

Abstract
The methods based on the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system have quickly gained popularity for genome editing and transcriptional regulation in many organisms, including yeast. This review aims to provide a comprehensive overview of CRISPR application for different yeast species: from basic principles and genetic design to applications.
Keywords: CRISPR/Cas, CRISPR interference, genome editing, yeasts, CRISPR transcriptional regulation, Saccharomyces cerevisiae
A comprehensive review on application of CRISPR technology in yeast.
INTRODUCTION
In 2003, Francisco Mojica and colleagues discovered that the spacer sequences from bacterial clustered regularly interspaced short palindromic repeats (CRISPR) loci match viral and conjugative plasmid sequences and hypothesized that CRISPR must be part of the bacterial immune system (Mojica et al.2005; Lander 2016). In the following years, multiple studies had been performed to unravel the mechanism of CRISPR functionality (Lander 2016) until, in 2012, two research groups managed to reprogram the targeting of CRISPR-associated nuclease (Cas9), so Cas9 would introduce double-strand DNA breaks (DSBs) in a sequence-specific manner in vitro (Gasiunas et al.2012; Jinek et al.2012). Following this, applications of CRISPR/Cas9 for in vivo genome editing in mammalian cells were published early in 2013 (Cong et al.2013; Mali et al.2013), followed by DiCarlo et al. (2013) reporting the usage of the system in the yeast Saccharomyces cerevisiae. Since then the technology has been optimized and adapted for numerous organisms, covering applications from industrial biotechnology (van Erp et al.2015) to plant breeding (Bortesi and Fischer 2015) and treatment of human diseases (Cai et al.2016).
In native type II CRISPR/Cas systems, Cas9 is guided to the target DNA region by a two-RNA molecule hybrid consisting of CRISPR RNA (crRNA) and transactivating crRNA (tracrRNA). Together with the tracrRNA, crRNA forms a secondary structure loop, which recruits Cas9. The crRNA guides the system to a genomic target of ∼20 bp through base pairing with the complementary DNA strand. The particular genomic target must be followed by the protospacer adjacent motif (PAM) NGG. The Cas9 nuclease domain HNH then cleaves the DNA-strand complementary to the crRNA-guide sequence, while RuvC-like domain cleaves the other DNA strand, thus resulting in a DSB. The DNA cleavage is performed three nucleotides upstream of the PAM site (Gasiunas et al.2012). For easier use in genome editing, the crRNA and tracrRNA can be fused tail to head via a linker into a single guiding RNA (gRNA) (Jinek et al.2012).
This review covers the technical details of the implementation of CRISPR/Cas-mediated genome editing in various yeast species, transcriptional regulation via the enzymatically inactive ‘dead’ dCas9, which binds but does not cut the DNA target (Jinek et al.2012), and presents examples of applying the technology for engineering of yeast cell factories.
CRISPR/Cas9 GENOME EDITING IN YEASTS
When Cas9 protein and gRNA are expressed in yeast cells, Cas9 introduces DSBs that must be repaired by the cells via non-homologous end joining (NHEJ) or homologous recombination (HR) (Liu et al.2017). By supplying a DNA repair template for use in HR, various DNA modifications can be obtained. In the case of efficient cutting, the generated DSBs serve as a negative selection. Thus, there is no need for using a selective marker as in non-CRISPR genome editing methods. Relatively precise and flexible targeting and elimination of the need for positive selection are the two key advantages of the CRISPR/Cas9 technology for yeast genome engineering. The method also allows engineering of diploid and polyploid industrial strains (Ryan et al.2014; Zhang et al.2014; Stovicek, Borodina and Forster 2015), which are challenging to manipulate genetically due to the difficulties with modifying multiple alleles and due to the lack of selection markers (Le Borgne 2012). Additionally, by combining several gRNAs, multiple sites can be targeted simultaneously allowing the unprecedented speed of multiple genetic edits (Ryan et al.2014; Bao et al.2015; Jakočiūnas et al.2015a). On the downside of CRISPR/Cas9, there is a considerable variation in efficiency when targeting different loci, perhaps due to a positional effect of the target region (Smith et al.2016). At the moment, there also seems to be an upper limit for the number of edits (up to six) that can be introduced simultaneously as every additional introduced DSB decreases the overall yield of surviving clones (Mans et al.2015; Jakočiūnas et al.2015a). Furthermore, CRISPR/Cas9 multiplexing still represents a significant increase in workload for finding correct clones.
Progress has been made on adapting the type II CRISPR/Cas system, described in Streptococcus pyogenes (Chylinski et al.2014), to various yeast species—Saccharomyces cerevisiae (Jakočiūnas, Jensen and Keasling 2016), Yarrowia lipolytica (Schwartz et al.2016a), Komagataella phaffii (formerly Pichia pastoris) (Weninger et al.2016), Kluyveromyces lactis (Horwitz et al.2015), Schizosaccharomyces pombe (Jacobs et al.2014), and the pathogenic yeast species Candida albicans (Vyas, Barrasa and Fink 2015) and Cryptococcus neoformans (Wang et al.2016). We first discuss the design of the targeting gRNA sequence as a critical aspect of all CRISPR/Cas9 applications. As the vast majority of the studies describing CRISPR/Cas9 genome editing in yeasts have focused on S. cerevisiae, the larger section dedicated to this model organism also details some of the more general issues related to the Cas9-mediated genome engineering. For clarity, the studies focusing on the other yeasts are discussed in a separate section.
COMPUTATIONAL TOOLS FOR gRNA DESIGN IN YEAST
Any ∼20-bp sequence proximal to the PAM site in the genome can serve as the gRNA targeting sequence. The rationale behind careful gRNA selection is to minimize the risk of Cas9-mediated cleavage at unwanted sites in the genome (off-target effects) and maximize the cutting efficiency at the selected site (on-target activity). Other factors may outweigh the best parameters and put additional constraints on the design, e.g. position of a target proximal to the beginning of the ORF for generating premature STOP codons or requirement of a target location in promoter/5΄ UTR region in case of gene repression/activation experiments (Mohr et al.2016). Several web-based tools have been developed to facilitate and automatize the design of gRNA targets (Table 1). Such tools aim mainly at providing guide sequences that minimize the likelihood of off-target effects, matching all possible targets within the given parameters against the reference genome. Some tools provide a list of targets with specified number of mismatches within the entire target sequence or the ‘seed’ sequence (8–12 bp adjacent to the PAM site) (CRISPy (Ronda et al.2014; Jakočiūnas et al.2015a); CRISPRdirect (Naito et al.2015)), filter out sequences with potential off-target effects (Yeastriction, Mans et al.2015) or introduce a specificity score based on number of mismatches within the target sequence and rank the targets accordingly (CRISPR-ERA, Liu et al.2015; Benchling, ATUM gRNA design). CHOPCHOP (Labun et al.2016) or E-CRISP (Heigwer, Kerr and Boutros 2014) provides the possibility for user-defined parameters of the off-target evaluation. Even though off-target effects are considered unlikely in such a small genome as yeast (Ryan et al.2014; Jakočiūnas et al.2015a), it is advisable to double check the design using yet another tool to avoid introduction of any undesired modifications. Although several potential requirements for gRNA design have been suggested to ensure efficient generation of DSB at the target site, it is still not easy to establish a set of golden rules that would guarantee a success until more experimental data have been acquired. Some of the tools highlight simple features that might influence gRNA efficiency, such as poly T presence in the sequence, GC content (CRISPRdirect) (Naito et al.2015), AT content or self-complementarity of a gRNA molecule and provide a score based on these parameters (Yeastriction, E-CRISP, CRISPR-ERA) (Heigwer, Kerr and Boutros 2014; Liu et al.2015; Mans et al.2015). Other tools such as Benchling have implemented more sophisticated efficiency scores based on an experimental evaluation of a large set of mammalian gRNAs and their sequence features (Doench et al.2014, 2016; Xu et al.2015). In some cases, users can even choose from several different algorithms of the on-target evaluation (CHOPCHOP, E-CRISP). A few tools also include information on the presence of a specific restriction site in the target sequence (CHOPCHOP, CRISPRdirect, Yeastriction) that might facilitate downstream validation of the cloned target molecule (Mans et al.2015). CRISPR-ERA or E-CRISP also facilitate designing of a gRNA molecule for engineering applications other than genome editing, e.g. gene repression or gene activation applications. While some of the tools support only one yeast genome, typically Saccharomyces cerevisiae reference genome, others provide gRNA design option for several yeast species or various strains of S. cerevisiae (Table 1). The CRISPy tool web server implementation, CRISPy-web (Blin et al.2016), allows for user upload of any GenBank format genome. CRISPRdirect is being frequently updated with new genomes, and CHOPCHOP offers an upload of new genomes on request.
Table 1.
