

Abstract
Introduction
Uremia results in a characteristic breath odor (uremic fetor) which is largely due to its high ammonia content. Earlier studies have shown a strong correlation between breath ammonia and blood urea levels and a 1 0-fold reduction in breath ammonia after hemodialysis in patients with chronic kidney disease. Potential sources of breath ammonia include: (i) local ammonia production from hydrolysis of urea in the oropharyngeal and respiratory tracts by bacterial flora, and (ii) release of circulating blood ammonia by the lungs. While the effects of uremia and hemodialysis on breath ammonia are well known their effects on blood ammonia are unknown and were explored here.
Methods
Blood samples were obtained from 23 hemodialysis patients (immediately before and after dialysis), 14 peritoneal dialysis patients, and 10 healthy controls. Blood levels of ammonia, creatinine, urea, and electrolytes were measured.
Findings
No significant difference was found in baseline blood ammonia between hemodialysis, peritoneal dialysis and control groups. Hemodialysis procedure led to a significant reduction in urea concentration (P<0.001) which was paradoxically accompanied by a modest but significant (P<0.05) rise in blood ammonia level in 10 of the 23 patients studied. Change in blood ammonia pre- and post-hemodialysis correlated with change in serum bicarbonate levels (r=0.61, P<0.01). On subgroup analysis of patients who had a rise in blood ammonia levels after dialysis, there was a strong correlation with drop in mean arterial pressure (r=0.88, P<0.01). The nadir intradialytic systolic blood pressure trended lower in the hemodialysis patients who had a rise in blood ammonia compared to the patients who manifested a fall in blood ammonia (124±8vs.136±6mmHg respectively, P=0.27).
Discussion
Fall in blood urea following hemodialysis in ESRD patients was paradoxically accompanied by a modest rise in blood ammonia levels in 43% of the patients studied, contrasting prior reported effects of hemodialysis on breath ammonia. In this subgroup of patients, changes in blood ammonia during hemodialysis correlated with rise in blood bicarbonate and fall in mean arterial blood pressure.
Keywords: Uremia, chronic kidney disease, urea, inflammation, dialysis
INTRODUCTION
Prior to the advent of modern laboratory techniques, characterization of the smell of patients’ breath was a common tool used by ancient clinicians for the diagnosis of various diseases. A prime example of a disease with a characteristic breath odor is renal failure. The fishy smell of these patients’ breath which is commonly described as uremic fetor is primarily due to the presence of large amounts of ammonia in their exhaled air.1 In an earlier study Narasimhan et al. found marked elevation of breath ammonia (NH3) concentration in a group of end-stage renal disease (ESRD) patients.2 Breath ammonia level in the study population directly correlated with their blood urea concentration. The fall in urea concentration was accompanied by a 10-fold drop in breath ammonia level, from 1500 to 2000 ppb before hemodialysis to 150–200 ppb after hemodialysis.2
Ammonia in the body is derived from two sources:3,4 (i) Cleavage of the amino groups of amino acids followed by its conversion to urea by the liver and to ammonium (NH4) by the renal tubular epithelial cells. Conversion of ammonia to ammonium in the renal tubules involves active secretion of H+ which is a critical step in the maintenance of acid-base balance by the kidney. (ii) Ammonia generated from the breakdown of urea by urease-possessing bacteria [NH2-CO-NH2+H2O → 2NH3+CO2] in the gastrointestinal tract. Most of the ammonia generated from hydrolysis of urea by the microbial flora is transported by the portal vein to the liver and converted back to urea for release in the circulation.
In patients with chronic kidney disease (CKD) urinary excretion of ammonia declines significantly in parallel with the fall in glomerular filtration rate (GFR).5,6 Both kidney and liver failure result in a significant increase in the breath ammonia level. In addition to limiting urinary excretion of ammonia as ammonium, accumulation of urea in the body fluids and its diffusion into the gastrointestinal tract in patients with renal failure greatly expands the substrate for generation and absorption of ammonia.
