Yu H, Chen K, Sun Y et al. Cytokines are markers of the Clostridium difficile-induced inflammatory response and predict disease severity. Clin Vaccine Immunol. 2017;24:e00037–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Zhang H, Cai C et al. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res 2015;25:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BZ, Cai J, Yu B et al. A DNA vaccine targeting TcdA and TcdB induces protective immunity against Clostridium difficile. BMC Infect Dis 2016;16:596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hamza T, Gao S et al. Masking autoprocessing of Clostridium difficile toxin A by the C-terminus combined repetitive oligo peptides. Biochem Bioph Res Co 2015;459:259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Park M, Tam J et al. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. P Natl Acad Sci USA 2014;111:3721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler MO, Jank T, Aktories K. et al. Conformational changes and reaction of clostridial glycosylating toxins. J Mol Biol 2008;377:1346–56. [DOI] [PubMed] [Google Scholar]