List of selected web-based bioinformatics tools for gRNA design in yeast.
| Name | Link | Reference | Input | Main features | Yeast species |
|---|---|---|---|---|---|
| CRISPy | http://staff.biosu stain.dtu.dk/laeb/ crispy_yeast/ | Ronda et al. (2014 ); Jakočiūnas et al. (2015a) | Gene name/ID | Off-target | S. cerevisiae reference, CEN.PK |
| CRISPy-web | http://crispy.second arymetabolites.org | Blin et al. (2016) | Gene name/ID, genomic coordinates | Off-target | Any user-submitted genome |
| CRISPR-ERA | http://crispr- era.stanford.edu/ | Liu et al. (2015) | Gene name, genomic coordinates, sequence | Off-target, efficiency score, gene repression/activation | S. cerevisiae reference |
| CHOPCHOP v2 | http://chopchop.cbu. uib.no | Labun et al. (2016) | Gene name, genomic coordinates, sequence | Off-target user defined, on-target algorithm, restriction sites | S. cerevisiae reference, |
| C. albicans, | |||||
| C. tropicalis, | |||||
| C. glabrata, | |||||
| P. pastoris | |||||
| CRISPRdirect | https://crispr. dbcls.jp/ | Naito et al. (2015) | Gene name, genomic coordinates, sequence | Off-target, GC content, poly T, restriction sites | S. cerevisiae, |
| Sch. pombe, | |||||
| K. lactis, | |||||
| Y. lipolytica, | |||||
| C. albicans, | |||||
| C. glabrata | |||||
| E-CRISPR | http://www.e- crisp.org/ | Heigwer, Kerr and Boutros (2014) | Gene symbol, sequence | Off-target, on-target algorithm, gene activation/repression | S. cerevisiae, |
| Sch. pombe | |||||
| Yeastriction | http://yeastriction. tnw.tudelft.nl | Mans et al. (2015) | Gene name | Off-target, AT content, self-complementarity, restriction sites | S. cerevisiae, several strains |
| Benchling | https://benchling. com/ crispr | Gene name, coordinates, sequence | Off-target, on-target algorithm | S. cerevisiae reference, | |
| Sch. pombe, | |||||
| C. albicans, | |||||
| Y. lipolytica | |||||
| ATUM gRNA Design Tool | https://www.atum. bio/eCommerce/ cas9/input | Gene name, coordinates, sequence | Off-target | S. cerevisiae reference |
CRISPR/Cas9 AND GENOME EDITING IN SACCHAROMYCES CEREVISIAE
Saccharomyces cerevisiae is an important eukaryotic model organism and also a widely used industrial host for production of fuels, chemicals and recombinant proteins (Borodina and Nielsen 2014; Li and Borodina 2015). Thanks to its excellent HR capability, S. cerevisiae is relatively easy to engineer genetically. Below we discuss the ways for delivering Cas9, gRNA and DNA repair templates to S. cerevisiae (summarized in Table 2).
Table 2.
List of available CRISPR/Cas9 tools for yeast.
| Reference | Availability | Organism and strain (ploidy) | Cas9 expression (vector, selection marker, promoter) | gRNA expression (vector, selection marker, promoter, terminator) | Application and efficiency |
|---|---|---|---|---|---|
| S. cerevisiae genome editing | |||||
| DiCarlo et al. (2013) | Addgene | S. cerevisiae BY4733 (n) | CEN/ARS, TRP1, PTEF1/GALL-Cas9 | 2μ, URA3, PSNR52/TSUP4 | Single-gene disruption/marker cassette insertion: 99% |
| Gao and Zhao (2014) | Addgene | S. cerevisiae LPY16936 (n) | 2μ, LEU2, PADH1-Cas9 | 2μ, URA3, PADH1-HH ribozyme/HDV ribozyme-TADH1 | Single-gene disruption: 100% |
| Ryan et al. (2014) | On request | S. cerevisiae S288C (n, 2n) ATCC4124 (poly n) | a 2μ, kanMX, PRNR2-Cas9b | tRNAPro -HDV ribozyme/TSNR52 | Single/multiple_gene disruption(s): 90%–100%/19%–85%, three-part marker cassette insertion: 70%–85% |
| Bao et al. (2015) | Addgene | S. cerevisiae BY4741 (n) CEN.PK2–1c (n) | a 2μ, truncated URA3, PTEF1-iCas9b | PSNR52-crRNA/TSUP4, PRPR1-tracrRNA/TRPR1 | Single/multiple-gene disruption: 27%–100% |
| Zhang et al. (2014) | Addgene | S. cerevisiae ATCC 4124 (poly n) | CEN/ARS, natMX, PTEF1-Cas9 | 2μ, hphMX, PSNR52/TSUP4 | Single-gene disruption: 15%–60% |
| Jakočiūnas et al. (2015a) | On request | S. cerevisiae CEN.PK2–1c (n) | CEN/ARS, TRP1, PTEF1-Cas9 | 2μ, LEU2, PSNR52/TSUP4 | Single/multiple-gene disruption(s): 100%/50%–100% |
| Mans et al. (2015) | Euroscarf | S. cerevisiae CEN.PK2–1c (n) CEN.PK113–7D (n) CEN.PK122 (2n) | integr. can1Δ::PTEF1-Cas9-natMX | 2μ, URA3, amdSYM/hphMX/ kanMX/LEU2/natMX/ HIS3/ TRP1, PSNR52/TSUP4 | Single-gene deletion: 25%–75%, multiple-gene deletions/multiple-gene cassette insertions: 65%–100% |
| Stovicek, Borodina and Forster (2015) | Addgene | S. cerevisiae CEN.PK113–7D (n) Ethanol Red, | CEN/ARS, kanMX, PTEF1/ADH1-Cas9 | 2μ, natMX, PSNR52/TSUP4 | Single-gene disruption and gene cassette insertion: 65%–97% |
| CLIB382, CBS7960 (2n) | |||||
| Horwitz et al. (2015) | On request | S. cerevisiae CEN.PK2–1c (n) | integr. gre3Δ::PFBA1-Cas9c-hphMX | 2μ, URA3/HIS3/natMX, PSNR52/TSUP4 | Single allele swap: 82%–100%, multiple-gene disruptions: 65%–91%, multiple-gene cassette integrations: 4.2% |
| Tsai et al. (2015) | On request | S. cerevisiae D452–2 (n) | CEN/ARS, natMX, PTEF1-Cas9 | 2μ, hphMX, PSNR52/TSUP4 | Two part-gene cassettes integration into a single-gene locus: 25%–100% |
| Laughery et al. (2015) | Addgene | S. cerevisiae BY4741 (n) | a 2μ, LEU2/URA3, PTDH3-Cas9 | PSNR52/TSUP4 | Single-gene disruption: 97%–98% |
| Lee et al. (2015) | Addgene | S. cerevisiae S288C (n) | a CEN/ARS, URA3, PPGK1-Cas9 | tRNAPhe-HDV ribozyme/TSNR52 | Single/multiple-gene disruption(s): 96%/21%–76% |
| Jakočiūnas et al. (2015b) | On request | S. cerevisiae CEN.PK111−27B (n) | CEN/ARS, TRP1, PTEF1-Cas9 | 2μ, LEU2, PSNR52/TSUP4 | Multiple part gene cassette integrations into multiple gene loci: 30%–97% |
| Ronda et al. (2015) | On request | S. cerevisiae CEN.PK2–1c (n) | CEN/ARS, TRP1, PTEF2-Cas9 | 2μ, natMX, PSNR52/TSUP4 | Gene cassette integration into multiple intergenic loci: 84–100% |
| Shi et al. (2016) | On request | S. cerevisiae HZ848 (n), CEN.PK2–1c (n) | a 2μ, truncated URA3, PTEF1-Cas9 | PSNR52-crRNA/TSUP4, PRPR1-tracrRNA/TRPR1 | Long gene fragment integration into multiple genomic loci: 75%–88% |
| Generoso et al. (2016) | Addgene | S. cerevisiae CEN.PK113–7D (n) Ethanol Red (2n) | a 2μ, kanMX/natMX, PROX3-Cas9c | PSNR52/TSUP4 | Single and double-gene disruptions: 91%–98% |
| Jessop-Fabre et al. (2016) | Addgene | S. cerevisiae CEN.PK113–7D (n) Ethanol Red (2n) | CEN/ARS, kanMX, PTEF1-Cas9 | 2μ, natMX, PSNR52/TSUP4 | Integration of a long gene fragment into a single locus: 95%–100%/multiple loci: 60–70% |
| Reider Apel et al. (2016) | On request | S. cerevisiae BY4742 (n) | a 2μ, URA3, LEU2, PADH1-Cas9c | tRNATyr -HDV ribozyme/TSNR52 | Three part-gene cassette integration into mutiple intergenic loci: 40%–95% |
| Garst et al. (2017) | On request | S. cerevisiae | a 2μ, truncated URA3, PTEF1-iCas9b | PSNR52-crRNA/TSUP4, PRPR1-tracrRNA/TRPR1 | Single-gene non-sense mutation: 70%–95% |
| BY4709 (n) | |||||
| RM11–1 (n) | |||||
| Liu et al. (2017) | On request | S. cerevisiae var. boulardii ATCC MYA-796 (n) | CEN/ARS, natMX, PTEF1-Cas9 | 2μ, hphMX, PSNR52/TSUP4 | Single/double-gene disruption(s):100%/N/A |