Elevation of breath ammonia level in patients with advanced liver disease is accompanied by elevated blood ammonia which contributes to hepatic encephalopathy. As noted above several studies have documented marked elevation of breath ammonia level in patients with chronic kidney disease not requiring dialysis5 and in ESRD patients maintained on hemodialysis and peritoneal dialysis,7–9 and its fall with hemodialysis procedure in ESRD patients.2 The origin of ammonia in the breath includes: (i) the bronchial and bronchiolar transfer of ammonia from the blood to the exhaled air via Rhesus glycoproteins (Rhesus BG and Rhesus CG) and passive diffusion across alveolar membrane,10 (ii) hydrolysis of urea contained in the saliva by oral bacterial flora,11 and (iii) possible increase in blood ammonia, due to its reduced urinary excretion as ammonium. To our knowledge the effects of renal failure and dialysis modalities on blood ammonia level have not been previously investigated. The present study was undertaken to address this issue.
METHODS
Patients and controls
Twenty-three stable hemodialysis patients (14 women and 9 men, aged 55.5±20.2 years) and 14 peritoneal dialysis patients (6 women and 8 men, aged 47.9±18.3 years) treated at the outpatient dialysis unit of the University California, Irvine (UCI) Medical Center were recruited for the study. The underlying causes of ESRD in the study population included diabetic nephropathy (12), hypertensive nephrosclerosis (9), IgA nephropathy (3), lupus nephritis (3), chronic glomerulonephritis (3), amyloidosis (1), multiple myeloma (1), focal segmental glomerulosclerosis (1) and CKD of unknown etiology (4). Patients received hemodialysis therapy for 3 h thrice weekly using polysulfone dialyzers. All patients received phosphate binders, erythropoiesis stimulating agents, iron supplements, and a multivitamin preparation. Details of dialysis treatments and patient vitals were obtained from chart review at the dialysis unit. None of the participating patients consumed food during the dialysis procedure. The highest and lowest intradialytic blood pressure values were recorded. Delta MAP represented the difference between highest and lowest MAP during a hemodialysis session. Ten healthy subjects (5 women and 5 men, aged 46.8±8.9 years) served as controls. The study protocol was approved by the Institutional Review Board of the University of California Irvine (HS# 2007-5572) and completed with the assistance of the UCI’s Institute for Clinical and Translational Science. Written informed consent was obtained from all participants.
Blood collection
Blood samples were obtained by venipuncture in the control group and peritoneal dialysis patients, and from the vascular access in the hemodialysis patients immediately before and after a hemodialysis session. Blood ammonia level was measured by the Clinical Laboratory center at UCI Medical Center within 30 minutes of collection. Blood creatinine, urea, electrolytes, hemoglobin, and white blood cell count were measured on automated bioanalyzers at the UCI Medical Center central laboratory.
Statistical analyses
One-way ANOVA, Tukey’s post-test, unpaired t-test, Pearson’s and Spearman’s correlation tests were used in analysis of the data which were expressed as mean± SEM. P-values less than 0.05 were considered significant.
RESULTS
General data
Data are summarized in Table 1. Compared with the control group the ESRD patients had significantly higher plasma urea, creatinine, and potassium levels and significantly lower blood hemoglobin levels. As expected, hemodialysis resulted in significant fall in serum urea, creatinine, and potassium concentrations and a significant rise in serum bicarbonate concentration. This was associated with a reduction in MAP (101±5 mmHg to 91±4 mmHg, P=0.12).
Table 1.
Blood urea nitrogen (BUN), creatinine, electrolytes including bicarbonate (HCO3), hemoglobin (Hb), white blood cell (WBC) counts and ammonia levels in the control (CTL, n=10) and peritoneal dialysis (PD, n=14) groups and in hemodialysis (HD, n=23) patients immediately before and after hemodialysis procedure
| CTL | PD | Pre-HD | Post-HD | |
|---|---|---|---|---|
| BUN (mg/dL) | 14.9±4.5 | 44.8±13.4* | 52±21.1* | 10±3.9** |
| Creatinine(mg/dL) | 0.8±0.1 | 12.2±2.9* | 10±2.3* | 3±0.5*,**,*** |
| Na (mEq/L) | 137.8±1.7 | 137.9±2.1 | 137±2.0 | 138±1.2 |
| K (mEq/L) | 4.3±0.4 | 4±0.5 | 5±0.6*,** | 3±0.4*,**,*** |
| Cl (mEq/L) | 103.6±2.0 | 97±3.0 | 95±2.8 | 95±1.4 |
| HCO3 (mEq/L) | 27.5±2.5 | 25.4±1.8 | 28±2.8 | 34±2.8*,**,*** |
| Hb (g/dL) | 14.2±0.9 | 10.7±1.0* | 9.8±0.6* | 10.4±0.7* |
| WBC (K/μL) | 7.3±3.0 | 6.8±2.1 | 7.4±2.1 | 7.8±1.7 |
| Ammonia (μmol/L) | 25.3±0.3 | 20.2±1.4 | 21.7±1.2 | 23.1±1.9 |
P<0.05 vs. control,
P<0.05 vs. PD,
P<0.05 vs. pre-hemodialysis.