| Nishida et al. (2016) | Addgene | S. cerevisiae | CEN/ARS, LEU2, PGAL1-nCas9 (840A)/nCas9 (D10A)/n(d)Cas9 (D10A/840A)-PmCDA1 | 2μ, URA3, PSNR52/TSUP4 | Cytidine deaminase-mediated single/double-gene disruption(s): 16%–54%/14%–31% |
| BY4741 (n) | |||||
| YPH501 (2n) | |||||
| Vanegas, Lehka and Mortensen (2017) | On request | S. cerevisiaeS288C (n)PJ69–4 (n) | Integr. intergenic X-3::PTEF1-Cas9-URA3 | CEN/ARS, LEU2PSNR52/TSUP4 | Integration of three-part multiple-gene fragment into an intergenic site: 100% |
| S. cerevisiae gene activation/repression | |||||
| Gilbert et al. (2013) | Addgene | S. cerevisiaeW303 | CEN/ARS, LEU2, PTDH3-dCas9(-Mxi1) | CEN/ARS, URA3, PSNR52/TSUP4 | Several 10-fold reporter gene transcription repression (CRISPRi) |
| Farzadfard, Perli and Lu (2013) | Addgene | S. cerevisiaeW303 (n) | integr. Δtrp:: PTPGI-dCas9c-VP64-TRP1 | 2μ, HIS3/LEU2, PRPR1/TRPR1 | Transcription activation (activator domain)/repression (CRISPRi) |
| Zalatan et al. (2015) | Addgene | S. cerevisiaeW303 (n) | integr. Δleu2/his3::PTDH3/GAL10-dCas9-CgLEU2/HIS3 | CEN/ARS, URA3, PSNR52-gRNA-sc(scaffold)RNA/TSUP4 | Multiple-gene transcription activation (RNA-binding chimeric activators)/repression (CRISPRi) |
| Chavez et al. (2015) | Addgene | S. cerevisiaeW303 (n) | CEN/ARS, TRP1, PTDH3-dCas9-VPR | 2μ, URA3, PSNR52/TSUP4 | Transcription activation (multiple activation domains) |
| Smith et al. (2016) | Addgene | S. cerevisiaeBY4741 (n) | a CEN/ARS, TRP1/URA3, PTEF1-dCas9-Mxi1 | PRPR1(TetO)/TRPR1 | Transcription repression (repression domain- CRISPRi) |
| Vanegas, Lehka and Mortensen (2017) | On request | S. cerevisiaeS288C (n)PJ69–4 (n) | Integr. Intergenic X-3::PTEF1-dCas9c/dCas9-VP64-URA3 | CEN/ARS, LEU2PSNR52/TSUP4 | Transcription activation (activator domain)/repression (CRISPRi) |
| Deaner and Alper (2017) | On request | S. cerevisiaeBY4741 (n) | a CEN/ARS, LEU2, PTDH3-dCas9-Mxi1/PTDH3-dCas9-VPR | PSNR52/TSUP4 | Graded gene activation/repression (fold transcription activation-gene silencing) |
| K. lactis | |||||
| Horwitz et al. (2015) | On request | K. lactis ATCC8585 (n) | integr. Klgal80::PFBA1-Cas9c-hphMX | pKD1, natMX, PSNR52/TSUP4 | Multiple-gene cassette insertion into multiple-gene loci: 2.1% |
| Y. lipolytica | |||||
| Schwartz et al. (2016a) | Addgene | Y. lipolytica ATCC MYA-2613 (n) | a CEN, LEU2, PUAS1B8-TEF-Cas9c | SCR'-tRNAGly/polyT | Single-gene disruptions (NHEJ/HR): 90%–100%/64%–88% (100% in KU mutant) |
| Gao et al. (2016) | Addgene | Y. lipolytica ATCC 201 249 ATCC MYA-2613 (n) | a CEN, LEU2/URA3, PTEFin-Cas9 | PTEFin-HH ribozyme/HDV ribozyme-TMIG1 | Single-gene disruption (NHEJ/HR): 62%–98%/72% (94% in KU mutant), multiple-gene disruptions (NHEJ): 19%–37% |
| Schwartz et al. (2016b) | Addgene | Y. lipolytica ATCC MYA-2613 (n) | a CEN, LEU2, PUAS1B8-TEF-Cas9c | SCR'-tRNAGly/polyT | Gene cassette integration into an intergenic locus: 48%–69% |
| Ko. phaffii (P. pastoris) | |||||
| Weninger et al. (2016) | On request | Ko. phaffii (P. pastoris) CBS7435 (n) | PARS1a, ZEO, PHTX1-Cas9 | PHTX1-HH ribozyme/HDV ribozyme-TAOX1 | Single-gene disruption: 87%–94%, double-gene disruptions: 69% |
| Sch. pombe | |||||
| Jacobs et al. (2014) | Addgene | Sch. pombe (n) | a ars, ura4, Padh1-Cas9 | Prrk1/HH ribozyme-Trrk1 | Single-gene disruption (allele swap): 85%–90% |
| Fernandez and Berro (2016) | On request | Sch. pombe FY527 (n) FY528 (n) | a ars, ura4/fex1, Padh1-Cas9 | Prrk1/HH ribozyme-Trrk1 | Single-gene deletion (ORF removal): 33% |
| C. albicans | |||||
| Vyas, Barrasa and Fink (2015) | On request | C. albicans SC5314 (2n) | a integr. ENO1 locus, natMX, PENO1-Cas9c | PSNR52/TENO1 | Single/multiple gene disruption(s): 60%–80%/20% |
| Min et al. (2016) | On request | C. albicans SC5314 (2n) SN152 (2n) | linear cassette, PENO1-Cas9c | linear cassette, PSNR52/TENO1 | Single-gene deletion (ORF replacement with marker cassette): 45%–67% |
| C. glabrata | |||||
| Enkler et al. (2016) | On request | C. glabrata CBS138 (n) | CEN, TRP1, PCgCYC1-Cas9 | CEN, LEU2, PCgRNAH1/TCgTY2 | Single-gene disruption |
| Cr. neoformans | |||||
| Wang et al. (2016) | On request | Cr. neoformans serotype D strain JEC21 (n) | a linear vector, URA5, PACT1-Cas9 | URA5, PCnU6/polyT | Single-gene disruption (NHEJ/HR): 40%–90%/20%–90% |
| Arras et al. (2016) | On request | Cr. neoformans serotype A strain H99 (n) | integr. ‘Safe Heaven’-PTEF1-Cas9 | linear vector, PACT1-HH ribozyme/HDV ribozyme-TTRP1 | Single-gene deletion (ORF replacement with marker cassette): 65%–70% |
The Cas9 gene is a human codon-optimized version unless otherwise marked. Addgene CRISPR/Cas9 plasmids for use in yeast are available at https://www.addgene.org/crispr/yeast/. Euroscarf deposited vectors can be ordered here www.euroscarf.de.
HH—Hammerhead ribozyme, HDV—hepatitis delta virus ribozyme, iCas9 – mutated ‘hyperactive’ variant, nCas9 – mutated ‘nicking’ variant causing single-strand DNA break, dCas9 – ‘dead’ nuclease activity-lacking variant, PmCDA1 – cytidine deaminase from sea lamprey (Petromyzon marinus), Mxi1 – mammalian transcriptional repressor, VP64 – mammalian transcriptional activator domain, VPR—VP64-p65-Rta tripartite activator domain.
Both components on a single expression element.
Native S. pyogenes Cas9.
Species codon-optimized Cas9.
Cas9 expression
The most commonly used Cas9 gene variant in S. cerevisiae has been Cas9 from Streptococcus pyogenes, fused with a nucleolar localization sequence. The DNA sequence of Cas9 can be either native (Ryan et al.2014; Bao et al.2015), human codon-optimized (DiCarlo et al.2013; Gao and Zhao 2014; Zhang et al.2014; Laughery et al.2015; Mans et al.2015; Stovicek, Borodina and Forster 2015; Jakočiūnas et al.2015a) or yeast codon-optimized (Horwitz et al.2015; Generoso et al.2016) (Table 2). Only Xu et al. (2015) reported the use of St. thermophilus CRISPR3 loci-encoded Cas9 (recognizing a different PAM site), albeit with much lower engineering efficiency. The Cas9 gene was most commonly expressed under the control of constitutive promoters of different strengths from self-replicating low-copy centromeric vectors (DiCarlo et al.2013; Zhang et al.2014; Stovicek, Borodina and Forster 2015; Jakočiūnas et al.2015a) or high-copy 2μ vectors (Gao and Zhao 2014; Ryan et al.2014; Bao et al.2015; Horwitz et al.2015; Laughery et al.2015; Generoso et al.2016) or integrated into the genome (Mans et al.2015) (Table 2). Expression of Cas9 on a high-copy vector from a strong constitutive promoter led to a negative influence on the growth of some yeast strains (Ryan et al.2014; Generoso et al.2016). However, this problem was not observed in other studies that used the same mode of Cas9 expression (Gao and Zhao 2014; Bao et al.2015; Laughery et al.2015). The toxicity of Cas9 nuclease could be avoided by using weaker promoters for Cas9 expression (Ryan et al.2014; Generoso et al.2016). Overall, the form of Cas9 expression does not seem to be a critical parameter in CRISPR/Cas9 engineering strategies for S. cerevisiae.
Guide RNA expression
Design, expression and delivery of the gRNA components are crucial parameters for successful CRISPR/Cas9 engineering. In S. cerevisiae, the most common strategy has been to express a chimeric gRNA molecule from a high-copy vector to ensure its abundant expression (Table 2). Both ends of the gRNA molecule must be precisely defined to create a functional Cas9/gRNA complex. Functional gRNA transcription has been achieved using (i) an RNA polymerase III (Pol III) promoter that provides a transcript with a leader sequence cleaved during molecule maturation (DiCarlo et al.2013; Farzadfard, Perli and Lu 2013); (ii) Pol III promoters containing cis-regulatory elements within the mature RNA molecule (tRNA) combined with a ribozyme, cleaving the transcript on its 5΄ end (Ryan et al.2014); and (iii) an RNA polymerase II (Pol II) promoter, if the gRNA molecule is flanked with two ribozymes executing cleavage on both ends of the molecule (Gao and Zhao 2014) (Fig. 1). Besides the chimeric gRNA approach, separate expression of a targeting crRNA array driven by a Pol III promoter, processed by native RNA processing enzymes, and tracrRNA transcribed from another Pol III promoter has been reported (Bao et al.2015). The expression cassette containing SNR52 promoter and SUP4 terminator, an approach shown to produce prokaryotic tRNA molecules in yeast (Wang and Wang 2008), was successfully used for targeting a single gene in haploid or diploid laboratory strains with engineering efficiencies reaching 100% (DiCarlo et al.2013; Horwitz et al.2015; Laughery et al.2015; Mans et al. 2015; Jakočiūnas et al.2015a; Generoso et al.2016) (Table 2). It is important to mention that engineering efficiencies discussed in this review are defined as the number of clones with the desired genomic edit per number of clones surviving after the transformation. Such values should not be mistaken with transformation efficiency values used traditionally in non-CRISPR engineering studies as these relate to the number of viable cells in the transformation reaction (Storici et al.2003; Alexander, Doering and Hittinger 2014). Although some studies also provide transformation efficiency values that reflect the number of cells not surviving the transformation (DiCarlo et al.2013; Stovicek, Borodina and Forster 2015), many others do not, leaving the engineering efficiency as the only relevant benchmark. The SNR52 promoter/SUP4 terminator setup also allowed for gene deletion in various diploid industrial strains with efficiencies ranging between 65% and 78% (Stovicek, Borodina and Forster 2015) and even polyploid strains with 15%–60% efficiency (Zhang et al.2014) (Table 2). The expression of a gRNA fused to a Hepatitis delta virus (HDV) ribozyme controlled by a tRNA promoter and SNR52 terminator led to almost 100% gene deletion efficiency in a diploid laboratory strain and more than 90% in a polyploid industrial strain (Ryan et al.2014). As for the third mentioned approach, a gRNA molecule flanked with Hammerhead (HH) and HDV ribozymes on the 5΄ and 3΄ end, respectively, expressed from ADH1 promoter also enabled efficient gene disruption in a laboratory strain (Gao and Zhao 2014). The crRNA array method achieved efficiencies of 76%–100% in a laboratory strain after several days of outgrowth of the transformed cells (Bao et al.2015).
Figure 1.

Overview of CRISPR/Cas9-mediated genome editing in yeast. (A) Illustration of Cas9 expression and various means of gRNA expression. (B) Mechanism of Cas9/gRNA ribonucleoprotein complex action, NGG (PAM site) highlighted in orange letters. (C) Different donor DNA templates for DSB repair. Pol II/III—RNA Polymerase II/III, NLS—nucleolar localization sequence, cis—cis regulatory element (tRNA), L—self-cleaved leader sequence (SNR52), cr—crRNA, tracr—tracrRNA, HH—hammerhead ribozyme, HDV—hepatitis delta virus ribozyme, *—STOP codon.
When a researcher decides to engineer a targeted genomic locus, only the ∼20 bp recognition sequence of a gRNA molecule needs to be modified to redirect the Cas9/gRNA complex to a particular target site. Several ways of obtaining an expression vector with a customized gRNA molecule have been described (Fig. 2). Several studies exchanged the recognition sequence of a gRNA vector using whole vector amplification with primers containing a new target-specific 20-bp region. Vector circularization was achieved via PCR with a phosphorylated primer, followed by ligation (Stovicek, Borodina and Forster 2015; Jakočiūnas et al.2015a), in vivo in yeast or in vitro Gibson assembly using two oligos overlapping at the target sequence (Generoso et al.2016), or via restriction-free cloning (van den Ent and Löwe 2006) with two 60-bp complementary oligos containing a target sequence (Ryan and Cate 2014). In other studies, two target-specific complementary oligos containing sequences overlapping with the gRNA cassette were cloned into a vector using Gibson assembly (Reider Apel et al.2016) or restriction sites located between promoter and the gRNA structural part (Laughery et al.2015; Lee et al.2015), or transformed directly into yeast along with the digested expression vector (Mans et al.2015). Alternatively, the gRNA cassette was amplified using two-step fusion PCR and cloned via Gibson assembly (DiCarlo et al.2013) and standard restriction cloning (Chin et al.2016) or transformed along with a digested expression vector for in vivo vector gap repair in yeast (Horwitz et al.2015). To omit the PCR amplification step, customized gRNA cassettes can be synthesized as gene blocks and integrated into a vector via restriction cloning (Zhang et al.2014) or USER assembly (Ronda et al.2015; Jakočiūnas et al.2015b). Lastly, Golden gate cloning of synthetic parts of the crRNA array has also been shown (Bao et al.2015).
Figure 2.

Generation of specific gRNA expression cassettes. (A) Vector can be circularized via ligation (one oligo phosphorylated) (Jakočiūnas et al.2015a; Stovicek, Borodina and Forster 2015), ligation-free primer extension reaction (Tsai et al.2015; Ryan, Poddar and Cate 2016), Gibson assembly or recombination in vivo (pair of oligos overlapping at the specific gRNA target sequence) (Generoso et al.2016). (B) Short synthetic oligos are cloned via e.g. Gibson assembly (oligos with overhangs homologous to the ends of the digested vector) (Reider Apel et al.2016), restriction cloning (oligos with overhangs complementary to a particular restriction site) (Laughery et al.2015), modular cloning (seamless assembly using type IIS restriction enzymes, oligos with overhangs complementary to a particular restriction site) (Lee et al.2015; Vyas, Barrasa and Fink 2015) or in vivo in yeast (Mans et al.2015). (C) Cloning of the two-step PCR generated gRNA cassette via Gibson assembly (DiCarlo et al.2013) or restriction cloning (Chin et al.2016). (D) Several single gRNA cassettes cloned via Gibson assembly (Weninger et al.2016), restriction cloning (Ryan et al.2014) or modular assembly (Lee et al.2015). Alternatively, two-gRNA cassette fragments in opposite orientation can be amplified in one reaction and cloned (Mans et al.2015; Generoso et al.2016). (E) Pool of several single gRNA cassettes transformed to yeast cells with a gapped vector for in vivo recombination (Horwitz et al.2015). (F) crRNA array is cloned via Golden gate assembly of short synthetic fragments with homologous overlaps (Bao et al.2015).
In summary, researchers can choose from a number of cloning systems for generation of a target gRNA molecule and can also benefit from online tools facilitating the particular cloning design (Laughery et al.2015; Mans et al.2015) or detailed (Ryan, Poddar and Cate 2016) and straightforward protocols (Jakočiūnas et al.2015a). However, even in its simplest version, the CRISPR/Cas9 engineering relies on a gRNA vector construction, which can be laborious and costly. The gap repair approach developed by Horwitz et al. (2015) skips the cloning step. However, it requires longer DSB repair templates, high efficiency of HR in the strain and may result in a non-equimolar expression of the gRNAs when multiplexing. A lower efficiency of engineering with vectors based on in vivo assembly has been documented (Mans et al.2015; Generoso et al.2016).
One can also choose to express Cas9 and gRNA from a single vector (Ryan et al.2014; Bao et al.2015; Laughery et al.2015; Generoso et al.2016). However, due to the large size of the Cas9 gene, generation of a gRNA via the whole plasmid PCR amplification (Ryan and Cate 2014) might be difficult. Such a system is also not compatible with the gap repair gRNA generation as this one requires expression of Cas9 prior the transformation with the gRNA vector (Walter, Chandran and Horwitz 2016).
Multiplexing gRNA expression
In S. cerevisiae, efficient HR system allows creating multiple genomic changes simultaneously using CRISPR/Cas9. For each genome edit, an individual gRNA must be expressed and a repair template delivered into the cells. The multiple gRNA expression has been achieved using (i) several vectors with different selection markers containing up to two different gRNA expression cassettes (Mans et al.2015), (ii) a single expression vector carrying several gRNA cassettes (Ryan et al.2014; Lee et al.2015; Jakočiūnas et al.2015a), (iii) an array of different interspaced crRNAs (Bao et al.2015) or (iv) different linear gRNA expression cassettes transformed along with a single gapped expression vector (Horwitz et al.2015). When up to three different vectors, each carrying two gRNA expression cassettes were transformed, 100%, 70% and 65% efficiency of two, four or six gene deletions was achieved, respectively (Mans et al.2015). The expression of five individual gRNAs from one vector provided target efficiencies ranging between 50% and 100% (Jakočiūnas et al.2015a). Ryan et al. (2014) reported successful gene deletion of two or three genes with efficiencies of 86% and 81% in haploid and 43% and 19% in diploid strains using HDV-gRNA expression cassettes in a single expression vector, respectively. Cloning of crRNA arrays targeting three different genes achieved engineering efficiencies ranging between 27% and 100% (Bao et al.2015). The gap repair approach using the transformation of three different gRNA cassettes and a single open vector enabled recovery of 64% positive three-gene deletion mutants (Horwitz et al.2015) (Table 2).
As described above, all setups enabled successful marker-free multiplexed genome editing in S. cerevisiae. However, despite the reported encouraging results, yeast strains can differ in engineering efficiencies given rather by their nature than differences in the described procedures. Diploid or polyploid industrial strains can be especially difficult to engineer (Zhang et al.2014; Stovicek, Borodina and Forster 2015; Generoso et al.2016), and multiplexing can create more work on the other end to identify the correct clones. The CRISPR/Cas9 system also greatly facilitates the sequential introduction of multiple genomic edits. For repeated rounds of editing, the strain is cultivated in the absence of selection pressure for gRNA vector, while maintaining selection pressure for Cas9 vector. Then a new gRNA vector can be introduced to accomplish the next round of genetic modifications. In the final strain, both vectors can be removed in the absence of selection pressure to generate a strain free of selection markers (Stovicek, Borodina and Forster 2015; Jessop-Fabre et al.2016).