Blood ammonia data
Data are depicted in Table 1. There was no significant difference in baseline blood ammonia levels between the hemodialysis, peritoneal dialysis and healthy control groups. Change in blood ammonia (difference between pre- and post-hemodialysis levels) directly correlated with the degree of increase in the serum bicarbonate concentration; Figure 1).
Figure 1.
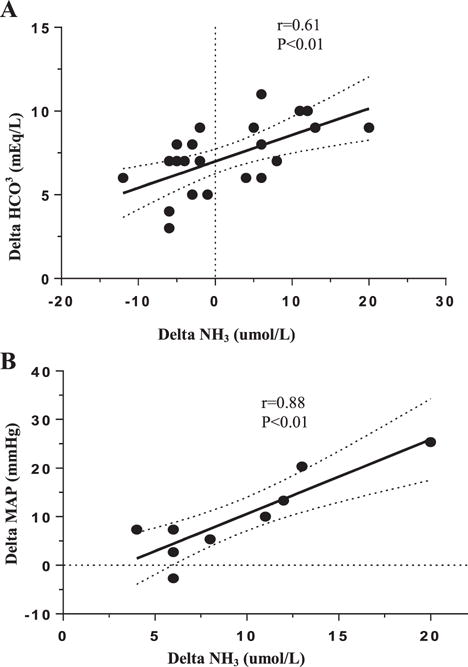
Correlation between hemodialysis-induced delta ammonia (NH3) with (A) changes in serum bicarbonate (HCO3) in all hemodialysis subjects, and (B) delta MAP (reflecting hemodynamic fluctuations) in hemodialysis patients who had a positive delta NH3.
No significant difference was found in predialysis and post-dialysis blood ammonia levels in the hemodialysis group as a whole. However, comparison of the pre- with the post-dialysis data revealed a significant (P<0.05) rise in blood ammonia level in 10 of 23 hemodialysis patients (43% of the cohort) despite significant (P<0.001) fall in serum urea concentration. In this subgroup, the increased blood ammonia following hemodialysis was inversely related to change in MAP (r=0.88) (Figure 1). Upon analysis of the lowest recorded intradialytic SBPs, there was a trend for lower values in the hemodialysis patients who had a rise in blood ammonia (124±8 vs. 136±6 mmHg in the patients who had a drop in blood ammonia, P=0.27, Table 2).
Table 2.
Comparison of hemodialysis subgroups who demonstrated rise vs. fall in blood ammonia (NH3) during dialysis
| Hemodialysis subgroup | n | Delta NH3 (μmol/L) |
Delta HCO3 (mEq/L) |
Lowest recorded SBP (mmHg) |
Lowest MAP (mmHg) |
|---|---|---|---|---|---|
| Positive delta NH3 | 10 | 9.1±1.6 | 8.5±0.5 | 124±8 | 86±7 |
| Negative delta NH3 | 13 | −4.5±0.8 | 6.2±0.5 | 136±6 | 94±6 |
| P<0.01 | P=0.27 | P=0.38 |
Delta ammonia and bicarbonate (HCO3) were calculated from pre- and post-dialysis biochemistries. Delta MAP was calculated from the highest and lowest recorded intradialytic blood pressures.