DNA repair templates
As mentioned above, the dominant mode of DSB repair in S. cerevisiae is HR when a homologous donor template is available. NHEJ response in S. cerevisiae provides unpredictable results at target sites and severely decreases overall yield of surviving cells (DiCarlo et al.2013; Mans et al.2015; Stovicek, Borodina and Forster 2015). It has been shown that short single-strand (Generoso et al.2016) or double-strand DNA donor oligos (DiCarlo et al.2013) sharing homology with a target site can serve as the simplest repair template. The donor oligo can be of various lengths, ranging between 80 and 120 bp, and can introduce various changes such as a premature STOP codon (DiCarlo et al.2013), a heterologous disrupting sequence (Horwitz et al.2015), a barcode (Ryan et al.2014) for easier genotyping or an entire ORF deletion (Mans et al.2015) (Fig. 1). The PAM site should always be removed from the donor sequence to prevent the cutting by Cas9 (DiCarlo et al.2013). The repair template can also be delivered as a part of an expression vector (Bao et al.2015; Garst et al.2017). Longer gene expression cassettes with at least 40-bp homology to the target site can also be used as repair templates. They enable integration of larger DNA fragments, e.g. carrying gene expression cassettes (DiCarlo et al.2013; Stovicek, Borodina and Forster 2015). The CRISPR/Cas9 approach has also been combined with in vivo assembly of several overlapping DNA parts (Fig. 1). Ryan et al. (2014) reported 70%–85% efficiency for assembly of a gene expression cassette consisting of three parts with 50 bp overlaps to a targeted locus in a diploid or polyploid strain. In another study, one transformation event enabled integration of six overlapping gene expression cassettes into a single-gene locus while another gene was deleted using a short oligo simultaneously (Mans et al.2015). A multiplex approach CasEMBLR demonstrated assembly of five overlapping DNA parts per locus in up to three different loci simultaneously with an efficiency ranging between 30% and 97% (Jakočiūnas et al.2015b). A metabolic pathway consisting of 11 genes on six DNA parts flanked with 500 bp arms homologous to three independent loci was used as a DSB repair template in the gap-repair gRNA delivery approach resulting in 4% efficiency of the pathway assembly (Horwitz et al.2015). Tsai et al. (2015) integrated two copies of a multigene pathway consisting of six genes on four DNA parts with 300 bp homologous arms into two different gene loci with 25%–100% efficiency. As the integration of a gene expression cassette into an ORF may influence expression of the heterologous gene (Stovicek, Borodina and Forster 2015), several toolkits targeting intergenic regions providing reliable level of gene expression have been developed. Three-DNA part gene expression cassettes with 1 kbp homologous arms used as a donor template resulted in 40%–95% integration efficiency depending on the particular site targeted (Reider Apel et al.2016). Although versatile, in vivo CRISPR/Cas9-mediated assembly requires tedious multiplex genotyping. Thus, preassembly of donor templates with sufficiently long homologous arms might be an alternative option to omit this. Using the system developed by Bao et al. (2015), preassembled large metabolic pathways were integrated into transposable Ty elements in multiple copies with efficiencies more than 80% (Shi et al.2016). Ronda et al. (2015) targeted multiple validated intergenic loci with preassembled gene expression cassettes reaching efficiencies of 84% with three simultaneous integrations. When using the marker-free variant of previously designed integrative vectors (Jensen et al.2014; Stovicek et al.2015) targeting intergenic loci, integration of up to six heterologous genes was achieved with 70% efficiency (Jessop-Fabre et al.2016). In summary, due to the high efficiency of HR, large pathways can be assembled directly in vivo omitting in vitro cloning steps. However, the preassembly of donor DNA fragments always leads to higher integration efficiencies and does not require subsequent extensive genotyping.
Taken together, mainly linear DNA fragments of different length have been successfully used for efficient DSB repair. However, a recent study demonstrated that episomal vectors that contain both a gRNA expression cassette and a DNA repair template could also be used in the yeast S. cerevisiae (Garst et al. 2017). In a proof-of-concept experiment, ADE2 gene was mutated with 95% efficiency in a laboratory strain and with 70% efficiency in a wine strain. A particular advantage of this method is that the combined DNA elements, which contain a gRNA and a corresponding repair template, are small enough (∼200 bp) to be synthesized by high-throughput oligomer synthesis on arrays. Combined with a high transformation efficiency of episomal vectors into yeast, this enables generation of large strain libraries.
Direct DNA editing using CRISPR-cytidine deaminase fusion
A method for CRISPR-based targeted DNA mutagenesis was described by taking advantage of an activation-induced cytidine deaminase (AID), which is normally responsible for somatic hypermutation of the variable regions of antibodies (Nishida et al.2016). When AID was expressed as a fusion with dCas9 in S. cerevisiae, AID deaminated deoxycytidine to deoxyuridine 15–19 bases upstream of the PAM sequence on the non-complementary strand to gRNA, effectively creating C→G/T point mutations. The efficiency of gene inactivation using this approach was 16%–47%, depending on the chosen target site. The advantage of the CRISPR-AID method is a reduced toxicity in comparison to the nuclease-based CRISPR approaches (Nishida et al.2016).
CRISPR/Cas9 GENOME EDITING IN DIFFERENT YEAST SPECIES
Kluyveromyces lactis
Kluyveromyces lactis is used industrially for the production of recombinant proteins, fermented dairy products and some metabolites (Spohner et al.2016). Horwitz et al. (2015) demonstrated CRISPR/Cas9 genome editing in an industrial strain of K. lactis. The 2μ element in the expression vector was exchanged for the pKD1 vector-stabilizing element. To decrease the NHEJ activity in K. lactis, the authors deleted YKU80 gene. Although with low efficiency (2.3%), the method allowed integration of three six-gene DNA parts into three individual chromosomal loci (Horwitz et al.2015).
Yarrowia lipolytica
Yarrowia lipolytica is the most studied oleaginous yeast and is applied in the biotechnology industry for the production of lipase, citric acid, lactone fragrances and recently also ω-3 fatty acids (Thevenieau, Nicaud and Gaillardin 2009; Xue et al.2013). Several recent studies have demonstrated the potential of the CRISPR/Cas9 system in this yeast. Schwartz et al. (2016a) constructed Yarrowia codon-optimized Cas9 and hybrid SCR1΄-tRNA promoter for gRNA expression on a centromeric vector (Schwartz et al.2016a). It enabled efficient NHEJ-generated gene deletions. More than 50% or 90% of the cells acquired a gene deletion after 2 or 4-day outgrowth of the transformed cells, respectively. HR-mediated gene deletions with a donor fragment with 1-kbp homologous arms were also obtained with high efficiency. The HR-mediated repair was pronounced in KU70 mutant, lacking NHEJ-mediated response (Schwartz et al.2016a). A possibility of multiplex gene deletion in Y. lipolytica was also demonstrated (Gao et al.2016). Here a vector was designed to carry Yarrowia codon-optimized Cas9 gene driven by the strong, endogenous TEF1 promoter, and also gRNAs flanked with the HH and HDV ribozymes expressed from the TEF1 promoter. In the absence of donor DNA, NHEJ-mediated gene nonsense mutations occurred with efficiencies of 85%, 36% or 19% for one, two or three targeted genes, respectively, after 4 days of outgrowth of the transformed cells. Furthermore, HR-mediated gene disruption was shown when the donor template was delivered on the Cas9/gRNA vector, with higher rates in KU70/80 mutants (Gao et al.2016). CRISPR/Cas9 also allowed the development of a toolkit for integration of donor cassettes which were delivered into the cells by a separate replicative vector requiring an additional selection during the transformation (Schwartz et al.2016b). In an NHEJ-positive strain, 5 out of 17 tested locations were targeted with integration efficiencies from 48% to 69%, while 3 sites showed <6% and the remaining 9 sites did not show any positive integration. Sequential markerless integration of a metabolic pathway into the described loci was shown (Schwartz et al.2016b).
Komagataella phaffii (formerly Pichia pastoris)
Komagataella phaffii (P. pastoris) is an important recombinant protein producer due to its excellent folding and secretion capability. However, it is poor in HR, which makes it very hard to engineer. Weninger et al. (2016) extensively tested different modes of expression of the Cas9 gene and gRNA molecules. Use of a low-copy ARS element vector with bidirectional native HXT1 promoter driving the expression of human codon-optimized Cas9 and HH/HDV-ribozyme-flanked gRNA transcript resulted in up to 90% of single-gene nonsense mutations. When two genes were targeted, nonsense mutations in both ORFs were observed with a frequency of 69%. Although a donor template with 1-kbp homologous arms was provided, only very low integration efficiency (2%) occurred suggesting that NHEJ remained the dominant way of DSB repair (Weninger et al.2016).
Schizosaccharomyces pombe
The fission yeast Sch. pombe is an important model organism for the study of eukaryotic cellular biology and in particularly cell cycle regulation (Hoffman, Wood and Fantes 2015). Jacobs et al. (2014) used rrk1 promoter for expression of gRNA molecule as it provides a defined 5΄-leader, cleaved during maturation. The 3΄-end of the gRNA molecule was fused to the HH ribozyme, as rrk1 is a Pol II promoter resulting in polyadenylation of mature RNAs. Expression of gRNA and Cas9 separately on two low-copy ARS-containing vectors (or together on one vector to minimize the observed negative influence of Cas9 expression on cell growth) led to the 85%–98% efficiency of the target modification when a PCR-amplified mutated allele was used as donor template (Jacobs et al.2014). A similar system enabled construction of a single-gene deletion with 33% efficiency (Fernandez and Berro 2016).
Pathogenic yeasts
Targeted gene deletions are necessary for the study of gene functions in virulence models. In the most prevalent yeast pathogen—Candida albicans—the absence of haploid state and frequent aneuploidy of clinical isolates makes gene deletions very tedious. In the absence of autonomously replicating vectors, CRISPR/Cas9 was implemented via integrating Cas9 controlled by ENO1 promoter and gRNA expressed from SNR52 promoter into C. albicans genome (Vyas, Barrasa and Fink 2015). The Cas9 gene was codon-optimized for CTG clade yeasts. In ‘solo’ approach, gRNA expression cassette was integrated into a strain already expressing Cas9. In the ‘duet’ approach, both expression cassettes were integrated in a single transformation. Both ‘solo’ and ‘duet’ systems resulted in an acceptable gene deletion efficiency of 60%–80% and 20%–40%, respectively. The more efficient ‘solo’ system was then used for generation of deletions in several genes or deletion of two homologous genes with a single targeting gRNA molecule. Moreover, successful nonsense mutations in three different loci combining the solo and duet system for delivery of two different gRNA cassettes were documented (Vyas, Barrasa and Fink 2015). A possibility of transient expression of linear cassettes carrying both components was shown. A single gene was replaced with a linear marker gene cassette reaching more than 50% efficiency, while the linear gRNA and Cas9 cassettes were lost at the same time (Min et al.2016).
Another pathogenic yeast Cryptococcus neoformans exhibits a low rate of HR that hampers its manipulation and thus functional gene analysis. Two studies have demonstrated the CRISPR/Cas9 system capacity to generate nonsense mutations and to stimulate HR response in different serotypes of Cr. neoformans (Arras et al.2016; Wang et al.2016). As circular molecules are not stable in Cr. neoformans, linear DNA vectors were used for expression of Cas9 nuclease and gRNAs. gRNAs were expressed from CnU6 promoter and terminated by 6T terminator (Wang et al.2016). Alternatively, the Cas9 gene was integrated into the genome and a linear vector was used for expression of a gRNA molecule flanked with HH and HDV ribozymes from a Pol II promoter (Arras et al.2016). The introduction of nonsense mutations was achieved without donor DNA with efficiency above 80%. Mutated allele used as a donor template resulted in HR-mediated allele exchange when selecting for a particular phenotype. Full removal of an ORF occurred with frequencies of 20%–90% when a donor marker gene was fused to the Cas9/gRNA cassette followed by spontaneous loss of the Cas9/gRNA part eliminating thus the persistence of the CRISPR/Cas9 system (Wang et al.2016). Gene deletions were obtained in different serotypes of Cr. neoformans by using a marker cassette with homologous arms to the given ORF. Stimulation of HR led to 70% success rate for obtaining the mutants (Arras et al.2016).
Another pathogenic yeast with a dominant NHEJ pathway, C. glabrata was demonstrated to be amenable to the CRISPR/Cas9-mediated engineering (Enkler et al.2016). Here two centromeric vectors carrying Cas9 and gRNA expression cassette were used. Although adoption of Saccharomyces cerevisiae system (DiCarlo et al.2013) for expression of a gRNA appeared to be feasible, specific C. glabrata adjustments (RNAH promoter, tRNA terminator) led to better performance of the system (Enkler et al.2016). Besides efficient generation of indels by NHEJ, deletion of a reporter gene using a donor marker cassette with relatively short homologous arms was achieved with increased HR rates (Enkler et al.2016).
TRANSCRIPTIONAL REGULATION VIA CRISPR
Targeted regulation of gene expression is important both in the context of metabolic engineering and functional genomics. The CRISPR method has been adapted both for activation and repression of gene transcription in Saccharomyces cerevisiae, but so far not in other yeast species.
Qi et al. (2013) generated an enzymatically inactive variant of Cas9 by mutation of both nuclease sites (D10A and H840A) and showed that this null-nuclease dCas9 when targeted to a coding region of a gene caused transcriptional repression in Escherichia coli. In this approach, termed CRISPR interference (CRISPRi), dCas9 sterically blocks the binding and action of RNA polymerase. In a follow-up study, dCas9 was guided to a promoter region, resulting in efficient gene repression in S. cerevisiae (Gilbert et al.2013). The repression could be further enhanced by fusing a repressor domain to dCas9 (Fig. 3). GFP fluorescence was reduced 18-fold when the TEF1 promoter driving the GFP expression was targeted by dCas9, and the fluorescence decreased 53-fold when the same region was targeted by dCas9 fused to a mammalian transcriptional repressor domain Mxi1 (Gilbert et al.2013).
Figure 3.
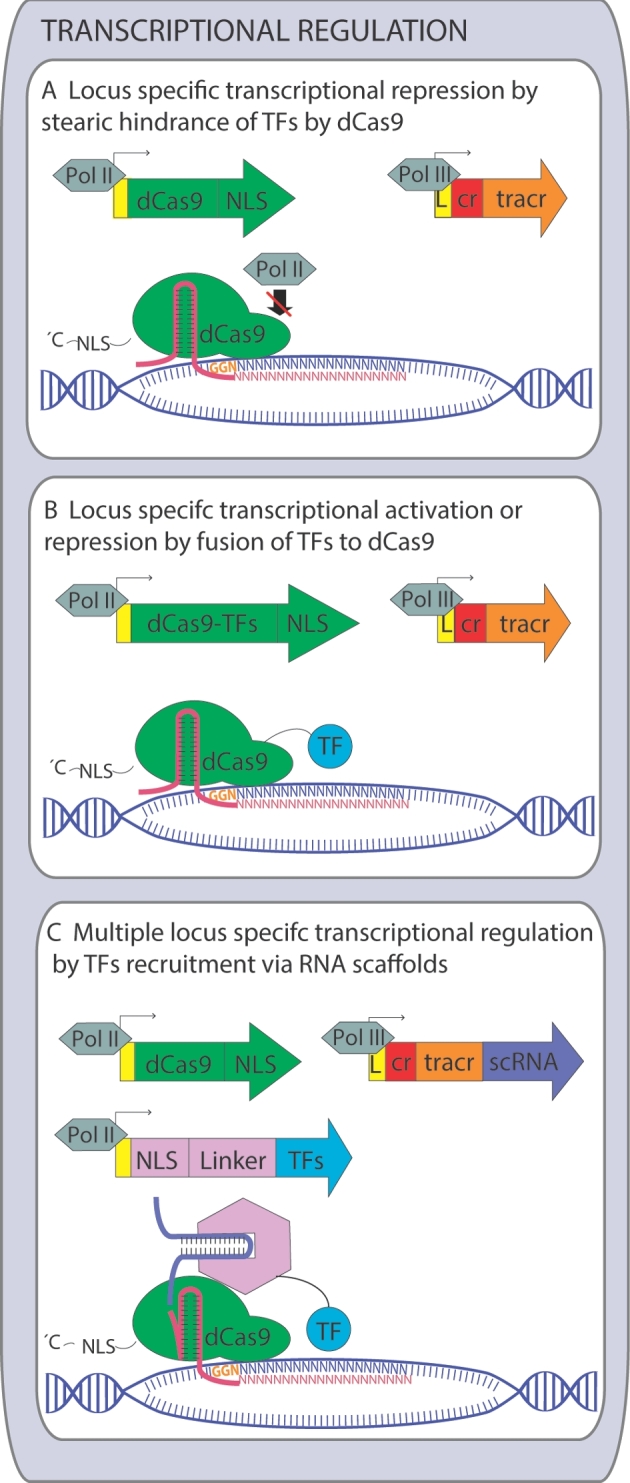
Overview of transcriptional control via CRISPR/Cas9 in yeast. (A) Steric block of transcriptional initiation/elongation by catalytically inactive (‘dead’) dCas9 bound in the promoter region. (B) Transcriptional activation/repression using dCas9 fused to transcriptional activator/repressor domains. (C) Multiple transcriptional regulation action using effector proteins recruited by RNA scaffolds. Pol III—RNA Polymerase III, NLS—nucleolar localization sequence, L—self-cleaved leader sequence (e.g. SNR52), cr—crRNA, tracr—tracrRNA, TF—transcription factor, scRNA—scaffold RNA, Linker—scaffold RNA-binding linker protein domain.
Farzadfard, Perli and Lu (2013) fused dCas9 to an activator domain (VP64) instead. The resulting chimeric protein could both repress and activate gene expression depending on the targeting site in the promoter region. When dCas9-VP64 was targeted to the region upstream the TATA box of the minimal CYC1m promoter, the promoter was activated. Targeting the sites immediately adjacent to the TATA box or transcriptional start site repressed the expression from PCYC1m. The obtained activation level was not very high, max 2.5-fold. To achieve a higher level of activation, the authors created a synthetic promoter by arraying multiple operators upstream the PCYC1m. The activation level increased proportionally to the number of operators, reaching 70-fold activation for 12 operators (Farzadfard, Perli and Lu 2013).
Fusion of dCas9 to a tripartite activator (VPR) composed of three strong activation domains (VP64, p65 and Rta) resulted in 38 and 78-fold activation of promoters PHED1 and PGAL7, respectively. Fusion of dCas9 with VP64 only gave 9 and 14-fold activation of the same promoters (Chavez et al.2015).