DISCUSSION
Retention of urea in renal failure leads to the rise in its concentration in the body fluids and its heavy influx into the gastrointestinal tract via passive diffusion and incorporation in glandular secretions.12 The heavy influx of urea in the gastrointestinal tract is accompanied by marked alteration of the gut microbial flora and dominance of urease-possessing bacteria in ESRD patients.13,14 Hydrolysis of urea by microbial urease leads to formation of large quantities of ammonia [NH2-CO-NH2+H2O → CO2+2NH3]. Most of the ammonia generated in the gut is converted to ammonium hydroxide [NH3+H2O → NH4OH], which accounts for the elevated pH of the intestinal milieu in patients with renal failure.15,16 There is growing evidence indicating the role of intestinal barrier dysfunction and increased intestinal permeability as a cause of systemic inflammation in advanced renal disease.17–20 However, until recently, the mechanism by which uremia increases intestinal epithelial permeability was not known. Earlier studies conducted in our laboratories demonstrated massive losses of claudin-1, occludin, and zonula occludens-1 (the key protein constituents of the epithelial tight junction) in the colon, stomach, jejunum, and ileum of rats with CKD.21,22 Our subsequent studies identified the role of urea-derived ammonium hydroxide in the disruption of the intestinal epithelial barrier structure.23,24 These observations clearly demonstrated the contribution of urea-derived ammonia to the pathogenesis of the leaky gut and systemic inflammation in advanced CKD.25,26
The present study revealed that despite massive generation of ammonia and its damaging effect on the gastrointestinal epithelial barrier, baseline blood ammonia in ESRD patients maintained on hemodialysis and peritoneal dialysis is comparable with that found in the control group. These observations confirm the local production of ammonia from hydrolysis of urea by the microbial flora in the oral, and pharyngeal, cavities of uremic patients as the primary cause of their elevated breath ammonia level. It was previously reported that the fall in blood urea level following hemodialysis procedure correlates with a 10-fold drop in breath ammonia level in ESRD patients.2 Our study demonstrates that in contrast to its effect on breath ammonia, hemodialysis not only failed to lower blood ammonia but instead significantly raised blood ammonia levels in almost half of the subjects. The mechanisms underlying the observed post-dialysis rise in blood ammonia in this population are currently unknown. However, it may be in part due to the acute change in acid-base status from mild acidosis in which the ammonia is held as nonvolatile ammonium to the normal or alkalotic states where it exists partly in a volatile state (NH3). Indeed, we noted a significant correlation between change in blood ammonia and the magnitude of rise in blood bicarbonate level following hemodialysis procedure in our ESRD subjects.
A second possible mechanism is the potential contribution of acute reduction of hepatic perfusion in the setting of ultrafiltration and decreased arterial pressures during hemodialysis. This transient liver ischemia may limit its ability to convert gut-derived ammonia to urea, thus raising systemic ammonia concentrations. This assumption is supported by direct correlation between the extent of the fall in MAP with the rise in posthemodialysis blood ammonia levels in a subset of patients. Similarly, there was a trend for lower nadir SBP in the patients that manifested a rise in blood ammonia compared to those exhibiting a fall in blood ammonia. The small sample sizes in our study likely limited detection of statistical significance.
In conclusion, the fall in blood urea concentration following hemodialysis is paradoxically accompanied by a modest rise in blood ammonia in almost 50% of hemodialysis subjects, contrasting the reported effect on breath ammonia. The mechanism of the posthemodialysis rise in blood ammonia is unknown and requires investigation. The observed rise in blood ammonia level was directly related to the rise in blood bicarbonate and with drop in mean arterial pressure during hemodialysis treatments.
Acknowledgments
Disclosure of grants or other funding: None.
Footnotes
Conflict of Interest: Neither author has any conflict of interest.
References
- 1.Jankowski J, Westhof T, Vaziri ND, Ingrosso D, Perna AF. Gases as uremic toxins: Is there something in the air? Semin Nephrol. 2014;34:135–150. doi: 10.1016/j.semnephrol.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Narasimhan LR, Goodman W, Patel CK. Correlation of breath ammonia with blood urea nitrogen and creatinine during hemodialysis. Proc Natl Acad Sci U S A. 2001;98:4617–4621. doi: 10.1073/pnas.071057598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Compr Physiol. 2013;3:201–220. doi: 10.1002/cphy.c120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergen WG, Wu G. Intestinal nitrogen recycling and utilization in health and disease. J Nutr. 2009;139:821–825. doi: 10.3945/jn.109.104497. [DOI] [PubMed] [Google Scholar]