Zalatan et al. (2015) undertook a different approach to achieve targeted upregulation and downregulation. Instead of fusing activation or repression domains to dCas9, they included effector protein recruitment domains into the guide RNA (Fig. 3). In the same strain, they expressed dCas9 and regulation proteins, fused with RNA-binding domains. They termed the resulting gRNAs with protein recruitment capabilities ‘scaffold RNA’ (scRNA). Gene activation using scRNA binding VP64 activation domain was 20 to 50-fold, much higher than the activation achieved with dCas9-VP64 fusion. Several hairpins could be combined in a single scRNA, which allowed amplification of activation or combination of repression and activation of different sites (Zalatan et al.2015).
In addition to the studies mentioned above focusing on the on/off states of gene expression, grade modulation of gene expression using dCas9 fused to either an activation or repression domain was shown (Deaner and Alper 2017). This was achieved by changing the gRNA target location and thus recruiting the dCas9-activator/repressor complex to different positions in gene promoters. It resulted in a dynamic range of gene expression from almost silenced gene to its several 10-fold overexpression related to the proximity of the dCas9-based regulators to the core of the promoter. The graded gene expression enabled tuning of metabolic pathways and optimization of the desired phenotypes in several metabolic engineering applications (Deaner and Alper 2017).
APPLICATION OF CRISPR/Cas9 FOR ENGINEERING OF YEAST CELL FACTORIES
CRISPR/Cas genome editing and transcriptional regulation are particularly suitable for developing yeast cell factories. As the strain development usually proceeds through iterative design-build-test cycles, the CRISPR technology facilitates this process because the strains can repeatedly be edited in a flexible multiplex way. As far as transcriptional regulation is concerned, CRISPR also enables relatively easy multiplexing. We will illustrate this with four brief examples (Fig. 4).
Figure 4.

Application of CRISPR/Cas9 systems for engineering of yeast cell factories. (A) Production of (R,R)-2,3-butanediol from xylose. Multicopy one-step integration of the xylose utilization and (R,R)-2,3-butanediol pathways into Ty-element delta sites in the genome (The figure is reprinted with permission from Elsevier: Shi et al. A highly efficient single-step, markerless strategy for multicopy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae. Metab Eng 2016;33:19–27.). (B) Production of lactic acid from glucose in an industrial yeast strain, one-step disruption of two genes in diploid strain and simultaneous integration of lactate dehydrogenase genes from L. plantarum (ldhL) (Stovicek, Borodina and Forster 2015). (C) Production of deoxyviolacein, violacein, prodeoxyviolacein and proviolacein from glucose. Transcriptional regulation (activation/repression) of different genes in violacein pathway leads to production of different violacein derivatives (The figure is reprinted with permission from Elsevier: Zalatan et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 2015;160:339–50.): VP64-activator domain, PP7/MS2 – RNA hairpin structures, PCP/MCP—RNA binding proteins. (D) Production of naringenin from glucose. Cas9-mediated one-step integration of the naringenin pathway into an intergenic locus. Downregulation of TSC13 mediated by catalytically inactive (‘dead’) dCas9 (CRISPRi) to avoid the formation of by-products (The figure adapted from Vanegas, Lehka and Mortensen 2017).
Shi et al. (2016) applied CRISPR/Cas9 to engineer Saccharo-myces cerevisiae towards production of a non-native product (R,R)-2,3-butanediol (BDO) from a non-native substrate xylose in a single transformation step. A 24-kb integration construct consisting of six gene expression cassettes (three for the xylose consumption pathway and three for the BDO biosynthesis pathway) was integrated into the delta sequence of the Ty transposon elements. Introducing Cas9-mediated DSBs at the delta sites allowed the integration of 10 copies of the 24-kb DNA fragment. A higher copy number of the pathways resulted in both higher xylose consumption rate and higher BDO production, where 0.31 g/L of BDO was produced from 20 g/L xylose (Shi et al.2016).
Stovicek et al. (2015) engineered diploid industrial S. cerevisiae strain Ethanol Red, used in many first generation ethanol plants, to produce lactic acid by replacing both alleles of pyruvate decarboxylase genes PDC1 and PDC5 with L-lactate dehydrogenase encoding gene (ldhL) from Lactobacillus plantarum. The genetic modification was accomplished in a single transformation event, leading to a strain producing 2.5 g/L lactic acid with the yield of 0.49 g of lactic acid/g of glucose (Stovicek, Borodina and Forster 2015).
Transcriptional regulation via CRISPRi was demonstrated for the production of the bacterial pigment violacein in S. cerevisiae. Here CRISPR RNA scaffolds were used to recruit transcriptional activators and repressors, alone or simultaneously, to a promoter site, which allowed tight control of transcriptional activation and repression. By simply changing the RNA scaffolds, the same strain could be reprogrammed to produce different ratios of the pathway products, deoxyviolacein, violacein, prodeoxyviolacein and proviolacein. Combining these RNA-encoded circuits with conditional expression of Cas9, a system for switching from growth to production phase was obtained (Zalatan et al.2015).
Recently, a combination of Cas9 genome editing and dCas9 transcriptional regulation was demonstrated by engineering S. cerevisiae for production of flavonoid precursor naringenin. First, Cas9 was used for integration of a multigene pathway into an intergenic locus leading to production of naringenin from phenylalanine. Next, the naringenin production was increased through dCas9-mediated downregulation of an essential gene TSC13 to prevent the formation of by-product phloretic acid (Vanegas, Lehka and Mortensen 2017).
OUTLOOK
This review summarizes the recent developments of CRISPR-based systems for genome editing and transcriptional regulation in various yeast species. The CRISPR/Cas9 technology has advantages over conventional marker-based genome editing in several aspects. It enables fast strain engineering of prototrophic wild and industrial yeast strains. Furthermore, it allows performing multiple genome edits simultaneously and is independent of marker cassette integration. For transcriptional regulation, the CRISPR offers an advantage of relatively easy design and implementation, the possibility of multiplexing and orthogonality. However, to enable the wide adaptation of CRISPR, the current limitations need to be addressed. These include (i) design of efficient and specific targeting for different yeast species, (ii) elimination of cloning necessity, (iii) enabling large-scale multiplexing and, finally, (iv) resolving the IP issues. The uncertainty about the ownership of the CRISPR technology delays its adaptation for industrial biotechnology and pharmaceutical applications and must be resolved as soon as possible so the technology can unfold its true potential.
Acknowledgments
The authors are grateful to Behrooz Darbani Shirvanehdeh and Michael Krogh Jensen for critical reading of the manuscript and Marie Blatt Bendtsen and Sidsel Ettrup Clemmensen for additional comments.
FUNDING
The work was financially supported by the Novo Nordisk Foundation [grant no. 16592] and by the European Union the 7th Framework Programme [BioREFINE-2G, grant no. FP7-613771].
Conflict of interest. None declared.
REFERENCES
- Alexander WG, Doering DT, Hittinger CT. High-efficiency genome editing and allele replacement in prototrophic and wild strains of saccharomyces. Genetics 2014;198:859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arras SDM, Chua SMH, Wizrah MSI et al. . Targeted genome editing via CRISPR in the pathogen Cryptococcus neoformans. PLoS One 2016;11:e0164322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Xiao H, Liang J et al. . Homology-integrated CRISPR–Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth Biol 2015;4:585–94. [DOI] [PubMed] [Google Scholar]
- Blin K, Pedersen LE, Weber T et al. . CRISPy-web: an online resource to design sgRNAs for CRISPR applications. Synth Syst Biotechnol 2016;1:118–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodina I, Nielsen J. Advances in metabolic engineering of yeast Saccharomyces cerevisiae for production of chemicals. Biotechnol J 2014;9:609–20. [DOI] [PubMed] [Google Scholar]
- Bortesi L, Fischer R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv 2015;33:41–52. [DOI] [PubMed] [Google Scholar]
- Cai L, Fisher AL, Huang H et al. . CRISPR-mediated genome editing and human diseases. Genes Dis 2016;3:244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S et al. . Highly efficient Cas9-mediated transcriptional programming. Nat Methods 2015;12:326–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin Y-W, Kang W-K, Jang HW et al. . CAR1 deletion by CRISPR/Cas9 reduces formation of ethyl carbamate from ethanol fermentation by Saccharomyces cerevisiae. J Ind Microbiol Biot 2016;43:1517–25. [DOI] [PubMed] [Google Scholar]
- Chylinski K, Makarova KS, Charpentier E et al. . Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res 2014;42:6091–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D et al. . Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaner M, Alper HS. Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae. Metab Eng 2017;40:14–22. [DOI] [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P et al. . Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 2013;41:4336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M et al. . Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB et al. . Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 2014;32:1262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkler L, Richer D, Marchand AL et al. . Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci Rep 2016;6:35766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadfard F, Perli SD, Lu TK. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth Biol 2013;2:604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez R, Berro J. Use of a fluoride channel as a new selection marker for fission yeast plasmids and application to fast genome editing with CRISPR/Cas9. Yeast 2016;33:549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Tong Y, Wen Z et al. . Multiplex gene editing of the Yarrowia lipolytica genome using the CRISPR-Cas9 system. J Ind Microbiol Biot 2016;43:1085–93. [DOI] [PubMed] [Google Scholar]
- Gao Y, Zhao Y. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol 2014;56:343–9. [DOI] [PubMed] [Google Scholar]
- Garst AD, Bassalo MC, Pines G et al. . Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat Biotechnol 2017;35:48–55. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P et al. . Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. P Natl Acad Sci USA 2012;109:E2579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Generoso WC, Gottardi M, Oreb M et al. . Simplified CRISPR-Cas genome editing for Saccharomyces cerevisiae. J Microbiol Methods 2016;127:203–5. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L et al. . CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013;154:442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat Methods 2014;11:122–3. [DOI] [PubMed] [Google Scholar]
- Hoffman CS, Wood V, Fantes PA. An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics 2015;201:403–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz AA, Walter JM, Schubert MG et al. . Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas. Cell Syst 2015;1:88–96. [DOI] [PubMed] [Google Scholar]
- Jacobs JZ, Ciccaglione KM, Tournier V et al. . Implementation of the CRISPR-Cas9 system in fission yeast. Nat Commun 2014;5:5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakočiūnas T, Bonde I, Herrgård M et al. . Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab Eng 2015a;28:213–22. [DOI] [PubMed] [Google Scholar]
- Jakočiūnas T, Rajkumar AS, Zhang J et al. . CasEMBLR: Cas9-facilitated multiloci genomic integration of in Vivo Assembled DNA Parts in Saccharomyces cerevisiae. ACS Synth Biol 2015b;4:1226–34. [DOI] [PubMed] [Google Scholar]
- Jakočiūnas T, Jensen MK, Keasling JD. CRISPR/Cas9 advances engineering of microbial cell factories. Metab Eng 2016;34:44–59. [DOI] [PubMed] [Google Scholar]
- Jensen NB, Strucko T, Kildegaard KR et al. . EasyClone: method for iterative chromosomal integration of multiple genes Saccharomyces cerevisiae. FEMS Yeast Res 2014;14:238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop-Fabre MM, Jakočiūnas T, Stovicek V et al. . EasyClone-markerfree: a vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol J 2016;11:1110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I et al. . A programmable dual-RNA–Guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Gagnon JA et al. . CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 2016;44:W272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES. The heroes of CRISPR. Cell 2016;164:18–28. [DOI] [PubMed] [Google Scholar]
- Laughery MF, Hunter T, Brown A et al. . New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast 2015;32:711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne S. Genetic engineering of industrial strains of Saccharomyces cerevisiae. In: Lorence A. (ed.). Recombinant Gene Expression. Totowa, NJ: Humana Press, 2012:451–65. [DOI] [PubMed] [Google Scholar]
- Lee ME, DeLoache WC, Cervantes B et al. . A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth Biol 2015;4:975–86. [DOI] [PubMed] [Google Scholar]
- Li M, Borodina I. Application of synthetic biology for production of chemicals in yeast Saccharomyces cerevisiae. FEMS Yeast Res 2015;15:1–12. [DOI] [PubMed] [Google Scholar]
- Liu H, Wei Z, Dominguez A et al. . CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics 2015;31:3676–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Liang Y, Ang EL et al. . A new era of genome integration—simply cut and paste! ACS Synth Biol 2017, DOI: 10.1021/acssynbio.6b00331. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM et al. . RNA-guided human genome engineering via Cas9. Science 2013;339:823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mans R, van Rossum HM, Wijsman M et al. . CRISPR/Cas9: a molecular swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res 2015;15, DOI: 10.1093/femsyr/fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K, Ichikawa Y, Woolford CA et al. . Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere 2016;1:e00130–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Hu Y, Ewen-Campen B et al. . CRISPR guide RNA design for research applications. FEBS J 2016;283:3232–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJM, Díez-Villaseñor C, García-Martínez J et al. . Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 2005;60:174–82. [DOI] [PubMed] [Google Scholar]
- Naito Y, Hino K, Bono H et al. . CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015;31:1120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Arazoe T, Yachie N et al. . Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016:aaf8729. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA et al. . Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013;152:1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reider Apel A, d’Espaux L, Wehrs M et al. . A Cas9-based toolkit to program gene expression in Saccharomyces cerevisiae. Nucleic Acids Res 2016:gkw1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronda C, Maury J, Jakočiūnas T et al. . CrEdit: CRISPR mediated multi-loci gene integration in Saccharomyces cerevisiae. Microb Cell Fact 2015;14:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronda C, Pedersen LE, Hansen HG et al. . Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol Bioeng 2014;111:1604–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan OW, Cate JHD. Chapter twenty-two - multiplex engineering of industrial yeast genomes using CRISPRm. In: Doudna JA, Sontheimer EJ (eds). Methods in Enzymology. Vol. 546. Academic Press, 2014:473–89. [DOI] [PubMed] [Google Scholar]
- Ryan OW, Poddar S, Cate JHD. CRISPR-Cas9 Genome engineering in Saccharomyces cerevisiae Cells. Cold Spring Harb Protoc 2016;2016:pdb.prot086827. [DOI] [PubMed] [Google Scholar]
- Ryan OW, Skerker JM, Maurer MJ et al. . Selection of chromosomal DNA libraries using a multiplex CRISPR system. eLife 2014;3:e03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz CM, Hussain MS, Blenner M et al. . Synthetic RNA polymerase III promoters facilitate high-efficiency CRISPR-Cas9-mediated genome editing in Yarrowia lipolytica. ACS Synth Biol 2016a;5:356–9. [DOI] [PubMed] [Google Scholar]
- Schwartz C, Shabbir-Hussain M, Frogue K et al. . Standardized markerless gene integration for pathway engineering in Yarrowia lipolytica. ACS Synth Biol 2016b, DOI: 10.1021/acssynbio.6b00285. [DOI] [PubMed] [Google Scholar]
- Shi S, Liang Y, Zhang MM et al. . A highly efficient single-step, markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae. Metab Eng 2016;33:19–27. [DOI] [PubMed] [Google Scholar]
- Smith JD, Suresh S, Schlecht U et al. . Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design. Genome Biol 2016;17:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spohner SC, Schaum V, Quitmann H et al. . Kluyveromyces lactis: An emerging tool in biotechnology. J Biotechnol 2016;222:104–16. [DOI] [PubMed] [Google Scholar]
- Storici F, Durham CL, Gordenin DA et al. . Chromosomal site-specific double-strand breaks are efficiently targeted for repair by oligonucleotides in yeast. P Natl Acad Sci USA 2003;100:14994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovicek V, Borja GM, Forster J et al. . EasyClone 2.0: expanded toolkit of integrative vectors for stable gene expression in industrial Saccharomyces cerevisiae strains. J Ind Microbiol Biot 2015;42:1519–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovicek V, Borodina I, Forster J. CRISPR–Cas system enables fast and simple genome editing of industrial Saccharomyces cerevisiae strains. Metab Eng Commun 2015;2:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenieau F, Nicaud J-M, Gaillardin C. Applications of the non-conventional yeast Yarrowia lipolytica. In: Satyanarayana T, Kunze G (eds.). Yeast Biotechnology: Diversity and Applications. Springer: Netherlands, 2009:589–613. [Google Scholar]
- Tsai C-S, Kong II, Lesmana A et al. . Rapid and marker-free refactoring of xylose-fermenting yeast strains with Cas9/CRISPR. Biotechnol Bioeng 2015;112:2406–11. [DOI] [PubMed] [Google Scholar]
- van den Ent F, Löwe J. RF cloning: A restriction-free method for inserting target genes into plasmids. J Biochem Bioph Meth 2006;67:67–74. [DOI] [PubMed] [Google Scholar]
- van Erp PB, Bloomer G, Wilkinson R et al. . The history and market impact of CRISPR RNA-guided nucleases. Curr Opin Virol 2015;12:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanegas KG, Lehka BJ, Mortensen UH. SWITCH: a dynamic CRISPR tool for genome engineering and metabolic pathway control for cell factory construction in Saccharomyces cerevisiae. Microb Cell Fact 2017;16:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas VK, Barrasa MI, Fink GR. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 2015;1, DOI: 10.1126/sciadv.1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JM, Chandran SS, Horwitz AA. CRISPR-cas-assisted multiplexing (CAM): simple same-day multi-locus engineering in yeast. J Cell Physiol 2016;231:2563–9. [DOI] [PubMed] [Google Scholar]
- Wang Q, Wang L. New methods enabling efficient incorporation of unnatural amino acids in yeast. J Am Chem Soc 2008;130:6066–7. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wei D, Zhu X et al. . A “suicide” CRISPR-Cas9 system to promote gene deletion and restoration by electroporation in Cryptococcus neoformans. Sci Rep 2016;6, DOI: 10.1038/srep31145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger A, Hatzl A-M, Schmid C et al. . Combinatorial optimization of CRISPR/Cas9 expression enables precision genome engineering in the methylotrophic yeast Pichia pastoris. J Biotechnol 2016;235:139–49. [DOI] [PubMed] [Google Scholar]
- Xu K, Ren C, Liu Z et al. . Efficient genome engineering in eukaryotes using Cas9 from Streptococcus thermophilus. Cell Mol Life Sci 2015;72:383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z, Sharpe PL, Hong S-P et al. . Production of omega-3 eicosapentaenoic acid by metabolic engineering of Yarrowia lipolytica. Nat Biotechnol 2013;31:734–40. [DOI] [PubMed] [Google Scholar]
- Zalatan JG, Lee ME, Almeida R et al. . Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015;160:339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Kong II, Kim H et al. . Construction of a quadruple auxotrophic mutant of an industrial polyploidy Saccharomyces cerevisiae using RNA-guided Cas9 nuclease. Appl Environ Microb 2014;80:7694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]