- 5.Pagonas N, Vautz W, Seifert L, et al. Volatile organic compounds in uremia. PLoS One. 2012;7:e46258. doi: 10.1371/journal.pone.0046258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vallet M, Metzger M, Haymann JP, et al. Urinary ammonia and long-term outcomes in chronic kidney disease. Kidney Int. 2015;88:137–145. doi: 10.1038/ki.2015.52. [DOI] [PubMed] [Google Scholar]
- 7.Scholze A, Jankowski V, Henning L, et al. Phenylacetic acid and arterial vascular properties in patients with chronic kidney disease stage 5 on hemodialysis therapy. Nephron Clin Pract. 2007;107:c1–c6. doi: 10.1159/000105137. [DOI] [PubMed] [Google Scholar]
- 8.Endre ZH, Pickering JW, Storer MK, et al. Breath ammonia and trimethylamine allow real-time monitoring of haemodialysis efficacy. Physiol Meas. 2011;32:115–130. doi: 10.1088/0967-3334/32/1/008. [DOI] [PubMed] [Google Scholar]
- 9.Neri G, Lacquaniti A, Rizzo G, Donato N, Latino M, Buemi M. Real-time monitoring of breath ammonia during haemodialysis: Use of ion mobility spectrometry (IMS) and cavity ring-down spectroscopy (CRDS) techniques. Nephrol Dial Transplant. 2012;27:2945–2952. doi: 10.1093/ndt/gfr738. [DOI] [PubMed] [Google Scholar]
- 10.Han KH, Mekala K, Babida V, et al. Expression of the gas-transporting proteins, Rh B glycoprotein and Rh C glycoprotein, in the murine lung. Am J Physiol Lung Cell Mol Physiol. 2009;297:L153–L163. doi: 10.1152/ajplung.90524.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simenhoff ML, Burke JF, Saukkonen JJ, Ordinario AT, Doty R. Biochemical profile or uremic breath. N Engl J Med. 1977;297:132–135. doi: 10.1056/NEJM197707212970303. [DOI] [PubMed] [Google Scholar]
- 12.Lee YT. Urea concentration in intestinal fluids in normal and uremic dogs. J Surg Oncol. 1971;3:163–168. doi: 10.1002/jso.2930030210. [DOI] [PubMed] [Google Scholar]
- 13.Vaziri ND, Wong J, Pahl M, et al. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83:308–315. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 14.Wong J, Piceno YM, Desantis TZ, Pahl M, Andersen GL, Vaziri ND. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am J Nephrol. 2014;39:230–237. doi: 10.1159/000360010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourke E, Milne MD, Stokes GS. Caecal pH and ammonia in experimental uraemia. Gut. 1966;7:558–561. doi: 10.1136/gut.7.5.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swales JD, Tange JD, Evans DJ. Intestinal ammonia in uraemia: The effect of a urease inhibitor, acetohydroxamic acid. Clin Sci. 1972;42:105–112. doi: 10.1042/cs0420105. [DOI] [PubMed] [Google Scholar]
- 17.Cachofeiro V, Goicochea M, de Vinuesa SG, Oubiña P, Lahera V, Luño J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int Suppl. 2008;111:S4–S9. doi: 10.1038/ki.2008.516. [DOI] [PubMed] [Google Scholar]
- 18.Stenvinkel P. Inflammation in end-stage renal disease: The hidden enemy. Nephrology (Carlton) 2006;11:36–41. doi: 10.1111/j.1440-1797.2006.00541.x. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013;83:1029–1041. doi: 10.1038/ki.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaziri ND. CKD impairs barrier function and alters microbial flora of the intestine: A major link to inflammation and uremic toxicity. Curr Opin Nephrol Hypertens. 2012;21:587–592. doi: 10.1097/MNH.0b013e328358c8d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaziri ND, Yuan J, Rahimi A, Ni Z, Said H, Subramanian VS. Disintegration of colonic epithelial tight junction in uremia: A likely cause of CKD- associated inflammation. Nephrol Dial Transplant. 2012;27:2686–2693. doi: 10.1093/ndt/gfr624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaziri ND, Yuan J, Nazertehrani S, Ni Z, Liu S. Chronic kidney disease causes disruption of gastric and small intestinal epithelial tight junction. Am J Nephrol. 2013;38:99–103. doi: 10.1159/000353764. [DOI] [PubMed] [Google Scholar]
- 23.Vaziri ND, Yuan J, Norris K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol. 2013;37:1–6. doi: 10.1159/000345969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaziri ND, Yuan J, Khazaeli M, Masuda Y, Ichii H, Liu S. Oral activated charcoal adsorbent (AST-120) ameliorates chronic kidney disease-induced intestinal epithelial barrier disruption. Am J Nephrol. 2013;37:518–525. doi: 10.1159/000351171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau WL, Kalantar-Zadeh K, Vaziri ND. The gut as a source of inflammation in chronic kidney disease. Nephron. 2015;130:92–98. doi: 10.1159/000381990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaziri ND, Zhao YY, Pahl MV. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol Dial Transplant. 2016;31:737–746. doi: 10.1093/ndt/gfv095. [DOI] [PubMed] [Google Scholar]